

Summary:
Many enveloped viruses require the endosomal sorting complexes required for transport (ESCRT) pathway to exit infected cells. This highly conserved pathway mediates essential cellular membrane fission events, which restricts the acquisition of adaptive mutations to counteract viral co-option. Here, we describe duplicated and truncated copies of the ESCRT-III factor CHMP3 that block ESCRT-dependent virus budding and that arose independently in New World monkeys and mice. When expressed in human cells, these retroCHMP3 proteins potently inhibit release of retroviruses, paramyxoviruses, and filoviruses. Remarkably, retroCHMP3 proteins have evolved to reduce interactions with other ESCRT-III factors and to have little effect on cellular ESCRT processes, revealing routes for decoupling cellular ESCRT functions from viral exploitation. The repurposing of duplicated ESCRT-III proteins thus provides a mechanism to generate broad-spectrum viral budding inhibitors without blocking highly conserved essential cellular ESCRT functions.
Keywords: ESCRT pathway, HIV-1, virus budding, retrocopy, antiviral protein
Graphical Abstract
In Brief:
Truncated retrocopies of ESCRT-III factor CHMP3, which have independently evolved in New World monkey and mouse species, block ESCRT-dependent virus budding with little effect on critical cellular ESCRT processes in human cells.
Introduction
The endosomal sorting complexes required for transport (ESCRT) pathway mediates essential cellular membrane fission events such as multivesicular body formation, cytokinetic abscission, and resealing of the post-mitotic nuclear envelope. Loss of ESCRT function is therefore highly detrimental in many cultured cell lines. Early-acting ESCRT factors are recruited to cellular sites of membrane fission by specific adapter proteins that, in turn, recruit core ESCRT-III charged multivesicular body proteins (CHMPs, e.g., CHMP2, CHMP3, and CHMP4), which form membrane-associated heteropolymeric filaments. These ESCRT-III protein filaments then recruit and are remodeled by the AAA ATPase VPS4, which is required for membrane constriction and fission (Christ et al., 2017; Henne et al., 2013; McCullough et al., 2018; Scourfield and Martin-Serrano, 2017).
To facilitate budding from cellular membranes, the structural proteins of enveloped viruses, including the human immunodeficiency virus type 1 (HIV-1), contain ESCRT recruitment motifs, termed late assembly domains, that mimic the motifs employed by host ESCRT adapter proteins (Votteler and Sundquist, 2013). Such piracy can generate evolutionary pressure to select for mutations in host proteins that abrogate the interaction with viral proteins (Daugherty and Malik, 2012). In this case, however, mutations in ESCRT proteins that abrogate viral pathway recruitment would likely also interfere with essential cellular ESCRT functions and be selected against (Elde and Malik, 2009). Therefore, the host has limited potential for adaptations of the ESCRT pathway that prevent exploitation by viruses while still maintaining interactions necessary for cellular processes.
Retrocopies are gene duplications that arise by reverse transcription and genomic insertion of mRNAs by retrotransposons such as long interspersed nuclear elements (LINEs). In rare cases, retrocopies evolve new functions that differ from the parental gene (Kaessmann et al., 2009; Kubiak and Makalowska, 2017). Here, we describe the independent evolution of retrocopies of the ESCRT-III protein CHMP3 from New World monkey and mouse species that can potently block ESCRT-dependent virus budding. Remarkably, these retroCHMP3 proteins have evolved to spare cellular ESCRT functions in abscission, and therefore specifically counteract the hijacking of the ESCRT pathway by viruses.
Results
RetroCHMP3 proteins inhibit HIV-1 budding
We searched for CHMP3 retrocopies by manual BLAST-like Alignment Tool (BLAT) analyses of genomes in the University of California Santa Cruz (UCSC) genome browser (Kent, 2002) or by manual National Center for Biotechnology Information (NCBI) BLAST analyses (Altschul et al., 1990) of whole-genome shotgun contigs. Our searches identified two independent duplications of the gene encoding the ESCRT-III protein CHMP3 in ancestors of New World monkeys and house mice. These gene duplications appear to be LINE-1-mediated, as evidenced by the absence of introns and the presence of flanking target site duplications, which are hallmarks of LINE-1 retrotransposition (see Table S1 for details). Importantly, the parental CHMP3 gene (~222 amino acids) remains intact in all species, which presumably allowed the retrocopies to diverge in function. These retrocopies acquired independent premature stop codons, removing approximately 70 carboxy-terminal amino acids from the predicted protein product, in two New World monkeys; Guyanese squirrel monkey (Saimiri sciureus sciureus, retroCHMP3SS, 155 amino acids) and black-handed spider monkey (Ateles geoffroyi, retroCHMP3AG, 157 amino acids), and in house mice (Mus musculus, retroCHMP3MM, 147 amino acids, Fig. 1A). We verified the expected RETROCHMP3 sequences by genomic DNA sequencing (Table S1, Fig. S1A, see Methods for details). RETROCHMP3 sequences predicted to express similar truncated proteins have also been detected in other species, and are described elsewhere (Rheinemann et al., 2021).
Fig. 1: RetroCHMP3 proteins inhibit HIV-1 budding.

(A) Schematic of the C-terminally truncated protein products of retrocopies of CHMP3 (light green) found in squirrel monkey (retroCHMP3SS), spider monkey (retroCHMP3AG), and mouse (retroCHMP3MM). Green circles indicate the acquisition of retrocopies. Parental full-length copies (dark green) are also present in all species. The CHMP3 retrocopy in capuchin is homologous to retroCHMP3SS, but degraded. (B) Schematic of full-length, parental squirrel monkey CHMP3 (CHMP3SS), C-terminally truncated parental CHMP3 (CHMP3SS(155)), and retroCHMP3SS. Black arrowheads indicate amino acid substitutions. (C), (D) Transient overexpression of HA-tagged retroCHMP3SS (C) and retroCHMP3MM (D) in human embryonic kidney (HEK293T) cells inhibits the release of HIV-1 proteins (CA, MA) and reduces viral titers in the supernatant. Titer graphs show mean ± SD from 3 experimental replicates. **** p<0.0001, *** p<0.001 by one-way ANOVA followed by Tukey's multiple comparisons test. For a complete set of pairwise comparisons, see Table S3. (E) Transmission electron micrographs of cells transfected with an HIV-1 expression vector and retroCHMP3SM or retroCHMP3MM. Scale bar 100nm. See also Fig. S1.
A truncated human CHMP3 construct lacking the C-terminal 72 amino acids can potently inhibit HIV-1 budding (Zamborlini et al., 2006). This truncated construct presumably functions as a dominant-negative inhibitor of ESCRT pathway functions because it retains the core filament assembly domain but lacks the C-terminal elements required for autoinhibition and recruitment of VPS4 and its filament depolymerization activity (Bajorek et al., 2009; Lata et al., 2008; Muziol et al., 2006; Shim et al., 2007; Stuchell-Brereton et al., 2007) (Fig. 1B). We hypothesized that the naturally occurring retroCHMP3 proteins might therefore also exhibit antiviral activity, and we tested this idea by co-expressing either HA-tagged retroCHMP3SS (Fig. 1C), retroCHMP3MM (Fig. 1D), or retroCHMP3AG (Fig. S1B) together with a proviral HIV-1 expression construct in human embryonic kidney (HEK293T) cells. In all three cases, retroCHMP3 expression potently inhibited virus release, as measured by reductions in the levels of viral MA (matrix) and CA (capsid) proteins released into the culture supernatant and by reductions in infectious titers. RetroCHMP3 expression also caused two additional phenotypes characteristic of HIV-1 budding defects or delays: 1) accumulation of the Gag processing intermediate, CA-SP1, relative to fully processed CA protein (Gottlinger et al., 1991), and 2) concomitant accumulation of budding virions seen at the plasma membrane by thin-section transmission electron microscopy (Fig. 1E). Therefore, retroCHMP3 proteins encoded by three different species can potently inhibit HIV-1 budding, presumably by dominantly inhibiting ESCRT pathway functions.
RetroCHMP3 proteins broadly inhibit ESCRT-dependent budding
To test whether retroCHMP3 inhibition can be generalized to other ESCRT-dependent viruses, we co-expressed either retroCHMP3SS or retroCHMP3MM with proviral expression constructs for Friend murine leukemia virus (FrMLV, Fig. 2A, S2A) or equine infectious anemia virus (EIAV, Fig. 2B, S2B) in HEK293T cells. Both of these retroviruses require the ESCRT pathway for budding (Garrus et al., 2001; Strack et al., 2003). RetroCHMP3 proteins reduced the release of FrMLV MA and CA proteins and viral titers in the supernatant without significantly altering the expression of viral proteins in the cell (Fig. 2A, S2A). EIAV titers were also reduced by retroCHMP3 protein expression (Fig. 2B, S2B), but in this case, the amount of EIAV CA protein in the supernatant was not substantially reduced. These observations are similar to our previous report that inhibiting ESCRT functions by depleting the ESCRT-III protein CHMP4B during EIAV budding also reduced viral titers but did not decrease virion-associated CA protein release (Sandrin and Sundquist, 2013). In that report, we also observed that EIAV virions released in the absence of CHMP4B exhibited aberrant tubular or multi-lobed virion morphologies, suggesting that Gag polymerization or bud neck closure defects led to the release of aberrant, non-infectious virions. In the case of retroCHMP3SS co-expression, we similarly observed a large number of immature EIAV virions that were either budding or were closely associated with the cell surface, and a subset displayed aberrant morphologies (Fig. 2C, black arrowheads). Hence, retroCHMP3 inhibition resembled the phenotype of depletion of an essential ESCRT-III protein during EIAV budding, although the number of tubular extracellular EIAV virions was larger in the absence of CHMP4B than in the presence of retroCHMP3.
Fig. 2: RetroCHMP3 proteins broadly inhibit ESCRT-dependent budding.

(A), (B) Transient overexpression of HA-tagged retroCHMP3SS in HEK293T cells reduces the release of viral proteins and viral titers in the supernatant of Friend murine leukemia virus (FrMLV, A) and equine infectious anemia virus (EIAV, B). Titer graphs show mean ± SD from 3 experimental replicates. (C) Transmission electron micrographs of cells transfected with EIAV expression vectors and empty control vector or retroCHMP3SS. Black arrowheads point to tubular and multi-lobed virions. Scale bar 100nm. (D), (E) Transient overexpression of HA-tagged retroCHMP3SS in HEK293T cells reduces the release of Ebola virus VP40 protein (D) and parainfluenza virus 5 VLPs (E) into the supernatant. Western blots show representative results from 3 experimental replicates. Bar graphs show integrated intensities of viral protein bands in supernatant normalized to cellular fraction from 3 experimental replicates. **** p<0.0001, *** p<0.001, * p<0.05 by one-way ANOVA followed by Tukey's multiple comparisons test. Only comparisons with the empty vector control (lane 1) are shown for clarity. For a complete set of pairwise comparisons, see Table S3. See also Fig. S2.
We also tested the inhibitory effect of retroCHMP3 proteins on the budding of an ESCRT-dependent filovirus (Ebola virus, EBOV, Fig. 2D, S2C) and an ESCRT-dependent paramyxovirus (parainfluenza virus 5, PIV 5, Fig. 2E, S2D) (Martin-Serrano et al., 2001; Schmitt et al., 2005). In both cases, retroCHMP3SS and retroCHMP3MM proteins inhibited virion release. Specifically, release of virus-like particles (VLPs) formed by the EBOV VP40 protein was inhibited by both retroCHMP3 proteins but not by parental full-length CHMP3 proteins. The effect was comparable to that of a positive control mutation in the VP40 late assembly domain (VP40 P11L) that abrogates ESCRT recruitment (Martin-Serrano et al., 2004). Release of PIV5 VLPs produced by the co-expression of the PIV5 M, NP, and HN proteins was similarly reduced by the retroCHMP3 proteins. This effect was comparable to the positive control for loss of ESCRT activity, in this case induced by overexpression of a dominant-negative mutant of VPS4 (VPS4A E228Q). Taken together, these data demonstrate that retroCHMP3 proteins can broadly block the release of highly divergent virus families by inhibiting ESCRT functions during budding.
RetroCHMP3 proteins evolved to lose cytotoxicity
The ESCRT pathway performs a series of important cellular functions, including cytokinetic abscission. Loss of ESCRT function is therefore typically cytotoxic. Indeed, overexpression of parental CHMP3 proteins that were artificially truncated to the same length as retroCHMP3 proteins dramatically reduced cell viability after 24 hours, consistent with dominant-negative inhibition of ESCRT functions (Fig. 3A, S3A, S3B). Strikingly, however, overexpression of retroCHMP3SS only very mildly reduced cell viability and was nearly as non-toxic as overexpression of the full-length, parental CHMP3SS protein (Fig. 3A). RetroCHMP3SS acquired 22 amino acid substitutions in comparison to the truncated parental CHMP3SS(155) (black arrowheads, Fig. 1B), suggesting that these amino acid substitutions, or a subset of them, cause the reduced cytotoxicity of retroCHMP3SS. RetroCHMP3MM and retroCHMP3AG also displayed lower cytotoxicity than their C-terminally truncated parental CHMP3 counterparts. In these cases, however, the proteins were moderately more cytotoxic than the corresponding full-length CHMP3 proteins (Figs. S3A, B). Accordingly, cellular expression of HIV-1 Gag, which was also assessed after 24 hours and is dependent on cell viability, was reduced moderately in samples transfected with retroCHMP3MM (Fig. 1C) and retroCHMP3AG (Fig. S1B), but not retroCHMP3SS (Fig. 1B). RetroCHMP3MM and retroCHMP3AG acquired 12 and 14 amino acid substitutions, respectively, compared to their truncated parental proteins. Notably, none of the retroCHMP3 proteins share common mutations (Fig. S1A), demonstrating that different combinations of amino acid substitutions can lead to a reduction of cytotoxicity.
Fig. 3: RetroCHMP3SS evolved to lose cytotoxicity.

(A) Viability of HEK293T cells transiently transfected with HA-tagged CHMP3SS variants at 24 h after transfection, as determined in a luminescent ATP cell viability assay. Mean ± SD from 5 experimental replicates with n≥ 6 each. (B) Immunofluorescence showing the dispersed (CHMP3SS and retroCHMP3SS) and punctate (CHMP3SS(155)) subcellular localization phenotypes in HeLa N cells. Scale bar 10 μm. Insets show percentages of cells with the two depicted phenotypes (diffuse or punctate: n≥ 450 cells/condition). White arrowhead points to a midbody. (C) Number of midbody-linked cells in unsynchronized cultures transiently transfected with the designated HA-tagged CHMP3SS variants 20 h after transfection. Mean ± SD from 3 experimental replicates, each with 150 HA-positive cells/condition. (D) Number of midbody-linked cells in synchronized cultures transiently transfected with the designated HA-tagged CHMP3SS variants. Times are postthymidine release. Mean ± SD from 3 experimental replicates with > 290 HA-positive cells total/timepoint. (E) Pulldown of myc-tagged human CHMP4B with STrEP/FLAG-tagged human CHMP3 (CHMP3HS), C-terminally truncated human CHMP3 (CHMP3HS(150)), or CHMP3SS variants. Cell lysis and pulldowns were performed in RIPA or NP-40 buffer, as indicated. Representative Western blot from 3 experimental repeats. **** p<0.0001, ** p<0.01, * p<0.05 by one-way ANOVA followed by Tukey's multiple comparisons test comparisons with p>0.05 omitted for clarity. See also Fig. S3.
Cell imaging experiments revealed that cytotoxicity levels correlated with the subcellular localization patterns of the different CHMP3 proteins (Fig. 3B): Specifically, the two non-toxic constructs, parental CHMP3SS and retroCHMP3SS, dispersed homogeneously throughout the cytoplasm, with some enrichment in the nucleus. In contrast, the toxic CHMP3SS(155) protein formed perinuclear punctae. Similarly, non-toxic parental CHMP3MM was dispersed, whereas CHMP3MM(147) showed a punctate localization pattern (Fig. S3C). RetroCHMP3MM displayed a mixed pattern with dispersed and punctate staining, consistent with incomplete detoxification. These data suggest that C-terminally truncated parental CHMP3 proteins formed insoluble aggregates due to the lack of the autoinhibitory and VPS4 recruitment regions, but that additional amino acid substitutions in retroCHMP3SS and retroCHMP3MM allowed these proteins to fully or partially restore their solubility to that of their respective parental CHMP3 proteins.
To test whether the cytotoxicity of the different CHMP3 proteins could be explained by ESCRT-dependent cell division phenotypes, we examined the effects of the different constructs on midbody resolution during cytokinetic abscission. Both truncated and retroCHMP3 proteins were recruited to the midbody, as was the full-length parental protein (Fig. S3D), in good accord with a previous report (Dukes et al., 2008). Consistent with their relative cellular toxicities, and in line with previously observed negative effects of C-terminally truncated human CHMP3 on cytokinesis (Dukes et al., 2008), we found that CHMP3SS(155) overexpression dramatically increased the number of unresolved midbodies as compared to either CHMP3SS or retroCHMP3SS (Fig. 3C). Midbody numbers were much lower in the presence of retroCHMP3SS, but were still slightly elevated over the control construct, consistent with the slight cytotoxicity observed for this construct (Fig. 3A). To determine the effect of retroCHMP3SS on the kinetics of abscission, we synchronized cells by addition of thymidine, which arrests cells in the G1/S phase. Release of the thymidine block then allows for synchronous progression through the remaining phases of the cell cycle. Cultures transfected with an empty control plasmid accumulated cells connected by midbodies until a peak was reached around 14 hours after thymidine release, after which the number of unresolved midbodies dropped. Most cells had completed abscission at 20 hours after thymidine release (Fig. 3D). Cells transfected with CHMP3SS(155) had elevated numbers of unresolved midbodies at all timepoints, consistent with inhibition of midbody resolution already seen in asynchronous cultures. Cultures transfected with retroCHMP3SS also exhibited elevated midbody numbers, and the peak of accumulation was delayed by about two hours relative to control cells. Eventually, the number of midbodies dropped as cells completed abscission. These observations suggest that midbodies are eventually resolved in cells transfected with retroCHMP3SS, albeit more slowly than in control cells. Thus, both the defects in abscission and reductions in cell viability were much more severe for CHMP3SS(155) than for retroCHMP3SS and CHMP3SS, suggesting that abscission defects are a major driver of cytotoxicity.
We also tested how retroCHMP3 affected another ESCRT-dependent cellular process; degradation of the epidermal growth factor receptor (EGFR). In response to stimulation with EGF, EGFR is ubiquitylated at the plasma membrane, sorted into intraluminal vesicles (ILVs) at endosomes in an ESCRT-dependent manner, and delivered to lysosomes for degradation (Eden et al., 2009; Raiborg and Stenmark, 2009). Overexpression of retroCHMP3SS delayed the EGF-mediated degradation of EGFR to the same extent as overexpression of a well-characterized inhibitor of the ESCRT pathway, an ATPase-deficient mutant of VPS4A, VPS4A E228Q (Fig. S3E). Therefore, in contrast to abscission, which was inhibited to a lesser extent by retroCHMP3SS compared to the dominant-negative CHMP3SS(155), sorting of plasma membrane proteins into ILVs was equally affected by retroCHMP3SS and the dominant-negative VPS4A E228Q construct. These observations suggest that multiple ESCRT processes are impaired by retroCHMP3, albeit with differential susceptibilities.
We next sought to determine how amino acid mutations functionally differentiate retroCHMP3SS from CHMP3SS(155). The punctae formed by CHMP3SS(155) suggested that this engineered construct might inhibit the ESCRT pathway by co-polymerizing with other ESCRT-III partners and thereby functioning as a dominant-negative inhibitor of the pathway. We therefore examined the interaction of retroCHMP3SS with the ESCRT-III protein CHMP4B, a major CHMP3 binding partner (Effantin et al., 2013). In pulldown experiments, full-length human CHMP3 (CHMP3HS) and full-length CHMP3SS bound CHMP4B under both stringent (RIPA buffer, Fig. 3E, top panel) and mild (NP-40 buffer, Fig. 3E, bottom panel) conditions. C-terminally truncated CHMP3HS(150) and CHMP3SS(155) also bound strongly to CHMP4B under both conditions. In contrast, retroCHMP3SS pulled down CHMP4B in NP-40 buffer, but not in RIPA buffer. This result demonstrates that mutations present in retroCHMP3SS substantially weaken interactions with CHMP4B, perhaps by inhibiting formation of stable heterocopolymeric filaments that remain intact under the more stringent RIPA buffer binding conditions (Bajorek et al., 2009; McCullough et al., 2015). Although the reduction in CHMP4B binding can explain why retroCHMP3SS toxicity is attenuated, it leaves open the question of how retroCHMP3SS can still inhibit ESCRT functions during virus budding.
RetroCHMP3 alters CHMP4B and VPS4A recruitment to HIV-1 Gag assembly sites
To examine the effects of retroCHMP3SS on the behavior of the ESCRT machinery at sites of virion assembly and budding, we used total internal reflection fluorescence microscopy (TIRF) to visualize how mEGFP-labeled Gag-Pol proteins co-assembled with mCherry-labeled versions of CHMP4B and VPS4A. (Fig. 4). This subviral construct includes the native Gag-Pol ribosomal frameshift site and therefore produces Gag-mEGFP and Gag-Pol-mEGFP proteins at normal viral ratios. The mEGFP fluorophore is inserted between the MA and CA domains of Gag so that both Gag and Gag-Pol proteins are fluorescently labeled, and the GFP is flanked by duplicated MA/CA cleavage sites for the viral protease (PR). Thus, assembling VLPs have the structural Gag protein and the enzymatic activities associated with Gag-Pol (including PR), and both Gag and Gag-Pol can undergo normal proteolytic maturation.
Fig. 4: RetroCHMP3 alters CHMP4B recruitment to HIV-1 Gag assembly sites.

(A), (B) Sample trace of mCherry-CHMP4B recruitment to Gag-Pol-mEGFP assembly sites in the absence (A) or presence (B) of retroCHMP3SS. Each numbered shaded region highlights one recruitment event and the corresponding sequence of images from that recruitment. The width of each image is 1.27 μm. (C) Histogram showing the number of times that CHMP4B was recruited to assembly sites in the absence (n= 46 assemblies) or presence (n=41 assemblies) of retroCHMP3SS. p<0.001 by one-tailed T-test. (D) Histogram showing the duration of each CHMP4B recruitment in the absence (n = 126 recruitments from 46 assemblies) or presence (n = 59 recruitments from 41 assemblies) of retroCHMP3SS. p<0.005 by one-tailed T-test. (E) Overlaid plots of average Gag recruitment for Gag-Pol-mEGFP without (black) or with (red) retroCHMP3SS and Gag-Pol-mEGFP-D25A with retroCHMP3 SS (blue). The D25A mutation inactivates protease function. Solid line indicates average and shaded overlay represents the 95% confidence interval. (F) Plot of average final intensity of Gag-Pol-mEGFP for the final three frames for each trace that visibly recruited mCherry-CHMP4B in the absence (black) or presence (red) of retroCHMP3SS or Gag-Pol-mEGFP-D25A with retroCHMP3SS (blue). Colored bars indicate median, n = 25 assemblies per condition. *** p<0.001, * p<0.05 by two-tailed T-test. ns – not significant. See also Fig. S4.
We and others previously reported that in a majority of VLP assembly events, ESCRT-III proteins and VPS4 are recruited once immediately prior to productive VLP budding, although multiple rounds of recruitment were observed in a minority of cases (Baumgartel et al., 2011; Bleck et al., 2014; Johnson et al., 2018; Jouvenet et al., 2011). Likewise, we observed that in the absence of retroCHMP3SS, mCherry-CHMP4B was recruited only once to mEGFP-marked virion assembly sites in 70% of the cases (Fig. 4A, C). In the presence of retroCHMP3SS, however, CHMP4B was recruited and lost multiple times at more than 60% of individual virion assembly sites (Fig. 4B, C). In extreme cases, more than seven cycles of CHMP4B recruitment to the same virion assembly site were observed. RetroCHMP3SS also increased the duration of each cycle of recruitment of CHMP4B to the VLP (Fig. 4D). In the absence of retroCHMP3SS, 83% of CHMP4B recruitment events lasted between 5 and 30 seconds, with occasional longer intervals. In contrast, when retroCHMP3SS was present, half of the visualized recruitment events lasted at least 30 seconds. These behaviors are consistent with multiple unproductive waves of ESCRT recruitment and failed membrane fission (Johnson et al., 2018). Thus, retroCHMP3SS interferes with productive ESCRT-III filament assembly and function.
In complementary experiments, we found that retroCHMP3SS did not alter the frequency of mCherry-VPS4A recruitment (Fig. S4A-C) but did reduce the residence time of VPS4A at the VLP, with nearly half of all VPS4A recruitments lasting ≤5 seconds in the presence of retroCHMP3SS (Fig. S4D). Thus, retroCHMP3SS also reduced the ability of ESCRT-III assemblies to bind and retain VPS4A, again suggesting that ESCRT-III filament assembly is compromised in the presence of retroCHMP3SS.
RetroCHMP3SS also altered the behavior of Gag-Pol-mEGFP virion assembly (Fig. 4E). Under control conditions, Gag-Pol-mEGFP fluorescence rose sharply during the initial virion assembly phase and then remained at a stable plateau until the endpoint of the experiment. In contrast, when retroCHMP3SS was present, Gag-Pol-mEGFP fluorescence at each VLP steadily decreased after reaching the initial plateau, so that the average mEGFP fluorescence intensity during the last three frames of each trace was lowered significantly (Fig. 4F). During viral assembly and maturation, the internal mEGFP fluorophore is released from Gag and Gag-Pol by PR processing. We found that the decrease in mEGFP-fluorescence required a functional protease because a construct with an inactive protease (Gag-Pol-D25A) did not lose fluorescence over time, even when retroCHMP3SS was present (Fig. 4E, F). Previous work has shown that precise temporal control of the sequential steps of virion assembly and budding is essential for viral infectivity (Bendjennat and Saffarian, 2016). In particular, when budding is delayed by ESCRT pathway inactivation, PR auto-activation and Gag-Pol processing precede budding, the processed Gag and Gag-Pol components escape through the open bud neck, and viral infectivity is reduced. A similar process appears to be occurring in the presence of retroCHMP3SS, where protease activation during delayed budding liberates mEGFP to diffuse back into the cytoplasm. Taken together, these results suggest that retroCHMP3SS impairs ESCRT-III filament assembly and delays membrane fission during budding.
Expression of mouse retroCHMP3 can be induced by interferon signaling
To determine whether retroCHMP3 is expressed in mice and squirrel monkeys, we first surveyed a panel of squirrel monkey and mouse cell lines as well as mouse tissues (Fig. S5). We observed detectable levels of Retrochmp3 RNA in several mouse and squirrel monkey cell lines as well as mouse testes and heart, although RNA abundance appeared to be low under basal conditions. Intriguingly, the murine Retrochmp3 ORF contains candidate upstream promoter elements, including two putative interferon-responsive STAT1/STAT3 binding sites, and a murine retroviral related sequence (MuRRS) element (Fig. 5A). Endogenous retroviruses are known to encode elements that can regulate expression through interferon-induced signaling (Chuong et al., 2017), suggesting that retroCHMP3MM might be regulated through immune signaling.
Fig. 5: Expression of mouse retroCHMP3 may be controlled by interferon signaling.

(A) Schematic of putative regulatory elements upstream of retroCHMP3 ORF in Mus musculus identified by transcription factor binding site prediction tools. Numbers indicate nucleotides relative to retroCHMP3 start codon. LTR – long terminal repeat, MuRRS – murine retrovirus-related sequence, ERV – endogenous retrovirus. (B) RT-PCR detection of retroCHMP3 RNA in mouse cardiac endothelial cells (MCECs) with and without interferon stimulation. RetroCHMP3 bands were excised, cloned, and sequenced to verify a match with the retroCHMP3 sequence. Representative gel from three independent repeats. RT – reverse transcriptase. (C) Droplet digital PCR (ddPCR) detection of retroCHMP3 (red) and RNAse L (grey) RNA in mouse cardiac endothelial cells (MCECs) with and without interferon stimulation. The housekeeping gene SDHA was used for normalization. Values were normalized to t=0, which was set to 1. Mean ± SD from 3 experimental replicates. See also Fig. S5.
Based on our observation of Retrochmp3 RNA expression in the mouse heart, we treated mouse cardiac endothelial cells with interferon and determined RNA expression levels by qRT-PCR (Fig. 5B) and droplet digital PCR (Fig. 5C). Interferon treatment modestly induced Retrochmp3 expression over 20 hours, compared to low levels of expression under basal conditions. Retrochmp3 RNA levels in squirrel monkey B cells were not increased by interferon treatment (Fig. S5B). These results support a model in which in mice, the retrotransposed Retrochmp3 coding sequence landed in a region of the genome where it could be regulated by immune signaling.
Discussion
We have identified RETROCHMP3 copies in three different mammalian species, each of which independently gained a premature stop codon that removed ~70 C-terminal amino acids. The frequency and retention of these independently evolved copies strongly suggest that they confer a selective advantage. When overexpressed in human cells, retroCHMP3 proteins potently block budding of a broad range of ESCRT-dependent viruses, yet they do not drastically inhibit the ESCRT-dependent cellular pathway of cytokinetic abscission. Their broad antiviral activity is consistent with the idea that retroCHMP3 proteins act by impeding a cellular function that is hijacked by a variety of viruses rather than targeting a specific viral protein.
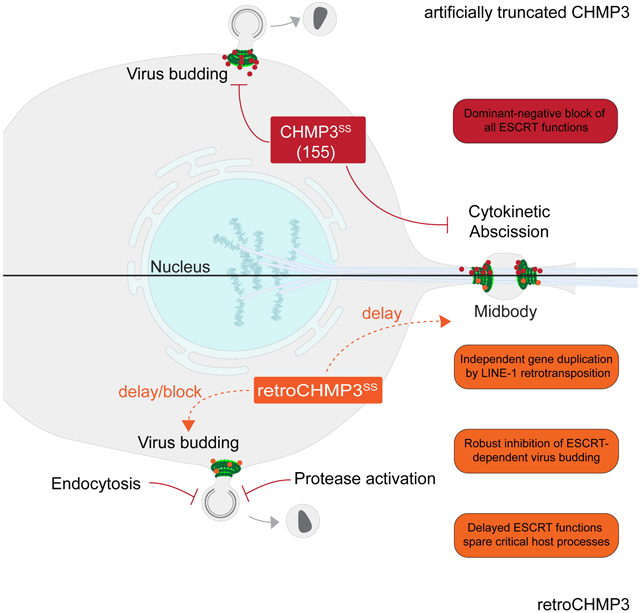
A model explaining how retroCHMP3 can inhibit virus budding, yet differs from genetically engineered truncations of CHMP3 is shown in the graphical abstract. Previous work has shown that truncated parental CHMP3 proteins can potently inhibit HIV-1 budding (Zamborlini et al., 2006), and also inhibit cellular ESCRT functions (Dukes et al., 2008). In this simple case, truncation activates the CHMP3 proteins for stable heteropolymeric filament formation, retains tight binding to CHMP4B (and possibly also other ESCRT-III proteins), and promotes cytoplasmic punctae formation. Truncated CHMP3 proteins could thus sequester ESCRT-III subunits away from functional sites such as the midbody and, even when recruited to the correct sites of action, create inert filaments that cannot undergo the VPS4-dependent subunit turnover required for membrane fission (Mierzwa et al., 2017). In contrast, retroCHMP3 proteins have evolved to reduce CHMP4B binding and apparently no longer form highly stable, inert ESCRT-III heteropolymers. We hypothesize that retroCHMP3 proteins therefore impair functional ESCRT-III filament assembly to a lesser degree than truncated CHMP3, thereby slowing down ESCRT-mediated membrane fission rather than blocking it entirely.
We envision that retroCHMP3 proteins achieve selective antiviral activity without undue cellular toxicity by exerting a disproportionate impact on the rapid and time-sensitive process of virus budding. After the initial assembly of virions at the plasma membrane, ESCRT-III proteins and VPS4 are recruited transiently for seconds or minutes, followed by membrane fission (Baumgartel et al., 2011; Bleck et al., 2014; Johnson et al., 2018). HIV-1 assembly, budding, and maturation are highly coordinated processes, and budding delays that disturb the proper sequence thus also reduce infectivity by allowing processed viral components to escape from the virion prior to bud neck closure (Bendjennat and Saffarian, 2016). Furthermore, virion particles that remain attached to the plasma membrane can be reinternalized and degraded by endocytosis (Neil et al., 2006). In contrast, at least some essential cellular ESCRT-mediated processes, such as abscission, occur over much longer time scales of 1-2 hours (Elia et al., 2011; Guizetti et al., 2011; Hesse et al., 2012; Mackay and Ullman, 2015) and can tolerate very significant delays (Nahse et al., 2017), thereby sparing them from the most detrimental effects of retroCHMP3. In line with this, EGF-mediated degradation of the EGFR receptor, which displays similar dynamics as virus budding (Wenzel et al., 2018), is susceptible to inhibition by retroCHMP3 proteins. However, inhibition of receptor degradation presumably contributes less to acute cytotoxicity than inhibition of cytokinesis, and therefore can be better tolerated.
Inhibition of ESCRT activity during infection has been described in other contexts: Intracellular bacteria such as Mycobacteria and Salmonella species replicate in membrane-bound vacuoles after their uptake into cells by phagocytosis or receptor-mediated endocytosis but eventually gain access to the host cytosol by breaching endolysosomal membranes (Bianchi and van den Bogaart, 2020; Friedrich et al., 2012). ESCRT activity can restrict the growth of mycobacteria, presumably by mediating repair of endosomal membrane damage (Lopez-Jimenez et al., 2018; Philips et al., 2008; Portal-Celhay et al., 2016; Skowyra et al., 2018). Mycobacterium tuberculosis secretes the effector proteins EsxG and EsxH to prevent recruitment of host ESCRT-III proteins to phagolysosomes and promote virulence (Mehra et al., 2013; Mittal et al., 2018; Portal-Celhay et al., 2016). Similarly, Shigella flexneri IcsB suppresses antibacterial autophagy by interaction with the ESCRT-III protein CHMP5 (Liu et al., 2018). While the exact mechanism for ESCRT inhibition remains to be determined in these cases, they highlight the importance of pathogen engagement and modulation of the host ESCRT pathway.
Although we cannot yet say whether the potent inhibition of virus budding that we observe in cultured cells occurs in vivo, Retrochmp3 mRNA is expressed in squirrel monkey and mouse cell lines and in mouse testes and heart (Fig. S5). Retrochmp3 mRNA levels appear to be low under normal conditions, but are modestly induced by type I interferon in mice (Fig. 5), raising the possibility that retroCHMP3 levels could be regulated in specific cell types and/or during viral infection.
LINE1-mediated retrocopies are generally considered to be defective pseudogenes since the promoter and regulatory elements of parental genes are not retrocopied. Additionally, retrocopies are often truncated, either at the 5’ end due to incomplete reverse transcription during the retrotransposition event or due to premature stop codons (Casola and Betran, 2017). In some cases, however, retrocopies can acquire new expression patterns or functions (Kaessmann et al., 2009; Kubiak and Makalowska, 2017). Another ESCRT-III protein, CHMP1B, was retrotransposed in a common ancestor of placental mammals. In primates, this retrocopy replaced the parental gene and became the predominantly expressed form, demonstrating the potential of ESCRT-III retrocopies to gain regulatory elements and become functional (Ciomborowska et al., 2013). Retrotransposition events have also contributed to the evolution of other antiviral proteins: The restriction factor TRIM5α blocks replication of HIV-1 in Old World monkeys, but TRIM5α variants from New World monkeys restrict HIV-1 less efficiently (Song et al., 2005; Stremlau et al., 2004). In the New World owl monkey, however, retrotranposition of the coding sequence of the HIV-1 capsid binding factor cyclophilin A between coding exons of TRIM5α created the chimeric TRIMCyp gene that potently blocks HIV-1 replication (Nisole et al., 2004; Sayah et al., 2004). Further, some Old World macaque species encode a chimeric TRIMCyp protein that evolved by an independent retrotransposition event and restricts replication of HIV-2, feline immunodeficiency virus, and simian immunodeficiency virus SIVAGMtan (Brennan et al., 2008; Liao et al., 2007; Newman et al., 2008; Virgen et al., 2008; Wilson et al., 2008). Similarly, duplication of the retroviral restriction factors Fv1 in wild mice (Yap et al., 2020) and APOBEC3 in primates (Yang et al., 2020) by retrotransposition may have expanded the capacity of these proteins to restrict retroviruses by increasing the number of gene copies per genome. Retroduplication of CHMP3 in the presence of an intact parental gene appears to be another variation on this theme that allowed RETROCHMP3 genes to be repurposed as virus budding inhibitors.
RetroCHMP3 proteins arose independently in at least three mammalian species by convergent evolution. We have also recently identified multiple full-length and truncated CHMP3 retrocopies in other rodent and primate genomes, including two retrocopies in mouse lemurs and dwarf lemurs that are truncated to approximately 150 amino acids (Rheinemann et al., 2021). We hypothesize that duplicated ESCRT-III proteins, and CHMP3 in particular, are especially suited to become budding inhibitors. ESCRT-III proteins consist of an N-terminal core domain that mediates filament assembly and a C-terminal autoinhibitory region. This architecture is particularly conducive to creating dominantly inhibitory ESCRT-III proteins simply by introducing a premature stop codon that removes the sites required for autoinhibition and recycling (Bajorek et al., 2009; Lata et al., 2008; Muziol et al., 2006; Shim et al., 2007; Stuchell-Brereton et al., 2007). According to our model, detoxification requires weakening of the interaction of CHMP3 with other ESCRT-III proteins. ESCRT-III proteins form highly interlocked polymers (Bertin et al., 2020; McCullough et al., 2015; Moser von Filseck et al., 2020; Nguyen et al., 2020) that can be disturbed by different combinations of amino acid substitutions along the many interaction interfaces. Therefore, attenuation of ESCRT-III filament strength can be achieved in many different ways, as evidenced by the fact that retroCHMP3 proteins in different species do not share amino acid substitutions.
The ortholog of CHMP3 in yeast, Vps24, forms an essential bridge between two paralogous ESCRT-III proteins and binding partners Vps2 (CHMP2) and Snf7 (CHMP4) and is required for MVB formation (Babst et al., 2002; Teis et al., 2008). Interestingly, recent work demonstrates that increased concentrations of Vps2 or a Vps2 version with three amino acid substitutions in the N-terminal basic region can functionally replace Vps24 during MVB formation (Banjade et al., 2021). In mammalian cells, CHMP3 is dispensable for retrovirus budding (Bartusch and Prange, 2016; Morita et al., 2011). However, CHMP3 depletion leads to abscission defects (Morita et al., 2010) and impairs EGFR degradation (Bache et al., 2006), and CHMP3 is therefore not dispensable for cellular ESCRT functions. Thus, CHMP2 and CHMP4 appear to be able to interact and form filaments in the absence of CHMP3 during certain ESCRT-mediated processes. We speculate that this may have rendered CHMP3 more tolerant to the accumulation of mutations and therefore particularly suitable to be repurposed as a virus budding inhibitor. In support of this idea, retrocopies of CHMP2A and CHMP4B are much rarer in primate and rodent genomes than CHMP3 retrocopies and the retrotransposed genes that are identifiable are highly degraded in the majority of cases (Rheinemann et al., 2021). These observations indicate that CHMP3 retrocopies are either more frequently generated and/or are retained more frequently than CHMP2A and CHMP4B retrocopies. We cannot presently say whether this higher frequency of CHMP3 retrocopies is due to reduced CHMP3 essentiality, and therefore a higher tolerance to mutations, or whether other factors such as retroduplication frequency also play a role.
In summary, we have identified retroCHMP3 proteins as broad, potent inhibitors of ESCRT-dependent virus budding. A priori, it might appear that viruses that co-opt essential cellular processes like the ESCRT pathway have found an Achilles heel that cannot easily be overcome by the host. However, retroCHMP3 proteins modify ESCRT activity to exploit a key difference in the ways that cells and viruses use the pathway. Additionally, the observation that retroCHMP3 alters ESCRT pathway function instead of targeting a viral protein raises the intriguing possibility that retroCHMP3 may be more resistant to viral counter-adaptations than other antiviral proteins that directly inhibit viral replication. Thus, retroCHMP3 proteins reveal a possibility for blocking virus budding while maintaining cellular ESCRT functions.
Limitations of the study
We characterized the effect of retroCHMP3 proteins on viral budding and on a subset of cellular ESCRT-dependent processes in cultured cells. The independent evolution and retention of RETROCHMP3 copies in multiple different mammals indicates that retroCHMP3 proteins also provide a selective advantage in vivo, but the roles of endogenous retroCHMP3 proteins in inhibiting virus budding remains to be determined. Future studies characterizing endogenous retroCHMP3 expression in multiple mammalian species, and testing whether endogenous retroCHMP3 proteins can inhibit budding in vivo without inducing undue toxicity, will be critical for determining the natural antiviral roles of retroCHMP3 proteins.
We also do not yet fully understand how retroCHMP3 evolved to reduce toxicity while retaining antiviral activity. Our data indicate that reduced toxicity reflects weakening of the interaction of retroCHMP3 with other ESCRT-III proteins, and a corresponding attenuation of dominant-negative effects. However, further studies are required to assess fully the interactions of retroCHMP3 proteins with other known CHMP3 binding partners, including CHMP4, CHMP2, and CHMP3 itself. Defining whether/how retroCHMP3 is recruited to sites of viral budding and cellular ESCRT-mediated processes might also reveal details helping to better define the mechanism of action of retroCHMP3.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and request for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nels C. Elde (nelde@genetics.utah.edu).
Materials Availability
Plasmids generated in this study have been deposited to Addgene.
Data and Code Availability
Sequencing data of CHMP3 retrocopies is provided in Table S1.
This study did not generate any new code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cells
HEK293T cells (CRL-3216, Human embryonic kidney endothelial cells, sex: female, RRID: CVCL_0063), GSML cells (CRL-2699, squirrel monkey B lymphoblast cell line, sex: male, RRID: CVCL_3691), SML cells (CRL-2311, squirrel monkey lymphocytes cell line, clone 4D8, sex: male, RRID: CVCL_6446), RAW 264.7 cells (TIB-71, mouse macrophage cell line, sex: male, RRID: CVCL_0493) and J774A.1 cells (TIB-67, mouse monocyte/macrophage cells, sex: female, RRID: CVCL_0358) were obtained from ATCC. MCEC cells (mouse cardiac endothelial cells, CLU510, sex: unknown, RRID: CVCL_D355) were obtained from Cedarlane. MT-4 cells (Human T cells, NIH-ARP 120, sex: male, RRID: CVCL_2632) were obtained from the NIH AIDS Reagent program. HeLa N cells (human cervical epithelial cells, sex: female) were a kind gift from Katharine Ullman (University of Utah). DFJ8 cells (DF-1 chicken embryonic fibroblasts stably expressing MCAT-1, sex: unknown) were a kind gift from Walther Mothes (Yale University). MES cells (mouse embryonic stem cells, line G4-56, passage 14, sex: unknown) were a kind gift from Mario Capecchi (University of Utah). MEF cells (mouse embryonic fibroblasts, sex: unknown) were isolated from WT E13.5 C57BL/6 embryos and immortalized by lentiviral transduction with large T antigen (Zhu et al., 1991).
HEK293T, DFJ8, HeLa N, MCEC and MEFs were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Thermo Fisher Scientific) containing 10% FBS at 37°C and 5% CO2. RAW 264.7 cells and J774A.1 cells were maintained in DMEM (Gibco, Thermo Fisher Scientific) containing 5% FBS at 37°C and 5% CO2. MT-4 and squirrel monkey B lymphoblast cells were maintained in RPMI-1640 medium (Gibco, Thermo Fisher Scientific) containing 10% FBS (Atlanta Biologicals) at 37°C and 5% CO2. MES cells were maintained as previously described (George et al., 2007).
Cells were tested for mycoplasma contamination every 3 months using the PCR Mycoplasma Detection Kit (abm). Cells have not been authenticated.
Genomic DNA sources
Sources for genomic DNA were as follows: Blood samples from Cebus apella (tufted capuchin, three male individuals, 8, 8 and 12 years old) were obtained from the NIH Animal Center (Dickerson MD, USA). Blood samples from Ateles geoffroyi (black-handed spider monkey, male, 44 years old) and Ateles fusciceps (brown-headed spider monkey, female, 45 years old) were obtained from Utah’s Hogle Zoo (Salt Lake City, UT, USA). Blood samples from Saimiri boliviensis boliviensis (Bolivian squirrel monkey, 3 male and 3 female individuals, ages unknown), Saimiri boliviensis peruviensis (Peruvian squirrel monkey, 6 female individuals, ages unknown) and Saimiri sciureus sciureus (Guyanese squirrel monkey, 3 male and 3 female individuals, ages unknown) were obtained from the Institute Pasteur de la Guyane (Cayenne Cedex, French Guiana). Primary fibroblast cell lines and genomic DNA from Saimiri sciureus (common squirrel monkey, AG05311, female, age unknown) were obtained from Coriell Cell Repositories. Blood samples from Mus musculus (house mouse, C57BL/6, male, 20 weeks old) were obtained from the University of Utah animal facility (Salt Lake City, UT, USA).
METHOD DETAILS
Identification, sequencing and cloning of CHMP3 retrocopies
Retrocopies of CHMP3 were identified using BLAT on the UCSC genome browser (http://genome.ucsc.edu/index.html) or NCBI BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Specifically, the full-length CHMP3 cDNA (5’ UTR, coding sequence and 3’UTR) encoded by Homo sapiens (ENST00000263856.9, https://www.ncbi.nlm.nih.gov/nuccore/NM_016079.4) or Mus musculus (ENST00000263856.9, https://www.ncbi.nlm.nih.gov/nuccore/NM_025783.4) were used as queries against mammalian genomes on the UCSC browser or whole genome shotgun contigs via NCBI BLAST to retrieve retrogenes present in the corresponding genomes. Candidate retrocopies were identified as CHMP3 sequences lacking introns – a key structural characteristic of processed pseudogenes. In all cases, we have identified the target site duplication flanking each retrocopy.
Retrogene insertions in New World monkeys and Mus genomes were validated by PCR amplification of genomic DNA using primers positioned in flanking sequences upstream and downstream of the target-site duplication. For those species where no genomic DNA was available, sequences are from publicly available whole genome shotgun contigs on NCBI BLAST. Primer sequences used are listed in Table S2. PCR products were resolved on 2% agarose gels followed by extraction and purification of candidate DNA amplicons using Zymoclean Gel DNA Recovery Kits (Zymo Research). Purified PCR products were cloned using the TOPO TA cloning kit (Invitrogen), following the manufacturer’s instructions. Following transformation and plating, bacterial colonies were selected and grown in overnight cultures for plasmid purification using the Zyppy Plasmid Miniprep kit (Zymo Research). Sanger sequencing of clones was performed using M13F and M13R primers. RetroCHMP3 sequences are reported in Table S1.
Plasmids
The expression constructs used in this study are described in the Key Resources Table. All newly generated expression constructs have been submitted to the Addgene plasmid repository (https://www.addgene.org/). The HIV-1 expression construct NLENG1-IRES-GFP was a kind gift from David N. Levy (New York University) (Levy et al., 2004). The FrMLV expression vector pLRB303 FrMLV PRR Env(273)GFP was a kind gift from Walther Mothes (Yale University) (Lehmann et al., 2005; Sherer et al., 2003). The pLNCX2-mCherry-CHMP4B and pLNCX2-mCherry-VPS4A plasmids were previously described (Bleck et al., 2014; Johnson et al., 2018). pCRV1-NL4.3GagPol-mEGFP and GagPol-mEGFP-D25A were created from the pCRV1-NL4.3-GagPol plasmid, which was a kind gift from Paul Bieniasz (Rockefeller University), by inserting mEGFP in the matrix domain with a matrix/capsid cleavage site on either side of the mEGFP.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-HIV CA | Covance | UT415 |
| Rabbit anti-HIV MA | Covance | UT556 |
| Rabbit anti-EIAV CA | Covance | UT418 |
| Friend murine leukemia virus antiserum from goat | ATCC | VR-1537 |
| Mouse monoclonal anti-PIV5 M | (Randall et al., 1987) | M-f |
| Mouse monoclonal anti-PIV5 NP | (Randall et al., 1987) | Np-a |
| Mouse monoclonal anti-beta actin | ThermoFisher Scientific | AM4302; RRID: AB_2536382 |
| Mouse monoclonal anti-GAPDH | EMD Millipore | MAB374; RRID: AB_2107445 |
| Rabbit anti-HA tag | Sigma-Aldrich | H6908; RRID: AB_260070 |
| Rabbit anti-HA tag | Proteintech | 51064-2-AP; RRID: AB_11042321 |
| Mouse monoclonal anti-myc tag, clone 4A6 | EMD Millipore | 05-724; RRID: AB_309938 |
| Rabbit anti-GFP | ThermoFisher Scientific | A-6455; RRID: AB_221570 |
| Mouse monoclonal anti-GFP | Roche | 11814460001; RRID:AB_390913 |
| Mouse monoclonal anti-FLAG M2 | Sigma-Aldrich | F1804; RRID: AB_262044 |
| Mouse monoclonal anti-alpha-tubulin | Sigma-Aldrich | T5168; RRID: AB_477579 |
| Rabbit EGF Receptor (D38B1) XP | Cell Signaling Technology | 4267: RRID: AB_2246311 |
| Goat-anti-rabbit IgG Alexa Fluor 488 | Thermo Fisher Scientific | A-11034; RRID: AB_2576217 |
| Goat-anti-mouse IgG Alexa Fluor 594 | Thermo Fisher Scientific | A-11032; RRID: AB_2534091 |
| Goat-anti-Rabbit IgG HRP conjugate | EMD Millipore | AP132; RRID: AB_92486 |
| Goat-anti-Mouse IgG/IgM HRP conjugate | EMD Millipore | AP130P; RRID: AB_91266 |
| IRDye® 800CW Goat anti-Rabbit IgG Secondary Antibody | LI-COR | 926-32211; RRID: AB_621843 |
| IRDye® 680RD Goat anti-Mouse IgG Secondary Antibody | LI-COR | 926-68070; RRID: AB_10956588 |
| Alkaline phosphatase-conjugated AffiniPure Goat Anti-Mouse IgG | Jackson ImmunoResearch | 115-055-062 |
| Alkaline phosphatase-conjugated AffiniPure Goat Anti-Rabbit IgG | Jackson ImmunoResearch | 111-055-003 |
| Bacterial and Virus Strains | ||
| HIV-1 NLENG1-IRES-GFP | David N. Levy, New York University (Levy et al., 2004) | N/A |
| pLRB303 FrMLV PRR Env(273)GFP | Walther Mothes, Yale School of Medicine (Lehmann et al., 2005; Sherer et al., 2003) | N/A |
| Biological Samples | ||
| Cebus apella (tufted capuchin) blood sample | NIH Animal Center (Dickerson MD, USA) | N/A |
| Ateles geoffroyi (black-handed spider monkey) blood sample | Utah’s Hogle Zoo (Salt Lake City UT, USA) | N/A |
| Ateles fusciceps (brown-headed spider monkey) blood sample | Utah’s Hogle Zoo (Salt Lake City UT, USA) | N/A |
| Saimiri boliviensis boliviensis (Bolivian squirrel monkey) blood sample | Institute Pasteur de la Guyane (Cayenne Cedex, French Guiana) | N/A |
| Saimiri boliviensis peruviensis (Peruvian squirrel monkey) blood sample | Institute Pasteur de la Guyane (Cayenne Cedex, French Guiana) | N/A |
| Saimiri sciureus sciureus (Guyanese squirrel monkey) blood sample | Institute Pasteur de la Guyane (Cayenne Cedex, French Guiana) | N/A |
| Saimiri sciureus (common squirrel monkey) primary fibroblasts and genomic DNA | Coriell Cell Repositories | AG05311 |
| Mus musculus (house mouse, C57BL/6) blood and tissue samples | University of Utah animal facility (Salt Lake City, UT, USA) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| e. coli-derived mouse IFN-gamma protein | R&D Systems | Cat# 485-MI-100 |
| EK293-derived mouse IFN-beta protein | R&D Systems | Cat# 8234-MB-010 |
| Universal Type I Interferon | pbl assay science | Cat# 11200-2 |
| Lipofectamine 2000 | Thermo Fisher Scientific | Cat# 11668019 |
| Lipofectamine LTX with Plus Reagent | Thermo Fisher Scientific | Cat# 15338100 |
| FuGENE 6 | Promega | Cat# E2691 |
| EMbed-812 Resin | Electron Microscopy Sciences | Cat# 14120 |
| DAPI Fluoromount-G Mounting Medium | Southern Biotech | Cat# 0100-20 |
| Strep-Tactin Sepharose 50% suspension | IBA Lifesciences | Cat# 2-1201-010 |
| Vistra ECF substrate | GE Healthcare | Cat# RPN5785 |
| Critical Commercial Assays | ||
| Zymoclean Gel DNA Recovery Kit | Zymo Research | Cat# D4001 |
| TOPO TA cloning kit | ThermoFisher Scientific | Cat# 450071 |
| Zyppy Plasmid Miniprep kit | Zymo Research | Cat# D4036 |
| CellTiter-Glo Luminescent cell viability assay | Promega | Cat# G7570 |
| Zymo Quick-RNA Miniprep kit | Zymo Research | Cat# R1054 |
| Maxima First Strand Synthesis Kit | ThermoFisher Scientific | Cat# K1672 |
| Phusion Flash High-Fidelity PCR Master Mix | ThermoFisher Scientific | Cat# F548L |
| ddPCR Supermix for Probes (no dUTP) | BioRad | Cat# 1863024 |
| Experimental Models: Cell Lines | ||
| Human: 293T | ATCC | CRL-3216; RRID: CVCL_0063 |
| Human: HeLa N | Katharine S. Ullman (University of Utah) | N/A |
| Human: MT4 | NIH AIDS Reagent Program | Cat# 120; RRID: CVCL_2632 |
| Squirrel monkey: GSML | ATCC | CRL-2699; RRID: CVCL_3691 |
| Squirrel monkey: SML, clone 4D8 | ATCC | CRL-2311; RRID: CVCL_6446 |
| Mouse: RAW 264.7 | ATCC | TIB-71; RRID: CVCL_0493 |
| Mouse: J774A.1 | ATCC | TIB-67; RRID: CVCL_0358 |
| Mouse: mouse embryonic stem cells, passage 14 | Mario Capecchi (University of Utah) | G4-56 |
| Mouse: mouse embryonic fibroblasts | E13.5 C57BL/6 embryo | N/A |
| Mouse: mouse cardiac endothelial cells (MCEC) | Cedarlane | CLU510; RRID: CVCL_D355 |
| Chicken: DFJ8 | Walther Mothes (Yale University) | N/A |
| Oligonucleotides | ||
| Primers | This study | See Table S1 |
| Recombinant DNA | ||
| pEV53 | (Olsen, 1998) | N/A |
| pSIN6.1CeGFPW | (Olsen, 1998) | N/A |
| phCMV- VSV-G | (Yee et al., 1994) | N/A |
| pCR3.1-myc-EbVp40 | Paul Bieniasz, Rockefeller University, (Martin-Serrano et al., 2001) | N/A |
| pCR3.1-myc-EbVp40 P11L | Paul Bieniasz, Rockefeller University, (Martin-Serrano et al., 2001) | N/A |
| pCAGGS-PIV5 M | Anthony Schmitt, Pennsylvania State University (Schmitt et al., 2002) | N/A |
| pCAGGS-PIV5 HN | Anthony Schmitt, Pennsylvania State University (Schmitt et al., 2002) | N/A |
| pCAGGS-PIV5 NP | Anthony Schmitt, Pennsylvania State University (Schmitt et al., 2002) | N/A |
| pEGFP-VPS4 | (Garrus et al., 2001) | DNASU HsCD00587768 |
| pEGFP-VPS4 E228Q | (Garrus et al., 2001) | Addgene #80351 |
| pCMV(Δ3)-squirrel-monkey-full-length-CHMP3-HA | This study | Addgene #154169 |
| pCMV(Δ2)-squirrel-monkey-full-length-CHMP3-HA | This study | Addgene #154170 |
| pCMV(Δ1)-squirrel-monkey-full-length-CHMP3-HA | This study | Addgene #154171 |
| pEF1α-squirrel-monkey-full-length-CHMP3-HA | This study | Addgene #154172 |
| pEF1α-squirrel-monkey-CHMP3(155)-HA | This study | Addgene #154173 |
| pEF1α-squirrel-monkey-retroCHMP3-HA | This study | Addgene #154174 |
| pCMV(WT)-mouse-full-length-CHMP3-HA | This study | Addgene #154175 |
| pEF1α-mouse-full-length-CHMP3-HA | This study | Addgene #154176 |
| pEF1α-mouse-CHMP3(147)-HA | This study | Addgene #154177 |
| pEF1α-mouse-retroCHMP3-HA | This study | Addgene #154178 |
| pEF1α-spider-monkey-retroCHMP3-HA | This study | Addgene #154179 |
| pCAG-OSF-PP-human-CHMP3 | This study | Addgene #154180 |
| pCAG-OSF-PP-human-CHMP3(150) | This study | Addgene #154181 |
| pCAG-OSF-PP-squirrel-monkey-CHMP | This study | Addgene #154182 |
| pCAG-OSF-PP-squirrel-monkey-CHMP3(155) | This study | Addgene #154183 |
| pCAG-OSF-PP-squirrel-monkey-retroCHMP3 | This study | Addgene #154184 |
| pCRV1-HIV-1NL4-3 GagPol | Paul Bieniasz, Rockefeller University | N/A |
| pCRV1-HIV-1NL4-3 GagPol-mEGFP | Sanford M. Simon, Rockefeller University | N/A |
| pCRV1-HIV-1NL4-3 GagPol-D25A | Sanford M. Simon, Rockefeller University | N/A |
| pEF1α-squirrel-monkey-retroCHMP3-T2A-H2B-BFP | This study | Addgene #154185 |
| pLNCX2-mCherry-CHMP4B | Sanford M. Simon, Rockefeller University, (Bleck et al., 2014; Johnson et al., 2018) | Addgene #116923 |
| pLNCX2-mCherry-VPS4A | Sanford M. Simon, Rockefeller University, (Bleck et al., 2014; Johnson et al., 2018) | Addgene #115334 |
| Software and Algorithms | ||
| GraphPad Prism 8 | www.graphpad.com | N/A |
| BLAST-like Alignment Tool (BLAT) | (Kent, 2002) | https://genome.ucsc.edu/cgi-bin/hgBlat |
| NCBI BLAST analyses (Altschul et al., 1990) | (Altschul et al., 1990) | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| MetaMorph Software 7.10.3.279 | https://www.moleculardevices.com/products/cellular-imaging-systems/acquisition-and-analysis-software/metamorph-microscopy | N/A |
| NSite | (Shahmuradov and Solovyev, 2015) | N/A |
| CiiiDER | (Gearing et al., 2019) | N/A |
HIV-1 budding assays
0.8 × 106 HEK293T cells were seeded in 6-well plates 18-24 h before transfection. Cells were co-transfected with 1 μg HIV-1 NLENG1-IRES-GFP expression vector and increasing amounts of expression vectors for full-length CHMP3 or retroCHMP3 to generate an expression gradient using 10 μl Lipofectamine 2000 (Thermo Fisher Scientific) according to manufacturer’s instructions. Plasmid DNA amounts were as follows: 2 μg pCMV(Δ3)-squirrel-monkey-full-length-CHMP3-HA, 2 μg pCMV(Δ2)-squirrel-monkey-full-length-CHMP3-HA, 2 μg pCMV(Δ1)-squirrel-monkey-full-length-CHMP3-HA; 500 ng, 1 μg and 1.5 μg pEF1α-squirrel-monkey-retroCHMP3-HA; 100 ng, 250 ng and 500 ng pCMV(WT)-mouse-full-length-CHMP3-HA; 500 ng, 1 μg and 1.5 μg pEF1α-mouse-retroCHMP3-HA, 2 μg pEF1α-spider-monkey-retroCHMP3-HA. If necessary, plasmid amounts were adjusted with empty pCMV(WT) vector. The medium was replaced with 2 ml DMEM 4-6 h later. Cells were harvested for western blot analysis and supernatants were harvested for titer measurements and western blot analysis as described below 24 h post-transfection.
For western blot analyses, virions from 1 ml supernatant were pelleted by centrifugation through a 200 μl 20% sucrose cushion (90 min, 15,000×g, 4°C) and denatured by adding 50 μl 1x Laemmli SDS-PAGE loading buffer and boiling for 5 min. Cells were washed in 1 ml PBS, lysed for 5min on ice in 200 μl Triton lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% Triton X-100) supplemented with mammalian protease inhibitor (Sigma-Aldrich). 150 μl 2× Laemmli SDS-PAGE loading buffer supplemented with 10% 2-mercaptoethanol (Sigma-Aldrich) were added, and samples were boiled for 10 min. Proteins were separated by SDS-PAGE, transferred onto PVDF membranes and probed with antibodies. Primary antibodies were as follows: anti-HIV CA (Covance, UT415), anti-HIV MA (Covance, UT556), anti-HA (Sigma-Aldrich, H6908, RRID: AB_260070), anti-GAPDH (EMD Millipore, MAB374, RRID: AB_2107445). Antibodies with UT numbers were raised against recombinant proteins purified in the Sundquist laboratory. Bands were visualized by probing the membrane with fluorescently labeled secondary antibodies (Li-Cor Biosciences) and scanning with an Odyssey Imager (Li-Cor Biosciences).
HIV-1 titers were assayed in MT-4 cells by quantifying GFP expression in target cells. Briefly, MT-4 cells (100,000 cells/well, 96-well plate) were infected with three different dilutions of virus-containing culture media in duplicate. After 16 h, dextran sulfate was added to a final concentration of 100 μg/ml. Cells were harvested after 48 h, washed in PBS three times and fixed with 2% paraformaldehyde for 20 min at room temperature. Percentages of GFP-positive cells were determined by flow cytometry on a FACSCanto (BD Biosciences).
FrMLV, EIAV and EBOV Vp40 budding assays
5 × 105 HEK293T cells (FrMLV, EIAV) or 8 × 105 HEK293T cells (EBOV Vp40) were seeded in 6-well plates 18-24 h before transfection. Cells were co-transfected with 2 μg FrMLV-GFP expression vector pLRB303 FrMLV PRR Env(273)GFP (Lehmann et al., 2005; Sherer et al., 2003) or an EIAV vector system (Olsen, 1998; Yee et al., 1994) comprising 0.2 μg pEV53, 0.2 μg pSIN6.1CeGFPW and 0.075 μg phCMV-VSV-G, or 1 μg Myc-EbVp40-WT (Martin-Serrano et al., 2001) or Myc-EbVp40-P11L (Martin-Serrano et al., 2004) expression vector and increasing amounts of expression vectors for full-length CHMP3 or retroCHMP3 to generate an expression gradient using 10 μl Lipofectamine 2000 (Thermo Fisher Scientific) according to manufacturer’s instructions. Plasmid DNA amounts were as follows: 2 μg pCMV(Δ3)-squirrel-monkey-full-length-CHMP3-HA, 2 μg pCMV(Δ2)-squirrel-monkey-full-length-CHMP3-HA, 2 μg pCMV(Δ1)-squirrel-monkey-full-length CHMP3-HA; 500 ng, 1 μg and 1.5 μg pEF1α-squirrel-monkey-retroCHMP3-HA; 100 ng, 250 ng and 500 ng pCMV(WT)-mouse-full-length-CHMP3-HA; 500 ng, 1 μg and 1.5 μg pEF1α-mouse-retroCHMP3-HA. If necessary, plasmid amounts were adjusted with empty pCMV(WT) vector. The medium was replaced with 2 ml DMEM 4-6 h later. Cells were harvested for western blot analysis and supernatants were harvested for titer measurements and western blot analysis as described below 48 h (EIAV and FrMLV) or 18 h (EbVp40) post-transfection.
Western blot analyses were carried out as described for HIV-1. Primary antibodies were as follows: anti-EIAV CA (UT418), anti-FriendMLV (ATCC, VR-1537), anti-HA (Sigma- Aldrich, H6908, RRID: AB_260070), anti-myc (EMD Millipore, 05-724, RRID: AB_309938), anti-GAPDH (EMD Millipore, MAB374, RRID: AB_2107445). Bands were visualized by probing the membrane with fluorescently labeled secondary antibodies (Li-Cor Biosciences) and scanning with an Odyssey Imager (LI-COR Biosciences).
Titers for the EIAV vector and FrMLV were assayed in HEK293T or DFJ cells, respectively, by quantifying GFP expression in target cells. Briefly, cells (5 × 104 cells/well, 24-well plate) were infected with three different dilutions of virus-containing culture media. For FrMLV, 4 μg/ml polybrene was added during infection. After 16 h, fresh growth medium was added. Cells were harvested after 48 h using 0.25% trypsin-EDTA (Gibco, ThermoFisher Scientific) and fixed with 2% paraformaldehyde for 20 min at RT. Percentages of GFP-positive cells were determined by flow cytometry on a FACSCanto (BD Biosciences).
For EBOV Vp40 release, band intensities from Western Blots of supernatants were quantified using LI-COR Image Studio software and normalized to band intensities of cellular fractions.
PIV-5 budding assays
For PIV5 budding assays, HEK293T cells in 6-well plates were transfected at 70-80% confluency using Lipofectamine-Plus reagents (Thermo Fisher Scientific) according to manufacturer’s instructions. Cells were transfected with plasmids encoding PIV5 M, NP, and HN proteins for generation of virus-like particles as described previously (Schmitt et al., 2002), together with plasmids encoding full-length CHMP3, retroCHMP3, VPS4, or VPS4-E228Q. Plasmid amounts were as follows: 300 ng pCAGGS-PIV5 M, 100 ng pCAGGS-PIV5 NP, 750 ng pCAGGS-PIV5 HN, 100 ng pGFP-VPS4A, 100 ng pGFP-VPS4A E228Q, 2 μg pCMV(Δ3)-squirrel-monkey-full-length-CHMP3-HA, 2 μg pCMV(Δ2)-squirrel-monkey full-length-CHMP3-HA, 2 μg pCMV(Δ1)-squirrel-monkey-full-length-CHMP3-HA; 500 ng, 1 μg and 1.5 μg pEF1α-squirrel-monkey-retroCHMP3-HA. Total plasmid quantities were equalized using pCMV empty vector. After overnight incubation, the culture medium was replaced with DMEM supplemented with 2% fetal bovine serum. 40 h post-transfection, cell and culture media fractions were collected. Cells were pelleted and lysed in SDS-PAGE loading buffer containing 2.5% (wt/vol) DTT. Virus-like particles from culture media fractions were pelleted through 20% sucrose cushions, resuspended, floated to the tops of sucrose flotation gradients, pelleted again, and then resuspended in SDS-PAGE loading buffer containing 2.5% (wt/vol) DTT. Proteins from cell lysates and virus-like particles were fractionated using 10% or 15% SDS-PAGE and transferred to PVDF membranes for immunodetection. Primary antibodies were as follows: anti-PIV5 M (M-f) (Randall et al., 1987), anti-PIV5 NP (NP-a) (Randall et al., 1987), anti-HA (Proteintech 51064-2-AP, RRID: AB_11042321), anti-GFP (Thermo Fisher Scientific, A-6455, RRID: AB_221570). Bands were visualized by probing with alkaline phosphatase-conjugated secondary antibodies followed by incubation with Vistra ECF substrate (GE Healthcare) and imaging using a Fuji FLA-7000 phosphorimager. VLP production efficiency was calculated as the quantity of M protein in purified VLPs divided by the quantity of M protein in the corresponding cell lysate fraction, normalized to the value obtained in the absence of CHMP3 or VPS4A expression.
Transmission electron microscopy (TEM)
For thin sectioning, 0.8 × 106 HEK293T cells were seeded in 6-well plates 18-24 h before transfection. Cells were co-transfected with the designated expression plasmids as described above. 24 h post-transfection, the media was removed and cells were harvested in 1 ml fixation buffer (2.5% glutaraldehyde/1% paraformaldehyde in sodium cacodylate buffer [50 mM sodium cacodylate, 18 mM sucrose, 2 mM CaCl2, pH 7.4]). This and all subsequent steps were performed at 23°C. Cells were fixed for 20 min, washed three times for 5 min in 1 ml sodium cacodylate buffer, and stained with 50 μl 2% OsO4 for 1 h. The pellet was washed three times in 1 ml water for 5 min, followed by incubation in 1-2 drops of 4% uranyl acetate solution for 30 min. Stained cells were dehydrated in a graded ethanol series followed by acetone, and embedded in epoxy resin EMBed-812 (Electron Microscopy Sciences). Thin sections (80–100 nm) were cut, post-stained with saturated uranyl acetate for 20 min, rinsed with water, dried, stained with Reynolds' lead citrate for 10 min, and dried again. TEM images were collected on a JEOL-JEM 1400 Plus transmission electron microscope equipped with a LaB6 filament and operated at an accelerating voltage of 120 kV. Images were recorded using a charge-coupled device camera (Gatan Orius SC1000B).
Toxicity assays
For cytotoxicity assays, 1.5 × 104HEK293T cells were seeded in 96-well plates. After 24 h, cells were transfected with Lipofectamine 2000 (Thermo Fisher Scientific) according to manufacturer’s instructions. Each well received varying amounts of HA-tagged CHMP3 construct, shown to result in equal expression across all constructs by Western blot. After 48 hours, cellular toxicity was analyzed using the CellTiter-Glo Luminescent cell viability assay (Promega) following manufacturer’s instructions. Luminescence was detected using a Biotek Synergy microplate reader.
To confirm equal expression levels, 5 × 105 HEK293T cells were seeded and transfected in parallel in 6-well plates. After 48 hours, cells were harvested, pelleted and lysed in Pierce RIPA Buffer (Pierce) containing Halt Protease Inhibitor (Thermo Fisher Scientific). Samples were mixed with equal amounts 2x Laemmli Sample Buffer containing 10% 2-mercaptoethanol and heated at 95°C for 30 minutes. Samples were run on Mini-Protean TGX™ Precast Protein Gels (Biorad) and transferred to Immobilon-P PDVF Membrane (Millipore). Membranes were blocked in PBS + 0.1% Tween 20 + 5% powdered milk for 1 hour and then incubated with anti-HA (Sigma-Aldrich, H6908, RRID: AB_260070) or anti-Actin monoclonal antibody made in mouse (ThermoFisher Scientific, AM4302, RRID: AB_2536382) for 2 hours at room temperature. After three washes in PBS + 0.1% Tween 20, membranes were incubated in anti-Rabbit IgG HRP conjugate (Millipore #AP132, RRID: AB_92486) or anti-Mouse IgG/IgM HRP conjugate (Millipore #AP130P, RRID: AB_91266) for 2 hours, washed three times in PBS + 0.1% Tween 20 and developed with Advansta WesternBright ECL HRP substrate. Blots were imaged using a LI-COR blot scanner.
Immunofluorescence imaging
1 × 105 HeLa N cells were seeded on chambered coverslips (μ-Slide 4 well, ibidi). After 24 h, cells were transfected with Lipofectamine 2000 (Thermo Fisher Scientific) according to manufacturer’s instructions. Each well received varying amounts of HA-tagged CHMP3 construct, shown to result in equal expression across all constructs. 20 h after transfection, cells were washed once in PBS and fixed in 4% paraformaldehyde for 30 min at room temperature, then washed three times with PBS. Cells were permeabilized with 0.1% Triton-100 for 30 min, then washed three times with PBS. Cells were blocked with sterile-filtered 2% BSA + 0.1% saponin for one hour. Cells were incubated with primary antibody (mouse anti-α-tubulin (Sigma-Aldrich T5168, RRID: AB_477579), rabbit anti-HA (Sigma-Aldrich H6908, RRID: AB_260070) diluted 1:250 in blocking buffer) overnight at 4°C. The coverslips were washed three times with PBS and incubated in secondary antibody (goat-anti-rabbit Alexa Fluor 488 (Thermo Fisher Scientific A-11034, RRID: AB_2576217), goat-anti-mouse Alexa Fluor 594 (Thermo Fisher Scientific A-11032, RRID: AB_2534091) diluted 1:1000 in blocking buffer) for 2 h at room temperature. Cells were washed three times with PBS and covered with DAPI Fluoromount-G (Southern Biotech). Images were captured with a Nikon A1R laser scanning confocal microscope.
Midbody scoring
To assess abscission defects in unsynchronized cultures, 1 × 105HeLa N cells were seeded on chambered coverslips (μ-Slide 4 well, ibidi) and 24-well plates 18-24 h before transfection. Cells were transfected with 500 ng empty vector, 25 ng pEF-1alpha-squirrel-monkey-full-length CHMP3-HA, 250 ng pEF-1alpha-squirrel-monkey-CHMP3-HA(155) or 500 ng pEF-1alpha-squirrel-monkey-retroCHMP3-HA using 5 μl Lipofectamine 2000 (Thermo Fisher Scientific) according to manufacturer’s instructions. Total plasmid quantities were equalized using empty vector. Transfection mixes were prepared as 2.5fold master mix and distributed between coverslips and 24-wells. The medium was replaced with 1 ml DMEM 4-6 h later. 20 h post-transfection, cells in 24-well plates were washed once with PBS, detached using 1x TrypLE Express (Gibco, ThermoFisher Scientific) and lysed for 5 min on ice in 50 μl Triton lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% Triton X-100) supplemented with mammalian protease inhibitor (Sigma-Aldrich). 50 μl 2× Laemmli SDS-PAGE loading buffer supplemented with 10% 2-mercaptoethanol (Sigma-Aldrich) were added and samples were boiled for 10 min. Proteins were separated by SDS-PAGE, transferred onto PVDF membranes and probed with antibodies to confirm equal expression levels. Cells on coverslips were fixed in 4% paraformaldehyde for 20 min at room temperature and processed for immunofluorescence as described above.
For synchronization experiments, 1 × 105 HeLa N cells were seeded on chambered coverslips (μ-Slide 4 well, ibidi) 18-24 h before transfection. Cells were transfected with 500 ng empty vector, 250 ng pEF-1alpha-squirrel-monkey-CHMP3-HA(155) or 500 ng pEF-1alpha-squirrel-monkey-retroCHMP3-HA using 5 μl Lipofectamine 2000 (Thermo Fisher Scientific) according to manufacturer’s instructions. Total plasmid quantities were equalized using empty vector. 8h after transfection, the medium was replaced with DMEM supplemented with 2mM thymidine (Sigma Aldrich) to synchronize cells. 24 h later, cells were released from thymidine arrest by washing three times with 1x PBS and addition of fresh DMEM. At indicated timepoints, cells on coverslips were fixed in 4% paraformaldehyde for 20 min at room temperature and processed for immunofluorescence as described above.
Images were captured with an Axio Imager M2 (Carl Zeiss) equipped with a Colibri7 LED light source (Carl Zeiss), a pco.edge 4.2 CMOS camera microscope, and a 63X oil-immersion objective (Carl Zeiss, Plan Apochromat, NA 1.4). Laser settings for each channel were confirmed with the untransfected well to ensure no false positives in the green channel. 200 nm optical Z-sections were captured for each field and the z-stack was cycled through. Numbers of midbodies in transfected, HA-positive cells were assessed by a blinded reviewer.
EGF receptor degradation
0.6 × 106 HEK293T cells/well were seeded in 6-well plates 24 h before transfection. Cells were transfected with 1 μg empty vector, 1 μg pEF-1alpha-squirrel-monkey-retroCHMP3-HA or 500 ng pGFP-VPS4A E228Q using 10 μl Lipofectamine 2000 (Thermo Fisher Scientific) according to manufacturer’s instructions. Total plasmid quantities were equalized using empty vector. 5-6 h after transfection, media was aspirated and replaced with DMEM without FBS to starve cells. 20h after transfection, starvation media was replaced with DMEM supplemented with 10% FBS, 100 ng/ml recombinant human EGF protein (Thermo Fisher Scientific) and 10 μg/ml cycloheximide (Sigma Aldrich). Cells were harvested at indicated timepoints by detaching in TrypLE Express (Gibco, ThermoFisher Scientific) and lysed in RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1%NP-40, 0.5% Na-deoxycholate, 0.1% SDS, 0.1 mM EDTA) supplemented with mammalian protease inhibitor (Sigma-Aldrich) for 15 minutes on ice. The protein concentration in lysates was determined using the Pierce BCA Protein Assay Kit (Thermo Scientific) and concentration was adjusted using RIPA buffer. Equal amounts of 2× Laemmli SDS-PAGE loading buffer supplemented with 10% 2-mercaptoethanol (Sigma-Aldrich) were added, and samples were boiled for 10 min. Proteins were separated by SDS-PAGE, transferred onto nitrocellulose membranes and probed with antibodies. Primary antibodies were as follows: anti-EGF receptor (Cell Signaling Technology, D38B1, 4267: RRID: AB_2246311), anti-tubulin (Sigma Aldrich, T5168, RRID: AB_477579), anti-HA (Sigma-Aldrich, H6908, RRID: AB_260070), anti-GFP (Roche, 11814460001, RRID:AB_390913). Bands were visualized by probing the membrane with goat-anti-rabbit or anti-mouse HRP-conjugated (Sigma Aldrich, ) and enhanced chemiluminescence substrate (Thermo Fisher), and detected with a ChemiDoc MP imaging system (Bio-rad). Band intensities were quantified using LI-COR Image Studio software and normalized to band intensities at t = 0h.
Pulldown experiments
For pulldown experiments utilizing the One-STrEP-FLAG (OSF) tag, HEK293T cells were seeded in a 6-well plate at 0.8 × 106 cells/well and co-transfected 18–24 h later using 10 μl Lipofectamine 2000 (Thermo Fisher Scientific) with 1.5 μg CHMP4B and the following amounts of CHMP3-expressing plasmids: 1 μg each of pCAG-OSF-PP-human-CHMP3, pCAG-OSF-PP-human-CHMP3(150), pCAG-OSF-PP-squirrel-monkey-CHMP and pCAG-OSF-PP-squirrel-monkey-CHMP3(155) and 3 μg pCAG-OSF-PP-squirrel-monkey-retroCHMP3. Total plasmid quantities were equalized using pCAG-OSF-PP empty vector. The media was changed to fresh DMEM 6-8 h post-transfection. Cells were harvested 24 h post-transfection and lysed in 200 μl RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1%NP-40, 0.5% Na-deoxycholate, 0.1% SDS, 0.1 mM EDTA) or NP-40 buffer (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% NP-40) supplemented with mammalian protease inhibitor (Sigma-Aldrich) on ice for 15 min. Lysates were clarified by centrifugation at 16,100×g for 10 min at 4°C. Cell lysates were diluted to 1 ml with RIPA buffer without SDS or NP-40 buffer and incubated for 45 min at 4°C with 20 μl pre-equilibrated Strep-Tactin Sepharose resin (IBA Lifesciences). The resin was washed three times with RIPA buffer without SDS or with NP-40 buffer. After the final wash, the streptactin beads were aspirated to near dryness, and bound proteins were eluted by boiling in 40 μl 2× Laemmli SDS-PAGE loading buffer supplemented with 10% 2-mercaptoethanol (Sigma-Aldrich), resolved by SDS-PAGE, transferred onto PVDF membranes and probed with primary antibodies (anti-myc (EMD Millipore, 05-724, RRID: AB_309938), anti-FLAG (Sigma-Aldrich, F1804, RRID: AB_262044). Bands were visualized by probing the membrane with fluorescently labeled secondary antibodies (Li-Cor Biosciences) and scanning with an Odyssey Imager (Li-Cor Biosciences).
Live-cell imaging
HeLa and HEK293T cells for live cell imaging were seeded onto MatTek dishes with no. 1.5 coverslips coated with fibronectin (Invitrogen). HeLa cells were transfected 6 hours before imaging with Fugene 6 (Promega) and 1000 ng of DNA. HEK293T cells were transfected with Lipofectamine 2000 (Thermo Fisher) and 1000 ng of DNA 5 h before imaging. Cells were transfected with 600 ng untagged pCRV1-HIV-1NL4.3 GagPol and 150 ng pCRV1-HIV-1NL4.3 GagPol-mEGFP and 250 ng pLNCX-mCherry-CHMP4B or VPS4A. Samples with retroCHMP3 additionally received 250 ng pEF1α-squirrel-monkey-retroCHMP3-T2A-H2B-tagBFP, which encodes contains a self-cleaving H2B-BFP fusion protein for identification of transfected cells.
Live cell imaging was performed with a custom-built microscope that is based on an Olympus IX-81 frame and equipped with a custom-built through-the-objective polarized TIRFM illuminator (Johnson et al., 2014). A 100X PLANAPO 1.50NA Olympus Objective was used. A 488 nm (100 mW LuxX diode laser, Omicron), a 594 nm laser (100 mW iode-pumped solid-state laser, Cobolt AB), and a 405 nm laser (100 mW LuxX diode laser, Omicron) were used at 25 mW, 30 mW, and 2 mW respectively. For live cells, the temperature was maintained at 37°C throughout imaging using custom-built housing for the microscope. TIR light was azimuthally scanned at 100 Hz with mirror galvanometers (Nutfield Technology). Emission was collected sequentially using a Chroma ZET405/488/594m filter and a CMOS camera (Flash-4.0, Hamamatsu). Image acquisition was done with MetaMorph software. All exposure times are 200 ms. GagPol-mEGFP images were taken every 20 s and ESCRT images were taken every 5 s.
HeLa and HEK293T cell assembly videos were analyzed with MetaMorph software. The camera has 100 units added to each pixel and this we subtracted from all frames prior to analysis. The images of GagPol assembly with excitation at 488 nm were analyzed first and all assemblies were marked with circular regions that encompassed the entire assembling particle. Assemblies were confirmed by the shape of the intensity curve over time which shows accumulation and plateau as shown previously (Jouvenet et al., 2008; Jouvenet et al., 2006). After all assemblies were selected, the regions from the assemblies were overlaid onto the ESCRT images taken with excitation at 594 nm of the same cell. Any spikes in intensity from the ESCRT regions were checked to confirm that they correspond to puncta with a Gaussian distribution that overlapped with the GagPol puncta. The number of frames that a single ESCRT puncta stayed and the number of recruitments per assembly was recorded.
To plot assemblies, each assembly was selected via a circular region that encompassed the whole puncta and was trimmed to the first time the assembling puncta was visible and the last time the puncta was visible. This automatically set the first frame of the assembly as time zero. To account for the differing intensities between different cells, the intensities for each assembly trace were rescaled by the following equation: . After rescaling, a rolling filter was applied to the traces. The rescaled max intensity for each frame of the assemblies were plotted together using python. For plotting ESCRT recruitment, the same circular region was used as the assembly the ESCRT was recruited to and the trace was trimmed to the same first and last time as the puncta. No filters or rescaling were applied to the ESCRT traces.
To plot overall number of repeat recruitments and length of recruitment, histograms were made which plotted the percent of total counts for either CHMP4B or VPS4A with or without retroCHMP3. Statistical differences between histograms were calculated using a one-tailed T-test. To plot the final gag assembly intensity, the average of the final 3 frames of each trace was plotted. Statistical significance was calculated using a two-tailed T-test.
Detection of retroCHMP3 RNA
Total RNA was isolated from cultured squirrel monkey and mouse cell lines according to the manufacturer’s instructions using the Zymo Quick-RNA Miniprep kit (Zymo Research), including the In-column DNAse 1 treatment. For expression analysis in mouse tissues, dissected tissues from male C57BL/6J mice (supplied by the Transgenic Gene-Targeting Mouse Facility at the University of Utah) were homogenized in 1 ml TRIzol reagent (Thermo Fisher Scientific) in a bead homogenizer and RNA was extracted according to manufacturer’s instructions. In order to remove any contaminating genomic DNA, total RNA was treated with TURBO DNase (Thermo Fisher Scientific) prior to cDNA synthesis.
For cDNA synthesis, 2 μg total RNA was reverse transcribed according to the manufacturer’s instructions using the Maxima First Strand Synthesis Kit with DNase treatment (Thermo Fisher Scientific). Thus, each sample underwent two rounds of DNase treatment for removal of genomic DNA. Additionally, all samples were synthesized in duplicate, minus or plus reverse transcriptase.
PCR amplification of products was performed with Phusion Flash High-Fidelity PCR Master Mix (Thermo Scientific). For each amplification, the −/+ RT samples were treated equally. The minus RT serves as a control to ensure complete removal of genomic DNA from samples; any product in the +RT sample is the result of RNA transcript, as opposed to the −RT control, which indicates genomic DNA contamination. Retrocopies of CHMP3 were amplified 30 to 34 cycles due to their low abundance. Primer sequences used are listed in Table S2. All bands were excised, cloned and sequenced to verify a complete match with the retroCHMP3 sequence.
Promoter analysis
The upstream region of the mouse retroCHMP3 sequence was examined for regulatory elements on the USCS genome browser (GRCm38/mm10). The DNA sequence 15kb upstream of the start codon was extracted and run through the NSite (Shahmuradov and Solovyev, 2015) and CiiiDER (Gearing et al., 2019) prediction tools.
Interferon stimulation
For interferon stimulation, 2 x 106 mouse cardiac endothelial cells (MCEC) or 6 x 106 squirrel monkey B cells were induced for 6, 12, or 20 h with the addition of 1000 U/mL IFN mix (e. coli-derived mouse IFN-gamma protein (R&D Systems, 485-MI-100), HEK293-derived mouse IFN-beta protein (R&D Systems, 8234-MB-010 and Universal Type I Interferon (pbl assay science, 11200-2)), or none as a control. Cells were trypsinized and pelleted, resuspended in RNA lysis buffer; the control flask was collected together with the 6hr IFN treated flask. Pellets were stored at −80°C until RNA extraction. Total RNA was extracted using the Quick-RNA Miniprep Kit (Zymo) and processed as described above. RetroCHMP3, ISG15, and β-actin or CTCF were amplified from the cDNA products (+/− RT) using Phusion Flash High-Fidelity Master Mix (Thermo Scientific). Primers are listed in Table S2.
Droplet digital PCR
Freshly synthesized cDNA was frozen at −80°C until submission. Succinyl dehydrogenase (SDHA) was used as a low copy housekeeping transcript and RNaseL was used as a positive control for interferon stimulation (Malathi et al., 2007). In all cases, 11 μL of BioRad ddPCR Supermix for Probes (no dUTP) were mixed with 1.1 μL of specific 20x assays and 9.9 μL cDNA template. For detection of retroCHMP3, 9.9 μL cDNA was added neat. To compare SDHA to retroCHMP3, 9.9 μL of 1:100 dilution was added to the SDHA primer/probe assay. For RNaseL and SDHA duplexes, 9.9 μL of 1:10 dilution was added to the assay. Droplets were generated and PCR was performed using the BioRad QX200 AutoDG Droplet Digital PCR System. A single cycle of 10 min at 95°C was followed by 50 cycles of 95°C for 15 seconds and 60°C for 60 seconds, followed by a single cycle of 98°C for 10 min. Primers and probes are listed in Table S2.
Data were analyzed using BioRad Quantasoft Analysis Pro software. Copies per 20 μL were calculated after manual gating. The fold change in retroCHMP3 or RNaseL expression was calculated for each time point (6, 12 and 20 h) by dividing copies/20 μL at each time point by the number of copies at time 0. Finally, the relative fold change was calculated by dividing the fold change (as described above) by the ratio of SDHA expression at time 6, 12 or 20 by SDHA expression at time 0.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was determined using the GraphPad Prism 8 software package. Statistical tests used, exact values of n, number of replicates and definition of center, dispersion and precision measures for each experiment are noted in the figure legends. All samples were analyzed without blinding, except for phenotype scoring in Fig. 3B and midbody scoring in Fig. 3C and 3D, which were analyzed by a blinded reviewer.
ADDITIONAL RESOURCES
This study did not generate any additional resources.
Supplementary Material
Fig. S1: RetroCHMP3 genes in three mammalian species. Related to Fig. 1. (A) Alignment of parental CHMP3 and retroCHMP3 protein sequences. Note that retroCHMP3 proteins do not share amino acid substitutions. Red box in retroCHMP3AG indicates sequence derived from an Alu element which was removed to enable expression. (B) Transient overexpression of HA-tagged retroCHMP3AG inhibits release of HIV-1 proteins (CA, MA) from producer cells and reduces viral titers in supernatant. Titer graph shows mean ± SD from 3 experimental replicates. **** p<0.0001, *** p<0.001, ** p<0.01, *p<0.05 by one-way ANOVA followed by Tukey’s multiple comparisons test comparisons with p>0.05 omitted for clarity.
Fig. S2: RetroCHMP3MM broadly inhibits ESCRT-dependent budding. Related to Fig. 2. (A), (B) Transient overexpression of HA-tagged retroCHMP3MM in HEK293T cells reduces the release of viral proteins and viral titers in culture supernatant of Friend murine leukemia virus (FrMLV, A) and equine infectious anemia virus (EIAV, B). Titer graphs show mean ± SD from 3 experimental replicates. (C), (D) Transient overexpression of HA-tagged retroCHMP3MM in HEK293T cells reduces the release of Ebola virus VP40 protein (C) and parainfluenza virus 5 VLPs (D) into the supernatant. Western blots show representative results from 3 experimental replicates. Bar graphs show integrated intensities of viral protein bands in supernatant normalized to cellular fraction from 3 experimental replicates. **** p<0.0001, *** p<0.001, * p<0.05 by one-way ANOVA followed by Tukey's multiple comparisons test. Only comparisons with the empty vector control (lane 1) are shown for clarity. For a complete set of pairwise comparisons, see Table S3.
Fig. S3: RetroCHMP3MM and RetroCHMP3AG evolved to lose cytotoxicity. Related to Fig. 3 (A), (B) Cell viability of cells transiently transfected with HA-tagged CHMP3MM variants (A) and retroCHMP3AG 24 h after transfection. (B). Mean ± SD from 5 experimental replicates with n≥4 each. (C) Immunofluorescence of subcellular localization of HA-tagged CHMP3MM variants. Scale bar 10 μm. (D) Localization of HA-tagged CHMP3SS variants to the midbody. White arrowhead points to CHMP3SS variants at the Flemming body. Scale bar 10 μm. (E) Transient overexpression of HA-tagged retroCHMP3SS delays EGF-induced degradation of the EGF receptor (EGFR) to a comparable degree as dominant-negative VPS4A (VPS4A-E228Q). Western blot shows representative result from 3 experimental replicates. Graph shows integrated intensities of EGFR bands normalized to intensity at t = 0h. Mean ± SD from 3 experimental replicates.
Fig. S4: RetroCHMP3 does not affect VPS4A recruitment to HIV-1 Gag assembly sites. Related to Fig. 4. (A), (B) Sample trace of mCherry-VPS4A recruitment to Gag-Pol-mEGFP assembly sites in the absence (A) or presence (B) of retroCHMP3SS. Each numbered shaded region highlights one recruitment event and the corresponding sequence of images from that recruitment. Width of each image is 1.27 μm. (C) Histogram showing the number of times that VPS4A was recruited to assembly sites in the absence (n= 48 assemblies) or presence (n=42 assemblies) of retroCHMP3SS. p=0.349 by one-tailed T-test (D) Histogram showing the duration of each VPS4A recruitment in the absence (n = 62 recruitments from 48 assemblies) or presence (n = 59 recruitments from 42 assemblies) of retroCHMP3SS. p<0.005 by one-tailed T-test.
Fig. S5: Detection of retroCHMP3 RNA. Related to Fig. 5. (A) Detection of retroCHMP3 RNA in mouse cell lines. MEF – mouse embryonic fibroblasts, MES – mouse embryonic stem cells, RAW – mouse macrophage cell line, J774 – mouse macrophage/monocyte cell line. (B) Detection of retroCHMP3 RNA in squirrel monkey B cells with and without interferon (IFN) treatment. (C) Detection of retroCHMP3 RNA in mouse tissues. IFN – interferon, RT – reverse transcriptase. All retroCHMP3 bands were excised, cloned and sequenced to verify a perfect match with the retroCHMP3 sequence.
Table S3: P-values for pairwise comparison of means. Related to Figs. 1C, 1D, 2C, S2C, 2D and S2D. P-values were calculated using an ordinary one-way ANOVA followed by a Tukey's multiple comparisons test.
Truncated retrocopies of CHMP3 arose independently in New World monkeys and mice.
RetroCHMP3 proteins broadly inhibit budding of ESCRT-dependent viruses.
RetroCHMP3 proteins evolved to lose cytotoxicity associated with ESCRT inhibition.
Delays in ESCRT function inhibit virus budding but spare critical host pathways.
Acknowledgments:
We thank Benoit de Thoisy and Christophe Duplais for squirrel monkey blood samples from the Institut Pasteur de la Guyane, Annika Paukner and Angela Ruggiero for capuchin monkey blood samples from the Laboratory of Comparative Ethology, NICHD, and Utah’s Hogle Zoo for spider monkey blood samples. We thank Dustin Hancks for helpful discussion, Janet Iwasa for artwork, Linda Nikolova and David Belnap at the University of Utah Electron Microscopy Core for assistance with thin-section electron microscopy, Douglas Mackay and Katharine Ullman for helpful discussion on cytokinesis experiments, Marina Bleck for advice on live-cell imaging, and Daniel Johnson and Joan Pulupa for maintenance of the TIRF microscope at Rockefeller University. Immunofluorescence microscopy was performed at the University of Utah Cell Imaging Core. Flow cytometry was performed at the University of Utah Flow Cytometry Core. This work was supported by NIH grants R37 AI 51174 (to W.I.S), P50 AI150464 (to N.C.E) and AI121880 (to A.P.S), NIH/NIGMS grant 5R01GM119585 (to S.M.S), USDA NIFA grant 1010021 (to A.P.S.), a Burroughs Wellcome Fund Investigators in the Pathogenesis of Infectious Disease award (to N.C.E) ), and a Pew Charitable Trusts Innovation Fund Award (to W.I.S and N.C.E).
Footnotes
Declaration of Interests: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. J Mol Biol 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Babst M, Katzmann DJ, Estepa-Sabal EJ, Meerloo T, and Emr SD (2002). Escrt-III: an endosome-associated heterooligomeric protein complex required for mvb sorting. Dev Cell 3, 271–282. [DOI] [PubMed] [Google Scholar]
- Bache KG, Stuffers S, Malerod L, Slagsvold T, Raiborg C, Lechardeur D, Walchli S, Lukacs GL, Brech A, and Stenmark H (2006). The ESCRT-III subunit hVps24 is required for degradation but not silencing of the epidermal growth factor receptor. Mol Biol Cell 17, 2513–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajorek M, Schubert HL, McCullough J, Langelier C, Eckert DM, Stubblefield WM, Uter NT, Myszka DG, Hill CP, and Sundquist WI (2009). Structural basis for ESCRT-III protein autoinhibition. Nat Struct Mol Biol 16, 754–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banjade S, Shah YH, Tang S, and Emr SD (2021). Design principles of the ESCRT-III Vps24-Vps2 module. Elife 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartusch C, and Prange R (2016). ESCRT Requirements for Murine Leukemia Virus Release. Viruses 8, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartel V, Ivanchenko S, Dupont A, Sergeev M, Wiseman PW, Krausslich HG, Brauchle C, Muller B, and Lamb DC (2011). Live-cell visualization of dynamics of HIV budding site interactions with an ESCRT component. Nat Cell Biol 13, 469–474. [DOI] [PubMed] [Google Scholar]
- Bendjennat M, and Saffarian S (2016). The Race against Protease Activation Defines the Role of ESCRTs in HIV Budding. PLoS Pathog 12, e1005657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertin A, de Franceschi N, de la Mora E, Maity S, Alqabandi M, Miguet N, di Cicco A, Roos WH, Mangenot S, Weissenhorn W, et al. (2020). Human ESCRT-III polymers assemble on positively curved membranes and induce helical membrane tube formation. Nat Commun 11, 2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi F, and van den Bogaart G (2020). Vacuolar escape of foodborne bacterial pathogens. J Cell Sci 134. [DOI] [PubMed] [Google Scholar]
- Bleck M, Itano MS, Johnson DS, Thomas VK, North AJ, Bieniasz PD, and Simon SM (2014). Temporal and spatial organization of ESCRT protein recruitment during HIV-1 budding. Proceedings of the National Academy of Sciences 111, 12211–12216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan G, Kozyrev Y, and Hu SL (2008). TRIMCyp expression in Old World primates Macaca nemestrina and Macaca fascicularis. Proc Natl Acad Sci U S A 105, 3569–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casola C, and Betran E (2017). The Genomic Impact of Gene Retrocopies: What Have We Learned from Comparative Genomics, Population Genomics, and Transcriptomic Analyses? Genome Biol Evol 9, 1351–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ L, Raiborg C, Wenzel EM, Campsteijn C, and Stenmark H (2017). Cellular Functions and Molecular Mechanisms of the ESCRT Membrane-Scission Machinery. Trends Biochem Sci 42, 42–56. [DOI] [PubMed] [Google Scholar]
- Chuong EB, Elde NC, and Feschotte C (2017). Regulatory activities of transposable elements: from conflicts to benefits. Nat Rev Genet 18, 71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciomborowska J, Rosikiewicz W, Szklarczyk D, Makalowski W, and Makalowska I (2013). "Orphan" retrogenes in the human genome. Mol Biol Evol 30, 384–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugherty MD, and Malik HS (2012). Rules of Engagement: Molecular Insights from Host-Virus Arms Races. Annual Review of Genetics 46, 677–700. [DOI] [PubMed] [Google Scholar]
- Dukes JD, Richardson JD, Simmons R, and Whitley P (2008). A dominant-negative ESCRT-III protein perturbs cytokinesis and trafficking to lysosomes. Biochem J 411, 233–239. [DOI] [PubMed] [Google Scholar]
- Eden ER, White IJ, and Futter CE (2009). Down-regulation of epidermal growth factor receptor signalling within multivesicular bodies. Biochem Soc Trans 37, 173–177. [DOI] [PubMed] [Google Scholar]
- Effantin G, Dordor A, Sandrin V, Martinelli N, Sundquist WI, Schoehn G, and Weissenhorn W (2013). ESCRT-III CHMP2A and CHMP3 form variable helical polymers in vitro and act synergistically during HIV-1 budding. Cellular Microbiology 15, 213–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elde NC, and Malik HS (2009). The evolutionary conundrum of pathogen mimicry. Nat Rev Microbiol 7, 787–797. [DOI] [PubMed] [Google Scholar]
- Elia N, Sougrat R, Spurlin TA, Hurley JH, and Lippincott-Schwartz J (2011). Dynamics of endosomal sorting complex required for transport (ESCRT) machinery during cytokinesis and its role in abscission. Proc Natl Acad Sci U S A 108, 4846–4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich N, Hagedorn M, Soldati-Favre D, and Soldati T (2012). Prison break: pathogens' strategies to egress from host cells. Microbiol Mol Biol Rev 76, 707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, et al. (2001). Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 107, 55–65. [DOI] [PubMed] [Google Scholar]
- Gearing LJ, Cumming HE, Chapman R, Finkel AM, Woodhouse IB, Luu K, Gould JA, Forster SC, and Hertzog PJ (2019). CiiiDER: A tool for predicting and analysing transcription factor binding sites. PLoS One 14, e0215495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George SH, Gertsenstein M, Vintersten K, Korets-Smith E, Murphy J, Stevens ME, Haigh JJ, and Nagy A (2007). Developmental and adult phenotyping directly from mutant embryonic stem cells. Proc Natl Acad Sci U S A 104, 4455–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlinger HG, Dorfman T, Sodroski JG, and Haseltine WA (1991). Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc Natl Acad Sci U S A 88, 3195–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guizetti J, Schermelleh L, Mantler J, Maar S, Poser I, Leonhardt H, Muller-Reichert T, and Gerlich DW (2011). Cortical constriction during abscission involves helices of ESCRT-III-dependent filaments. Science 331, 1616–1620. [DOI] [PubMed] [Google Scholar]
- Henne WM, Stenmark H, and Emr SD (2013). Molecular mechanisms of the membrane sculpting ESCRT pathway. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse M, Raulf A, Pilz GA, Haberlandt C, Klein AM, Jabs R, Zaehres H, Fugemann CJ, Zimmermann K, Trebicka J, et al. (2012). Direct visualization of cell division using high-resolution imaging of M-phase of the cell cycle. Nat Commun 3, 1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Bleck M, and Simon SM (2018). Timing of ESCRT-III protein recruitment and membrane scission during HIV-1 assembly. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Toledo-Crow R, Mattheyses AL, and Simon SM (2014). Polarization-controlled TIRFM with focal drift and spatial field intensity correction. Biophys J 106, 1008–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenet N, Bieniasz PD, and Simon SM (2008). Imaging the biogenesis of individual HIV-1 virions in live cells. Nature 454, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenet N, Neil SJ, Bess C, Johnson MC, Virgen CA, Simon SM, and Bieniasz PD (2006). Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol 4, e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouvenet N, Zhadina M, Bieniasz PD, and Simon SM (2011). Dynamics of ESCRT protein recruitment during retroviral assembly. Nature Cell Biology 13, 394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaessmann H, Vinckenbosch N, and Long M (2009). RNA-based gene duplication: mechanistic and evolutionary insights. Nature Reviews Genetics 10, 19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ (2002). BLAT--the BLAST-like alignment tool. Genome Res 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubiak MR, and Makalowska I (2017). Protein-Coding Genes' Retrocopies and Their Functions. Viruses 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lata S, Roessle M, Solomons J, Jamin M, GQttlinger HG, Svergun DI, and Weissenhorn W (2008). Structural Basis for Autoinhibition of ESCRT-III CHMP3. Journal of Molecular Biology 378, 818–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann MJ, Sherer NM, Marks CB, Pypaert M, and Mothes W (2005). Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J Cell Biol 170, 317–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DN, Aldrovandi GM, Kutsch O, and Shaw GM (2004). Dynamics of HIV-1 recombination in its natural target cells. Proc Natl Acad Sci U S A 101, 4204–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao CH, Kuang YQ, Liu HL, Zheng YT, and Su B (2007). A novel fusion gene, TRIM5-Cyclophilin A in the pig-tailed macaque determines its susceptibility to HIV-1 infection. AIDS 21 Suppl 8, S19–26. [DOI] [PubMed] [Google Scholar]
- Liu W, Zhou Y, Peng T, Zhou P, Ding X, Li Z, Zhong H, Xu Y, Chen S, Hang HC, et al. (2018). N(epsilon)-fatty acylation of multiple membrane-associated proteins by Shigella IcsB effector to modulate host function. Nat Microbiol 3, 996–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Jimenez AT, Cardenal-Munoz E, Leuba F, Gerstenmaier L, Barisch C, Hagedorn M, King JS, and Soldati T (2018). The ESCRT and autophagy machineries cooperate to repair ESX-1-dependent damage at the Mycobacterium-containing vacuole but have opposite impact on containing the infection. PLoS Pathog 14, e1007501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay DR, and Ullman KS (2015). ATR and a Chk1-Aurora B pathway coordinate postmitotic genome surveillance with cytokinetic abscission. Mol Biol Cell 26, 2217–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malathi K, Dong B, Gale M Jr., and Silverman RH (2007). Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature 448, 816–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Serrano J, Perez-Caballero D, and Bieniasz PD (2004). Context-dependent effects of L domains and ubiquitination on viral budding. J Virol 78, 5554–5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Serrano J, Zang T, and Bieniasz PD (2001). HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat Med 7, 1313–1319. [DOI] [PubMed] [Google Scholar]
- McCullough J, Clippinger AK, Talledge N, Skowyra ML, Saunders MG, Naismith TV, Colf LA, Afonine P, Arthur C, Sundquist WI, et al. (2015). Structure and membrane remodeling activity of ESCRT-III helical polymers. Science (New York, NY) 350, 1548–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough J, Frost A, and Sundquist WI (2018). Structures, Functions, and Dynamics of ESCRT-III/Vps4 Membrane Remodeling and Fission Complexes. Annu Rev Cell Dev Biol 34, 85–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehra A, Zahra A, Thompson V, Sirisaengtaksin N, Wells A, Porto M, Koster S, Penberthy K, Kubota Y, Dricot A, et al. (2013). Mycobacterium tuberculosis type VII secreted effector EsxH targets host ESCRT to impair trafficking. PLoS Pathog 9, e1003734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierzwa BE, Chiaruttini N, Redondo-Morata L, von Filseck JM, Konig J, Larios J, Poser I, Muller-Reichert T, Scheuring S, Roux A, et al. (2017). Dynamic subunit turnover in ESCRT-III assemblies is regulated by Vps4 to mediate membrane remodelling during cytokinesis. Nat Cell Biol 19, 787–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal E, Skowyra ML, Uwase G, Tinaztepe E, Mehra A, Koster S, Hanson PI, and Philips JA (2018). Mycobacterium tuberculosis Type VII Secretion System Effectors Differentially Impact the ESCRT Endomembrane Damage Response. mBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Colf LA, Karren MA, Sandrin V, Rodesch CK, and Sundquist WI (2010). Human ESCRT-III and VPS4 proteins are required for centrosome and spindle maintenance. Proceedings of the National Academy of Sciences 107, 12889–12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Sandrin V, McCullough J, Katsuyama A, Baci Hamilton I, and Sundquist Wesley I. (2011). ESCRT-III Protein Requirements for HIV-1 Budding. Cell Host & Microbe 9, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser von Filseck J, Barberi L, Talledge N, Johnson IE, Frost A, Lenz M, and Roux A (2020). Anisotropic ESCRT-III architecture governs helical membrane tube formation. Nat Commun 11, 1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muziol T, Pineda-Molina E, Ravelli RB, Zamborlini A, Usami Y, Gottlinger H, and Weissenhorn W (2006). Structural basis for budding by the ESCRT-III factor CHMP3. Dev Cell 10, 821–830. [DOI] [PubMed] [Google Scholar]
- Nahse V, Christ L, Stenmark H, and Campsteijn C (2017). The Abscission Checkpoint: Making It to the Final Cut. Trends Cell Biol 27, 1–11. [DOI] [PubMed] [Google Scholar]
- Neil SJ, Eastman SW, Jouvenet N, and Bieniasz PD (2006). HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog 2, e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman RM, Hall L, Kirmaier A, Pozzi LA, Pery E, Farzan M, O'Neil SP, and Johnson W (2008). Evolution of a TRIM5-CypA splice isoform in old world monkeys. PLoS Pathog 4, e1000003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HC, Talledge N, McCullough J, Sharma A, Moss FR 3rd, Iwasa JH, Vershinin MD, Sundquist WI, and Frost A (2020). Membrane constriction and thinning by sequential ESCRT-III polymerization. Nat Struct Mol Biol 27, 392–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisole S, Lynch C, Stoye JP, and Yap MW (2004). A Trim5-cyclophilin A fusion protein found in owl monkey kidney cells can restrict HIV-1. Proc Natl Acad Sci U S A 101, 13324–13328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JC (1998). Gene transfer vectors derived from equine infectious anemia virus. Gene Ther 5, 1481–1487. [DOI] [PubMed] [Google Scholar]
- Philips JA, Porto MC, Wang H, Rubin EJ, and Perrimon N (2008). ESCRT factors restrict mycobacterial growth. Proc Natl Acad Sci U S A 105, 3070–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portal-Celhay C, Tufariello JM, Srivastava S, Zahra A, Klevorn T, Grace PS, Mehra A, Park HS, Ernst JD, Jacobs WR Jr., et al. (2016). Mycobacterium tuberculosis EsxH inhibits ESCRT-dependent CD4(+) T-cell activation. Nat Microbiol 2, 16232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg C, and Stenmark H (2009). The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 458, 445–452. [DOI] [PubMed] [Google Scholar]
- Randall RE, Young DF, Goswami KK, and Russell WC (1987). Isolation and characterization of monoclonal antibodies to simian virus 5 and their use in revealing antigenic differences between human, canine and simian isolates. J Gen Virol 68 (Pt 11), 2769–2780. [DOI] [PubMed] [Google Scholar]
- Rheinemann L, Downhour DM, Davenport KA, McKeown AN, Necessary CR, Sundquist WI, and Elde NC (2021). Recurrent Emergence of an Antiviral Defense through Repeated Retrotransposition and Truncation of CHMP3. bioRxiv, 2021.2004.2027.441704. [Google Scholar]
- Sandrin V, and Sundquist WI (2013). ESCRT requirements for EIAV budding. Retrovirology 10, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayah DM, Sokolskaja E, Berthoux L, and Luban J (2004). Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430, 569–573. [DOI] [PubMed] [Google Scholar]
- Schmitt AP, Leser GP, Morita E, Sundquist WI, and Lamb RA (2005). Evidence for a new viral late-domain core sequence, FPIV, necessary for budding of a paramyxovirus. J Virol 79, 2988–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AP, Leser GP, Waning DL, and Lamb RA (2002). Requirements for budding of paramyxovirus simian virus 5 virus-like particles. J Virol 76, 3952–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scourfield EJ, and Martin-Serrano J (2017). Growing functions of the ESCRT machinery in cell biology and viral replication. Biochem Soc Trans 45, 613–634. [DOI] [PubMed] [Google Scholar]
- Shahmuradov IA, and Solovyev VV (2015). Nsite, NsiteH and NsiteM computer tools for studying transcription regulatory elements. Bioinformatics 31, 3544–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer NM, Lehmann MJ, Jimenez-Soto LF, Ingmundson A, Horner SM, Cicchetti G, Allen PG, Pypaert M, Cunningham JM, and Mothes W (2003). Visualization of retroviral replication in living cells reveals budding into multivesicular bodies. Traffic 4, 785–801. [DOI] [PubMed] [Google Scholar]
- Shim S, Kimpler LA, and Hanson PI (2007). Structure/function analysis of four core ESCRT-III proteins reveals common regulatory role for extreme C-terminal domain. Traffic 8, 1068–1079. [DOI] [PubMed] [Google Scholar]
- Skowyra ML, Schlesinger PH, Naismith TV, and Hanson PI (2018). Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song B, Javanbakht H, Perron M, Park DH, Stremlau M, and Sodroski J (2005). Retrovirus restriction by TRIM5alpha variants from Old World and New World primates. J Virol 79, 3930–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack B, Calistri A, Craig S, Popova E, and Gottlinger HG (2003). AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114, 689–699. [DOI] [PubMed] [Google Scholar]
- Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, and Sodroski J (2004). The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427, 848–853. [DOI] [PubMed] [Google Scholar]
- Stuchell-Brereton MD, Skalicky JJ, Kieffer C, Karren MA, Ghaffarian S, and Sundquist WI (2007). ESCRT-III recognition by VPS4 ATPases. Nature 449, 740–744. [DOI] [PubMed] [Google Scholar]
- Teis D, Saksena S, and Emr SD (2008). Ordered assembly of the ESCRT-III complex on endosomes is required to sequester cargo during MVB formation. Dev Cell 15, 578–589. [DOI] [PubMed] [Google Scholar]
- Virgen CA, Kratovac Z, Bieniasz PD, and Hatziioannou T (2008). Independent genesis of chimeric TRIM5-cyclophilin proteins in two primate species. Proc Natl Acad Sci U S A 105, 3563–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votteler J, and Sundquist WI (2013). Virus budding and the ESCRT pathway. Cell Host Microbe 14, 232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel EM, Schultz SW, Schink KO, Pedersen NM, Nahse V, Carlson A, Brech A, Stenmark H, and Raiborg C (2018). Concerted ESCRT and clathrin recruitment waves define the timing and morphology of intraluminal vesicle formation. Nat Commun 9, 2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson SJ, Webb BL, Ylinen LM, Verschoor E, Heeney JL, and Towers GJ (2008). Independent evolution of an antiviral TRIMCyp in rhesus macaques. Proc Natl Acad Sci U S A 105, 3557–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Emerman M, Malik HS, and McLaughlin RNJ (2020). Retrocopying expands the functional repertoire of APOBEC3 antiviral proteins in primates. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap MW, Young GR, Varnaite R, Morand S, and Stoye JP (2020). Duplication and divergence of the retrovirus restriction gene Fv1 in Mus caroli allows protection from multiple retroviruses. PLoS Genet 16, e1008471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee JK, Miyanohara A, LaPorte P, Bouic K, Burns JC, and Friedmann T (1994). A general method for the generation of high-titer, pantropic retroviral vectors: highly efficient infection of primary hepatocytes. Proc Natl Acad Sci U S A 91, 9564–9568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamborlini A, Usami Y, Radoshitzky SR, Popova E, Palu G, and Gottlinger H (2006). Release of autoinhibition converts ESCRT-III components into potent inhibitors of HIV-1 budding. Proc Natl Acad Sci U S A 103, 19140–19145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JY, Abate M, Rice PW, and Cole CN (1991). The ability of simian virus 40 large T antigen to immortalize primary mouse embryo fibroblasts cosegregates with its ability to bind to p53. J Virol 65, 6872–6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1: RetroCHMP3 genes in three mammalian species. Related to Fig. 1. (A) Alignment of parental CHMP3 and retroCHMP3 protein sequences. Note that retroCHMP3 proteins do not share amino acid substitutions. Red box in retroCHMP3AG indicates sequence derived from an Alu element which was removed to enable expression. (B) Transient overexpression of HA-tagged retroCHMP3AG inhibits release of HIV-1 proteins (CA, MA) from producer cells and reduces viral titers in supernatant. Titer graph shows mean ± SD from 3 experimental replicates. **** p<0.0001, *** p<0.001, ** p<0.01, *p<0.05 by one-way ANOVA followed by Tukey’s multiple comparisons test comparisons with p>0.05 omitted for clarity.
Fig. S2: RetroCHMP3MM broadly inhibits ESCRT-dependent budding. Related to Fig. 2. (A), (B) Transient overexpression of HA-tagged retroCHMP3MM in HEK293T cells reduces the release of viral proteins and viral titers in culture supernatant of Friend murine leukemia virus (FrMLV, A) and equine infectious anemia virus (EIAV, B). Titer graphs show mean ± SD from 3 experimental replicates. (C), (D) Transient overexpression of HA-tagged retroCHMP3MM in HEK293T cells reduces the release of Ebola virus VP40 protein (C) and parainfluenza virus 5 VLPs (D) into the supernatant. Western blots show representative results from 3 experimental replicates. Bar graphs show integrated intensities of viral protein bands in supernatant normalized to cellular fraction from 3 experimental replicates. **** p<0.0001, *** p<0.001, * p<0.05 by one-way ANOVA followed by Tukey's multiple comparisons test. Only comparisons with the empty vector control (lane 1) are shown for clarity. For a complete set of pairwise comparisons, see Table S3.
Fig. S3: RetroCHMP3MM and RetroCHMP3AG evolved to lose cytotoxicity. Related to Fig. 3 (A), (B) Cell viability of cells transiently transfected with HA-tagged CHMP3MM variants (A) and retroCHMP3AG 24 h after transfection. (B). Mean ± SD from 5 experimental replicates with n≥4 each. (C) Immunofluorescence of subcellular localization of HA-tagged CHMP3MM variants. Scale bar 10 μm. (D) Localization of HA-tagged CHMP3SS variants to the midbody. White arrowhead points to CHMP3SS variants at the Flemming body. Scale bar 10 μm. (E) Transient overexpression of HA-tagged retroCHMP3SS delays EGF-induced degradation of the EGF receptor (EGFR) to a comparable degree as dominant-negative VPS4A (VPS4A-E228Q). Western blot shows representative result from 3 experimental replicates. Graph shows integrated intensities of EGFR bands normalized to intensity at t = 0h. Mean ± SD from 3 experimental replicates.
Fig. S4: RetroCHMP3 does not affect VPS4A recruitment to HIV-1 Gag assembly sites. Related to Fig. 4. (A), (B) Sample trace of mCherry-VPS4A recruitment to Gag-Pol-mEGFP assembly sites in the absence (A) or presence (B) of retroCHMP3SS. Each numbered shaded region highlights one recruitment event and the corresponding sequence of images from that recruitment. Width of each image is 1.27 μm. (C) Histogram showing the number of times that VPS4A was recruited to assembly sites in the absence (n= 48 assemblies) or presence (n=42 assemblies) of retroCHMP3SS. p=0.349 by one-tailed T-test (D) Histogram showing the duration of each VPS4A recruitment in the absence (n = 62 recruitments from 48 assemblies) or presence (n = 59 recruitments from 42 assemblies) of retroCHMP3SS. p<0.005 by one-tailed T-test.
Fig. S5: Detection of retroCHMP3 RNA. Related to Fig. 5. (A) Detection of retroCHMP3 RNA in mouse cell lines. MEF – mouse embryonic fibroblasts, MES – mouse embryonic stem cells, RAW – mouse macrophage cell line, J774 – mouse macrophage/monocyte cell line. (B) Detection of retroCHMP3 RNA in squirrel monkey B cells with and without interferon (IFN) treatment. (C) Detection of retroCHMP3 RNA in mouse tissues. IFN – interferon, RT – reverse transcriptase. All retroCHMP3 bands were excised, cloned and sequenced to verify a perfect match with the retroCHMP3 sequence.
Table S3: P-values for pairwise comparison of means. Related to Figs. 1C, 1D, 2C, S2C, 2D and S2D. P-values were calculated using an ordinary one-way ANOVA followed by a Tukey's multiple comparisons test.
Data Availability Statement
Sequencing data of CHMP3 retrocopies is provided in Table S1.
This study did not generate any new code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.