

Abstract
Diabetic cardiovascular complications and impaired cardiac function are considered to be the main causes of death in diabetic patients worldwide, especially patients with type 2 diabetes mellitus (T2DM). An increasing number of studies have shown that melatonin, as the main product secreted by the pineal gland, plays a vital role in the occurrence and development of diabetes. Melatonin improves myocardial cell metabolism, reduces vascular endothelial cell death, reverses microcirculation disorders, reduces myocardial fibrosis, reduces oxidative and endoplasmic reticulum stress, regulates cell autophagy and apoptosis, and improves mitochondrial function, all of which are the characteristics of diabetic cardiomyopathy (DCM). This review focuses on the role of melatonin in DCM. We also discuss new molecular findings that might facilitate a better understanding of the underlying mechanism. Finally, we propose potential new therapeutic strategies for patients with T2DM.
Keywords: autophagy, diabetic cardiomyopathy, melatonin, mitochondrial function, myocardial fibrosis, myocardial metabolism, oxidative stress
In this article, we show the main mechanisms of melatonin in the treatment of diabetic cardiomyopathy, including: (1) Melatonin regulates glucose and fatty acid metabolism in diabetic cardiomyopathy. (2) Melatonin improves endothelial dysfunction and microcirculation disorders in diabetic cardiomyopathy. (3) Melatonin inhibits myocardial fibrosis in diabetes and improves ventricular remodeling. (4) Melatonin regulates autophagy in diabetic cardiomyopathy. (5) Melatonin in regulating oxidative stress. (6) Melatonin inhibits cell death in diabetic cardiomyopathy. (7) Melatonin improves endoplasmic reticulum stress in diabetes. (8) Melatonin improves mitochondrial function. (9) Melatonin rescues cardiomyocytes from MI/R injury. (10) Melatonin reduces hyperglycemia, improves insulin resistance, and protects pancreatic β‐cells. (11) Melatonin decreases plasma uric acid levels. (12) Melatonin inhibits the expression of AGEs‐related receptors.
![]()
Abbreviations
- AGEs
advanced glycation end products
- ATF‐6
activated transcription factor 6
- Bax
Bcl‐2‐associated X protein
- Bcl‐2
B‐cell lymphoma‐2
- DCM
diabetic cardiomyopathy
- DOX
doxorubicin
- Drp1
dynamin‐related protein 1
- EPC
endothelial progenitor cell
- ER
endoplasmic reticulum
- ET‐1
endothelin‐1
- G6PD
glucose‐6‐phosphate dehydrogenase
- GRP78
glucose‐regulated protein 78
- GSH
glutathione
- GSIS
glucose‐stimulated insulin secretion
- HO‐1
heme oxygenase 1
- iNOS
inducible nitric oxide synthase
- JAK/STAT
Janus kinase/signal transducer and activator of transcription
- MAPK
mitogen‐activated protein kinase
- MDA
malondialdehyde
- MI/R
myocardial ischemia/reperfusion
- NF‐κB
nuclear factor kappa B
- NLRP3
NOD‐like receptor 3
- NR
Nuclear hormone receptor
- NR4A1
nuclear receptor subfamily 4, group A, member 1
- NRF1
nuclear respiratory factor 1
- PKC
protein kinase C
- RAGE
receptor for AGE
- RORα
related orphan receptor alpha
- ROS
reactive oxygen species
- SAFE
survivor activating factor enhancement
- SOD
superoxide dismutase
- STAT3
signal transducer and activator of transcription 3
- STZ
streptozocin
- T1DM
type 1 diabetes mellitus
- T2DM
type 2 diabetes mellitus
- taVNS
transcutaneous vagus nerve stimulation
- TFAM
mitochondrial transcription factor A
- TGF‐β
transforming growth factor β
- TLR4
toll‐like receptor 4
- TNFα
tumor necrosis factor alpha
- UCP
uncoupling protein
- UPR
unfolded protein response
- WT
wild‐type
- ZDF
Zucker diabetic fatty
1. INTRODUCTION
Thirty years ago, the prevalence of diabetic patients was only one‐fourth the current prevalence. In fact, the current number of diabetic patients has soared to 1 diabetic patient in every 11 people globally. 1 Moreover, diabetes has become the ninth leading cause of death worldwide. According to statistics from the World Health Organization, as of 2000, the number of diabetes patients worldwide had exceeded 170 million, and it is still increasing at an extremely fast rate. It is predicted that the number of diabetic‐related deaths will double between 2000 and 2030. 2 Among diabetic patients, type 2 diabetes mellitus (T2DM) is most common, accounting for approximately 90% of adult patients with diabetes. Unlike type 1 diabetes mellitus (T1DM), which is an autoimmune disease, the pathogenesis underlying T2DM is insulin resistance. Obesity, age, diet, a sedentary lifestyle with little exercise, and environmental factors are all susceptible factors for the onset of T2DM. An elevated blood glucose level and insulin resistance often lead to diabetic complications, including cardiovascular disease, diabetic neuropathy, diabetic retinopathy, and diabetic nephropathy.
Diabetic cardiomyopathy (DCM) is a diabetic complication independent of coronary artery disease and hypertension. The basic pathologic changes of DCM include abnormal carbohydrate and fatty acid metabolism, microvascular disease and endothelial dysfunction, myocardial necrosis and fibrosis, excessive cardiomyocyte programmed death, increased oxidative stress, endoplasmic reticulum (ER) stress, mitochondrial dysfunction, insulin resistance, glycotoxicity, and other related pathologic changes in the myocardium. 5 , 6 , 7 , 8 These adverse reactions, driven by increased blood glucose, will largely lead to structural remodeling and impaired function of the heart. According to reports, DCM significantly increases the risk of heart failure and can lead to a preserved or reduced ejection fraction. 9 Experiments conducted using a DCM model showed that diastolic dysfunction is more common than systolic dysfunction in T2DM‐related cardiomyopathy, whereas the opposite occurs in patients with T1DM. 10 Thus, the different types of diabetes exert different effects on myocardial function.
Melatonin, which is mainly produced in the pineal gland of most mammals, is a hormone that regulates the circadian rhythm. Melatonin can also be synthesized locally in a variety of cells and tissues. 11 Studies have shown that melatonin plays an important role in anti‐aging, anti‐oxidation, immunomodulation, curing cancer, treatment of cardiovascular disease, and treatment of diabetes and diabetic complications. 12 , 13 , 14 , 15 There is also evidence showing the therapeutic effect of melatonin in patients with diabetes‐associated cardiac dysfunction. Therefore, in this review we focused on the role of melatonin in the pathogenesis underlying DCM with the aim of collating evidence for this potential medical treatment for DCM (Figure 1).
FIGURE 1.
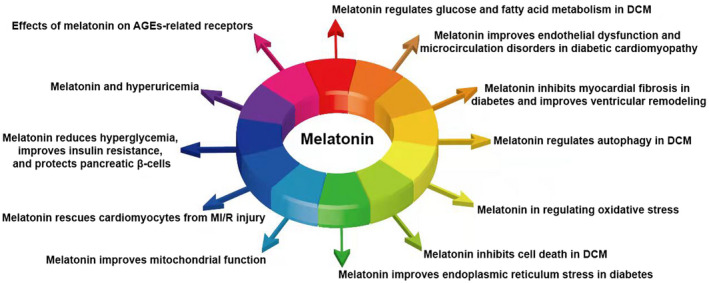
The role of melatonin in the treatment of diabetic cardiomyopathy. AGE, advanced glycation end products; DCM, diabetic cardiomyopathy
2. MELATONIN AND RELATIVE RECEPTORS
2.1. Melatonin and the nuclear melatonin receptor, retinoic acid–related orphan receptor alpha
Nuclear hormone receptors (NRs) comprise a superfamily of transcription factors. NRs have diverse physiologic functions, including regulation of metabolism and stress responses. 16 , 17 , 18 In addition, NRs are molecular targets for drug therapy, highlighting the potential role in therapeutic intervention. Related orphan receptor alpha (RORα), also known as the nuclear melatonin receptor, is a unique receptor in cardiomyocytes. 19 RORα is involved in the development and regulation of DCM. Zhao et al. 20 reported that RORα deficiency contributes to myocardial apoptosis, autophagy dysfunction, and oxidative stress by disrupting antioxidant gene expression. They observed that in a T1DM mouse model induced by streptozocin (STZ) injection, mice with higher RORα expression due to genetic modification or taking melatonin had improved cardiac function and structure compared with the control group after 8 weeks. This effect, induced by activation of RORα, was attenuated by SR3335, an RORα inhibitor. 20 RORα deficiency can also significantly worsen pathologic myocardial hypertrophy, 21 which is related to a decrease in transactivation of the manganese‐dependent superoxide dismutase (SOD) induced by RORα. Defects in intracellular protective signaling is also a feature of heart dysfunction and myocardial damage caused by diabetes. Ischemic preconditioning or ischemic postconditioning are weakened in patients with diabetes. 22 , 23 He et al. 24 concluded that melatonin prevents myocardial ischemia/reperfusion (MI/R) injury by activation of RORα. These experimental results suggest that regulating the nuclear melatonin receptor, RORα, may delay the development of DCM.
Diabetes can also significantly increase the risk of cardiovascular events. 25 And many evidences indicates that RORα plays an important role in preventing cardiovascular disease in diabetic patients. A number of reports have shown that RORα is distributed on blood vessels, and melatonin can act on these receptors. Specifically, melatonin activates RORα in endothelial cells to produce a vascular protective effect, including a reduction in reactive oxygen species (ROS) production and an increase in NO production. 26 Melatonin can also effectively inhibits the differentiation of macrophages in the atherosclerotic plaque to the pro‐inflammatory M1 phenotype by the RORα‐dependent AMP kinase (AMPK) α‐STATs pathway to improve inflammation in the plaque, stabilize rupture‐prone vulnerable plaques, and reduce the risk of cardiovascular events. 27
2.2. Melatonin and membranous melatonin receptors
A number of studies have demonstrated that melatonin receptors (MTs) have potential to protect against DCM. There are two melatonin membrane receptors, MT1 and MT2, which are expressed on cardiomyocytes. 19 It has been reported that MT2, but not MT1, is upregulated after melatonin administration and ameliorates myocardial injury and improves cardiac function after MI/R. 28 , 29 This finding may be because upregulation of MT2 activates the cGMP‐PKG signaling pathway, affects the subordinate signaling pathways, inhibits myocardial oxidative stress and nitrification stress induced by MI/R, reduces ER stress and mitochondrial damage, and inhibits cardiomyocyte apoptosis. In another set of experiments, Li et al. 30 reported that melatonin activates the mitogen‐activated protein kinase (MAPK)‐ERK signaling pathway activated by MTs, then maintains the mitochondrial membrane potential in a model of oxidative stress induced by hydrogen peroxide. This finding indicates that activation of MTs stabilizes mitochondrial function in cardiomyocytes treated with oxidative stress.
There are also MTs expressed in the cardiovascular system, including MT1 and MT2, 31 , 32 and melatonin has an effect on these MTs. Experiments show that the mRNA for both the MT1 and MT2 was localized in the smooth muscle layer. Melatonin activates MT1 on vascular smooth muscle, which enhances vasoconstriction. Alternatively, melatonin activates the MT2 on vascular smooth muscle, which leads to vasodilation. 33 , 34
2.3. Melatonin acts on the cardiovascular system through nonreceptors or other receptors
Melatonin also affects the cardiovascular system through other receptor or nonreceptor pathways. Melatonin has a role on other receptors in the myocardium, such as activating toll‐like receptor 4 (TLR4) and further initiating the survivor activating factor enhancement (SAFE) pathway which is mediated by tumor necrosis factor alpha (TNFα) and signal transducer and activator of transcription 3 (STAT3). 35 TNFα has been shown to protect cardiomyocytes from hypoxic damage (There are also many findings that prove that TNFα increase the damage of cardiomyocytes. It is currently believed that this contradiction may be related to different receptors or concentration differences of TNFα 36 ), and this protective effect is speculated to be produced by activating the JAK/STAT pathway. 36 STAT3 is then activated to reduce cardiomyocyte apoptosis during ischemia‐reperfusion.
Zhao et al. 37 , 38 reported that melatonin passes through the cell membrane and activates the large‐conductance Ca2+‐activated K+ channel on smooth muscle cells directly to cause cell membrane hyperpolarization. This process relaxes the artery.
2.4. Melatonin receptor ligands
Activation of melatonin receptors is involved in multiple cytoprotective pathways; however, in addition to melatonin alone, melatonin receptors also bind to a variety of other ligands. Understanding these ligands may help to understand the prospects of melatonin replacement therapy in the treatment of DCM. The well‐known, non‐selective MT1‐MT2 receptor ligands with agonist properties include ramelteon, agomelatine, and tasimelteon, and these drugs are all MT receptor‐selective drugs. In addition, there are many non‐selective MT receptor agonists, such as 6‐chloromelatonin, 6‐hydroxymelatonin, 2‐iodomelatonin, and GR 196429. 39 It has been confirmed that by activating the MT receptor, especially MT2, some of the above drugs exert a myocardial protective effect. For example, ramelteon improves the reperfusion injury of cardiomyocytes. 40 The relevant mechanism involves activation of the downstream signaling pathways (Akt and PKG) caused by the activation of MTs, and eventually causes opening of mitochondrial calcium‐sensitive potassium channels and release of ROS. 41 Release of ROS to a certain degree is beneficial by inhibiting the opening of mitochondrial permeability transition pores, thereby reducing the influx of cytochrome C into the cytoplasm and preventing the initiation of the cytochrome C‐mediated apoptosis program. 42
3. MELATONIN REGULATES GLUCOSE AND FATTY ACID METABOLISM IN DCM
Metabolic disorders are distinguishing feature of diabetic patients that mainly manifest as glucose utilization disorders and increased mitochondrial fatty acid intake. 43 Indeed, the main energy substrates of cardiomyocytes are glucose and fatty acids. The ATP production‐to‐oxygen consumption ratio is lower for fatty acids than glucose (2.33 vs. 2.58) in cardiomyocytes. Therefore, increased fat oxidation and decreased glucose oxidation caused by diabetes will reduce the efficiency of the myocardium. 44
The main cause of increased fat uptake by cardiomyocytes is the excessive activation of transcription factor peroxisome proliferator‐activated receptor‐α (PPAR‐α), which is regulated by the level of fatty acids in the myocardium. When the insulin effect is insufficient, body fat synthesis will be weakened, lipolysis will increase, and blood lipids and fatty acids in different tissues will increase. Increased fatty acids in the myocardium, however, will cause increased expression of PPAR‐α in the myocardium. PPAR‐α regulates a variety of enzymes related to β oxidation in the myocardium, 45 thereby upregulating the utilization of fatty acids and inhibiting the utilization of glucose. 44 At present, there are few studies that have focused on the effect of melatonin on fatty acid metabolism in the myocardium. In an experiment designed to determine whether melatonin affects glucose and lipid metabolism disorders caused by light, the levels of blood glucose, blood lipids, and PPAR‐α mRNA expression in guinea pigs were determined when guinea pigs were treated differently. 46 After 10 weeks of feeding, experiments showed that a high‐fat diet combined with 24 h light/day (Light, especially night light, will severely inhibit the secretion of melatonin 47 ) leads to increased blood glucose and lipids, including total cholesterol and low‐density lipoprotein‐cholesterol, and a decreased level PPAR‐α mRNA in the liver and adipose tissue of guinea pigs; however, melatonin reversed the above findings. Subcutaneous injection of melatonin for 3 weeks increased the level of PPAR‐α mRNA and decreased the plasma concentration of lipids and glucose in guinea pigs. 46 In another similar experiment with hamsters, melatonin also increased the expression of PPAR‐α in the liver and reduced blood glucose and lipids. 48 The expression of carnitine palmitoyl transferase I in the liver of hamsters was also increased, thus β oxidation in the liver was increased. 48 It seems that melatonin increases the expression of PPAR‐α and enhances the utilization of fatty acids by cells, which did not meet our expectations. Indeed, this experiment detected PPAR‐α in liver and adipose tissue instead of the myocardium. There is also detection of PPAR‐α in the myocardium. Melatonin reduces myocardial PPAR‐α mRNA in hyperthyroidism. 49 Thus, melatonin can reduce blood lipids and glucose in guinea pigs with abnormal metabolism, and in theory, either directly or by reducing the concentration of blood lipids to inhibit the activation of myocardial PPAR‐α, 50 normalizing myocardial fatty acid metabolism.
Melatonin also has an effect on abnormal glucose metabolism in tissues. For example, existing experiments have confirmed that melatonin increases the activity of glucose‐6‐phosphate dehydrogenase (G6PD) in red blood cells, 51 which effectively increases NADPH and the synthesis of glutathione. In a STZ‐induced diabetic rat model, the activity of hexokinase and G6PD in the rat liver was significantly downregulated; however, whether pre‐ or post‐treated with melatonin, the hexokinase and G6PD in the liver tissues of diabetic rats were effectively restored. 52
An impaired AKT/glucose transporter 4 (GLUT4) signaling pathway is considered to be the main cause of insufficient heart glucose uptake in diabetic patients. 53 Existing studies have shown that the expression and membrane translocation of GLUT4 is decreased in the myocardium in TM1D and TM2D 54 , 55 ; however, melatonin has a very significant rescue effect on this pathway. Melatonin successfully restores the decrease in GLUT4 mRNA in myocardium caused by hyperthyroidism, 49 and the myocardial glucose uptake rate is consistent with the change in GLUT4 mRNA. Taken together, the use of melatonin is beneficial to the uptake of glucose by cardiomyocytes.
4. MELATONIN IMPROVES ENDOTHELIAL DYSFUNCTION AND MICROCIRCULATION DISORDERS IN DCM
Endothelial dysfunction in diabetic patients is primarily caused by the reduced release of endothelium‐derived relaxing factors or the impairment of NO bioavailability caused by hyperglycemia, 56 , 57 , 58 which is considered to be a cause of various cardiovascular diseases. Both endothelium‐dependent and endothelium‐independent vasodilatation impairment in the micro‐ and macro‐circulation are associated with diabetes mellitus. 59 Hence, if the impaired endothelium‐derived relaxing factors secretory function of endothelial cells can be improved or the response of blood vessels to dilators can be increased, the cardiovascular complications of diabetes may be improved.
In a set of controlled experiments, an animal model of T2DM was used to determine the effect of L‐carnitine and melatonin treatment. 60 After melatonin treatment, T2DM mice had better vascular responses to acetylcholine relaxation compared with the untreated diabetic group. 60
Endothelial progenitor cell (EPC) apoptosis significantly reduces the production of antioxidant NO, the ability to repair vascular endothelium, and affects the function of vascular endothelial cells. This finding may be related to the impaired EPC autophagy mediated by advanced glycation end products (AGEs) in diabetic patients; however, melatonin improves the impaired autophagy flux and protects EPCs from apoptosis in diabetic patients. 61 In addition, melatonin alleviates atherosclerosis by inhibiting the expression of inducible nitric oxide synthase (iNOS), 62 which differs from endothelial (e) and neuronal (n)NOS. Inducible nitric oxide synthase promotes the generation of peroxynitrite, a proatherosclerosis oxidant, and promotes the formation of atherosclerosis. 63
It is thought that calcium overload is the cause of endothelial cell apoptosis under oxidative stress conditions. 64 One of the main signals of damage is inositol 1,4,5‐triphate receptor (IP3R) 65 ; however, microvascular endothelial cell oxidative damage is a characteristic of cardiac microvascular reperfusion injury and is closely related to no reflow. 66 Based on the above characteristics, melatonin has been shown to have a good effect in regulating the calcium balance of vascular endothelial cells and preventing endothelial cell apoptosis under conditions of oxidative stress. 67 H2O2 treatment simulates a vascular endothelial cell model of oxidative damage in which calcium overload and cell apoptosis were noted. H2O2 activates the ER calcium channel receptor, IP3R, which causes cytoplasmic calcium overload, then the calcium enters the mitochondria through a voltage‐dependent anion channel (VDAC), leading to mitochondrial calcium overload, impaired mitochondrial membrane potential, and opening of the mitochondrial permeability transition pores. All of these findings are prerequisites of cytochrome c release, which result in cell apoptosis. 68 , 69 Melatonin has an effect on IP3R and VDAC. Melatonin inhibits the activation of IP3R and VDAC to alleviate calcium overload in the cytoplasm and mitochondria, and prevents cardiac microvascular endothelial cell death under oxidative stress conditions.
Microcirculation disorders are also an important characteristic of DCM. Focal necrosis of the myocardium due to microcirculation disorders reduces myocardial contractility. The coronary blood flow reserve of diabetic patients is significantly less than healthy people. 70 It has also been shown in animal experiments that the occurrence of diabetes leads to a decrease of arterioles in the heart. 71 The reason for this phenomenon may be partly related to the increased secretion of endothelin‐1 (ET‐1) which is thought to induce potent vasoconstriction through increasing the expression of protein kinase C (PKC). 72 , 73 High levels of ET‐1 also impair endothelial NO production via an isoform‐specific PKC‐mediated inhibition of eNOS expression. 74 These changes greatly enhance the contractile function of the vascular endothelium and cause microcirculation disorders. In addition, endothelin also increases the proliferation and migration of vascular smooth muscle cells, which is related to the development of atherosclerosis. 75 , 76
The inhibitory effect of melatonin on ET‐1 has been demonstrated in several experiments. For example, melatonin inhibits edn‐1 mRNA (the first step in ET‐1 synthesis) expression in colon cancer by inhibiting edn‐1 promoter activity regulated by FoxO1 and nuclear factor kappa B (NF‐κB), therefore reducing the production of ET‐1. 77 Melatonin also inhibits the concentration of ET‐1 in the blood of cigarette smokers, 78 and ET‐1 concentration, as well as an increased expression of endothelin receptor mRNA in hepatic ischemia/reperfusion. 79
5. MELATONIN INHIBITS MYOCARDIAL FIBROSIS IN DIABETES AND IMPROVES VENTRICULAR REMODELING
Myocardial fibrosis is one of the common complications in patients with diabetes, and is widely observed in patients with type 1 and 2 diabetes 80 , 81 The occurrence of this complication will largely lead to increased myocardial stiffness and decreased ventricular diastolic function. The causes of myocardial fibrosis include the formation and accumulation of AGEs, activation of the receptor for AGE (RAGE) by AGEs, and stimulation of hyperglycemia. All of this ultimately leads to the proliferation, differentiation, and collagen production of cardiac fibroblasts. 82 , 83 , 84
Therefore, it is meaningful to discuss whether melatonin can inhibit myocardial fibrosis and reverse the diastolic dysfunction caused by myocardial fibrosis. Transforming growth factor β (TGF‐β) is one of the important pathways for regulating tissue fibrosis. The TGF‐β pathway not only promotes the occurrence of fibrosis but also inhibits the progress of fibrosis. SMAD proteins are the pivotal intracellular effectors of TGF‐β, and multiple subtypes of SMAD proteins have positive effects on the epithelial‐myofibroblast transition and collagen expression, such as SMAD2 and SMAD3, whereas some of the subtypes of SMAD proteins have negative effects, such as SMAD7. 85 , 86 NOD‐like receptor 3 (NLRP3)/IL‐1β pathway is related to TGF‐β/SMAD pathway and located upstream of TGF‐β/SMAD pathway. IL‐1β is a downstream factor of NLRP3 pathway, and can promote TGF‐β gene expression. 87 It has been shown that blocking the NLRP3 pathway can reduce myocardial fibrosis. 88 In addition, the NLRP3 protein monomer is involved in fibrosis but is independent of the fibrosis of the inflammation mediated by the NLRP3 complex. 89 At present, there is relevant evidence to provide the anti‐fibrosis effect of influencing these signaling pathways to DCM. It was found that tissue growth factor, fibronectin, type I collagen, and type III collagen of diabetic mice induced by STZ was significantly higher than normal mice. The diabetic mice were administered intragastric melatonin for 8 weeks had a significantly lower level of the above data compared with diabetic mice that did not receive melatonin treatment. 90 The subsequent detection of the TGF‐β1/SMADS and NLRP3 signaling pathways was also as expected. The levels of TGF‐β1, p‐SMAD2, p‐SMAD3, and NLRP3 inflammasome complexes, as well as the downstream NLRP3 factors, such as IL‐1β, IL‐18, and cleaved caspase‐1 were all increased in the myocardium of diabetic mice, and melatonin treatment significantly reversed these adverse effects by increasing the expression of miR‐141 and inhibiting the activity of lncR‐MALAT1 (there is a binding site of miR‐141 on lncR‐MALAT1, and the binding of the former with the latter inhibits the activity of the latter). 90 At the same time, melatonin can significantly improve ventricular dysfunction caused by diabetes. For example, compared with untreated diabetic mice, the mRNA levels of brain natriuretic peptide in the myocardium of diabetic mice treated with melatonin decreased significantly. Echocardiography also showed that the cardiac ejection fraction and fraction shortening of diabetic mice were improved by melatonin. 90 Taken together, the effect of melatonin in improving myocardial fibrosis and inhibiting ventricular remodeling was verified.
6. MELATONIN REGULATES AUTOPHAGY IN DCM
Autophagy is a process by which cellular organelles are engulfed by double‐membraned structures and subsequently targeted for lysosomal degradation. 91 Autophagy can eliminate redundant and structurally damaged cells, and recycle the intracellular substances to maintain the normal physiologic function of cells, so as to adapt to various adverse stimuli. 91 Normal autophagy has an important role in cell function and quality control of protein and organelles. 92 Excessive autophagy can degrade more important components in cells and induce cell apoptosis or death 93 ; however, recent findings indicate that abnormal autophagy of cardiomyocytes is related to insulin resistance and T2DM. 94 Thus, autophagy involvement in the pathophysiology underlying DCM might play a key role in the development of T2DM and its complications. Using a murine model, it was shown that myocardial autophagy is elevated in patients with T2DM, but suppressed in patients with T1DM. 95 , 96 , 97 In a study involving melatonin action in diabetic mice induced by STZ, the protein level of the specific autophagic substrate, sequestosome 1 (p62), was increased in diabetic sg/sg mice (mice expressing inactive RORα), 20 which represents a reduction in autophagy flux. The double membrane‐bound autophagic vesicles and phosphorylation levels of AMPK, a positive regulator of autophagy, 98 are decreased in the diabetic hearts of sg/sg mice, while the activity of mammalian target of rapamycin complex, a well‐recognized negative regulator of autophagy, is significantly increased. Melatonin used to treat a STZ‐induced rat model of T1DM promoted autophagy by inhibiting the mammalian Ste20‐like kinase 1 (Mst1), a protein that regulates autophagy, 99 phosphorylation, and activating silent information regulator 3 (SIRT3) expression, as well as reducing the level of mammalian target of rapamycin complex expression. 100 , 101 , 102 Inhibition of Mst1 is effective in improving cardiomyocyte autophagy. Melatonin treatment of Mst1 transgenic (Mst1 Tg) mice increased cardiomyocyte autophagy, reduced apoptosis, and improved mitochondrial function, but had no effect on Mst1 knockout (Mst1−/−) mice. In addition to the above, melatonin also increases the ratio of LC3‐II to LC3‐I (as a marker of autophagy, LC3‐II participates in the formation of autophagosomes 103 ), but attenuates the level of p62 in the cardiomyocytes and EPCs of diabetic patients, 61 , 104 indicating the increase in autophagy flux. In H9C2 cells, however, after anoxia/reoxygenation injury treatment, melatonin inhibits excessive mitochondrial autophagy, 105 suggesting that the effect of melatonin on autophagy depends on cell or organelle status. Melatonin or melatonin receptors play an important role in regulating autophagy of cardiomyocytes and EPCs. It is important to note that the level of autophagy needs to be balanced. The current consensus is that acute autophagy induction (e.g., acute myocardial infarction induced by permanent coronary artery occlusion) may be beneficial, while persistent autophagy induction (e.g., failing heart, DCM induced by T2DM, and cardiac anoxia/reoxygenation) may be harmful. 97 , 106 , 107 How to determine if the role of autophagy induction in DCM is more beneficial or harmful will require a more specific model.
7. MELATONIN IN REGULATING OXIDATIVE STRESS
Oxidative stress is caused by the loss of balance between oxidation and antioxidant systems in cells and tissues, which over‐produce oxidative‐free radicals and ROS. 108 , 109 The production of ROS in the human body mainly comes from NADPH oxidase, xanthine oxidase/oxidoreductase, the mitochondrial electron transport chain, uncoupled nitric oxide synthase, the arachidonic acid metabolic pathway, and microsomal enzymes. 110 The main ROS in organisms are free radicals, including nitric oxide (NO•), superoxide radical anion (O2•−), hydroxyl radical (OH•), carbonate radical anion (CO3•−), nitrogen dioxide (NO2•), alkoxyl/alkyl peroxyl (RO•/ROO•) and non‐free radicals, including hydrogen peroxide (H2O2), peroxynitrite (ONOO−)/peroxynitrous acid (ONOOH), and hypochlorous acid (HOCl). 111 A significant increase in oxidative stress and ROS production has been demonstrated in the myocardium of diabetics, 112 and the main site of ROS production is myocardial cells. If the glucose content in the cell increases, the flux of electron transfer donors into the mitochondrial respiratory chain will also increase. Then, the resulting hyperpolarization of the mitochondrial inner membrane potential will inhibit electron transport in complex III and accumulate electrons to ubisemiquinone, therefore generating ROS. 108 , 113 In contrast, NADPH oxidase produces a huge amount of superoxide O2•−. 114 These unstable superoxides will subsequently dismutate to stable H2O2. In fact, a large number of NADPH oxidase subtypes (Nox2 and Nox4) are expressed in cardiomyocytes and can be activated by activated PKC, angiotensin receptor AT1, and Ca2+/calmodulin‐dependent protein kinase II induced by high glucose. 115 , 116 , 117 , 118 , 119 The cardiovascular system also contributes to produce ROS in diabetes. Except for the increase in ROS caused by mitochondria and NADPH oxidase similar to cardiomyocytes, 120 hyperglycemia increases the expression of iNOS in vascular endothelium, which will form large quantities of superoxide and NO and further produce strong oxidizing peroxynitrite ONOO−. 121 , 122
The mechanism underlying subclinical inflammation and oxidative stress is now widely accepted to play an important role in the development of diabetes and its complications. 123 When ROS are excessive, DNA, protein, and lipids will be damaged, and the ROS will participate in various pathologic states in DCM, such as cardiac inflammation, myocardial hypertrophy, apoptosis, fibrosis, vascular endothelial dysfunction, atherosclerosis, and arterial stiffness. 124
In previous studies, it has been shown that melatonin is an immunomodulator that protects against pathologic inflammation. 125 , 126 In STZ‐induced diabetes animal model experiments, melatonin has been shown to be a strong antioxidant. Melatonin injected intraperitoneally increased the activity of antioxidant enzymes in mice and inhibits the release of superoxide‐free radicals. 127 A similar study showed that the increased glycemia, fructosamine, cholesterol, triglycerides, and lipoperoxides caused by STZ induction are reversed by a melatonin injection. 128 Additional experiments directly proved that melatonin effectively reduces inflammation and oxidative stress in mice with T2DM. 129 Using young male Zucker diabetic fatty (ZDF) rats in an experimental model of metabolic syndrome and T2DM, melatonin treatment reduced the levels of interleukin‐6, TNFα, C‐reactive protein, and plasma lipid peroxidation, a criterion for evaluating oxidative stress in ZDF rats, indicating that melatonin treatment attenuated the inflammatory state and oxidative stress. Melatonin administration also decreased the malondialdehyde (MDA) level, and increased the reduced glutathione (GSH) level and other antioxidant enzymes in the myocardium. 130 Experiments with rabbits yielded similar results. 131 Although early‐stage diabetes led to an increase in the serum GSH level in rabbits, prolonged diabetes resulted in a lower serum GSH level and higher serum oxidized glutathione level compared with controls 131 ; however, melatonin treatment maintained the level of serum GSH at an elevated level in diabetic rabbits. Clinical trials have generated corresponding evidence for the effect of melatonin on oxidative stress inhibition. In a double‐blind trial to study the effects of melatonin on diabetic patients, 132 the group treated with melatonin had an increased level of GSH and nitric oxide and a decreased level of MDA compared with the placebo‐treated group. Together, these data indicate that melatonin plays an important role in improving oxidative stress induced by diabetes.
To date, melatonin has been shown to inhibit oxidative stress via multiple pathways. RORα, as a transcription factor and an important mediator of melatonin‐exerted cardioprotection, has strong antioxidant activity in ischemic heart injuries. He et al. 24 assessed the role of RORα in regulating MI/R‐induced oxidative/nitrative stress. Specifically, MI/R induced oxidative stress and nitrification stress in wild‐type (WT) mice, 24 as evidenced by increased accumulation of ROS and nitrotyrosine. In sg/sg mice, these metabolites were increased compared with WT mice. Moreover, it was shown that RORα deficiency increases NADPH oxidase activity, 133 and increases the level of NADPH oxidase subunit gp91phox as well as iNOS, a proatherogenic enzyme that mediates nitrification stress. 63 , 121 Melatonin inhibits MI/R‐induced myocardial oxidative stress injury by activating RORα.
In addition to the MTs that protect mitochondrial function in the myocardium from oxidative stress via the MAPK‐ERK signaling pathway, as previously discussed, activation of melatonin receptors also reduces ROS in cardiomyocytes and reverses the inhibited activity of mitochondrial SOD and other anti‐oxidant substances in an oxidative stress model induced by hydrogen peroxide. 30 In the doxorubicin (DOX)‐induced oxidative stress model, melatonin also has a therapeutic effect, which may have an enlightening role for melatonin in the treatment of oxidative stress in DCM. Activation of the AMPK signaling pathway, silent information regulator 1 (SIRT1), LKB1, AMPK, and peroxisome proliferator‐activated receptor coactivator 1alpha (PGC‐1α) have the effect of protecting myocardial function 134 , 135 (Figure 2). The PGC‐1α downstream signaling molecules, including nuclear respiratory factor 1 (NRF1), mitochondrial transcription factor A (TFAM), and uncoupling protein (UCP), plays an important role in mitochondrial energy homeostasis. 136 Melatonin treatment reversed downregulation of the AMPK phosphorylation induced by oxidative stress, thus increasing the expression of PGC‐1α in hypoxic and ischemic cardiomyocytes. Compared with the control group, H9C2 cells induced by DOX had lower ROS levels and increased activity of the SOD after melatonin treatment, implying that melatonin alleviates the damage of oxidative stress in cardiomyocytes. 137 Another group used a STZ‐induced mouse model of T1DM and reported that melatonin treatment increases the level of PGC‐1α, SIRT3, and mitochondrial SOD2 in cardiomyocytes by activating AMPK‐PGC‐1α‐SIRT3 signaling and preserving mitochondrial function, thus significantly improving MI/R injury. It has been reported that activation of SIRT3 prevents myocardial mitochondrial oxidative stress and damage 138 , 139 ; however, these protective effects of melatonin are blunted by compound C (a special AMPK signal blocker). 140 Moreover, nuclear factor erythroid 2‐related factor 2 (Nrf2) participates in AMPK‐PGC‐1α pathway, together forming the PGC‐1α/Nrf2 signaling pathway. 141 The activation of Nrf2 has been shown to reduce simulated ischemia reperfusion‐induced oxidative stress and apoptosis. 142 , 143 The related mechanisms may be due to activation of Nrf2, which promotes the expression of anti‐oxidative proteins and phase II detoxifying enzymes, including heme oxygenase 1 (HO‐1), UCP2, and SOD in cardiomyocytes 144 ; however, melatonin also has an effect on the Nrf2 signaling pathway. It has been reported that melatonin pretreatment of H9c2 cells with simulated ischemia reperfusion significantly increases the expression of Nrf2 in the nuclei and whole cells, which reduces ischemia/reperfusion‐induced oxidative stress and apoptosis in H9c2 cell compared with controls. In addition, the expression of HO‐1, an important target gene of Nrf2, also increased after using melatonin. 145 , 146 , 147 , 148 HO‐1 has been shown to be an indispensable endogenous protective enzyme in response to inflammation and oxidative stress, and is an important target for the prevention and treatment of a variety of cardiovascular diseases. 149 Therefore, the antioxidative effect of melatonin depends on the activation of the PGC‐1α/Nrf2 signaling pathway.
FIGURE 2.
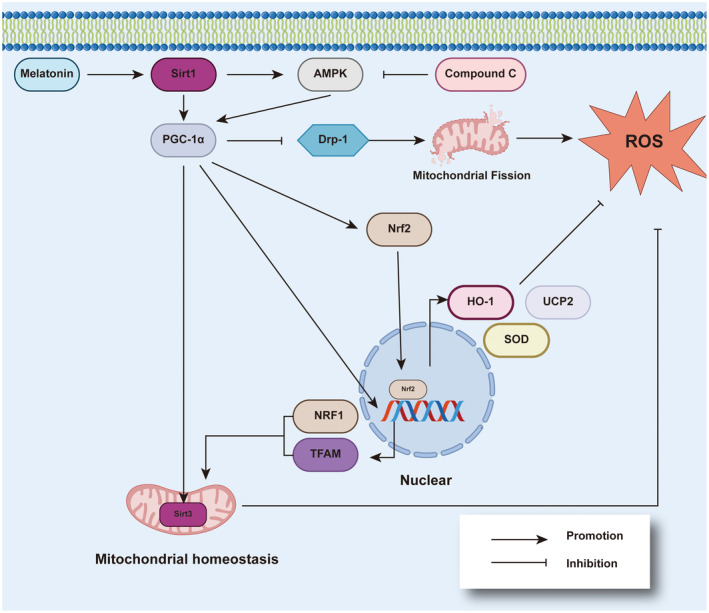
Melatonin relieves oxidative stress by activating PGC‐1α. Melatonin can activate Sirt1/PGC‐1α with or without AMPK activation, which produces anti‐oxidative stress effects. This effect is mainly achieved by four pathways. (1) PGC‐1α inhibits mitochondrial fission promoted by Drp1, maintaining the stability of mitochondrial structure and function, then reducing the production of ROS. (2) PGC‐1α activates Nrf2 and generates a variety of molecules with antioxidant effects, including HO‐1, SOD, and UCP2. (3) PGC‐1α also helps transcribe NRF1 and TFAM, these two substances are beneficial to mitochondrial copy and biogenesis. (4) PGC‐1α activates Sirt3 located on the mitochondria, which helps maintain mitochondrial homeostasis. AMPK, AMP kinase; Drp1, dynamin‐related protein 1; HO‐1, heme oxygenase 1; NRF1, nuclear respiratory factor 1; PGC‐1α, proliferator‐activated receptor coactivator 1 alpha; ROS, reactive oxygen species; SIRT1, silent information regulator 1; SOD, superoxide dismutase; TFAM, mitochondrial transcription factor A; UCP, uncoupling protein
8. MELATONIN INHIBITS CELL DEATH IN DCM
In addition to regulating autophagy and relieving oxidative stress, melatonin also reduces cell apoptosis. Programmed cell death usually occurs when the cells are damaged by non‐receptor‐mediated stimulation and is very common in normal life activities. As an important means for cell renewal, proper apoptosis always maintains the normal vitality of cells. 150 , 151 Once the intensity of this activity is too high, it will cause the original normal tissue and organ function to decline. Now, it has been proven that increased cell death, including both apoptotic‐ and necrotic‐related cell death, is a frequent characteristic in the myocardium of humans and rodent models with diabetes. 152 , 153 , 154 , 155 This type of cell death in T1DM is augmented by RORα deficiency due to a reduction in the anti‐apoptotic protein, B‐cell lymphoma‐2 (Bcl‐2), and an increase in the pro‐apoptotic protein, Bcl‐2‐associated X protein (Bax), and cytoplasmic cytochrome c. 20 By feeding melatonin to diabetic rats, Amin et al. 156 effectively reduced cardiomyocyte apoptosis. Amin et al. 156 noted that melatonin enhanced Bcl‐2 expression and blocked activation of CD95 and apoptotic proteins (caspases‐3, ‐8, and ‐9). Serum creatine kinase‐MB and lactate dehydrogenase were also significantly improved. 156 All these findings demonstrate the effect of melatonin in improving the diabetic myocardium. In addition, the use of melatonin therapy significantly reduces the level of JNK phosphorylation in the heart, reduces the p53 level, and inhibits cell apoptosis in post‐MI diabetic mice. 157
9. MELATONIN IMPROVES ER STRESS IN DIABETES
ER stress is characterized by a perturbation in Ca2+ or redox balance in the ER, and cause the accumulation of mis‐ or un‐folded proteins, 158 , 159 which exceed the processing capacity of the ER, resulting in disturbed homeostasis and cell dysfunction. In the early stage of ER stress, the unfolded protein response (UPR), a protective mechanism, is initiated to alleviate the protein load by reducing the rate of new protein synthesis and increasing chaperone protein expression, but if this mechanism is inadequate, UPR induces cellular apoptosis and related effects. 160 , 161 UPR is mediated by three important transducers, including protein kinase RNA‐like endoplasmic reticulum kinase (PERK), activated transcription factor 6 (ATF‐6), and inositol‐requiring protein‐1α (IRE1α). Usually, the transducers bind to ER chaperone protein glucose‐regulated protein 78 (GRP78), but with the accumulation of unfolded protein in the ER, GRP78 leave the transducers and participate in handling the accumulated protein. 162
Existing research shows that ER stress is very closely related to diabetes. 163 , 164 , 165 As a cellular function damaged by diabetes, ER stress participates in myocardial infarction, ischemia, pressure overload, DCM, heart failure, and hypertrophy. 166 ER stress is often accompanied by oxidative stress, but ER stress is independent of oxidative stress in diabetes. 167 Indeed, ER stress has been shown to intersect with many different signaling pathways and cause a variety of cell dysfunction complications.
It has been shown that if ER stress persists for a long time, it will lead to cell apoptosis in various ways. Activation of the downstream signaling pathway of PERK, ATF‐6 and IRE1α will downregulate the anti‐apoptotic protein Bcl‐2, and increase the expression of CHOP (As a transcription factor, CHOP can downregulate the expressions of BCL2, BCL‐XL, and MCL‐1, and upregulate the expression of B ak and Bax 168 , 169 ) and the caspase family of proapoptotic cysteine proteases, including caspase‐12, ‐9, ‐8, and ‐3. 170 , 171 , 172 , 173 , 174 , 175 It is also observed that the expression of GRP78, PERK, ATF‐6, CHOP, and pro‐apoptotic protein BAX, caspase‐3 were increased, the anti‐apoptotic protein Bcl‐2 was decreased in the myocardium of diabetic rats induced by STZ. 176 The percentage of apoptotic myocardial cells was also increased in diabetic rats compared with controls. These changes are caused, at least in part, by the reduced activation of SIRT1. It has been reported that SIRT1 plays an important role in DCM. Researchers have observed that activation of SIRT1 improves organ and tissue dysfunction induced by hyperglycemia, including cardiomyocyte apoptosis. 177 , 178 And melatonin can happen to activate SIRT1 (Figure 3). A related study showed that melatonin activates SIRT1, then suppresses PERK/eIF2α/ATF4/CHOP signaling pathway, thereby attenuates ER stress, and eventually protects the function of cardiomyocytes. 179 The increased expression of ATF‐6 caused by diabetes in the rat heart is also inhibited by melatonin. 176 In addition to cardiomyocytes, melatonin has a similar effect in osteoblastic cells. Melatonin reduces the expression of P‐PERK, P‐eIF2α, and P‐ATF4, then affects the expression of CHOP and attenuates apoptosis of osteoblastic cells. 180 Taken together, it is evident that melatonin inhibits ER stress, thereby protecting cells from apoptosis. It is noteworthy, however, that melatonin does not significantly reduce the increased expression of IRE1α caused by diabetes. 176 The effect of melatonin in renal tissues is different. 181 Specifically, melatonin restores the level of GRP78, IRE1α, and ATF6 expression, but has no significant effect on the level of PERK expression in diabetic obese rats, indicating that the effect of melatonin on ER stress has tissue or cellular differences.
FIGURE 3.
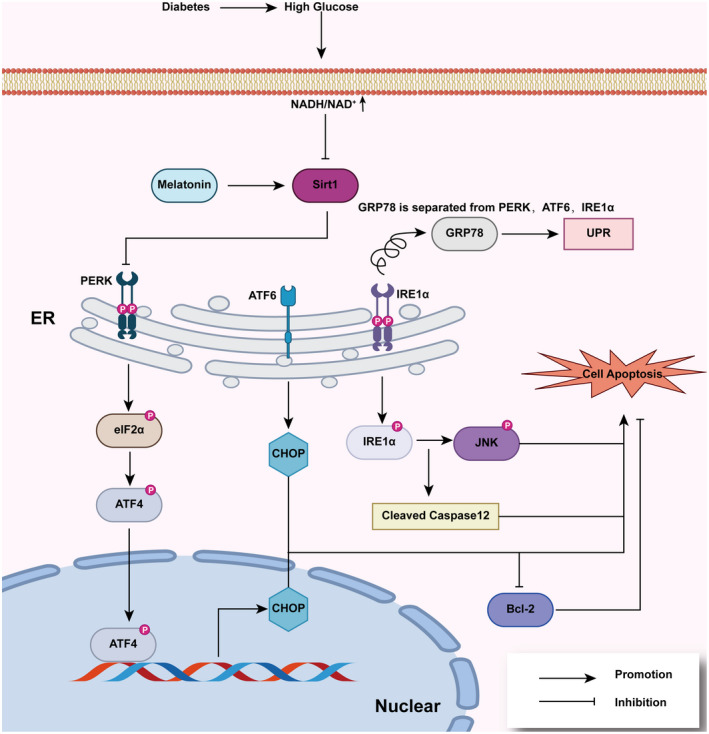
The inhibitory of melatonin on endoplasmic reticulum (ER) stress. When ER stress occurs, GRP78 is separated from PERK, ATF6, and IRE1α, which activates three signal pathways and leads to increased levels of downstream molecules, including p‐elf2α, p‐ATF4, CHOP, p‐IRE1α, p‐JNK, and cleaved caspase 12, which eventually leads to cell apoptosis. Melatonin activates Sirt1, which is inhibited by hyperglycemia in cardiomyocytes, and inhibits the PERK‐p‐elf2α‐ATF4‐CHOP signaling pathway, thus reducing ER stress and reducing cardiomyocyte apoptosis in diabetic patients. ATF‐6, activated transcription factor 6; IRE1α, inositol‐requiring protein‐1α; PERK, protein kinase RNA‐like endoplasmic reticulum kinase
Insulin resistance has an important effect on DCM. 182 Reversing insulin resistance may help treat DCM. According to some reports, inhibition of ER stress alleviates insulin resistance, 183 , 184 which may be one of the mechanisms by which melatonin restores insulin sensitivity. The Akt protein is an important downstream signaling molecule for insulin. ER stress reduces phosphorylated Akt in a severity‐dependent manner. 185 After skeletal muscle cells are exposed to insulin, tunicamycin, an ER stress activator, causes ER stress, inhibits Akt phosphorylation, and stimulates PERK phosphorylation; however, melatonin reverses the changes induced by tunicamycin. The phosphorylation of Akt was increased and the phosphorylation of PERK was decreased by melatonin pretreatment in this ER stress model. 186 Therefore, insulin resistance caused by inhibition of Akt phosphorylation is restored, in part, because of the alleviation of ER stress by melatonin.
Melatonin attenuates ER stress in hepatocytes by regulating microRNA, 187 but there are no studies regarding the effect on the myocardium. Therefore, a study to determine if melatonin regulates ER stress in the myocardium via microRNA is warranted.
10. MELATONIN IMPROVES MITOCHONDRIAL FUNCTION
In skeletal muscle cells of diabetic patients, increased fatty acid uptake promotes mitochondrial fission and the damage of electron transport chain activity, leading to impaired mitochondrial function. 188 A previous study confirmed that this kind of damage can be suppressed by metformin. 189 In recent years, it was shown that melatonin also has a similar effect in cardiomyocytes. 190 Specifically, SIRT1 is the target of melatonin, and activation of SIRT1 activates PGC‐1α, then inhibits dynamin‐related protein 1 (Drp1)‐mediated mitochondrial fission and reduces the level of superoxide produced by mitochondria 190 (Figure 2). Hyperglycemia increased Drp1 protein expression in H9c2 cells, causing mitochondria to become spherical and shorter, indicating mitochondrial fragmentation; however, hyperglycemia‐induced mitochondrial fragmentation was attenuated after melatonin treatment. 190 Moreover, melatonin treatment reduced mitochondria‐derived superoxide production induced by hyperglycemia in cells. 190
Activation of nuclear receptor subfamily 4, group A, member 1 (NR4A1) is also a potential target of melatonin in the treatment of DCM. It has been reported that NR4A1 contributes to cardiomyocyte injury in cardiac microvascular ischemia/reperfusion by fatal mitochondrial fission. 191 Increased NR4A1 expression activated serine/threonine kinase casein kinase 2α, which promotes phosphorylation of mitochondrial fission factor, enhances translocation of Drp1 to the mitochondria, thus leading to fatal mitochondrial fission. Other studies have confirmed this finding. 192 , 193 Indeed, it is possible that NR4A1 upregulation activates the DNA‐PKcs/p53 pathway, increases Drp1 expression in cells and translocation to mitochondria, and inhibits Bnip3‐required mitophagy, thereby aggravating mitochondrial fission and impairing mitochondrial homeostasis. Melatonin inhibits upregulation of NR4A1 to inhibit mitochondrial fission in the liver. 194 Nevertheless, direct clinical evidence is needed to confirm that melatonin also protects the myocardium by inhibiting NR4A1 in diabetes.
Melatonin also improves the activity of some enzymes and mitochondrial complexes in mitochondria. 195 The activity of citrate synthase and mitochondrial complexes Ⅰ, Ⅲ, and Ⅳ are reduced in the hepatic mitochondria of ZDF rats. Treatment with melatonin improves the activity of these important enzymes and complexes, especially complex Ⅳ. Melatonin increases the intermediate product of the tricarboxylic acid cycle in the mitochondria of an insulin resistance skeletal muscle cell model induced by palmitic acid. 196 The content of alpha‐ketoglutarate, citrate, and malate was decreased in this insulin resistance model; however, melatonin treatment reversed this change, improved the content of the three substances to a normal level, and improved mitochondrial function. 196 Notably, in normal cells, melatonin treatment only increases citrate content, but not alpha‐ketoglutarate and malate content. Thus, the effect of melatonin increasing the alpha‐ketoglutarate and malate content appears to be limited to cells in special states, such as insulin resistance.
Moreover, melatonin improves mitochondrial function and content in intramuscular pre‐adipocytes by increasing the gene expression of mitochondrial biogenesis. 197 After melatonin administration, the intracellular ATP generation in porcine intramuscular adipocytes is increased and the expression of mitochondrial‐related genes, including PGC‐1α, TFAM, carnitine palmitoyl transferase‐1β, and the thermogenesis gene is upregulated. Furthermore, the mitochondrial copy is also increased in intramuscular adipocytes.
Melatonin has a clear protective effect on cardiomyocyte mitochondrial dysfunction when the myocardium has an ischemia/reperfusion injury. 198 After melatonin pre‐treatment of rats with ischemia/reperfusion injury, mitochondrial SOD activity is increased and mitochondrial H2O2 and MDA are decreased, indicating that melatonin functions to preserve mitochondrial redox potential.
A recent study showed that melatonin is able to relieve ER dysfunction and mitochondrial calcium overload associated with mitochondrial dysfunction. Excessive production of mitochondrial ROS leads to peroxidation of ER‐related proteins and causes ER dysfunction. Then, the ER releases calcium into the cytoplasm, causing mitochondrial calcium overload in the cell, which in turn affects mitochondrial membrane potential and function. 64 , 67 , 199 Zhou et al. 200 reported that inhibition of spleen tyrosine kinase (Syk) by melatonin promotes cardiomyocyte survival by reducing mitochondria and ER‐related apoptosis. When activated by hyperglycemia, Syk weakens the expression of mitochondrial complex I, thereby increasing the production of ROS, resulting in peroxidation of sarcoplasmic reticulum Ca2+ ATPase (SERCA). Then, the ability of SERCA to transport calcium back to ER is impaired, which leads to cytoplasmic calcium overload. Eventually, mitochondrial dysfunction, ER dysfunction, and cardiomyocyte apoptosis occur 200 ; however, melatonin supplementation delays or even reverses these alterations by regulating the Syk/COX‐I/SERCA signaling pathway. 200
11. MELATONIN RESCUES CARDIOMYOCYTES FROM MI/R INJURY
Clinical evidence has shown that compared with non‐diabetic patients, diabetic patients with acute myocardial infarction have a larger infarct size after reperfusion therapy. 201 , 202 Similarly, the infarct size decreases after acute myocardial infarction in the short term, but always increases the infarct size with time in STZ‐induced diabetic mice compared with normal mice, 203 , 204 indicating that diabetes causes a deficiency in cardiomyocyte protective function. An earlier study showed that melatonin has a protective effect on MI/R. 205 Sahna et al. 206 verified that melatonin helps reduce the infarct size induced by MI/R. Sahna et al. 206 observed that melatonin treatment in diabetic mice reversed the increased level of MDA and decreased the GSH level caused by MI/R. The specific signaling pathway involving cardiomyocyte protection in MI/R has been studied in other experiments. For example, the Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway has been confirmed to play an important role in cardiomyocyte protection during MI/R. 207 There is evidence that melatonin activates the JAK/STAT signaling pathway in MI/R to produce cardiomyocyte protection. 198 , 208 In these experiments, melatonin pre‐treatment of the heart led to increased levels of p‐STAT3 and p‐JAK2 expression, which increased the level of Bcl‐2 expression and decreased the levels of Bax, caspase‐3, and C‐caspase‐3 expression after MI/R administration. 198 , 208 Eventually, the infarct size and cell apoptosis were reduced. 198 , 208 These effects are inhibited by the JAK2/STAT3 inhibitor, AG490. Yet, is activation of the JAK2/STAT3 signaling pathway always beneficial to cells and tissues? Does melatonin always activate the JAK2/STAT3 signaling pathway? Part of the existing evidence indicates that excessive activation of the JAK2/STAT3 signaling pathway has a damaging effect on cardiomyocytes and endothelial cells. For example, the JAK2/STAT3 signaling pathway is activated by H2O2 in endothelial cells, but the expression of caspase‐3 and Bax is also increased, the expression of Bcl‐2 is decreased, and melatonin reverses these changes. 209 Similarly, in septic myocardium, activation of the JAK2/STAT3 signaling pathway aggravates myocardial damage, which is attenuated by melatonin via inhibition of the JAK2/STAT3 signaling pathway. 210 In summary, although the effects of melatonin on the JAK2/STAT3 signaling pathway are different under specific circumstances, the main effects are to protect cells.
SIRT3‐dependent regulation is also a significant method in cardiomyocyte protection during MI/R. Zhai et al. 211 reported that compared with the sham operation group, MI/R caused a decrease in LVEF and LVFS in the IR group, which was reversed by melatonin treatment (20 mg/kg intraperitoneally 10 min before reperfusion). In the same experiments, melatonin also significantly reduced the infarct size and the level of serum lactate dehydrogenase after MI/R administration. 211 Related mechanisms may involve melatonin‐induced increased SIRT3 expression, resulting in activation of SOD2 and glutathione peroxidase (an important peroxide decomposing enzyme), upregulating the expression of Bcl‐2 and Nrf2, and downregulating the expression of Bax, caspase‐3, and gp91phox.
Moreover, reducing SIRT1 is another reason of worsening myocardial MI/R injury. 179 , 212 In the T2DM model, melatonin reduced the apoptosis of cardiomyocytes by increasing the expression of SIRT1 that was inhibited by diabetes, increased LVEF and LVFS, and improved cardiac function after MI/R, which is related to the inhibition of the PERK‐eIF2α‐ATF4‐CHOP signaling pathway by melatonin. This protective effect was suppressed by SIRT1 inhibition, which confirmed that melatonin relieves ER stress via the SIRT1 signaling pathway.
In addition, MT2 receptor‐dependent cGMP‐PKGIα signaling is another signaling pathway in which melatonin protects cardiomyocytes from I/R injury. 29 It has been reported that Nrf2‐HO‐1 and MAPK signaling, two key downstream targets of cGMP‐PKG signaling in the MI/R injured diabetic heart, have changed. Specifically, Nrf2/HO‐1 signaling is decreased, while the phosphorylation levels of ERK, JNK and p38 MAPK (three MAPK signal molecules) is increased; however, melatonin has a role in protecting cardiomyocytes in diabetics during ischemia/reperfusion via cGMP‐PKG signal by acting on the MT2 receptor. Melatonin enhances cGMP‐PKG signaling, upregulates Nrf2/HO‐1 signaling, downregulates MAPK pathway (mainly reduces p‐JNK/JNK and p‐p38 MAPK/p38 MAPK level, but increases p‐ERK/ERK level), and eventually reduces myocardial apoptosis.
In addition to the protective effect of melatonin on MI/R injury, melatonin can also directly promote myocardial proliferation of an infarction injury. 213 MicroRNA can be used as a target for melatonin to combat ischemic injury of cardiomyocytes. For example, inhibiting the expression of miR‐143‐3p, melatonin increases the protein expression of Yap and Ctnnd1, and eventually enhances cardiomyocyte proliferation in MI neonatal mice; however, this effect is limited to the neonatal heart. Furthermore, both MT1 and MT2 siRNA inhibit these alterations, indicating that the miR‐143‐3p/Yap/Ctnnd1 signaling pathway may be mediated by the MT receptors.
12. MELATONIN REDUCES HYPERGLYCEMIA, IMPROVES INSULIN RESISTANCE, AND PROTECTS PANCREATIC β‐CELLS
Hyperglycemia, hyperinsulinemia, and insulin resistance are risk factors for the development of DCM. The resulting series of abnormal reactions, such as metabolism, inflammation, and stress, lead to cardiac tissue interstitial fibrosis, cardiac stiffness/diastolic dysfunction, and later, systolic dysfunction. 6 Therefore, controlling the blood glucose level and improving insulin resistance are important measures in the treatment of DCM.
A recent clinic trial confirmed that melatonin reduces the fasting plasma glucose level and increases insulin sensitivity in diabetic hemodialysis patients after 12 weeks of treatment. 214 In animal experiments involving male Wistar rats with T2DM, the blood glucose level and insulin resistance were determined after 15 days of melatonin treatment. 215 It was found that the serum glucose level was significantly improved and insulin resistance was reduced. 215 In addition to melatonin injection, stimulating the vagus nerve to secrete melatonin effectively controls the blood glucose level. 216 Transcutaneous vagus nerve stimulation (taVNS) triggers extrapineal melatonin secretion in ZDF rats. Furthermore, once daily taVNS for 7 days reduces the glucose concentration to a normal level, and 5 consecutive weeks of taVNS maintains normal plasma glycosylated hemoglobin, thus verifying the ability of melatonin to lower and maintain the elevated blood glucose level.
It has been reported that treatment with melatonin combined with proper exercise contributes to the expression of PGC1α, NRF‐1, NRF‐2, TFAM, and GLUT4 gene in the myocardium of T2DM rats. 217 Melatonin and exercise increase mitochondrial biogenesis, alleviates insulin resistance, improves glucose metabolism and mitochondrial function, and the uptake and utilization of glucose by myocardial cells. 217 For other ways to treat insulin resistance, melatonin also plays a role. Akt at Ser473 is a downstream protein in insulin signaling, which can activate GLUT4, thereby promoting glucose transport. 218 In an insulin resistance model induced by palmitic acid, the level of Akt phosphorylation was decreased by palmitic acid treatment in rat skeletal muscle cells, which causes insulin resistance. When melatonin is used in insulin resistance cells, the level of Akt phosphorylation is increased, insulin resistance is alleviated, and glucose uptake is restored. 196
In contrast, hyperglycemia leads to oxidative stress, reduced production of insulin, ER stress independent of oxidative stress, and death of pancreatic β cells. 167 , 219 , 220 As in other cells, glucotoxicity significantly stimulates ROS generation in a rat pancreatic β‐cell line (INS‐1 cells) with increased phosphorylation of proapoptotic factors (p38‐MAPK and JNK) and an increased level of cleaved caspase‐3, indicating an increased apoptosis of INS‐1 cells. Hyperglycemia also upregulates the expression of ER stress‐related genes and reduces glucose‐stimulated insulin secretion (GSIS). 221 The activities of SOD, glutathione, and catalase decreased significantly in INS‐1 cells treated with high glucose, but melatonin reversed these changes. Melatonin reduces oxidative stress injury in INS‐1 cells exposed to high glucose levels, but does not have a significant effect on the levels of ER stress‐related gene expression (ATF4, CHOP, and GRP78) or the impaired GSIS in INS‐1 cells. 221 This finding suggested that the decreased viability of INS‐1 cells is due oxidative stress, while the attenuated GSIS is due to ER stress. The final conclusion is that melatonin can effectively alleviate the oxidative stress of pancreatic β cells, but cannot effectively improve GSIS.
13. MELATONIN AND HYPERURICEMIA
Hyperuricemia is an independent risk factor for T2DM. In the early stage of impaired glucose metabolism, uric acid levels increase. In diabetic patients, hyperuricemia is associated with micro‐ and macro‐vascular complications. 222 , 223 In contrast, the increase in uric acid levels also increases the risk of atrial fibrillation and conduction block in patients with T2DM. 224 , 225 In fact, obese patients with elevated uric acid levels have more severe insulin resistance and oxidative stress than patients with normal uric acid levels, and this relationship is closely related to the occurrence and development of DCM. 226 Xanthine oxidoreductase catalyzes hypoxanthine to uric acid, accompanied by the production of ROS. 227 Although uric acid is the main antioxidant in plasma, uric acid is a pro‐oxidant in cells and has a contributory role in the pathogenesis of various cardiovascular diseases. 228 Uric acid also has a toxic effect on vascular endothelial function, which may underlie the cause of cardiovascular disease. 229 Specifically, uric acid inhibits NO production in a dose‐dependent fashion and causes vascular endothelial dysfunction. Moreover, high serum uric acid levels are often associated with cardiovascular disease, such as coronary artery disease and hypertension. 230 Therefore, corollary studies involving the effect of melatonin on serum uric acid are warranted. A clinical study showed a significant negative correlation between the levels of endogenous melatonin and uric acid in healthy young males. 231 Another clinical study showed that endogenous melatonin may impact cardiovascular disease risk. 232 Use of exogenous melatonin might decrease the serum uric acid level, and may even reduce the risk of cardiovascular disease. Direct evidence has shown that melatonin decreases plasma uric acid levels and counteracts changes in the plasma LDL‐c, triglycerides, and cholesterol levels in rats with metabolic syndrome. 233
14. EFFECTS OF MELATONIN ON AGE‐RELATED RECEPTORS
AGEs are complex compounds with crosslinks. Unlike enzymatic glycosylation of proteins, AGEs are produced by glycated proteins undergoing a series of non‐enzymatic chemical rearrangements. The accumulation of AGEs increase in hyperglycemic states. 234 , 235 AGEs lead to reduced collagen degradation and increased fibrosis with increased myocardial stiffness and impaired cardiac relaxation, 236 which is related to the phenomenon of collagen cross‐linking in collagen resistant to hydrolytic turnover. 237 AGEs contribute to an increase in ROS during production of AGEs and activation of RAGE, leading to complications of diabetes. 109 , 238 Furthermore, AGEs activate TLR4 239 and RAGE increases the expression of myeloid differentiation factor 88 (MyD88) and NF‐κB activity, causing overstimulation of the immune system and hyperinflammation. 240 The excessive expression of TLR4 enhances formation of foam cells and increased MCP‐1 and IL‐8 in monocytes, leading to atherosclerosis. 241 , 242 By activating P38, one of the MAPK pathways, AGEs increase EPC apoptosis and decrease the production of NO. 243 Therefore, determining if melatonin has a role in the production of AGEs or AGE‐related receptors and signaling pathways has become a new research direction in the treatment of DCM.
According to related studies, melatonin has an effect on inhibiting the damage caused by AGEs related receptors. Melatonin treatment significantly reduced the RAGE overexpression and the levels of p‐IKKβ and NF‐κB expression in D‐galactose‐treated mice nervous system. 244 Other studies have also focused on the effect of melatonin on AGEs and related receptors. Specifically, the injection of melatonin significantly inhibits the overexpression of AGEs and TRL4. P38, c‐Jun N‐terminal kinases phosphorylation, MyD88, as well as NF‐κB translocation, are also suppressed by melatonin. And early growth response protein 1, messenger RNA of macrophage inflammatory protein 2 are increased by melatonin. These findings suggest that melatonin prevents liver fibrosis by inhibiting necroptosis‐associated inflammatory signaling. 245 , 246 However, as mentioned above, melatonin also has an activating effect on TLR4, and it can even rescue cardiomyocytes through the SAFE pathway. 35 So, which aspect is the main role? Are different effects related to tissue specificity? These questions still need a lot of experimental data to solve.
15. DISCUSSION
Globally, the main cause of non‐infectious deaths is heart disease, ranking first among all non‐infectious diseases. The prevalence of heart disease is on the rise in both developed and developing countries, 247 and diabetic patients with insulin resistance are at greater risk of heart disease. It has been confirmed that diabetes has a significant effect on heart function and the cardiovascular system. Compared with healthy patients, the risk of sudden cardiac death in diabetic patients is increased 2‐ to 10‐fold. 248 There are many reasons for this phenomenon, such as diastolic dysfunction, heart failure, increased sympathetic tone, and inflammation. 249 Melatonin has many advantages in improving diabetes‐related cardiac complications. Melatonin is mainly secreted by the pineal gland, as well as other tissues, and it acts on many tissues and organs. In the heart, melatonin is effective against diabetes‐induced cell apoptosis, excessive oxidative stress, ER stress, insulin resistance, sympathetic hypertonia, morbid cardiac microcirculation, and MI/R. In addition, melatonin also has a protective effect on the pancreatic β‐cell, which maintains insulin secretion and promotes normal blood glucose levels, thus slowing down the progression of diabetes and its complications.
In most animal experiments that this article refers to, melatonin was used by intraperitoneal injection. This method of use maximizes the utilization of melatonin, but the current use of melatonin in the market is mainly oral. According to some reports, oral melatonin has approximately 3% bioavailability compared with intravenous injection, 250 but there are reports that this value is 9%–33%. 251 Although it is accompanied by huge experimental data and individual differences in these two reports, it is undeniable that the bioavailability of oral melatonin is very low. The reason for this finding is largely due to hepatic first pass effect. 252
In addition to the exogenous administration method of directly supplementing melatonin, we may be able to find some ways to increase the production of endogenous melatonin. It would be great if these methods can also be used to treat diabetes and cardiomyopathy. So, what is the effect of traditional medicines for the treatment of diabetes and heart failure on the melatonin signal? Angiotensin receptor antagonists and β‐receptor blockers are first‐line drugs for the treatment of heart failure. In principle, noradrenergic stimulation and angiotensin can increase the secretion of melatonin. 253 , 254 Related experiments are indeed disappointing. Stoschitzky et al. proved in a set of clinical trials that β‐blockers decrease melatonin release via specific inhibition of adrenergic beta1‐receptors. 255 Baltatu et al. proved in animal experiments that using losartan to block the angiotensin AT1‐receptor significantly reduces the content of melatonin in the rat pineal gland. 254 As one of the core drugs for diabetes treatment, insulin potentiates norepinephrine‐mediated melatonin synthesis in cultured rat pineal glands through post‐transcriptional mechanisms. 256 In addition, there are also drugs that increase the secretion of melatonin, but are not related to traditional diabetes and heart failure treatments. For example, 5‐hydroxytryptamine, as the precursor of the melatonin precursor N‐acetylserotonin, increases melatonin secretion evoked by submaximal norepinephrine. 257 Corticosterone potentiates noradrenaline‐induced melatonin production through glucocorticoid receptor inhibition of NFkappaB nuclear translocation. 258 Thyroxine T3 can increase the melatonin levels in pineal under light conditions. 259 From this perspective, if it is necessary to show the beneficial effects of melatonin in the treatment of diabetic myocardium, then appropriate supplementation of exogenous melatonin or stimulation of endogenous melatonin production is of clinical value.
Due to the many benefits of melatonin, current research involving melatonin in the treatment of DCM is a dynamic field. In some ways, melatonin can be considered as a new treatment for DCM. In this regard, we look forward to the effective use of melatonin in the treatment of all diabetes‐related diseases and the research and development of new therapeutic mechanisms of melatonin.
16. NOMENCLATURE OF TARGETS AND LIGANDS
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS guide to PHARMACOLOGY, 260 and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/2020. 261 , 262
DISCLOSURE
The authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
Keming Huang and Xianling Luo conceived, designed, and planned the study. All authors collected and read the literature. Keming Huang drafted the manuscript. Jian Feng revised the manuscript. All authors read and approved the final manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
ACKNOWLEDGMENTS
The authors thank the fund of the Key Laboratory of Medical Electrophysiology, the Fund of Collaborative Innovation Center for Prevention and Treatment of Cardiovascular Disease of Sichuan Province, and Southwest Medical University for fund support.
Huang K, Luo X, Zhong Y, Deng L, Feng J. New insights into the role of melatonin in diabetic cardiomyopathy. Pharmacol Res Perspect. 2022;10:e00904. doi: 10.1002/prp2.904
Keming Huang and Xianling Luo contributed equally to this paper.
Funding information
This research was funded by the fund (KeyME‐KeyME‐2020‐004) of the Key Laboratory of Medical Electrophysiology, the Fund (xtcx2019‐13) of Collaborative Innovation Center for Prevention and Treatment of Cardiovascular Disease of Sichuan Province; Southwest Medical University.
DATA AVAILABILITY STATEMENT
Not applicable.
REFERENCES
- 1. Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018;14(2):88‐98. [DOI] [PubMed] [Google Scholar]
- 2. Hossain P, Kawar B, El Nahas M. Obesity and diabetes in the developing world–a growing challenge. N Engl J Med. 2007;356(3):213‐215. [DOI] [PubMed] [Google Scholar]
- 3. Wu Y, Ding Y, Tanaka Y, Zhang W. Risk factors contributing to type 2 diabetes and recent advances in the treatment and prevention. Int J Med Sci. 2014;11(11):1185‐1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Prasad RB, Groop L. Genetics of type 2 diabetes‐pitfalls and possibilities. Genes. 2015;6(1):87‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aneja A, Tang WH, Bansilal S, Garcia MJ, Farkouh ME. Diabetic cardiomyopathy: insights into pathogenesis, diagnostic challenges, and therapeutic options. Am J Med. 2008;121(9):748‐757. [DOI] [PubMed] [Google Scholar]
- 6. Jia G, Whaley‐Connell A, Sowers JR. Diabetic cardiomyopathy: a hyperglycaemia‐ and insulin‐resistance‐induced heart disease. Diabetologia. 2018;61(1):21‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Isfort M, Stevens SC, Schaffer S, Jong CJ, Wold LE. Metabolic dysfunction in diabetic cardiomyopathy. Heart Fail Rev. 2014;19(1):35‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia. 2014;57(4):660‐671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hölscher ME, Bode C, Bugger H. Diabetic cardiomyopathy: does the type of diabetes matter? Int J Mol Sci. 2016;17(12):2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Radovits T, Korkmaz S, Loganathan S, et al. Comparative investigation of the left ventricular pressure‐volume relationship in rat models of type 1 and type 2 diabetes mellitus. Am J Physiol Heart Circ Physiol. 2009;297(1):H125‐H133. [DOI] [PubMed] [Google Scholar]
- 11. Amaral FGD, Cipolla‐Neto J. A brief review about melatonin, a pineal hormone. Arch Endocrinol Metab. 2018;62(4):472‐479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karasek M. Melatonin, human aging, and age‐related diseases. Exp Gerontol. 2004;39(11–12):1723‐1729. [DOI] [PubMed] [Google Scholar]
- 13. Reiter R, Rosales‐Corral S, Tan D‐X, et al. Melatonin, a full service anti‐cancer agent: inhibition of initiation, progression and metastasis. Int J Mol Sci. 2017;18(4):843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sun H, Gusdon AM, Qu S. Effects of melatonin on cardiovascular diseases: progress in the past year. Curr Opin Lipidol. 2016;27(4):408‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mok JX, Ooi JH, Ng KY, Koh RY, Chye SM. A new prospective on the role of melatonin in diabetes and its complications. Horm Mol Biol Clin Investig. 2019;40(1). doi: 10.1515/hmbci-2019-0036 [DOI] [PubMed] [Google Scholar]
- 16. Yao T, Ying X, Zhao Y, et al. Vitamin D receptor activation protects against myocardial reperfusion injury through inhibition of apoptosis and modulation of autophagy. Antioxid Redox Signal. 2015;22(8):633‐650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marciano D, Chang Mi Ra, Corzo C, et al. The therapeutic potential of nuclear receptor modulators for treatment of metabolic disorders: PPARγ, RORs, and Rev‐erbs. Cell Metab. 2014;19(2):193‐208. [DOI] [PubMed] [Google Scholar]
- 18. Ito A, Hong C, Rong X, et al. LXRs link metabolism to inflammation through Abca1‐dependent regulation of membrane composition and TLR signaling. eLife. 2015;4:e08009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Peliciari‐Garcia RA, Zanquetta MM, Andrade‐Silva J, Gomes DA, Barreto‐Chaves ML, Cipolla‐Neto J. Expression of circadian clock and melatonin receptors within cultured rat cardiomyocytes. Chronobiol Int. 2011;28(1):21‐30. [DOI] [PubMed] [Google Scholar]
- 20. Zhao Y, Xu L, Ding S, et al. Novel protective role of the circadian nuclear receptor retinoic acid‐related orphan receptor‐α in diabetic cardiomyopathy. J Pineal Res. 2017;62(3):e12378. [DOI] [PubMed] [Google Scholar]
- 21. Xu L, Su Y, Zhao Y, et al. Melatonin differentially regulates pathological and physiological cardiac hypertrophy: crucial role of circadian nuclear receptor RORα signaling. J Pineal Res. 2019;67(2):e12579. [DOI] [PubMed] [Google Scholar]
- 22. Sivaraman V, Hausenloy DJ, Wynne AM, Yellon DM. Preconditioning the diabetic human myocardium. J Cell Mol Med. 2010;14(6b):1740‐1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Przyklenk K, Maynard M, Greiner DL, Whittaker P. Cardioprotection with postconditioning: loss of efficacy in murine models of type‐2 and type‐1 diabetes. Antioxid Redox Signal. 2011;14(5):781‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He B, Zhao Y, Xu L, et al. The nuclear melatonin receptor RORα is a novel endogenous defender against myocardial ischemia/reperfusion injury. J Pineal Res. 2016;60(3):313‐326. [DOI] [PubMed] [Google Scholar]
- 25. Danaei G, Finucane MM, Lu Y, et al. National, regional, and global trends in fasting plasma glucose and diabetes prevalence since 1980: systematic analysis of health examination surveys and epidemiological studies with 370 country‐years and 2·7 million participants. Lancet. 2011;378(9785):31‐40. [DOI] [PubMed] [Google Scholar]
- 26. Huang H, Liu X, Chen D, et al. Melatonin prevents endothelial dysfunction in SLE by activating the nuclear receptor retinoic acid‐related orphan receptor‐α. Int Immunopharmacol. 2020;83:106365. [DOI] [PubMed] [Google Scholar]
- 27. Ding S, Lin N, Sheng X, et al. Melatonin stabilizes rupture‐prone vulnerable plaques via regulating macrophage polarization in a nuclear circadian receptor RORα‐dependent manner. J Pineal Res. 2019;67(2):e12581. [DOI] [PubMed] [Google Scholar]
- 28. Han D, Wang Y, Chen J, et al. Activation of melatonin receptor 2 but not melatonin receptor 1 mediates melatonin‐conferred cardioprotection against myocardial ischemia/reperfusion injury. J Pineal Res. 2019;67(1):e12571. [DOI] [PubMed] [Google Scholar]
- 29. Yu L‐M, Di W‐C, Dong X, et al. Melatonin protects diabetic heart against ischemia‐reperfusion injury, role of membrane receptor‐dependent cGMP‐PKG activation. Biochim Biophys Acta. 2018;1864(2):563‐578. [DOI] [PubMed] [Google Scholar]
- 30. Li P, Hu F, Cao X, Luo L, Tu Q. Melatonin receptor protects cardiomyocyte against oxidative stress‐induced apoptosis through the MAPK‐ERK signaling pathway. J Recept Signal Transduct Res. 2020;40(2):117‐125. [DOI] [PubMed] [Google Scholar]
- 31. Ekmekcioglu C, Haslmayer P, Philipp C, et al. Expression of the MT1 melatonin receptor subtype in human coronary arteries. J Recept Signal Transduct Res. 2001;21(1):85‐91. [DOI] [PubMed] [Google Scholar]
- 32. Ekmekcioglu C, Thalhammer T, Humpeler S, et al. The melatonin receptor subtype MT2 is present in the human cardiovascular system. J Pineal Res. 2003;35(1):40‐44. [DOI] [PubMed] [Google Scholar]
- 33. Masana MI, Doolen S, Ersahin C, et al. MT(2) melatonin receptors are present and functional in rat caudal artery. J Pharmacol Exp Ther. 2002;302(3):1295‐1302. [DOI] [PubMed] [Google Scholar]
- 34. Doolen S, Krause DN, Dubocovich ML, Duckles SP. Melatonin mediates two distinct responses in vascular smooth muscle. Eur J Pharmacol. 1998;345(1):67‐69. [DOI] [PubMed] [Google Scholar]
- 35. Nduhirabandi F, Lamont K, Albertyn Z, Opie LH, Lecour S. Role of toll‐like receptor 4 in melatonin‐induced cardioprotection. J Pineal Res. 2016;60(1):39‐47. [DOI] [PubMed] [Google Scholar]
- 36. Lecour S. Activation of the protective Survivor Activating Factor Enhancement (SAFE) pathway against reperfusion injury: does it go beyond the RISK pathway? J Mol Cell Cardiol. 2009;47(1):32‐40. [DOI] [PubMed] [Google Scholar]
- 37. Zhao T, Zhang H, Jin C, Qiu F, Wu Y, Shi L. Melatonin mediates vasodilation through both direct and indirect activation of BK(Ca) channels. J Mol Endocrinol. 2017;59(3):219‐233. [DOI] [PubMed] [Google Scholar]
- 38. Xu Z, Wu Y, Zhang Y, Zhang H, Shi L. Melatonin activates BK(Ca) channels in cerebral artery myocytes via both direct and MT receptor/PKC‐mediated pathway. Eur J Pharmacol. 2019;842:177‐188. [DOI] [PubMed] [Google Scholar]
- 39. Jockers R, Delagrange P, Dubocovich ML, et al. Update on melatonin receptors: IUPHAR Review 20. Br J Pharmacol. 2016;173(18):2702‐2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Baltatu OC, Senar S, Campos LA, Cipolla‐Neto J. Cardioprotective melatonin: translating from proof‐of‐concept studies to therapeutic use. Int J Mol Sci. 2019;20(18):4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Torregroza C, Jalajel O, Raupach A, et al. Activation of PKG and Akt is required for cardioprotection by ramelteon‐induced preconditioning and is located upstream of mKCa‐channels. Int J Mol Sci. 2020;21(7):2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cao CM, Xia Q, Gao Q, Chen M, Wong TM. Calcium‐activated potassium channel triggers cardioprotection of ischemic preconditioning. J Pharmacol Exp Ther. 2005;312(2):644‐650. [DOI] [PubMed] [Google Scholar]
- 43. Stanley WC, Lopaschuk GD, McCormack JG. Regulation of energy substrate metabolism in the diabetic heart. Cardiovasc Res. 1997;34(1):25‐33. [DOI] [PubMed] [Google Scholar]
- 44. Amaral N, Okonko DO. Metabolic abnormalities of the heart in type II diabetes. Diab Vasc Dis Res. 2015;12(4):239‐248. [DOI] [PubMed] [Google Scholar]
- 45. Watanabe K, Fujii H, Takahashi T, et al. Constitutive regulation of cardiac fatty acid metabolism through peroxisome proliferator‐activated receptor alpha associated with age‐dependent cardiac toxicity. J Biol Chem. 2000;275(29):22293‐22299. [DOI] [PubMed] [Google Scholar]
- 46. Liu W, Zhang Y, Chen QI, et al. Melatonin alleviates glucose and lipid metabolism disorders in guinea pigs caused by different artificial light rhythms. J Diabetes Res. 2020;2020:4927403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Touitou Y, Reinberg A, Touitou D. Association between light at night, melatonin secretion, sleep deprivation, and the internal clock: health impacts and mechanisms of circadian disruption. Life Sci. 2017;173:94‐106. [DOI] [PubMed] [Google Scholar]
- 48. Ou TH, Tung YT, Yang TH, Chien YW. Melatonin improves fatty liver syndrome by inhibiting the lipogenesis pathway in hamsters with high‐fat diet‐induced hyperlipidemia. Nutrients. 2019;11(4):748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ghosh G, De K, Maity S, et al. Melatonin protects against oxidative damage and restores expression of GLUT4 gene in the hyperthyroid rat heart. J Pineal Res. 2007;42(1):71‐82. [DOI] [PubMed] [Google Scholar]
- 50. Ferré P. The biology of peroxisome proliferator‐activated receptors: relationship with lipid metabolism and insulin sensitivity. Diabetes. 2004;53(suppl 1):S43‐S50. [DOI] [PubMed] [Google Scholar]
- 51. Ciftçi M, Bilici D, Küfrevioğlu OI. Effects of melatonin on enzyme activities of glucose‐6‐phosphate dehydrogenase from human erythrocytes in vitro and from rat erythrocytes in vivo. Pharmacol Res. 2001;44(1):7‐11. [DOI] [PubMed] [Google Scholar]
- 52. Akmali M, Ahmadi R, Vessal M. Pre‐ and post‐treatment of streptozocin administered rats with melatonin: effects on some hepatic enzymes of carbohydrate metabolism. Arch Iran Med. 2010;13(2):105‐110. [PubMed] [Google Scholar]
- 53. Huang JP, Huang SS, Deng JY, Hung LM. Impairment of insulin‐stimulated Akt/GLUT4 signaling is associated with cardiac contractile dysfunction and aggravates I/R injury in STZ‐diabetic rats. J Biomed Sci. 2009;16(1):77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Desrois M, Sidell RJ, Gauguier D, King LM, Radda GK, Clarke K. Initial steps of insulin signaling and glucose transport are defective in the type 2 diabetic rat heart. Cardiovasc Res. 2004;61(2):288‐296. [DOI] [PubMed] [Google Scholar]
- 55. Camps M, Castelló A, Muñoz P, et al. Effect of diabetes and fasting on GLUT‐4 (muscle/fat) glucose‐transporter expression in insulin‐sensitive tissues. Heterogeneous response in heart, red and white muscle. Biochem J. 1992;282(3):765‐772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Loader J, Montero D, Lorenzen C, et al. Acute hyperglycemia impairs vascular function in healthy and cardiometabolic diseased subjects: systematic review and meta‐analysis. Arterioscler Thromb Vasc Biol. 2015;35(9):2060‐2072. [DOI] [PubMed] [Google Scholar]
- 57. Vanhoutte PM, Shimokawa H, Feletou M, Tang EH. Endothelial dysfunction and vascular disease—a 30th anniversary update. Acta Physiol. 2017;219(1):22‐96. [DOI] [PubMed] [Google Scholar]
- 58. Godo S, Shimokawa H. Endothelial functions. Arterioscler Thromb Vasc Biol. 2017;37(9):e108‐e114. [DOI] [PubMed] [Google Scholar]
- 59. Walther G, Obert P, Dutheil F, et al. Metabolic syndrome individuals with and without type 2 diabetes mellitus present generalized vascular dysfunction: cross‐sectional study. Arterioscler Thromb Vasc Biol. 2015;35(4):1022‐1029. [DOI] [PubMed] [Google Scholar]
- 60. Salmanoglu DS, Gurpinar T, Vural K, Ekerbicer N, Darıverenli E, Var A. Melatonin and L‐carnitin improves endothelial disfunction and oxidative stress in type 2 diabetic rats. Redox Biol. 2016;8:199‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jin H, Zhang Z, Wang C, et al. Melatonin protects endothelial progenitor cells against AGE‐induced apoptosis via autophagy flux stimulation and promotes wound healing in diabetic mice. Exp Mol Med. 2018;50(11):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Crespo E, Macías M, Pozo D, et al. Melatonin inhibits expression of the inducible NO synthase II in liver and lung and prevents endotoxemia in lipopolysaccharide‐induced multiple organ dysfunction syndrome in rats. FASEB J. 1999;13(12):1537‐1546. [PubMed] [Google Scholar]
- 63. Liu VW, Huang PL. Cardiovascular roles of nitric oxide: a review of insights from nitric oxide synthase gene disrupted mice. Cardiovasc Res. 2008;77(1):19‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhang Y, Zhou H, Wu W, et al. Liraglutide protects cardiac microvascular endothelial cells against hypoxia/reoxygenation injury through the suppression of the SR‐Ca2+‐XO‐ROS axis via activation of the GLP‐1R/PI3K/Akt/survivin pathways. Free Radic Biol Med. 2016;95:278‐292. [DOI] [PubMed] [Google Scholar]
- 65. Haorah J, Knipe B, Gorantla S, Zheng J, Persidsky Y. Alcohol‐induced blood‐brain barrier dysfunction is mediated via inositol 1,4,5‐triphosphate receptor (IP3R)‐gated intracellular calcium release. J Neurochem. 2007;100(2):324‐336. [DOI] [PubMed] [Google Scholar]
- 66. Kloner RA, King KS, Harrington MG. No‐reflow phenomenon in the heart and brain. Am J Physiol Heart Circ Physiol. 2018;315(3):H550‐H562. [DOI] [PubMed] [Google Scholar]
- 67. Zhu H, Jin Q, Li Y, et al. Melatonin protected cardiac microvascular endothelial cells against oxidative stress injury via suppression of IP3R‐[Ca2+]c/VDAC‐[Ca2+]m axis by activation of MAPK/ERK signaling pathway. Cell Stress Chaperones. 2018;23(1):101‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kagan VE, Borisenko GG, Tyurina YY, et al. Oxidative lipidomics of apoptosis: redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Radic Biol Med. 2004;37(12):1963‐1985. [DOI] [PubMed] [Google Scholar]
- 69. Lu M, Gong X. Upstream reactive oxidative species (ROS) signals in exogenous oxidative stress‐induced mitochondrial dysfunction. Cell Biol Int. 2009;33(6):658‐664. [DOI] [PubMed] [Google Scholar]
- 70. Nahser PJ Jr, Brown RE, Oskarsson H, Winniford MD, Rossen JD. Maximal coronary flow reserve and metabolic coronary vasodilation in patients with diabetes mellitus. Circulation. 1995;91(3):635‐640. [DOI] [PubMed] [Google Scholar]
- 71. Abdelrazik Soliman NG, Abdel‐Hamid AAM, El‐Hawwary AA, Ellakkany A. Impact of liraglutide on microcirculation in experimental diabetic cardiomyopathy. Acta Histochem. 2020;122(3):151533. [DOI] [PubMed] [Google Scholar]
- 72. Yamauchi T, Ohnaka K, Takayanagi R, Umeda F, Nawata H. Enhanced secretion of endothelin‐1 by elevated glucose levels from cultured bovine aortic endothelial cells. FEBS Lett. 1990;267(1):16‐18. [DOI] [PubMed] [Google Scholar]
- 73. Tickerhoof MM, Farrell PA, Korzick DH. Alterations in rat coronary vasoreactivity and vascular protein kinase C isoforms in Type 1 diabetes. Am J Physiol Heart Circ Physiol. 2003;285(6):H2694‐H2703. [DOI] [PubMed] [Google Scholar]
- 74. Ramzy D, Rao V, Tumiati LC, et al. Elevated endothelin‐1 levels impair nitric oxide homeostasis through a PKC‐dependent pathway. Circulation. 2006;114(1 suppl):I319‐I326. [DOI] [PubMed] [Google Scholar]
- 75. Rodriguez‐Vita J, Ruiz‐Ortega M, Rupérez M, et al. Endothelin‐1, via ETA receptor and independently of transforming growth factor‐beta, increases the connective tissue growth factor in vascular smooth muscle cells. Circ Res. 2005;97(2):125‐134. [DOI] [PubMed] [Google Scholar]
- 76. Bousette N, Giaid A. Endothelin‐1 in atherosclerosis and other vasculopathies. Can J Physiol Pharmacol. 2003;81(6):578‐587. [DOI] [PubMed] [Google Scholar]
- 77. León J, Casado J, Jiménez Ruiz SM, et al. Melatonin reduces endothelin‐1 expression and secretion in colon cancer cells through the inactivation of FoxO‐1 and NF‐κβ. J Pineal Res. 2014;56(4):415‐426. [DOI] [PubMed] [Google Scholar]
- 78. Wang Z, Ni L, Wang J, et al. The protective effect of melatonin on smoke‐induced vascular injury in rats and humans: a randomized controlled trial. J Pineal Res. 2016;60(2):217‐227. [DOI] [PubMed] [Google Scholar]
- 79. Park SW, Choi SM, Lee SM. Effect of melatonin on altered expression of vasoregulatory genes during hepatic ischemia/reperfusion. Arch Pharm Res. 2007;30(12):1619‐1624. [DOI] [PubMed] [Google Scholar]
- 80. Shimizu M, Umeda K, Sugihara N, et al. Collagen remodelling in myocardia of patients with diabetes. J Clin Pathol. 1993;46(1):32‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sutherland CG, Fisher BM, Frier BM, Dargie HJ, More IA, Lindop GB. Endomyocardial biopsy pathology in insulin‐dependent diabetic patients with abnormal ventricular function. Histopathology. 1989;14(6):593‐602. [DOI] [PubMed] [Google Scholar]
- 82. van Heerebeek L, Hamdani N, Handoko ML, et al. Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation. 2008;117(1):43‐51. [DOI] [PubMed] [Google Scholar]
- 83. Zhao J, Randive R, Stewart JA. Molecular mechanisms of AGE/RAGE‐mediated fibrosis in the diabetic heart. World J Diabetes. 2014;5(6):860‐867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Shamhart PE, Luther DJ, Adapala RK, et al. Hyperglycemia enhances function and differentiation of adult rat cardiac fibroblasts. Can J Physiol Pharmacol. 2014;92(7):598‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Xu F, Liu C, Zhou D, Zhang L. TGF‐β/SMAD pathway and its regulation in hepatic fibrosis. J Histochem Cytochem. 2016;64(3):157‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ma TT, Meng XM. TGF‐β/Smad and renal fibrosis. Adv Exp Med Biol. 2019;1165:347‐364. [DOI] [PubMed] [Google Scholar]
- 87. Zhang L‐L, Huang S, Ma X‐X, et al. Angiotensin(1–7) attenuated Angiotensin II‐induced hepatocyte EMT by inhibiting NOX‐derived H2O2‐activated NLRP3 inflammasome/IL‐1β/Smad circuit. Free Radic Biol Med. 2016;97:531‐543. [DOI] [PubMed] [Google Scholar]
- 88. Gao R, Shi H, Chang S, et al. The selective NLRP3‐inflammasome inhibitor MCC950 reduces myocardial fibrosis and improves cardiac remodeling in a mouse model of myocardial infarction. Int Immunopharmacol. 2019;74:105575. [DOI] [PubMed] [Google Scholar]
- 89. Alyaseer AAA, de Lima MHS, Braga TT. The role of NLRP3 inflammasome activation in the epithelial to mesenchymal transition process during the fibrosis. Front Immunol. 2020;11:883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Che H, Wang Y, Li H, et al. Melatonin alleviates cardiac fibrosis via inhibiting lncRNA MALAT1/miR‐141‐mediated NLRP3 inflammasome and TGF‐β1/Smads signaling in diabetic cardiomyopathy. FASEB J. 2020;34(4):5282‐5298. [DOI] [PubMed] [Google Scholar]
- 91. Levine B, Klionsky DJ. Development by self‐digestion: molecular mechanisms and biological functions of autophagy. Dev Cell. 2004;6(4):463‐477. [DOI] [PubMed] [Google Scholar]
- 92. Nakai A, Yamaguchi O, Takeda T, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13(5):619‐624. [DOI] [PubMed] [Google Scholar]
- 93. Fulda S, Kögel D. Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy. Oncogene. 2015;34(40):5105‐5113. [DOI] [PubMed] [Google Scholar]
- 94. Bhattacharya D, Mukhopadhyay M, Bhattacharyya M, Karmakar P. Is autophagy associated with diabetes mellitus and its complications? A review. EXCLI J. 2018;17:709‐720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Riehle C, Wende AR, Sena S, et al. Insulin receptor substrate signaling suppresses neonatal autophagy in the heart. J Clin Investig. 2013;123(12):5319‐5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sciarretta S, Maejima Y, Zablocki D, Sadoshima J. The role of autophagy in the heart. Annu Rev Physiol. 2018;80:1‐26. [DOI] [PubMed] [Google Scholar]
- 97. Mellor KM, Reichelt ME, Delbridge LM. Autophagy anomalies in the diabetic myocardium. Autophagy. 2011;7(10):1263‐1267. [DOI] [PubMed] [Google Scholar]
- 98. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhang M, Zhang L, Hu J, et al. MST1 coordinately regulates autophagy and apoptosis in diabetic cardiomyopathy in mice. Diabetologia. 2016;59(11):2435‐2447. [DOI] [PubMed] [Google Scholar]
- 100. Zhang M, Lin J, Wang S, et al. Melatonin protects against diabetic cardiomyopathy through Mst1/Sirt3 signaling. J Pineal Res. 2017;63(2):e12418. [DOI] [PubMed] [Google Scholar]
- 101. Wang S, Zhao Z, Feng X, et al. Melatonin activates Parkin translocation and rescues the impaired mitophagy activity of diabetic cardiomyopathy through Mst1 inhibition. J Cell Mol Med. 2018;22(10):5132‐5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kandemir YB, Tosun V, Güntekin Ü. Melatonin protects against streptozotocin‐induced diabetic cardiomyopathy through the mammalian target of rapamycin (mTOR) signaling pathway. Adv Clin Exp Med. 2019;28(9):1171‐1177. [DOI] [PubMed] [Google Scholar]
- 103. Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36(12):2503‐2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Zhang WX, He BM, Wu Y, Qiao JF, Peng ZY. Melatonin protects against sepsis‐induced cardiac dysfunction by regulating apoptosis and autophagy via activation of SIRT1 in mice. Life Sci. 2019;217:8‐15. [DOI] [PubMed] [Google Scholar]
- 105. Wu J, Yang Y, Gao Y, Wang Z, Ma J. Melatonin attenuates anoxia/reoxygenation injury by inhibiting excessive mitophagy through the MT2/SIRT3/FoxO3a signaling pathway in H9c2 cells. Drug Des Devel Ther. 2020;14:2047‐2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Kanamori H, Takemura G, Goto K, et al. Autophagy limits acute myocardial infarction induced by permanent coronary artery occlusion. Am J Physiol Heart Circ Physiol. 2011;300(6):H2261‐2271. [DOI] [PubMed] [Google Scholar]
- 107. Zhu H, Tannous P, Johnstone JL, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Investig. 2007;117(7):1782‐1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404(6779):787‐790. [DOI] [PubMed] [Google Scholar]
- 109. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813‐820. [DOI] [PubMed] [Google Scholar]
- 110. Varga ZV, Giricz Z, Liaudet L, Haskó G, Ferdinandy P, Pacher P. Interplay of oxidative, nitrosative/nitrative stress, inflammation, cell death and autophagy in diabetic cardiomyopathy. Biochem Biophys Acta. 2015;1852(2):232‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhang L, Wang X, Cueto R, et al. Biochemical basis and metabolic interplay of redox regulation. Redox Biol. 2019;26:101284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Anderson EJ, Kypson AP, Rodriguez E, Anderson CA, Lehr EJ, Neufer PD. Substrate‐specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J Am Coll Cardiol. 2009;54(20):1891‐1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Trumpower BL. The protonmotive Q cycle. Energy transduction by coupling of proton translocation to electron transfer by the cytochrome bc1 complex. J Biol Chem. 1990;265(20):11409‐11412. [PubMed] [Google Scholar]
- 114. Babior BM. NADPH oxidase: an update. Blood. 1999;93(5):1464‐1476. [PubMed] [Google Scholar]
- 115. Zhang M, Kho AL, Anilkumar N, et al. Glycated proteins stimulate reactive oxygen species production in cardiac myocytes: involvement of Nox2 (gp91phox)‐containing NADPH oxidase. Circulation. 2006;113(9):1235‐1243. [DOI] [PubMed] [Google Scholar]
- 116. Maalouf RM, Eid AA, Gorin YC, et al. Nox4‐derived reactive oxygen species mediate cardiomyocyte injury in early type 1 diabetes. Am J Physiol Cell Physiol. 2012;302(3):C597‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Nishio S, Teshima Y, Takahashi N, et al. Activation of CaMKII as a key regulator of reactive oxygen species production in diabetic rat heart. J Mol Cell Cardiol. 2012;52(5):1103‐1111. [DOI] [PubMed] [Google Scholar]
- 118. Bey EA, Xu B, Bhattacharjee A, et al. Protein kinase C delta is required for p47phox phosphorylation and translocation in activated human monocytes. J Immunol 2004;173(9):5730‐5738. [DOI] [PubMed] [Google Scholar]
- 119. Fontayne A, Dang PM, Gougerot‐Pocidalo MA, El‐Benna J. Phosphorylation of p47phox sites by PKC alpha, beta II, delta, and zeta: effect on binding to p22phox and on NADPH oxidase activation. Biochemistry. 2002;41(24):7743‐7750. [DOI] [PubMed] [Google Scholar]
- 120. Shen GX. Oxidative stress and diabetic cardiovascular disorders: roles of mitochondria and NADPH oxidase. Can J Physiol Pharmacol. 2010;88(3):241‐248. [DOI] [PubMed] [Google Scholar]
- 121. Berka V, Liu W, Wu G, Tsai AL. Comparison of oxygen‐induced radical intermediates in iNOS oxygenase domain with those from nNOS and eNOS. J Inorg Biochem. 2014;139:93‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Qi J, Wu Q, Cheng Q, Chen X, Zhu M, Miao C. High glucose induces endothelial COX2 and iNOS expression via inhibition of monomethyltransferase SETD8 expression. J Diabetes Res. 2020;2020:2308520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Ferreira L, Teixeira‐de‐Lemos E, Pinto F, et al. Effects of sitagliptin treatment on dysmetabolism, inflammation, and oxidative stress in an animal model of type 2 diabetes (ZDF rat). Mediators Inflamm. 2010;2010:592760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kayama Y, Raaz U, Jagger A, et al. Diabetic cardiovascular disease induced by oxidative stress. Int J Mol Sci. 2015;16(10):25234‐25263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Carrillo‐Vico A, Guerrero JM, Lardone PJ, Reiter RJ. A review of the multiple actions of melatonin on the immune system. Endocrine. 2005;27(2):189‐200. [DOI] [PubMed] [Google Scholar]
- 126. Radogna F, Diederich M, Ghibelli L. Melatonin: a pleiotropic molecule regulating inflammation. Biochem Pharmacol. 2010;80(12):1844‐1852. [DOI] [PubMed] [Google Scholar]
- 127. Klepac N, Rudes Z, Klepac R. Effects of melatonin on plasma oxidative stress in rats with streptozotocin induced diabetes. Biomed Pharmacother. 2006;60(1):32‐35. [DOI] [PubMed] [Google Scholar]
- 128. Montilla PL, Vargas JF, Túnez IF, et al. Oxidative stress in diabetic rats induced by streptozotocin: protective effects of melatonin. J Pineal Res. 1998;25(2):94‐100. [DOI] [PubMed] [Google Scholar]
- 129. Agil A, Reiter RJ, Jiménez‐Aranda A, et al. Melatonin ameliorates low‐grade inflammation and oxidative stress in young Zucker diabetic fatty rats. J Pineal Res. 2013;54(4):381‐388. [DOI] [PubMed] [Google Scholar]
- 130. Cruz A, Tasset I, Ramírez LM, et al. Effect of melatonin on myocardial oxidative stress induced by experimental obstructive jaundice. Rev Esp Enferm Dig. 2009;101(7):460‐463. [DOI] [PubMed] [Google Scholar]
- 131. Winiarska K, Fraczyk T, Malinska D, Drozak J, Bryla J. Melatonin attenuates diabetes‐induced oxidative stress in rabbits. J Pineal Res. 2006;40(2):168‐176. [DOI] [PubMed] [Google Scholar]
- 132. Raygan F, Ostadmohammadi V, Bahmani F, Reiter RJ, Asemi Z. Melatonin administration lowers biomarkers of oxidative stress and cardio‐metabolic risk in type 2 diabetic patients with coronary heart disease: a randomized, double‐blind, placebo‐controlled trial. Clin Nutr. 2019;38(1):191‐196. [DOI] [PubMed] [Google Scholar]
- 133. Serpillon S, Floyd BC, Gupte RS, et al. Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose‐6‐phosphate dehydrogenase‐derived NADPH. m J Physiol Heart Circ Physiol. 2009;297(1):H153‐H162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Wang S, Wang Y, Zhang Z, Liu Q, Gu J. Cardioprotective effects of fibroblast growth factor 21 against doxorubicin‐induced toxicity via the SIRT1/LKB1/AMPK pathway. Cell Death Dis. 2017;8(8):e3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Yuan H, Zhang Q, Guo J, et al. A PGC‐1α‐mediated transcriptional network maintains mitochondrial redox and bioenergetic homeostasis against doxorubicin‐induced toxicity in human cardiomyocytes: implementation of TT21C. Toxicol Sci. 2016;150(2):400‐417. [DOI] [PubMed] [Google Scholar]
- 136. Villena JA. New insights into PGC‐1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015;282(4):647‐672. [DOI] [PubMed] [Google Scholar]
- 137. Liu D, Ma Z, Di S, et al. AMPK/PGC1α activation by melatonin attenuates acute doxorubicin cardiotoxicity via alleviating mitochondrial oxidative damage and apoptosis. Free Radic Biol Med. 2018;129:59‐72. [DOI] [PubMed] [Google Scholar]
- 138. You J, Yue Z, Chen S, et al. Receptor‐interacting Protein 140 represses Sirtuin 3 to facilitate hypertrophy, mitochondrial dysfunction and energy metabolic dysfunction in cardiomyocytes. Acta Physiol. 2017;220(1):58‐71. [DOI] [PubMed] [Google Scholar]
- 139. Matsushima S, Sadoshima J. The role of sirtuins in cardiac disease. Am J Physiol Heart Circ Physiol. 2015;309(9):H1375‐H1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Yu L, Gong B, Duan W, et al. Melatonin ameliorates myocardial ischemia/reperfusion injury in type 1 diabetic rats by preserving mitochondrial function: role of AMPK‐PGC‐1α‐SIRT3 signaling. Sci Rep. 2017;7:41337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Zhi W, Li K, Wang H, Lei M, Guo Y. Melatonin elicits protective effects on OGD/R‐insulted H9c2 cells by activating PGC‐1α/Nrf2 signaling. Int J Mol Med. 2020;45(5):1294‐1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Chen RC, Sun GB, Wang J, Zhang HJ, Sun XB. Naringin protects against anoxia/reoxygenation‐induced apoptosis in H9c2 cells via the Nrf2 signaling pathway. Food Funct. 2015;6(4):1331‐1344. [DOI] [PubMed] [Google Scholar]
- 143. Shi S, Lei S, Tang C, Wang K, Xia Z. Melatonin attenuates acute kidney ischemia/reperfusion injury in diabetic rats by activation of the SIRT1/Nrf2/HO‐1 signaling pathway. Biosci Rep. 2019;39(1). doi: 10.1042/BSR20181614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Dludla P, Muller C, Joubert E, et al. Aspalathin protects the heart against hyperglycemia‐induced oxidative damage by up‐regulating Nrf2 expression. Molecules. 2017;22(1):129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Zhang Y, Qiao B, Gao F, Wang H, Miao S, Zhao H. Melatonin protects H9c2 cells against ischemia/reperfusion‐induced apoptosis and oxidative stress via activation of the Nrf2 signaling pathway. Mol Med Rep. 2018;18(3):3497‐3505. [DOI] [PubMed] [Google Scholar]
- 146. Sutradhar S, Deb A, Singh SS. Melatonin attenuates diabetes‐induced oxidative stress in spleen and suppression of splenocyte proliferation in laboratory mice. Arch Physiol Biochem. 2020;1‐12. [DOI] [PubMed] [Google Scholar]
- 147. Parada E, Buendia I, León R, et al. Neuroprotective effect of melatonin against ischemia is partially mediated by alpha‐7 nicotinic receptor modulation and HO‐1 overexpression. J Pineal Res. 2014;56(2):204‐212. [DOI] [PubMed] [Google Scholar]
- 148. Fernández‐Ortiz M, Sayed RKA, Fernández‐Martínez J, et al. Melatonin/Nrf2/NLRP3 Connection in Mouse Heart Mitochondria during Aging. Antioxidants. 2020;9(12):1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Durante W. Targeting heme oxygenase‐1 in the arterial response to injury and disease. Antioxidants. 2020;9(9):829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. D'Arcy MS. Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell Biol Int. 2019;43(6):582‐592. [DOI] [PubMed] [Google Scholar]
- 152. Frustaci A, Kajstura J, Chimenti C, et al. Myocardial cell death in human diabetes. Circ Res. 2000;87(12):1123‐1132. [DOI] [PubMed] [Google Scholar]
- 153. Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ. Hyperglycemia‐induced apoptosis in mouse myocardium: mitochondrial cytochrome C‐mediated caspase‐3 activation pathway. Diabetes. 2002;51(6):1938‐1948. [DOI] [PubMed] [Google Scholar]
- 154. Huynh K, Kiriazis H, Du X‐J, et al. Targeting the upregulation of reactive oxygen species subsequent to hyperglycemia prevents type 1 diabetic cardiomyopathy in mice. Free Radic Biol Med. 2013;60:307‐317. [DOI] [PubMed] [Google Scholar]
- 155. Chowdhry MF, Vohra HA, Galiñanes M. Diabetes increases apoptosis and necrosis in both ischemic and nonischemic human myocardium: role of caspases and poly‐adenosine diphosphate‐ribose polymerase. J Thorac Cardiovasc Surg. 2007;134(1):124‐131. [DOI] [PubMed] [Google Scholar]
- 156. Amin AH, El‐Missiry MA, Othman AI. Melatonin ameliorates metabolic risk factors, modulates apoptotic proteins, and protects the rat heart against diabetes‐induced apoptosis. Eur J Pharmacol. 2015;747:166‐173. [DOI] [PubMed] [Google Scholar]
- 157. Lu L, Ma J, Sun M, et al. Melatonin ameliorates MI‐induced cardiac remodeling and apoptosis through a JNK/p53‐dependent mechanism in diabetes mellitus. Oxid Med Cell Longev. 2020;2020:1535201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Prins D, Michalak M. Endoplasmic reticulum proteins in cardiac development and dysfunction. Can J Physiol Pharmacol. 2009;87(6):419‐425. [DOI] [PubMed] [Google Scholar]
- 159. Zhou H, Liu R. ER stress and hepatic lipid metabolism. Front Genet. 2014;5:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Sha H, He Y, Yang L, Qi L. Stressed out about obesity: IRE1α‐XBP1 in metabolic disorders. Trends Endocrinol Metab. 2011;22(9):374‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8(7):519‐529. doi: 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- 162. Ibrahim IM, Abdelmalek DH, Elfiky AA. GRP78: a cell's response to stress. Life Sci. 2019;226:156‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem. 2012;81:767‐793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Yoshida H. ER stress and diseases. FEBS J. 2007;274(3):630‐658. [DOI] [PubMed] [Google Scholar]
- 165. Li Z, Zhang T, Dai H, et al. Involvement of endoplasmic reticulum stress in myocardial apoptosis of streptozocin‐induced diabetic rats. J Clin Biochem Nutr. 2007;41(1):58‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Groenendyk J, Agellon LB, Michalak M. Coping with endoplasmic reticulum stress in the cardiovascular system. Annu Rev Physiol. 2013;75:49‐67. [DOI] [PubMed] [Google Scholar]
- 167. Mooradian AD, Haas MJ. Glucose‐induced endoplasmic reticulum stress is independent of oxidative stress: a mechanistic explanation for the failure of antioxidant therapy in diabetes. Free Radic Biol Med. 2011;50(9):1140‐1143. [DOI] [PubMed] [Google Scholar]
- 168. Iurlaro R, Muñoz‐Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016;283(14):2640‐2652. [DOI] [PubMed] [Google Scholar]
- 169. Tsukano H, Gotoh T, Endo M, et al. The endoplasmic reticulum stress‐C/EBP homologous protein pathway‐mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2010;30(10):1925‐1932. [DOI] [PubMed] [Google Scholar]
- 170. Hu H, Tian M, Ding C, Yu S. The C/EBP Homologous Protein (CHOP) transcription factor functions in endoplasmic reticulum stress‐induced apoptosis and microbial infection. Front Immunol. 2018;9:3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Kepp O, Semeraro M, Bravo‐San Pedro JM, et al. eIF2α phosphorylation as a biomarker of immunogenic cell death. Semin Cancer Biol. 2015;33:86‐92. [DOI] [PubMed] [Google Scholar]
- 172. Annis MG, Yethon JA, Leber B, Andrews DW. There is more to life and death than mitochondria: Bcl‐2 proteins at the endoplasmic reticulum. Biochem Biophys Acta. 2004;1644(2–3):115‐123. [DOI] [PubMed] [Google Scholar]
- 173. Nakagawa T, Zhu H, Morishima N, et al. Caspase‐12 mediates endoplasmic‐reticulum‐specific apoptosis and cytotoxicity by amyloid‐beta. Nature. 2000;403(6765):98‐103. [DOI] [PubMed] [Google Scholar]
- 174. Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress‐specific caspase cascade in apoptosis. Cytochrome c‐independent activation of caspase‐9 by caspase‐12. J Biol Chem. 2002;277(37):34287‐34294. [DOI] [PubMed] [Google Scholar]
- 175. Song L, De Sarno P, Jope RS. Central role of glycogen synthase kinase‐3beta in endoplasmic reticulum stress‐induced caspase‐3 activation. J Biol Chem. 2002;277(47):44701‐44708. [DOI] [PubMed] [Google Scholar]
- 176. Xiong FY, Tang ST, Su H, et al. Melatonin ameliorates myocardial apoptosis by suppressing endoplasmic reticulum stress in rats with long‐term diabetic cardiomyopathy. Mol Med Rep. 2018;17(1):374‐381. [DOI] [PubMed] [Google Scholar]
- 177. Kitada M, Koya D. SIRT1 in type 2 diabetes: mechanisms and therapeutic potential. Diabetes Metab J. 2013;37(5):315‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Guo R, Liu W, Liu B, Zhang B, Li W, Xu Y. SIRT1 suppresses cardiomyocyte apoptosis in diabetic cardiomyopathy: an insight into endoplasmic reticulum stress response mechanism. Int J Cardiol. 2015;191:36‐45. [DOI] [PubMed] [Google Scholar]
- 179. Yu L, Liang H, Dong X, et al. Reduced silent information regulator 1 signaling exacerbates myocardial ischemia‐reperfusion injury in type 2 diabetic rats and the protective effect of melatonin. J Pineal Res. 2015;59(3):376‐390. [DOI] [PubMed] [Google Scholar]
- 180. Zhou R, Ma Y, Tao Z, et al. Melatonin Inhibits glucose‐induced apoptosis in osteoblastic cell line through PERK‐eIF2α‐ATF4 pathway. Front Pharmacol. 2020;11:602307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Aouichat S, Navarro‐Alarcon M, Alarcón‐Guijo P, et al. Melatonin improves endoplasmic reticulum stress‐mediated IRE1α pathway in zücker diabetic fatty rat. Pharmaceuticals. 2021;14(3):232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Ouwens DM, Diamant M. Myocardial insulin action and the contribution of insulin resistance to the pathogenesis of diabetic cardiomyopathy. Arch Physiol Biochem. 2007;113(2):76‐86. [DOI] [PubMed] [Google Scholar]
- 183. Dong H, Huang HU, Yun X, et al. Bilirubin increases insulin sensitivity in leptin‐receptor deficient and diet‐induced obese mice through suppression of ER stress and chronic inflammation. Endocrinology. 2014;155(3):818‐828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Salvadó L, Palomer X, Barroso E, Vázquez‐Carrera M. Targeting endoplasmic reticulum stress in insulin resistance. Trends Endocrinol Metab. 2015;26(8):438‐448. [DOI] [PubMed] [Google Scholar]
- 185. Yung HW, Charnock‐Jones DS, Burton GJ. Regulation of AKT phosphorylation at Ser473 and Thr308 by endoplasmic reticulum stress modulates substrate specificity in a severity dependent manner. PLoS One. 2011;6(3):e17894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Quan X, Wang J, Liang C, Zheng H, Zhang L. Melatonin inhibits tunicamycin‐induced endoplasmic reticulum stress and insulin resistance in skeletal muscle cells. Biochem Biophys Res Comm. 2015;463(4):1102‐1107. [DOI] [PubMed] [Google Scholar]
- 187. Kim S‐J, Kang HS, Lee J‐H, et al. Melatonin ameliorates ER stress‐mediated hepatic steatosis through miR‐23a in the liver. Biochem Biophys Res Comm. 2015;458(3):462‐469. [DOI] [PubMed] [Google Scholar]
- 188. Jheng H‐F, Tsai P‐J, Guo S‐M, et al. Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol. 2012;32(2):309‐319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Wang Q, Zhang M, Torres G, et al. Metformin suppresses diabetes‐accelerated atherosclerosis via the inhibition of Drp1‐mediated mitochondrial fission. Diabetes. 2017;66(1):193‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Ding M, Feng NA, Tang D, et al. Melatonin prevents Drp1‐mediated mitochondrial fission in diabetic hearts through SIRT1‐PGC1α pathway. J Pineal Res. 2018;65(2):e12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Zhou H, Wang J, Zhu P, et al. NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1‐mediated mitophagy and promoting Mff‐required mitochondrial fission by CK2α. Basic Res Cardiol. 2018;113(4):23. [DOI] [PubMed] [Google Scholar]
- 192. Zhu Y, Han XQ, Sun XJ, Yang R, Ma WQ, Liu NF. Lactate accelerates vascular calcification through NR4A1‐regulated mitochondrial fission and BNIP3‐related mitophagy. Apoptosis. 2020;25(5–6):321‐340. [DOI] [PubMed] [Google Scholar]
- 193. Sheng J, Li H, Dai Q, et al. NR4A1 promotes diabetic nephropathy by activating mff‐mediated mitochondrial fission and suppressing parkin‐mediated mitophagy. Cell Physiol Biochem. 2018;48(4):1675‐1693. [DOI] [PubMed] [Google Scholar]
- 194. Zhou H, Du W, Li YE, et al. Effects of melatonin on fatty liver disease: the role of NR4A1/DNA‐PKcs/p53 pathway, mitochondrial fission, and mitophagy. J Pineal Res. 2018;64(1):e12450. [DOI] [PubMed] [Google Scholar]
- 195. Agil A, El‐Hammadi M, Jiménez‐Aranda A, et al. Melatonin reduces hepatic mitochondrial dysfunction in diabetic obese rats. J Pineal Res. 2015;59(1):70‐79. [DOI] [PubMed] [Google Scholar]
- 196. Teodoro BG, Baraldi FG, Sampaio IH, et al. Melatonin prevents mitochondrial dysfunction and insulin resistance in rat skeletal muscle. J Pineal Res. 2014;57(2):155‐167. [DOI] [PubMed] [Google Scholar]
- 197. Liu K, Yu W, Wei W, et al. Melatonin reduces intramuscular fat deposition by promoting lipolysis and increasing mitochondrial function. J Lipid Res. 2019;60(4):767‐782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Yang Y, Duan W, Jin Z, et al. JAK2/STAT3 activation by melatonin attenuates the mitochondrial oxidative damage induced by myocardial ischemia/reperfusion injury. J Pineal Res. 2013;55(3):275‐286. [DOI] [PubMed] [Google Scholar]
- 199. Zhou H, Wang J, Zhu P, Hu S, Ren J. Ripk3 regulates cardiac microvascular reperfusion injury: the role of IP3R‐dependent calcium overload, XO‐mediated oxidative stress and F‐action/filopodia‐based cellular migration. Cell Signal. 2018;45:12‐22. [DOI] [PubMed] [Google Scholar]
- 200. Zhou H, Yue Y, Wang J, Ma Q, Chen Y. Melatonin therapy for diabetic cardiomyopathy: a mechanism involving Syk‐mitochondrial complex I‐SERCA pathway. Cell Signal. 2018;47:88‐100. [DOI] [PubMed] [Google Scholar]
- 201. Alegria JR, Miller TD, Gibbons RJ, Yi QL, Yusuf S. Infarct size, ejection fraction, and mortality in diabetic patients with acute myocardial infarction treated with thrombolytic therapy. Am Heart J. 2007;154(4):743‐750. [DOI] [PubMed] [Google Scholar]
- 202. Marso SP, Miller T, Rutherford BD, et al. Comparison of myocardial reperfusion in patients undergoing percutaneous coronary intervention in ST‐segment elevation acute myocardial infarction with versus without diabetes mellitus (from the EMERALD trial). Am J Cardiol. 2007;100(2):206‐210. [DOI] [PubMed] [Google Scholar]
- 203. Ma G, Al‐Shabrawey M, Johnson JA, et al. Protection against myocardial ischemia/reperfusion injury by short‐term diabetes: enhancement of VEGF formation, capillary density, and activation of cell survival signaling. Naunyn Schmiedebergs Arch Pharmacol. 2006;373(6):415‐427. [DOI] [PubMed] [Google Scholar]
- 204. Marfella R, D'amico M, Filippo CD, et al. Myocardial infarction in diabetic rats: role of hyperglycaemia on infarct size and early expression of hypoxia‐inducible factor 1. Diabetologia. 2002;45(8):1172‐1181. [DOI] [PubMed] [Google Scholar]
- 205. Dobsak P, Siegelova J, Eicher JC, et al. Melatonin protects against ischemia‐reperfusion injury and inhibits apoptosis in isolated working rat heart. Pathophysiology. 2003;9(3):179‐187. [DOI] [PubMed] [Google Scholar]
- 206. Sahna E, Parlakpinar H, Turkoz Y, Acet A. Protective effects of melatonin on myocardial ischemia/reperfusion induced infarct size and oxidative changes. Physiol Res. 2005;54(5):491‐495. [PubMed] [Google Scholar]
- 207. Sheng M, Huang Z, Pan L, et al. SOCS2 exacerbates myocardial injury induced by ischemia/reperfusion in diabetic mice and H9c2 cells through inhibiting the JAK‐STAT‐IGF‐1 pathway. Life Sci. 2017;188:101‐109. [DOI] [PubMed] [Google Scholar]
- 208. Lan H, Su Y, Liu Y, et al. Melatonin protects circulatory death heart from ischemia/reperfusion injury via the JAK2/STAT3 signalling pathway. Life Sci. 2019;228:35‐46. [DOI] [PubMed] [Google Scholar]
- 209. Duan W, Yang Y, Yi W, et al. New role of JAK2/STAT3 signaling in endothelial cell oxidative stress injury and protective effect of melatonin. PLoS One. 2013;8(3):e57941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Zhen G, Liang W, Jia H, Zheng X. Melatonin relieves sepsis‐induced myocardial injury via regulating JAK2/STAT3 signaling pathway. Minerva Med. 2020. doi: 10.23736/S0026-4806.20.06626-4 [DOI] [PubMed] [Google Scholar]
- 211. Zhai M, Li B, Duan W, et al. Melatonin ameliorates myocardial ischemia reperfusion injury through SIRT3‐dependent regulation of oxidative stress and apoptosis. J Pineal Res. 2017;63(2):e12419. [DOI] [PubMed] [Google Scholar]
- 212. Hsu C‐P, Zhai P, Yamamoto T, et al. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation. 2010;122(21):2170‐2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Ma W‐Y, Song R‐J, Xu B‐B, et al. Melatonin promotes cardiomyocyte proliferation and heart repair in mice with myocardial infarction via miR‐143‐3p/Yap/Ctnnd1 signaling pathway. Acta Pharmacol Sin. 2021;42(6):921‐931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Ostadmohammadi V, Soleimani A, Bahmani F, et al. The effects of melatonin supplementation on parameters of mental health, glycemic control, markers of cardiometabolic risk, and oxidative stress in diabetic hemodialysis patients: a randomized, double‐blind, placebo‐controlled trial. J Ren Nutr. 2020;30(3):242‐250. [DOI] [PubMed] [Google Scholar]
- 215. Abdulwahab DA, El‐Missiry MA, Shabana S, Othman AI, Amer ME. Melatonin protects the heart and pancreas by improving glucose homeostasis, oxidative stress, inflammation and apoptosis in T2DM‐induced rats. Heliyon. 2021;7(3):e06474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Wang S, Zhai X, Li S, McCabe MF, Wang X, Rong P. Transcutaneous vagus nerve stimulation induces tidal melatonin secretion and has an antidiabetic effect in Zucker fatty rats. PLoS One. 2015;10(4):e0124195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Rahman MM, Kwon HS, Kim MJ, Go HK, Oak MH, Kim DH. Melatonin supplementation plus exercise behavior ameliorate insulin resistance, hypertension and fatigue in a rat model of type 2 diabetes mellitus. Biomed Pharmacother. 2017;92:606‐614. [DOI] [PubMed] [Google Scholar]
- 218. Luo W, Ai L, Wang BF, Zhou Y. High glucose inhibits myogenesis and induces insulin resistance by down‐regulating AKT signaling. Biomed Pharmacother. 2019;120:109498. [DOI] [PubMed] [Google Scholar]
- 219. Olson LK, Redmon JB, Towle HC, Robertson RP. Chronic exposure of HIT cells to high glucose concentrations paradoxically decreases insulin gene transcription and alters binding of insulin gene regulatory protein. J Clin Investig. 1993;92(1):514‐519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Hou ZQ, Li HL, Gao L, Pan L, Zhao JJ, Li GW. Involvement of chronic stresses in rat islet and INS‐1 cell glucotoxicity induced by intermittent high glucose. Mol Cell Endocrinol. 2008;291(1–2):71‐78. [DOI] [PubMed] [Google Scholar]
- 221. Park JH, Shim HM, Na AY, et al. Melatonin prevents pancreatic β‐cell loss due to glucotoxicity: the relationship between oxidative stress and endoplasmic reticulum stress. J Pineal Res. 2014;56(2):143‐153. [DOI] [PubMed] [Google Scholar]
- 222. Lv Q, Meng X‐F, He F‐F, et al. High serum uric acid and increased risk of type 2 diabetes: a systemic review and meta‐analysis of prospective cohort studies. PLoS One. 2013;8(2):e56864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Katsiki N, Papanas N, Fonseca VA, Maltezos E, Mikhailidis DP. Uric acid and diabetes: is there a link? Curr Pharm Des. 2013;19(27):4930‐4937. [DOI] [PubMed] [Google Scholar]
- 224. Mantovani A, Rigolon R, Pichiri I, et al. Relation of elevated serum uric acid levels to first‐degree heart block and other cardiac conduction defects in hospitalized patients with type 2 diabetes. J Diabetes Complications. 2017;31(12):1691‐1697. [DOI] [PubMed] [Google Scholar]
- 225. Mantovani A, Rigolon R, Pichiri I, et al. Hyperuricemia is associated with an increased prevalence of atrial fibrillation in hospitalized patients with type 2 diabetes. J Endocrinol Invest. 2016;39(2):159‐167. [DOI] [PubMed] [Google Scholar]
- 226. Fabbrini E, Serafini M, Colic Baric I, Hazen SL, Klein S. Effect of plasma uric acid on antioxidant capacity, oxidative stress, and insulin sensitivity in obese subjects. Diabetes. 2014;63(3):976‐981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Chen C, Lü JM, Yao Q. Hyperuricemia‐related diseases and xanthine oxidoreductase (XOR) inhibitors: an overview. Med Sci Monit. 2016;22:2501‐2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Sautin YY, Johnson RJ. Uric acid: the oxidant‐antioxidant paradox. Nucleosides, Nucleotides Nucleic Acids. 2008;27(6):608‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Khosla UM, Zharikov S, Finch JL, et al. Hyperuricemia induces endothelial dysfunction. Kidney Int. 2005;67(5):1739‐1742. [DOI] [PubMed] [Google Scholar]
- 230. Luo Q, Xia X, Li B, Lin Z, Yu X, Huang F. Serum uric acid and cardiovascular mortality in chronic kidney disease: a meta‐analysis. BMC Nephrol. 2019;20(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Yildiz BS, Sahin A, Aladag NB, Yildiz M. Association of endogenous melatonin with uric acid and traditional cardiovascular risk factors in healthy young male. Adv Clin Exp Med. 2015;24(2):233‐237. [DOI] [PubMed] [Google Scholar]
- 232. Masue T, Wada K, Hayashi M, et al. Associations of urinary 6‐sulfatoxymelatonin with biomarkers related to cardiovascular disease in Japanese women. Metab, Clin Exp. 2012;61(1):70‐75. [DOI] [PubMed] [Google Scholar]
- 233. Cardinali DP, Bernasconi PA, Reynoso R, Toso CF, Scacchi P. Melatonin may curtail the metabolic syndrome: studies on initial and fully established fructose‐induced metabolic syndrome in rats. Int J Mol Sci. 2013;14(2):2502‐2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Daroux M, Prévost G, Maillard‐Lefebvre H, et al. Advanced glycation end‐products: implications for diabetic and non‐diabetic nephropathies. Diabetes Metab. 2010;36(1):1‐10. [DOI] [PubMed] [Google Scholar]
- 235. Gugliucci A, Bendayan M. Histones from diabetic rats contain increased levels of advanced glycation end products. Biochem Biophys Res Comm. 1995;212(1):56‐62. [DOI] [PubMed] [Google Scholar]
- 236. Norton GR, Candy G, Woodiwiss AJ. Aminoguanidine prevents the decreased myocardial compliance produced by streptozotocin‐induced diabetes mellitus in rats. Circulation. 1996;93(10):1905‐1912. [DOI] [PubMed] [Google Scholar]
- 237. Verzijl N, DeGroot J, Thorpe SR, et al. Effect of collagen turnover on the accumulation of advanced glycation end products. J Biol Chem. 2000;275(50):39027‐39031. [DOI] [PubMed] [Google Scholar]
- 238. Chen J, Jing J, Yu S, et al. Advanced glycation endproducts induce apoptosis of endothelial progenitor cells by activating receptor RAGE and NADPH oxidase/JNK signaling axis. Am J Transl Res. 2016;8(5):2169‐2178. [PMC free article] [PubMed] [Google Scholar]
- 239. Wang YI, Luo WU, Han J, et al. MD2 activation by direct AGE interaction drives inflammatory diabetic cardiomyopathy. Nat Commun. 2020;11(1):2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Nielsen TB, Pantapalangkoor P, Yan J, et al. Diabetes exacerbates infection via hyperinflammation by signaling through TLR4 and RAGE. MBio. 2017;8(4). doi: 10.1128/mBio.00818-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Geng H, Wang A, Rong G, et al. The effects of ox‐LDL in human atherosclerosis may be mediated in part via the toll‐like receptor 4 pathway. Mol Cell Biochem. 2010;342(1–2):201‐206. [DOI] [PubMed] [Google Scholar]
- 242. Geng J, Liu H, Ge P, et al. PM2.5 promotes plaque vulnerability at different stages of atherosclerosis and the formation of foam cells via TLR4/MyD88/NFκB pathway. Ecotoxicol Environ Saf. 2019;176:76‐84. [DOI] [PubMed] [Google Scholar]
- 243. Shen C, Li Q, Zhang YC, et al. Advanced glycation endproducts increase EPC apoptosis and decrease nitric oxide release via MAPK pathways. Biomed Pharmacother. 2010;64(1):35‐43. [DOI] [PubMed] [Google Scholar]
- 244. Ali T, Badshah H, Kim TH, Kim MO. Melatonin attenuates D‐galactose‐induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NF‐K B/JNK signaling pathway in aging mouse model. J Pineal Res. 2015;58(1):71‐85. [DOI] [PubMed] [Google Scholar]
- 245. Choi HS, Kang JW, Lee SM. Melatonin attenuates carbon tetrachloride‐induced liver fibrosis via inhibition of necroptosis. Transl Res. 2015;166(3):292‐303. [DOI] [PubMed] [Google Scholar]
- 246. El‐Sisi AEE, Sokar SS, Shebl AM, Mohamed DZ, Abu‐Risha SE. Octreotide and melatonin alleviate inflammasome‐induced pyroptosis through inhibition of TLR4‐NF‐κB‐NLRP3 pathway in hepatic ischemia/reperfusion injury. Toxicol Appl Pharmacol. 2021;410:115340. [DOI] [PubMed] [Google Scholar]
- 247. Balakumar P, Maung UK, Jagadeesh G. Prevalence and prevention of cardiovascular disease and diabetes mellitus. Pharmacol Res. 2016;113(pt A):600‐609. [DOI] [PubMed] [Google Scholar]
- 248. Svane J, Pedersen‐Bjergaard U, Tfelt‐Hansen J. Diabetes and the risk of sudden cardiac death. Curr Cardiol Rep. 2020;22(10):112. [DOI] [PubMed] [Google Scholar]
- 249. Vasiliadis I, Kolovou G, Mavrogeni S, Nair DR, Mikhailidis DP. Sudden cardiac death and diabetes mellitus. J Diabetes Complicat. 2014;28(4):573‐579. [DOI] [PubMed] [Google Scholar]
- 250. Andersen LPH, Werner MU, Rosenkilde MM, et al. Pharmacokinetics of oral and intravenous melatonin in healthy volunteers. BMC Pharmacol Toxicol. 2016;17:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Fourtillan JB, Brisson AM, Gobin P, Ingrand I, Decourt JP, Girault J. Bioavailability of melatonin in humans after day‐time administration of D(7) melatonin. Biopharm Drug Dispos. 2000;21(1):15‐22. [DOI] [PubMed] [Google Scholar]
- 252. Di WL, Kadva A, Johnston A, Silman R. Variable bioavailability of oral melatonin. N Engl J Med. 1997;336(14):1028‐1029. [DOI] [PubMed] [Google Scholar]
- 253. Sugden D. Melatonin biosynthesis in the mammalian pineal gland. Experientia. 1989;45(10):922‐932. [DOI] [PubMed] [Google Scholar]
- 254. Baltatu O, Afeche SC, Santos SHJD, et al. Locally synthesized angiotensin modulates pineal melatonin generation. J Neurochem. 2002;80(2):328‐334. [DOI] [PubMed] [Google Scholar]
- 255. Stoschitzky K, Sakotnik A, Lercher P, et al. Influence of beta‐blockers on melatonin release. Eur J Clin Pharmacol. 1999;55(2):111‐115. [DOI] [PubMed] [Google Scholar]
- 256. Garcia RAP, Afeche SC, Scialfa JH, et al. Insulin modulates norepinephrine‐mediated melatonin synthesis in cultured rat pineal gland. Life Sci. 2008;82(1–2):108‐114. [DOI] [PubMed] [Google Scholar]
- 257. Lee BH, Hille B, Koh DS. Serotonin modulates melatonin synthesis as an autocrine neurotransmitter in the pineal gland. Proc Natl Acad Sci USA. 2021;118(43):e2113852118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Ferreira ZS, Fernandes PA, Duma D, Assreuy J, Avellar MC, Markus RP. Corticosterone modulates noradrenaline‐induced melatonin synthesis through inhibition of nuclear factor kappa B. J Pineal Res. 2005;38(3):182‐188. [DOI] [PubMed] [Google Scholar]
- 259. Catala MD, Quay WB, Timiras PS. Effects of thyroid hormone on light/dark melatonin synthesis and release by young and maturing rat pineal glands in vitro. Int J Dev Neurosci. 1988;6(3):285‐288. [DOI] [PubMed] [Google Scholar]
- 260. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 2018;46(D1):D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Alexander SPH, Cidlowski JA, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2021/22: nuclear hormone receptors. Br J Pharmacol. 2021;178(suppl 1):S246‐S263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Alexander SP, Kelly E, Mathie A, et al. The Concise Guide to PHARMACOLOGY 2021/22: introduction and other protein targets. Br J Pharmacol. 2021;178(suppl 1):S1‐S26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Alexander SP, Mathie A, Peters JA, et al. The Concise Guide to PHARMACOLOGY 2021/22: ion channels. Br J Pharmacol. 2021;178(suppl 1):S157‐S245. [DOI] [PubMed] [Google Scholar]
- 264. Alexander SP, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2021/22: catalytic receptors. Br J Pharmacol. 2021;178(suppl 1):S264‐S312. [DOI] [PubMed] [Google Scholar]
- 265. Alexander SP, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2021/22: enzymes. Br J Pharmacol. 2021;178(suppl 1):S313‐S411. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.