

Abstract
Neutral antagonists of GPCRs remain relatively rare—indeed, a large majority of GPCR antagonists are actually inverse agonists. The synthetic cannabinoid receptor agonist (SCRA) EG‐018 was recently reported as a low efficacy cannabinoid receptor agonist. Here we report a comparative characterization of EG‐018 and 13 analogues along with extant putative neutral antagonists of CB1. In HEK cells stably expressing human CB1, assays for inhibition of cAMP were performed by real‐time BRET biosensor (CAMYEL), G protein cycling was quantified by [35S]GTPγS binding, and stimulation of pERK was characterized by AlphaLISA (PerkinElmer). Signaling outcomes for the EG‐018 analogues were highly variable, ranging from moderate efficacy agonism with high potency, to marginal agonism at lower potency. As predicted by differing pathway sensitivities to differences in ligand efficacy, most EG‐018‐based compounds were completely inactive in pERK alone. The lowest efficacy analogue in cAMP assays, 157, had utility in antagonism assay paradigms. Developing neutral antagonists of the CB1 receptor has been a long‐standing research goal, and such compounds would have utility both as research tools and in therapeutics. Although these results emphasize again the importance of system factors in determining signaling outcomes, some compounds characterized in this study appear among the lowest efficacy agonists described to date and therefore suggest that development of neutral antagonists is an achievable goal for CB1.
Keywords: cannabinoids, CB1 receptor, EG‐018, signal transduction, structure‐activity relationships
Abbreviations
- BRET
bioluminescence resonance energy transfer
- CAMYEL
cAMP biosensor with YFP‐EPAC‐Rluc
- CB1
type one cannabinoid receptor
- GPCR
G‐protein‐coupled receptor
- pERK
phosphorylated extracellular signal‐regulated kinase
- SCRA
synthetic cannabinoid receptor agonist
1. INTRODUCTION
Generation of novel ligands with low efficacy has implicitly been perceived as a mere byproduct of drug discovery. However, low efficacy agonists can drive effects with greater subtlety than higher efficacy agonists: they exert less stimulus (i.e., responses may be smaller simply because they activate the receptor less strongly), they can behave as either agonists or antagonists depending on the context (hence Ariëns’ early designation of these compounds as “dualists”), 1 but also, critically, because they are less subject to receptor reserve. This facet of receptor theory refers to the fact that a drug's maximum biological effect may occur at partial receptor occupancy, and conversely, an agonist with low intrinsic efficacy (“stimulus per receptor”) 2 may be unable to saturate the activity pathway in question even at full receptor occupancy. Low agonist efficacy complements differing pathway coupling efficiencies to yield vast heterogeneity in how drugs manifest responses in different pathways.
The importance of this phenomenon manifests in surprising contexts. First, low efficacy agonists have been considered promising therapeutics due to their theoretically improved therapeutic window and ceiling effect. An example of this is nicotinic acetylcholine receptor drugs (nAChRs). Low stimulation of nAChRs by low efficacy agonists such as varenicline 3 and dianicline 4 is thought to temper nicotine‐mediated activation of pathways that cause addiction (mesolimbic‐dopaminergic system), without the severe withdrawal associated with smoking cessation (reviewed in Ref. [5]).
Second, low efficacy agonism overlaps with biased agonism (functional selectivity)—the concept that some drugs can preferentially activate certain activity pathways to a greater extent than would be predicted by their efficacy in another. 2 , 6 The prototypical example of biased agonism is the mu‐opioid receptor (MOR) system, and the hypothesis that the analgesic effects of MOR‐targeting opioids may be mediated by G protein pathways, while the adverse effects are β‐arrestin‐mediated. 7 , 8 More recently, however, it has been clarified that the prospective therapeutic advantages of “G protein‐biased” MOR agonists can be adequately explained by reduced efficacy. 9 Reduced β‐arrestin signaling with such compounds is likely merely a consequence of the absence of receptor reserve in this pathway, 10 consistent with coupling preferences for other GPCRs, including the CB1 cannabinoid receptor. 11 , 12
Finally, minimization of agonist efficacy abuts the concept of neutral antagonism; an assumption in some methodologies—including Schild analysis, which assumes that antagonist reactions do not impact basal activity. 13 Neutral antagonists have also been sought for potential utility in the clinic. For example the CB1 inverse agonist activity of rimonabant (which was developed as an appetite suppresant) may help explain its adverse psychological side effects, and underpins the development of CB1‐targeting neutral antagonists. 14 , 15
Relatively few low efficacy cannabinoid agonists have been reported. Undoubtedly, the best known and best characterized example is (‐)‐Δ 9 ‐tetrahydrocannabinol (THC) (reviewed in Ref. [16]). A less well known example of a low efficacy cannabinoid agonist is BAY59‐3074; a synthetic compound with apparently similar in vitro efficacy to THC. 11 We recently characterized the synthetic cannabinoid receptor agonist (SCRA) EG‐018. 17 This compound is an analogue of the better‐known earlier‐generation SCRA JWH‐018, and appears to have originated in the illicit drug marketplace. Notably, we showed that EG‐018 is a low efficacy agonist (in contrast to the high efficacy at both CB1 and CB2 associated with its “parent,” JWH‐018). 18 , 19
The structural similarity of EG‐018 to JWH‐018, together with its contrasting and unusual pharmacology, prompted the current study. We set out to synthesize and perform a systematic in vitro pharmacological characterization of a family of novel analogues of EG‐018 at CB1, on the basis that structural features of this compound appear to be critical determinants of high and low efficacy. Although further work at CB2 is undoubtedly merited, characterization of compound activities at CB1 has been of initial interest because our previous study 17 indicated that the efficacy of EG‐018 at CB1 is lower than that of THC.
2. MATERIALS AND METHODS
Details of the chemical synthesis for all novel compounds reported in this manuscript are provided in the Supplementary Material.
2.1. Drugs and drug preparation
Forskolin (FSK, F), CP55940, 2‐arachidonoyl glycerol (2‐AG), NESS‐0327, and SR141716A were purchased from Cayman Chemical Company; AM4113 was purchased both from Cayman Chemical Company and Sigma Aldrich.
FSK, and EG‐018 and all analogues were constituted at 31.6 mM in dimethyl sulfoxide (DMSO, Sigma Aldrich); NESS‐0327, AM4113 and SR141716A were constituted at 10 mM in DMSO. FSK was stored in large aliquots at room temperature and reused for multiple experiments, while all other drugs were aliquoted for single use in 0.2 ml tubes and stored at −80°C until use. Vehicle (DMSO and ethanol) content was controlled within each experiment, at 0.1% for each agonist in either DMSO or ethanol, with an additional 0.015% DMSO (for FSK) and an additional 0.1% ethanol for coelenterazine H in cAMP assays (see below).
2.2. Competition binding assays
HEK293 cell membrane preparations expressing the human CB1 (hCB1) receptor (PerkinElmer) were incubated at 30°C for 1 h in binding buffer (50 mM Tris‐HCl pH 7.4, 1 mM EDTA, 3 mM MgCl2, 5 mg/ml bovine serum albumin, BSA) with 1 nM [3H]CP55940 (Perkin Elmer) in a total volume of 0.5 ml. Nonspecific binding was determined by the inclusion of 10 μM unlabeled CP55940. Binding was terminated by vacuum filtration through GF/C glass fiber filter plates (PerkinElmer) pretreated in 0.1% (w/v) polyethyleneimine for at least 1 h. Reaction vessels were washed three times with ~2 ml ice‐cold rinse buffer (50 mM Tris‐HCl, 1 mg/ml BSA). The filter plates were air‐dried and sealed on the bottom. Liquid scintillate was added to the wells, and the top was sealed. Liquid scintillation spectrometry was used to measure radioactivity after incubating the plates in cocktail for at least 30 min. Assays were done in duplicate, and results represent combined data from three independent displacement curves.
Heterologous competition binding assays for AM4113 were performed using P2 membrane preparations made from pplss‐3HA‐hCB1 HEK cells, as previously described. 11
2.3. Cyclic AMP assays
Assays for cyclic AMP (cAMP) signaling were performed using the CAMYEL biosensor 20 in HEK293 cells stably expressing human CB1 with three N‐terminal hemagglutinin tags (3HA‐hCB1 HEK, first reported in Ref. [21]). All culturewares were purchased from Corning unless otherwise specified,media were purchased from Hyclone Laboratories (Cyvita) and all other culture reagents were purchased from Thermo Fisher Scientific.
Cells were lifted from a semi‐confluent cell culture flask with 0.05% trypsin/EDTA, then plated into 100‐mm cell culture dishes in normal growth medium (high glucose DMEM supplemented with 10% New Zealand‐origin fetal bovine serum, Moregate Biotech) and cultured overnight in a humidified cell culture incubator (37°C, 5% CO2). After 24 h, growth medium was replaced; cells were well‐adhered and approximately 50% confluent. A transfection mixture was prepared in 500 µl sterile isotonic saline, comprised of 5 µg of His‐CAMYEL plasmid and 30 µg of linear polyethyleneimine (PEI). Components were combined, mixed thoroughly, and incubated at room temperature for 10 min. The entire transfection mix was then gently dispensed to the dish, mixed, and the dish was returned to the incubator. The next day, cells were lifted with 0.05% trypsin/EDTA, and plated at high density (5e4 cells per well) in 96‐well, white CulturPlates (PerkinElmer) which had been pretreated with 0.05 mg/ml poly‐D‐lysine and then washed with sterile PBS. Plated cells were then returned to the incubator overnight.
On assay day, well contents were aspirated with a strip vacuum, wells were washed once with warm PBS, then warm assay medium was dispensed (phenol‐free high glucose DMEM, supplemented with 10 mM HEPES pH 7.4 and 1 mg/ml BSA, MP Biomedicals). Plates were returned to the incubator for 30 min equilibration prior to stimulation. Drugs were prepared at 10x concentration in assay medium, and were combined in equal quantities in a polypropylene, low‐binding V‐well dispensing plate (Hangzhou Gene Era Biotech Co Ltd). Coelenterazine H (Nanolight Technologies, prepared to 5 mM in absolute ethanol) was prepared at 10× concentration in assay medium immediately prior to dispensing (for a final in‐well concentration of 5 µM). Coelenterazine H was dispensed and the plate was incubated inside the 37°C LUMIstar Omega plate reader (BMG Labtech) in the dark for 5 min. Drugs were then dispensed into the assay plate (final stimulation volume of 100 µl), and the plate reader was immediately started. Reads used simultaneous BRET1 filters (475‐30 and 535‐30 nm), and cycles were set to run for approximately 20 min with a read interval time of 0.5 s per well.
Inverse BRET ratios (460/535) were plotted in GraphPad Prism v8 or v9 (GraphPad Software). Area‐under‐the‐curve analysis and subsequent normalizations were performed in GraphPad Prism, to create concentration–response plots.
2.4. Phospho‐ERK assays
3HA‐hCB1 HEK cells were lifted with 0.05% trypsin/EDTA and plated at relatively low density (2.5e4 cells per well) in normal growth medium in clear 96‐well assay plates, which had been pretreated with poly‐D‐lysine. Approximately 24 h after plating, medium was removed with a strip vacuum and replaced with 50 µl/well serum‐free high glucose DMEM supplemented with 1 mg/ml BSA. Cells were returned to the incubator to serum starve overnight. On assay day, cells were ~60%–80% confluent. Drugs were prepared at 2x concentration in serum‐free high glucose DMEM supplemented with 1 mg/ml BSA, dispensed into strip tubes (Axygen, Corning) and pre‐warmed in a 37°C waterbath. The assay plate was then moved into the waterbath, onto a barely submerged stage. Drugs were then dispensed into wells, beginning with the longest stimulation time point, so that the stimulation for the whole plate would end at the same time. The plate was then moved to a bed of ice, and well contents were quickly removed with a strip vacuum. Cells were immediately lysed with kit lysis buffer, and pERK was detected by AlphaLISA SureFire Ultra kit in a half‐area plate (PerkinElmer), and detected with a CLARIOstar Plus plate reader (BMG Labtech). Data were graphed in GraphPad Prism.
2.5. GTPγS assays
GTPγS assays were performed as previously described. 17 P2 membrane fractions (10 µg total protein) of HEK293 cells stably expressing the hCB1 receptor were pre‐equilibrated for 30 min at 30°C with 30 μM GDP and test compounds in a volume of 490 µL. After pre‐equilibration, 0.1 nM [35S]GTPγS was added to the reaction mix in a volume of 10 µL (2% final volume) in assay buffer (50 mM Tris Base, 100 mM NaCl, 3 mM MgCl2, 0.2 mM EGTA and 5 mg/ml BSA) and the reaction was incubated for 1 h. Non‐specific binding was determined by inclusion of 30 μM unlabeled GTPγS. Subsequent steps were the same as described for competition binding assays.
2.6. Data analysis
Figures presented in this manuscript are representative, in line with statistical recommendations, in order to avoid misestimation of response parameters from combination of data from independent experiments. 22 Concentration–response curves were fit in GraphPad Prism using the “log[agonist] versus response (three parameters)” equation. All curve parameters were obtained from individual fits (i.e., curves did not share constraints). Independent biological replicates were analyzed separately, and the reported mean potencies and efficacies were calculated by combining independent curve parameters (pEC50s and curve spans).
2.7. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, 23 the common portal for data from the IUPHAR/BPS Guide to Pharmacology. 24
3. RESULTS
Competition/displacement binding assays revealed that EG‐018 and the 13 analogues displace [3H]CP55940 with affinities ranging widely, from 4.79 nM (085) to 1.20 µM (142) (Table 1). Notably, all analogues except 085 showed lower affinity for hCB1 than the parent compound. Under these conditions, EG‐018 affinity was 16.6 nM. As all compound affinities were in a pharmacologically relevant range, all were taken forward for functional characterization—in this study, our primary interest was in compound efficacies which cannot be predicted from affinity data.
TABLE 1.
EG‐018 and analogue affinities at hCB1, from competition binding assays with 1 nM [3H]‐CP55940 (n = 3)

| Compound | R | Ar | pKi (±SEM), −log M | Rank order |
|---|---|---|---|---|
| JWH‐018 | CH3(CH2)3CH2 | 1‐naphthyl | 8.58 25 | n/a a |
| JWH‐018 contains the same R and Ar groups as EG‐018, but includes an indole core in place of the EG‐018 carbazole | ||||
| EG‐018 | CH3(CH2)3CH2 | 1‐naphthyl | 7.78 (±0.04) | 2 |
| 042 | CH3(CH2)2CH2 | 1‐naphthyl | 7.11 (±0.05) | 6 |
| 043 | CH3(CH2)4CH2 | 1‐naphthyl | 7.16 (±0.05) | 5 |
| 044 | CH3COO(CH2)4CH2 | 1‐naphthyl | 6.75 (±0.05) | 10 |
| 045 | HO(CH2)4CH2 | 1‐naphthyl | 6.78 (±0.05) | 9 |
| 150 | CH3(CH2)3CH2 | 4‐pyridinyl | 6.24 (±0.04) | 12 |
| 085 |

|
1‐naphthyl | 8.32 (±0.04) | 1 |
| 117 |

|
1‐naphthyl | 7.00 (±0.05) | 7 |
| 156 |

|
1‐naphthyl | 7.55 (±0.04) | 3 |
| 104 |

|
1‐naphthyl | 7.35 (±0.04) | 4 |
| 141 |

|
4‐methyl‐1‐naphthyl | 6.83 (±0.05) | 8 |
| 142 |

|
2‐indolyl | 5.92 (±0.05) | 14 |
| 146 |

|
2‐benzofuranyl | 6.53 (±0.04) | 11 |
| 157 |

|
4‐quinolinyl | 6.23 (±0.04) | 13 |
JWH‐018 data were not collected in the current study, and JWH‐018 was not included in the affinity ranking.
In HEK cells, CB1 cannabinoid receptors couple efficiently through Gαi to inhibit FSK‐stimulated cAMP with large receptor reserve, meaning that traditional low efficacy agonists such as THC and BAY59‐3074 appear virtually equi‐efficacious with compounds like CP55940. 11 Approximately half of the EG‐018 analogues assayed showed broadly similar profiles to EG‐018 in inhibiting FSK‐stimulated cAMP (Figure 1), including compounds 042, 045, 104, 117, 141, 150, and 156. Overall, however, the analogues activated this signaling pathway with exceptionally varying efficacies, ranging from 57.8% for analogue 085 (Figure 1C, Table 2), to a minimal 13.1% for analogue 157 (Figure 1D, Table 2)—a curve span of less than half that of the next lowest‐efficacy compound, 142 (Figure 1D, Table 2). Potencies were similarly diverse (Figure 1, Table 2), as predicted by affinity estimates.
FIGURE 1.
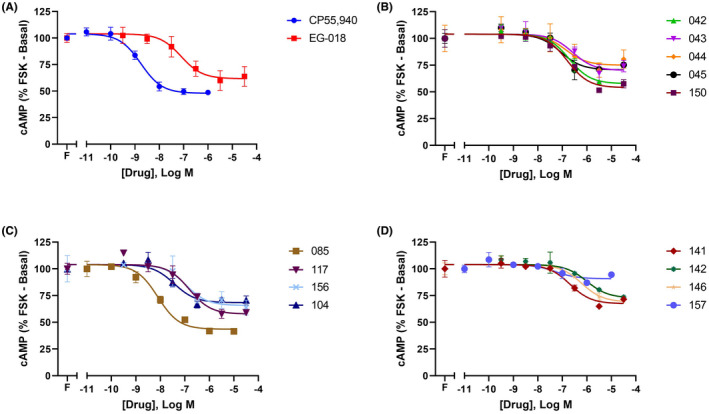
Inhibition of FSK‐stimulated cAMP signaling by CP55940 and EG‐018 (A), and thirteen EG‐018 analogues (B–D) in 3HA‐hCB1 HEK cells. Data are from a representative experiment, showing mean ± SD of conditions performed in technical duplicate. Curves are normalized to basal (vehicle only, 0%) and FSK only (100%). Combined n = 3 data are reported in Table 2
TABLE 2.
Potencies and efficacies for EG‐018 and analogues in 3HA‐hCB1 HEK cells to inhibit FSK‐stimulated cAMP (n = 3)
| Compound | pEC50 (±SEM), −log M | E MAX a (±SEM), % | Rank order efficacy |
|---|---|---|---|
| CP55940 | 8.98 (±0.16) | 58.7 (± 3.1) | n/a b |
| EG‐018 | 7.00 (±0.05) | 48.5 (±4.3) | 5 |
| 042 | 6.89 (±0.10) | 51.3 (±3.0) | 4 |
| 043 | 6.47 (±0.08) | 33.4 (±1.4) | 11 |
| 044 | 6.71 (±0.25) | 32.5 (±1.7) | 12 |
| 045 | 6.83 (±0.17) | 34.9 (±3.8) | 9 |
| 150 | 6.78 (±0.13) | 55.3 (±4.4) | 2 |
| 085 | 8.12 (±0.07) | 57.8 (±0.9) | 1 |
| 117 | 6.88 (±0.07) | 51.8 (±2.9) | 3 |
| 156 | 6.96 (±0.10) | 33.4 (±5.5) | 10 |
| 104 | 7.59 (±0.17) | 42.4 (±3.4) | 6 |
| 141 | 7.08 (±0.35) | 38.7 (±2.9) | 7 |
| 142 | 5.92 (±0.09) | 31.5 (±2.2) | 13 |
| 146 | 6.45 (±0.22) | 38.5 (±2.0) | 8 |
| 157 | 7.04 (±0.03) | 13.1 (±2.1) | 14 |
E MAX estimates were obtained from concentration–response curve spans (see Figure 2), where 100% is the difference between basal (vehicle only) and FSK‐alone. Larger values therefore represent greater efficacy.
CP55940 was not included in the cAMP inhibition efficacy ranking of the EG‐018 analogues.
The known large extent of receptor reserve associated with GPCR cAMP signaling 9 , 11 means that this pathway often is not optimal for comparing different compounds’ intrinsic efficacies. Assays for phosphorylation of ERK were an obvious follow‐on, as we have consistently observed widely ranging efficacies between compounds for this pathway, reflecting reduced receptor reserve. It was also of interest to test whether the reduced cAMP efficacy of EG‐018 and the analogues is preserved in other experimental endpoints, as this is necessary to identify potential agonist bias. Rather than undertaking concentration–response assays, time course experiments at single high concentrations of EG‐018 and analogues (31.6 µM) were performed, in comparison to several additional high efficacy reference compounds.
Figure 2 illustrates the ability of ERK assays to distinguish differing agonist efficacies. Importantly, even at a very high concentration of 31.6 µM, the parent compound EG‐018 (Figure 2A, red) shows negligible pERK activity at all timepoints—despite exhibiting only slightly reduced efficacy in the cAMP pathway compared to the common research compound CP55940 (Figure 1A). The ability of CP55940 to elicit a pERK response was also compared to 2‐AG, a major endocannabinoid often referred‐to as a “full” CB1 agonist (e.g., 11 , 26 —the clear window between the extents of pERK activation induced by saturating concentrations of these ligands underscores the sensitivity of the pERK pathway to varying ligand efficacies).
FIGURE 2.

Time course of phosphorylation of ERK in 3HA‐hCB1 HEK cells following treatment with 2‐AG (A, gray, 31.6 µM), CP55940 (B, blue, 1 µM), EG‐018 (A, red, 31.6 µM) or 13 EG‐018 analogues (B–D, all 31.6 µM). Vehicle (black) is shared between graphs. Data are from a representative experiment showing mean ± SD of conditions performed in technical duplicate or quadruplicate (vehicle and CP55940—these conditions were repeated on two plates in the assay). Curves are normalized to the peak CP55940 response at 4 min (100%)
Similar to the lack of response exhibited by EG‐018, most analogues were also unable to drive detectable pERK responses. Of the thirteen analogues, just three compounds were exceptions. Analogue 085 (the highest affinity analogue, Table 1) demonstrated very low but detectable pERK activation despite ranking as the highest efficacy analogue in cAMP inhibition (Table 2, Figure 1A,C). In the pERK activation assay, analogues 150 and 117 demonstrated somewhat greater efficacies (Figure 2B,C); the latter compound even approaching the extent of activation induced by CP55940. These two analogues also showed comparable efficacy to CP55940 in cAMP assays (Table 2), ranking 2 and 3, respectively.
Despite the ability of the pERK assays to distinguish different agonist efficacies—particularly in comparison to the higher efficacy, well‐known agonists CP55940 and 2‐AG—a different limitation is apparent in these data compared to the cAMP data: receptor‐coupling efficiency is sufficiently poor that, although there is no apparent receptor reserve, relatively few EG‐018 analogues produced responses with detectable efficacy. [35S]‐GTPγS binding assays were performed to overcome this issue. A major advantage of this assay type is that it is highly reductionist, being performed on semi‐purified cell membranes instead of live cells (reviewed in Ref. [27]). This means that assay conditions are highly adaptable, and can be optimized to maximize the signal window depending on the experimental question, by varying GDP concentration, Mg2+ and Na+ concentrations, and incubation time. [35S]‐GTPγS accumulation assays were therefore performed for EG‐018 and 12 of its analogues, under conditions that allowed maximum differentiation of efficacies.
As expected, [35S]‐GTPγS binding data revealed a large range of compound efficacies. Data were largely consistent with cAMP signaling data (Figure 1, Table 3), in that compounds grouped roughly by rank order efficacies were generally consistent between the two pathways.
TABLE 3.
Potencies and efficacies for EG‐018 and analogues in HEK cells expressing hCB1 to stimulate accumulation of [35S]‐GTPγS (n = 3)
| Compound | pEC50 (±SEM), −log M | E MAX a (±SEM), % | Rank order efficacy |
|---|---|---|---|
| EG‐018 | 7.77 (±0.21) | 107 (±8.86) | 7 |
| 042 | 6.99 (±0.12) | 163 (±10.7) | 4 |
| 043 | 7.42 (±0.07) | 82.2 (±11.4) | 11 |
| 044 | 7.26 (±0.46) | 69.7 (±9.85) | 13 |
| 045 | 6.59 (±0.14) | 81.9 (±9.45) | 12 |
| 150 | 5.78 (±0.08) | 256 (±27.6) | 1 |
| 085 | 7.89 (±0.07) | 216 (±17.7) | 2 |
| 117 | 6.58 (±0.13) | 171 (±33.2) | 3 |
| 156 | 7.18 (±0.04) | 96.5 (±16.6) | 9 |
| 104 | 6.99 (±0.16) | 130 (±25.9) | 5 |
| 141 | 6.45 (±0.23) | 106 (±15.5) | 8 |
| 142 | 5.21 (±0.23) | 123 (±29.2) | 6 |
| 146 | Not determined | Not determined | / |
| 157 | 6.59 (±0.05) | 88.2 (±10.1) | 10 |
E MAX estimates were obtained from concentration–response curve spans (curves not shown) and are reported as % net stimulation .
Data from the previously described signaling experiments suggest that several of the novel EG‐018 analogues described in this study possess exceptionally low efficacy. This led us to test the utility of compound 157, the lowest efficacy analogue in cAMP assays, as an antagonist in both cAMP inhibition and pERK stimulation assays. The ability of low efficacy agonists to antagonize the responses of higher efficacy compounds is well known—but the in vitro signaling data reported in this study suggests that some compounds are close to neutral antagonists. For the purposes of comparing the characteristics of analogue 157, cAMP assays were also performed for previously published CB1 neutral antagonists NESS‐0327 28 , 29 and AM4113, 30 , 31 in comparison to SR141716A (a well‐known CB1 inverse agonist).
These data indicate that, in a pathway with both constitutive activity and a large receptor reserve, putative “neutral” antagonists actually demonstrate substantial inverse agonism, with NESS‐0327 driving approximately equivalent inverse agonism to SR141716A (Figure 3A). However, the reported subnanomolar affinity of AM4113 (0.89 nM) 32 appeared incongruent with the extremely low potency inverse agonist‐like cAMP effect observed, so to confirm the specificity of AM4113 for CB1, cAMP experiments were performed using HEK wildtype (WT) cells that did not express CB1 (Figure 3B). A non‐specific effect of similar potency and efficacy resulted—indicating that high concentrations of AM4113 induce responses by acting at a non‐CB1 target in the HEK background. To rule out the possibility that non‐specific effect resulted from the drug having degraded, these assays were repeated with a different batch of AM4113 purchased from Sigma Aldrich—with unaltered results.
FIGURE 3.

Concentration–response experiments for cAMP (A–C) and pERK (D), showing the activity profiles of reported neutral CB1 antagonists in comparison to SR141716A (A), the activity of AM4113 in untransfected HEK WT cells (B), and the utility of analogue 157 (10 µM) to antagonize the activity of CP55940 (C, D). Data are from representative experiments showing mean ± SD of conditions performed in technical duplicate. cAMP data are normalized to basal (0%) and FSK only (100%); pERK data are not normalized
While these data are consistent with AM4113 acting as a neutral ligand at CB1, this conclusion is contingent on the ligand binding the receptor. We therefore attempted to replicate this published finding, by performing heterologous radioligand binding assays with [3H]‐CP55940, in P2 membrane preparations made from HEK cells expressing hCB1. These studies indicated that AM4113 has low affinity for CB1 (pKi 5.37 ± 0.10, n = 3), in disagreement with the original study. 32 Considering the differences between the affinities and inverse agonist potencies of SR141716A and NESS‐0327 in cAMP assays (where potency is approximately 2‐log units right‐shifted compared to affinity), it is unlikely that any AM4113 effects would be detectable under the current assay conditions.
In both cAMP and pERK assays, a high concentration of 157 resulted in an approximately 1‐log unit right‐shift in the potency of the CP55940 response, without reducing E MAX (Figure 3C,D), consistent with classical competitive antagonism. Data from both cAMP and pERK experiments were consistent with studies of 157 alone, with 10 µM 157 alone shifting the baseline of the CP55940 cAMP curve (agonism manifesting as inhibition of approximately 15% of the forksolin‐mediated response) while not driving an agonist response in pERK.
4. DISCUSSION
In the current study, we report a novel family of EG‐018 analogues which demonstrate among the lowest efficacies of any CB1 agonists reported to date. This study was spurred by our recent molecular pharmacology characterization of EG‐018, 17 which suggested that some of the features of the structure of EG‐018 might offer opportunity to further probe determinants of efficacy at CB1.
Some general structure‐activity relationships have emerged from the differences in cAMP and GTPγS efficacies, given that all the analogues share their core structure with EG‐018 and differ only in the N‐alkyl side‐chain and aromatic rings (Ar) attached to the C=O group (Table 1). Analogues 042, 043, 044, 045, 085, 104, 117 and 156 contain the same naphthyl ring group as the parent EG‐018, but different N‐alkyl side‐chains. Our data suggest that as EG‐018 analogue N‐alkyl side‐chain length increases, efficacy decreases (Tables 2 and 3); consistent with previous studies. 33 , 34 Similarly, analogues 085, 117 and 042 contain shorter N‐alkyl side‐chains than that of EG‐018, but induce greater efficacy. Other N‐alkyl side‐chain modifications also affected efficacy: for example, although 104 and 156 both contain 2‐carbon N‐alkyl side‐chains (shorter than that of EG‐018), they exhibit similar or slightly reduced efficacy. This may be due to incorporation of a 6‐member heteroatom ring at the far end of the N‐alkyl side‐chain. Electron effects likely also contribute to modulation of efficacy in this series of compounds: the reduced efficacy of compounds 044 and 045 (which share their 5‐carbon N‐alkyl side‐chains with EG‐018) is likely explained by the terminal OH and CH3COO groups.
Analogues 141, 142, 146 and 157 have different N‐alkyl side‐chains and Ar groups to EG‐018. 141 has a naphthyl ring attaching to the C=O group, while 142, 146, and 157 have fused ring heterocycles with basic unshared electron pairs. Electron effects from the unshared electron pairs of the heterocycles may help explain the particularly low efficacy of these compounds, and steric effects may also contribute. Analogue 150 contains the same linear 5‐carbon side‐chain as EG‐018, but differs in that the parental naphthyl ring was substituted for a pyridyl ring. Interestingly, this ring substitution resulted in substantially increased efficacy compared to EG‐018 (150 was among the highest efficacy compounds detected), likely due to altered steric properties.
It is easy to underestimate the extent of the low efficacy of the analogues reported because of the high efficiency of coupling between CB1 and the cAMP pathway (arguably the most extensively described in vitro effector pathway for CB1, for example). 11 , 21 , 35 , 36 , 37 For example, in cAMP inhibition assays, the efficacies of ligands that are known to vary substantially can manifest as similar—such as comparisons between THC (partial) and WIN55,212‐2. 11 In the current study, the lack of efficacy of most EG‐018 analogues in pERK assays reinforces this concept. The conclusion from these observations is that EG‐018 and a number of its analogues possess among the lowest efficacy of any CB1 agonists described to date, with analogue 157 approaching neutral activity in cAMP (with other neutral compounds in the pERK pathway). It is also important to note that efficacy in a given pathway is greatly influenced by receptor expression level. In the Operational Model, this is captured in the term R0 (sometimes, “RT”), and in turn τ, 38 , 39 and establishes the fact that systems with reduced receptor expression will more readily manifest no drug activity for low efficacy ligands (i.e., functionally neutral responses). Together with agonist efficacy, receptor number also determines the scale of antagonist‐mediated agonist potency shifts (as for 157 right‐shifting the potency of CP55940 by 1‐log unit, Figure 3D). Specifically, in assays with receptor reserve (relatively high receptor number and relatively high agonist efficacy), pharmacological blockade must be highly substantial in order for competitive antagonism to be observed. In the example of Figure 3D, CP55940 is a relatively efficacious CB1 agonist—meaning it is active at low receptor occupancy. Consequently, 157 receptor occupancy must be high before CP55940 potency is affected. For the same reason, 157 would theoretically induce a greater potency shift if the agonist used had possessed lower efficacy agonist than CP55940 (such as THC).
To date, two studies have included EG‐018 in a quantitative study of cannabinoid agonist bias. The first reported that EG‐018 was the only SCRA of the 21 included for which a 10‐fold bias “preference” was detected for G protein pathways (in comparison to β‐arrestin pathways, using CP55940 as the reference ligand). 40 The second study agreed with this finding, and concluded that EG‐018 was biased toward inducing CB1‐mediated G protein activity (cAMP inhibition) over β‐arrestin activity. 41 However, in the context of our own EG‐018 data, and considering the evolving understanding that “biased” MOR agonists may instead show reduced efficacy, 42 , 43 the notably low efficacy of EG‐018 17 draws into question the likelihood that the G protein bias finding is authentic. Indeed, we have previously shown that CB1 appears to be less efficiently coupled to arrestin pathways than G protein pathways (in the same cell model used in the current study) 12 , 36 , 44 —the activity profile of a low efficacy agonist at CB1, EG‐018, may be relatively underestimated when assessed in a pathway to which the receptor is already inefficiently coupled. However, a possible lack of “genuine” agonist bias does not detract from the therapeutic utility of reduced efficacy ligands. For example, if a MOR ligand induces responses with sufficient efficacy to drive G protein responses but not arrestin responses, the objective of developing G protein‐biased MOR ligands has effectively been met (even if the ligand is not biased). It is to be hoped that similar value may be derived from reduced efficacy CB1 ligands.
The AM4113 and NESS‐0327 data shown in the current study illustrate that many compounds reported as neutral antagonists must be viewed comparatively with inverse agonists before this categorization can be made with confidence. Due to their flexibility and high capacity for optimization, highly reductionist in vitro methods may offer greater opportunity than in vivo systems to detect inverse agonism when, often, the bulk of the data that leads to the description of compounds as “neutral” is from ex vivo (intact tissue) or even in vivo (whole animal) systems. An example of this might include an early study into the activity of NESS‐0327, which employed a vas deferens contraction paradigm, where the compound was described as having no effect on its own. 29 This has been subsequently interpreted by some researchers as being akin to a claim of neutral antagonism. However, despite our findings for NESS‐0327, other studies have differentiated NESS‐0327 from well‐known inverse agonists like SR141716A on the basis of its lower activity (e.g. [45]). Data currently do not exist to explain the disconnect between these contradictory observations. One attribute to consider is that neutral antagonism is a function of extent of constitutive activity. If the latter differs between two systems (e.g., due to differing effector abundances), then the closeness to neutrality that antagonist reactions can attain will also differ between these systems. In the current study, it is likely that our assay system has substantial constitutive activity because receptor expression is relatively high (as is typical for heterologous expression models), which means that efficacious inverse agonist responses (Figure 3A) may be amplified in comparison to endogenous or native assay systems, or in vivo. For very low efficacy agonists like 157, the highly sensitive assays system used throughout the current study will likely overestimate compound activity for the same reason.
Substantial knowledge of AM4113 originates from in vivo rodent data (mainly in appetite regulation paradigms), where the contribution of pharmacokinetics to the overall effect is not clear (though some in vitro data do exist that support the “neutral” designation). 30 , 31 , 32 Our data would suggest that further investigation of AM4113 remains necessary to corroborate earlier reports of CB1 binding. 32
Neutral antagonists are likely much rarer than implied by the use of the term. It has been estimated that about 85% of “antagonists” are actually inverse agonists, 46 though the ability to distinguish between these is as much associated with inherent constitutive activity, which is a characteristic of many GPCRs 47 including CB1, 48 , 49 as to do with the ligand. This is because inverse agonism is only detectable for receptors that are constitutively active, and in assay pathways where this activity is detectable. In the light of receptor theory and structural biology, perhaps this rarity is to be expected: two‐state receptor theory (reviewed in Ref. [39]) posits that ligands bind and stabilize the receptor conformation (active vs. inactive/R* vs. R) for which they have affinity, and that therefore ligand binding results in receptor populations “accumulating” into a dominant state. In this paradigm, neutral antagonists will bind receptors in such a fashion as to not alter the basal population R‐R* distribution. In structural terms, this appears to require that a neutral antagonist will specifically occupy its cognate receptor (to prevent agonist binding) but, simultaneously, either have approximately equal affinities for active and inactive receptor conformations, or allow the receptor to dynamically alternate between states even after binding. 50 This requirement seems exceptionally challenging, and suggests that aiming to minimize the efficacies of weakly partial agonists or inverse agonists may be the most fruitful approach for discovering functionally “neutral” compounds. Notably, however, this apparent requirement for neutral ligands to bind more than one receptor conformation (activity state) would appear to militate against these interactions being high affinity. As functional neutrality and high affinity are therefore unlikely to co‐vary, further optimization of the scaffolds reported in this study is likely to be challenging.
Though further understanding of EG‐018 and its analogues remains necessary (particularly in vivo pharmacokinetics), the molecular data presented in the current study underscore the unusual attributes associated with this family of compounds. It now seems that erstwhile “neutral antagonists” of CB1 must be carefully validated for their activity in individual assay endpoints—but at least one novel EG‐018 analogue (157) may be closer to a neutral antagonist in the experimental systems reported here than other compounds reported to date. The possible uses for a CB1 neutral antagonist—both in research and the clinic—means that this goal is worth pursuing.
DISCLOSURE
None.
ETHICS STATEMENT
All work on genetically modified organisms and uncleared biologicals that contributed to the current study was performed in Physical Containment Level 2 laboratory facilities, in compliance with and under license from the New Zealand Ministry of Primary Industries. No other ethics approvals were required; no experiments were performed on animals or humans.
AUTHORS’ CONTRIBUTIONS
Finlay, Nguyen, Gamage, Thomas, Wiley, Zhang, and Glass participated in research design. Finlay, Nguyen, Gamage, Chen, Barrus, and Patel conducted experiments. Nguyen and Zhang contributed to new reagents or analytic tools. Finlay, Nguyen, Gamage, Chen, Barrus, and Patel performed data analysis. Finlay, Nguyen, Gamage, Thomas, Wiley, Zhang, and Glass contributed to the writing of the manuscript.
Supporting information
Supplementary Material
ACKNOWLEDGMENT
None.
Finlay DB, Nguyen T, Gamage TF, et al. Exploring determinants of agonist efficacy at the CB1 cannabinoid receptor: Analogues of the synthetic cannabinoid receptor agonist EG‐018. Pharmacol Res Perspect. 2022;10:e00901. doi: 10.1002/prp2.901
Funding information
The authors gratefully acknowledge grants from Lottery Health Research (DBF); the Maurice and Phyllis Paykel Trust (MG), the Health Research Council of New Zealand (MG), and the National Institute on Drug Abuse, National Institutes of Health, U.S. (grants R01DA040693 [YZ], R01DA040460 [JLW], and K01DA045752 [TFG]).
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Ariens EJ. Affinity and intrinsic activity in the theory of competitive inhibition. I. Problems and theory. Arch Int Pharmacodyn Ther. 1954;99:32‐49. [PubMed] [Google Scholar]
- 2. Urban JD, Clarke WP, von Zastrow M, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2006;320:1‐13. [DOI] [PubMed] [Google Scholar]
- 3. Foulds J. The neurobiological basis for partial agonist treatment of nicotine dependence: varenicline. Int J Clin Pract. 2006;60:571‐576. [DOI] [PubMed] [Google Scholar]
- 4. Cohen C, Bergis OE, Galli F, et al. SSR591813, a novel selective and partial α4β2 nicotinic receptor agonist with potential as an aid to smoking cessation. J Pharmacol Exp Ther. 2003;306:407‐420. [DOI] [PubMed] [Google Scholar]
- 5. Hogg RC, Bertrand D. Partial agonists as therapeutic agents at neuronal nicotinic acetylcholine receptors. Biochem Pharmacol. 2007;73:459‐468. [DOI] [PubMed] [Google Scholar]
- 6. Kenakin T. Functional selectivity and biased receptor signaling. J Pharmacol Exp Ther. 2011;336:296‐302. [DOI] [PubMed] [Google Scholar]
- 7. Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta‐arrestin 2. Science. 1999;286:2495‐2498. [DOI] [PubMed] [Google Scholar]
- 8. Raehal KM, Walker JKL, Bohn LM. Morphine side effects in‐arrestin 2 knockout mice. J Pharmacol Exp Ther. 2005;314:1195‐1201. [DOI] [PubMed] [Google Scholar]
- 9. Gillis A, Gondin AB, Kliewer A, et al. Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci Signal. 2020;13:eaaz3140. [DOI] [PubMed] [Google Scholar]
- 10. Conibear AE, Kelly E. A biased view of μ‐Opioid receptors? Mol Pharmacol. 2019;96:542‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Finlay DB, Cawston EE, Grimsey NL, et al. Gαs signalling of the CB1 receptor and the influence of receptor number. Br J Pharmacol. 2017;174:2545‐2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ibsen MS, Finlay DB, Patel M, Javitch JA, Glass M, Grimsey NL. Cannabinoid CB1 and CB2 receptor‐mediated arrestin translocation: species, subtype, and agonist‐dependence. Front Pharmacol. 2019;10:350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Colquhoun D. Why the Schild method is better than Schild realised. Trends Pharmacol Sci. 2007;28:608‐614. [DOI] [PubMed] [Google Scholar]
- 14. Moreira FA, Crippa JAS. The psychiatric side‐effects of rimonabant. Rev Bras Psiquiatr. 2009;31:145‐153. [DOI] [PubMed] [Google Scholar]
- 15. Nguyen T, Thomas BF, Zhang Y. Overcoming the psychiatric side effects of the cannabinoid CB1 receptor antagonists: current approaches for therapeutics development. Curr Top Med Chem. 2019;19:1418‐1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pertwee RG. The diverse CB 1 and CB 2 receptor pharmacology of three plant cannabinoids: Δ 9 ‐tetrahydrocannabinol, cannabidiol and Δ 9 ‐tetrahydrocannabivarin. Br J Pharmacol. 2008;153:199‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gamage TF, Barrus DG, Kevin RC, et al. In vitro and in vivo pharmacological evaluation of the synthetic cannabinoid receptor agonist EG‐018. Pharmacol Biochem Behav. 2020;193:172918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Atwood BK, Huffman J, Straiker A, Mackie K. JWH018, a common constituent of “Spice” herbal blends, is a potent and efficacious cannabinoid CB1 receptor agonist. Br J Pharmacol. 2010;160:585‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tai S, Fantegrossi WE. Pharmacological and toxicological effects of synthetic cannabinoids and their metabolites. Curr Top Behav Neurosci. 2016;32:249‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jiang LI, Collins J, Davis R, et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1‐phosphate/G 13 pathway. J Biol Chem. 2007;282:10576‐10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cawston EE, Redmond WJ, Breen CM, Grimsey NL, Connor M, Glass M. Real‐time characterization of cannabinoid receptor 1 (CB1) allosteric modulators reveals novel mechanism of action. Br J Pharmacol. 2013;170:893‐907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hall DA, Langmead CJ. Matching models to data: a receptor pharmacologist’s guide. Br J Pharmacol. 2010;161:1276‐1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Res. 2018;46:D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alexander SPH, Christopoulos A, Davenport AP, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: G protein‐coupled receptors. Br J Pharmacol. 2021;178(S1):S27–S156. [DOI] [PubMed] [Google Scholar]
- 25. Aung MM, Griffin G, Huffman JW, et al. Influence of the N‐1 alkyl chain length of cannabimimetic indoles upon CB1 and CB2 receptor binding. Drug Alcohol Depend. 2000;60:133‐140. [DOI] [PubMed] [Google Scholar]
- 26. Savinainen JR, Järvinen T, Laine K, Laitinen JT. Despite sabstantial degradation, 2‐arachidonoylglycerol is a potent full efficacy agonist mediating CB1 receptor‐dependent g‐protein activation in rat cerebellar membranes. Br J Pharmacol. 2001;134:664‐672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Strange PG. Use of the GTPγS ([35S]GTPγS and Eu‐GTPγS) binding assay for analysis of ligand potency and efficacy at G protein‐coupled receptors. Br J Pharmacol. 2010;161:1238‐1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ruiu S, Pinna GA, Marchese G, et al. Synthesis and characterization of NESS 0327: a novel putative antagonist of the CB1 cannabinoid receptor. J Pharmacol Exp Ther. 2003;306:363‐370. [DOI] [PubMed] [Google Scholar]
- 29. Tambaro S, Mongeau R, Dessi C, Pani L, Ruiu S. Modulation of ATP‐mediated contractions of the rat vas deferens through presynaptic cannabinoid receptors. Eur J Pharmacol. 2005;525:150‐153. [DOI] [PubMed] [Google Scholar]
- 30. Cluny NL, Chambers AP, Vemuri VK, et al. The neutral cannabinoid CB1 receptor antagonist AM4113 regulates body weight through changes in energy intake in the rat. Pharmacol Biochem Behav. 2011;97:537‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. He X‐H, Jordan CJ, Vemuri K, et al. Cannabinoid CB1 receptor neutral antagonist AM4113 inhibits heroin self‐administration without depressive side effects in rats. Acta Pharmacol Sin. 2019;40:365‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sink KS, McLaughlin PJ, Wood JAT, et al. The novel cannabinoid CB1 receptor neutral antagonist AM4113 suppresses food intake and food‐reinforced behavior but does not induce signs of nausea in rats. Neuropsychopharmacology. 2008;33:946‐955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Compton DR, Rice KC, De Costa BR, et al. Cannabinoid structure‐activity relationships: correlation of receptor binding and in vivo activities. J Pharmacol Exp Ther. 1993;265:218‐226. [PubMed] [Google Scholar]
- 34. Thomas BF, Compton DR, Martin BR, Semus SF. Modeling the cannabinoid receptor: a three‐dimensional quantitative structure‐activity analysis. Mol Pharmacol. 1991;40:656‐665. [PubMed] [Google Scholar]
- 35. Eldeeb K, Leone‐Kabler S, Howlett AC. CB1 cannabinoid receptor‐mediated increases in cyclic AMP accumulation are correlated with reduced Gi/o function. J Basic Clin Physiol Pharmacol. 2016;27:311‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Finlay DB, Manning JJ, Ibsen MS, et al. Do toxic synthetic cannabinoid receptor agonists have signature in vitro activity profiles? A case study of AMB‐FUBINACA. ACS Chem Neurosci. 2019;10:4350‐4360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Howlett AC, Fleming RM. Cannabinoid inhibition of adenylate cyclase. Pharmacology of the response in neuroblastoma cell membranes. Mol Pharmacol. 1984;26:532‐538. [PubMed] [Google Scholar]
- 38. Black JW, Leff P. Operational models of pharmacological agonism. Proc R Soc London Ser B Biol Sci. 1983;220:141‐162. [DOI] [PubMed] [Google Scholar]
- 39. Finlay DB, Duffull SB, Glass M. 100 years of modelling ligand–receptor binding and response: a focus on GPCRs. Br J Pharmacol. 2020;177:1472‐1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wouters E, Walraed J, Robertson MJ, et al. Assessment of biased agonism among distinct synthetic cannabinoid receptor agonist scaffolds. ACS Pharmacol Transl Sci. 2019;3:285‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zagzoog A, Brandt AL, Black T, et al. Assessment of select synthetic cannabinoid receptor agonist bias and selectivity between the type 1 and type 2 cannabinoid receptor. Sci Rep. 2021;11:10611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gillis A, Sreenivasan V, Christie MJ. Intrinsic efficacy of opioid ligands and its importance for apparent bias, operational analysis, and therapeutic window. Mol Pharmacol. 2020;98:410‐424. [DOI] [PubMed] [Google Scholar]
- 43. Kliewer A, Gillis A, Hill R, et al. Morphine‐induced respiratory depression is independent of β‐arrestin2 signalling. Br J Pharmacol. 2020;177:2923‐2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Patel M, Manning JJ, Finlay DB, et al. Signalling profiles of a structurally diverse panel of synthetic cannabinoid receptor agonists. Biochem Pharmacol. 2020;175:113871. [DOI] [PubMed] [Google Scholar]
- 45. Meye FJ, Trezza V, Vanderschuren LJMJ, Ramakers GMJ, Adan RAH. Neutral antagonism at the cannabinoid 1 receptor: a safer treatment for obesity. Mol Psychiatry. 2013;18:1294‐1301. [DOI] [PubMed] [Google Scholar]
- 46. Greasley PJ, Clapham JC. Inverse agonism or neutral antagonism at G‐protein coupled receptors: a medicinal chemistry challenge worth pursuing? Eur J Pharmacol. 2006;553:1‐9. [DOI] [PubMed] [Google Scholar]
- 47. Seifert R, Wenzel‐Seifert K. Constitutive activity of G‐protein‐coupled receptors: cause of disease and common property of wild‐type receptors. Naunyn Schmiedebergs Arch Pharmacol. 2002;366:381‐416. [DOI] [PubMed] [Google Scholar]
- 48. Hanlon KE, Vanderah TW. Constitutive activity at the cannabinoid CB1 receptor and behavioral responses. Methods Enzymol. 2010;484:3‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leterrier C, Bonnard D, Carrel D, Rossier J, Lenkei Z. Constitutive endocytic cycle of the CB1 cannabinoid receptor. J Biol Chem. 2004;279:36013‐36021. [DOI] [PubMed] [Google Scholar]
- 50. Kenakin T. Inverse, protean, and ligand‐selective agonism: matters of receptor conformation. FASEB J. 2001;15:598‐611. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.