

Abstract
Neurons rely heavily on properly regulated mitochondrial and lysosomal homeostasis, with multiple neurodegenerative diseases linked to dysfunction in these two organelles. Interestingly, mitochondria-lysosome membrane contact sites have been identified as a key pathway mediating their crosstalk in neurons. Recent studies have further elucidated the regulation of mitochondria-lysosome contact dynamics via distinct tethering/untethering protein machinery. Moreover, this pathway has been shown to have additional functions in regulating organelle network dynamics and metabolite transfer between lysosomes and mitochondria. In this review, we highlight recent advances in the field of mitochondria-lysosome contact sites and their misregulation across multiple neurodegenerative disorders, which further underscore a potential role for this pathway in neuronal homeostasis and disease.
Keywords: Mitochondria, lysosomes, inter-organelle contact sites, Parkinson’s disease, Charcot-Marie-Tooth disease, lysosomal storage disorders
Roles for mitochondrial and lysosomal function in neuronal homeostasis
Properly regulated organelle dynamics and function are critical for maintaining neuronal homeostasis and have key implications for the etiology underlying neurodegeneration. Mitochondria rely on highly dynamic mitochondrial networks modulated by fission and fusion events as well as inter-mitochondrial tethering dynamics [1, 2] to perform a wide array of functions including oxidative phosphorylation, ATP production, regulation of reactive oxygen species, and metabolite storage such as calcium [3, 4]. Like mitochondria, lysosomes are highly dynamic organelles, and their dysfunction has been linked to many neurological disorders [5]. Lysosomes have multiple roles in protein degradation, protein turnover, and ion storage, along with additional functions in metabolic signaling and nutrient sensing [6]. However, the direct crosstalk between mitochondria and lysosomes at inter-organelle membrane contact sites (see Glossary) has only recently become appreciated as a significant pathway for regulating neuronal homeostasis and neurodegenerative disease pathogenesis. Mitochondria-lysosome contact sites can dynamically form in neurons [7–9], and are distinct from mitochondrial degradation by lysosomes via pathways such as mitophagy [10] or mitochondrial-derived vesicles [11]. Importantly, mitochondria-lysosome contacts allow for these organelles’ bidirectional regulation as well as the mechanistic modulation of organelle network dynamics, function and metabolite transfer at these sites. This Review highlights recent findings on the regulation and function of mitochondria-lysosome contact sites in neuronal homeostasis, and further summarizes defects in this pathway which have been associated with multiple neurodegenerative diseases.
The dynamic formation of mitochondria-lysosome contact sites
While indirect functional interactions between mitochondria and lysosomes have been demonstrated (reviewed in [12]), the direct interactions between mitochondria and lysosomes at membrane contact sites have only been recently investigated. Contact sites between mitochondria and lysosomes (Figure 1) were observed by electron microscopy in mammalian non-neuronal cell lines [13–15] and primary human progenitor cells [16], and had an average distance between membranes of ~10nm, consistent with other inter-organelle contacts. 3D super-resolution structured illumination microscopy (SIM) and confocal imaging of the lysosomal membrane and outer mitochondrial membrane further demonstrated the formation of mitochondria-lysosome contacts [13, 17]. Live confocal microscopy at high spatial and temporal resolutions in mammalian cell lines revealed that ~15% of lysosomes contacted a mitochondria at any given time, with contacts tethered for an average duration of ~60 sec [13] although tethering could last as long as 13 minutes [18]. Of note, mitochondria contacted lysosomes of varying sizes including both small (vesicle diameter <0.5μm) and larger (vesicle diameter >1μm) lysosomes [13]. In addition, lysosomes could also simultaneously contact multiple mitochondria [13].
Figure 1. Mitochondria-lysosome contact sites in neurons.
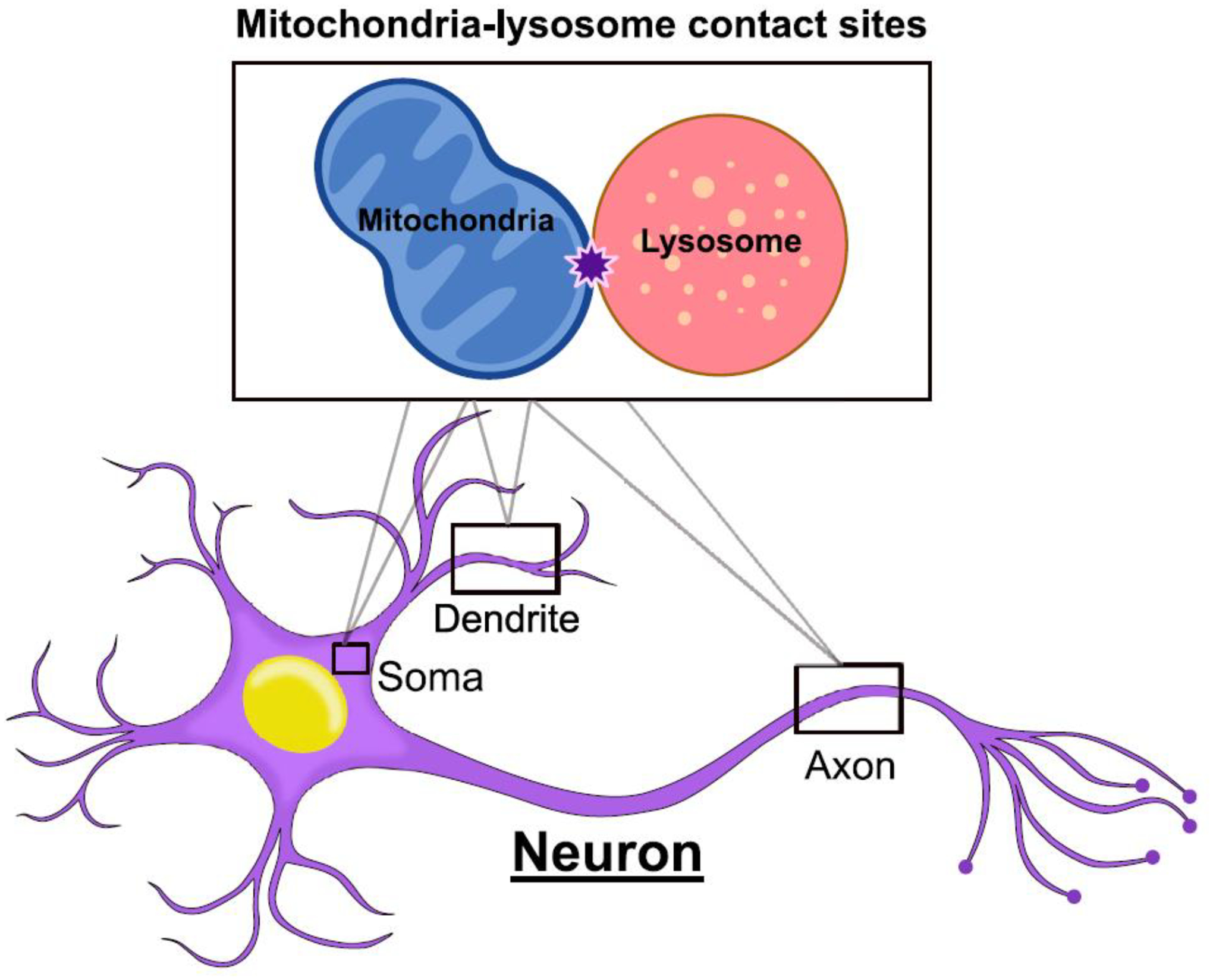
Mitochondria-lysosome contact sites are inter-organelle contacts which form between mitochondria and lysosomes, allowing for these organelles’ bidirectional crosstalk. Mitochondria-lysosome contact sites have been shown to form in the soma, axons and dendrites of neurons including human iPSC-derived neurons [7, 8].
Further imaging using correlative light electron microscopy (CLEM) of LysoTracker-positive vesicles contacting mitochondria [13], and CLEM combined with focused ion beam scanning electron microscopy (FIB-SEM) [19] also revealed mitochondria-lysosome contact site formation in mammalian cells lines. Additional studies using lattice light sheet spectral imaging [20] have also demonstrated the formation of mitochondria-lysosome contacts, as well as SIM imaging of organelles labeled with mitochondrial [21] and lysosomal dyes [18]. Finally, sensitized emission fluorescence resonance energy transfer (SE-FRET) between outer mitochondrial membranes and lysosomal membranes at mitochondria–lysosome contacts further confirmed the formation of these contacts in mammalian cells [13].
Importantly, mitochondria-lysosome contacts were shown to be distinct from pathways involving mitochondrial degradation by lysosomes. Of note, mitochondria that formed contacts with lysosomes were not mitochondrial-derived vesicles (MDV) as they were substantially larger (over 500 nm) than previously described MDVs (about 100 nm) [11]. In addition, intermembrane space mitochondrial proteins and mitochondrial matrix proteins were not bulk transferred into lysosomes, and lysosomal lumen content was not transferred into mitochondria at contact sites, demonstrating that bulk transfer of whole organelles did not occur through this pathway [13]. Moreover, mitochondria in contact with lysosomes did not undergo mitophagy as they were not engulfed by LC3-positive autophagosomes and were negative for autophagosome biogenesis markers [13]. Indeed, knockout of five autophagy receptors (NDP52, OPTN, NBR1, TAX1BP1, and p62) did not prevent mitochondria–lysosome contact formation, further demonstrating that contact formation is independent of mitophagy pathways [21].
More recently, neuronal mitochondria-lysosome contact sites have also been shown to form in the soma, axons, and dendrites of human-derived neurons (Figure 1). Using human induced pluripotent stem cells (iPSC), mitochondria-lysosome contact sites were found to dynamically form in iPSC-derived midbrain dopaminergic neurons [7]. 3D SIM and electron microscopy showed neuronal contact sites were stably tethered together, exhibiting similar dynamics to mammalian non-neuronal cells. While contacts formed at similar rates throughout the soma, axons, and dendrites, mitochondria-lysosome contacts in axons demonstrated increased tethering durations compared to those in the soma or dendrites [7]. Neuronal contacts between mitochondria and late endosomes/lysosomes have also been observed in the axons of Xenopus retinal ganglion cells [8], as well as in the soma and axons of mouse embryonic motor neurons [9]. Future studies on mitochondria-lysosome contact formation in neurons in vivo will consequently also be highly informative. Thus, mitochondria-lysosome contacts can dynamically form throughout neurons to potentially mediate mitochondrial and lysosomal crosstalk in neuronal homeostasis.
Mitochondria-lysosome contact site regulation and tethering machinery
The formation and dynamics of mitochondria–lysosome contacts are tightly regulated by multiple proteins on the mitochondrial and lysosomal membrane. In particular, this pathway is dependent on Rab7, a small GTPase which localizes to lysosomes/late endosomes upon GTP-binding and acts as a master regulator of lysosomal dynamics by binding Rab7 effector proteins in its GTP-bound state. Rab7 GTP hydrolysis was found to regulate mitochondria–lysosome contact untethering [13], whereby GTP-bound Rab7 promotes contact formation, while subsequent Rab7 GTP hydrolysis from a GTP-bound to GDP-bound state mediates the untethering of mitochondria from lysosomes at contact sites (Figure 2A). Indeed, expression of the constitutively active Rab7(Q67L)-GTP mutant which is unable to undergo Rab7 GTP hydrolysis resulted in an increased percentage of lysosomes in contacts, and prolonged contact tethering duration compared to wildtype Rab7 [13]. Importantly, Rab7 GTP hydrolysis at mitochondria–lysosome contact sites was driven by TBC1D15, a Rab7 GTPase activating protein (GAP) which localizes to the outer mitochondrial membrane through its binding to Fis1 [22–24]. Of note, inhibition of Rab7 GTP hydrolysis by TBC1D15 GAP-domain mutants (D397A, R400K) or mutant Fis1(LA) (unable to recruit TBC1D15 to mitochondria) led to prolonged mitochondria–lysosome contact durations due to inefficient untethering events [13]. In addition, constitutively active Rab7(Q67L)-GTP mutant also led to prolonged contact durations between mitochondria and late endosomes/lysosomes in the axons of Xenopus retinal ganglion cells [8].
Figure 2. Regulation of mitochondria-lysosome contact site dynamics.
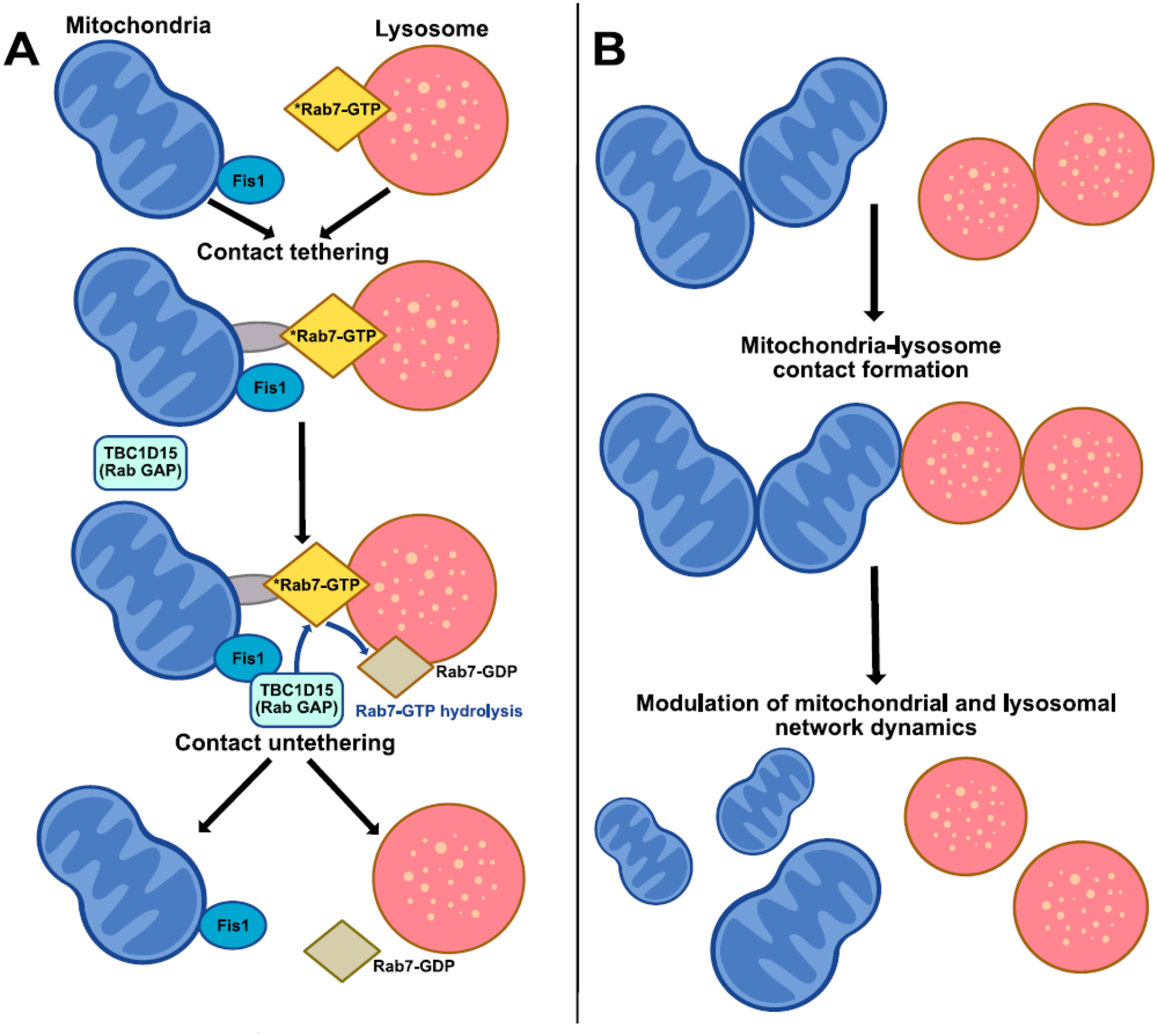
(A) Regulation of contact sites: Mitochondria and lysosomes undergo contact formation and tethering regulated by Rab7 on the lysosomal membrane. Active GTP-bound Rab7 may tether the lysosome to the mitochondria via various Rab7 effector proteins or protein complexes (gray). Subsequently, TBC1D15 (Rab7 GAP) which is recruited to the mitochondria by the outer mitochondrial membrane protein Fis1 drives Rab7 GTP hydrolysis from an active Rab7 GTP-bound state into an inactive Rab7 GDP-bound state at mitochondria-lysosome contacts. This transition from a GTP-bound state to a GDP-bound state leads to GDP-bound Rab7 leaving the lysosomal membrane, resulting in the subsequent untethering of mitochondria-lysosome contacts.
(B) Modulation of organelle dynamics: Mitochondria-lysosome contact sites can simultaneously bidirectionally regulate both mitochondrial and lysosomal network dynamics, including mitochondrial fission events, inter-mitochondrial contact untethering and lysosomal tethering.
Additional studies have also highlighted other regulators of mitochondria–lysosome contact site tethering. GDAP1 is a glutathione S-transferase on the outer mitochondrial membrane which has been suggested to directly interact with LAMP1, a lysosomal membrane protein, from co-immunoprecipitation and proximity ligation assays in human neuroblastoma SHSY5Y cells [9]. Knockdown of GDAP1 increased the distance between mitochondria and lysosomes, decreased the number of mitochondria-lysosome contacts, and also decreased contact duration [9]. The outer mitochondrial membrane protein Mitofusin2 (MFN2) has also been implicated in regulating mitochondria–lysosome contacts, as knockdown of MFN2 reduced the numbers of contacts between mitochondria and lysosomes in primary human erythroid progenitors [16]. It remains to be examined whether there are other protein complexes that act together to modulate contact tethering dynamics, in addition to mitochondria–lysosome contact regulation by Rab7 GTP hydrolysis.
Mitochondria-lysosome contact functions in modulating organelle dynamics
Mitochondria-lysosome contacts have been shown to play key roles in regulating the dynamics of both mitochondria and lysosomes (Figure 2B). Interestingly, lysosomes were found to mark the majority of mitochondrial fission events via mitochondria-lysosome contact sites [13]. Other vesicles such as early endosomes or peroxisomes were not observed at these fission events [13], and lysosomes preferentially marked mitochondrial fission but not fusion events [2]. Importantly, disruption of mitochondria-lysosome contact dynamics by inhibiting Rab7 GTP hydrolysis led to significantly reduced rates of mitochondrial fission [13]. As previous studies have also demonstrated roles for the endoplasmic reticulum (ER) and actin in mitochondrial fission [25–31], fission events in the midzone of cells were proposed to rely more heavily on ER and actin-mediated pre-constriction, while fission events in the periphery may be more dependent on lysosomal contacts [32]. In addition, other studies have also suggested that the ER may help recruit lysosomes to fission sites through the interaction of VAMP-associated proteins (VAPs) with the lysosomal lipid transfer protein ORP1L to induce three-way contacts between the ER, lysosome and mitochondria to regulate phosphatidylinositol 4-phosphate (PI(4)P) levels at mitochondrial fission events [33]. However, PI(4)P levels may also be regulated by Golgi-derived vesicles recruited at the final steps of mitochondrial division [34]. Thus, multiple organelles including lysosomal contacts may be involved in regulating the machinery involved in mitochondrial fission.
Tethering between two mitochondria at inter-mitochondrial contacts has also been found to be regulated by lysosomal contacts. Super-resolution imaging showed that inter-mitochondrial contact formation played a crucial role in mitochondrial networks by restricting mitochondrial motility [2]. Inter-mitochondrial contact formation occurred ten times more frequently than mitochondrial fission or fusion events. Time-lapse confocal imaging further revealed that lysosomes also preferentially marked sites of inter-mitochondrial contact untethering which were also modulated by Rab7 GTP hydrolysis [2], a key regulator of mitochondria-lysosome contact dynamics [13]. In addition, studies using grazing incidence structured illumination microscopy (GI-SIM) showed that lysosomes in contact with mitochondria could lead to large invaginations in mitochondria, move mitochondria as much as ~4 μm, and pull mitochondrial tubules from their parent mitochondria [35]. Consequently, multiple aspects of mitochondrial network dynamics may be directly regulated by mitochondria-lysosome contact sites.
Conversely, lysosomal networks may also be regulated by this pathway through the modulation of Rab7, via its known roles in late endosomal/lysosomal dynamics [36]. As Rab7 GTP hydrolysis occurs on late endosomal/lysosomal membranes mediated by mitochondrial TBC1D15 (Rab7 GAP) at mitochondria-lysosome contact sites [13], this further allows for mitochondrial regulation of Rab7’s GTP state. In particular, while active Rab7-GTP promotes its interaction with different binding partners (Rab7 effectors) [36], subsequent Rab7 GTP hydrolysis coupled to mitochondria-lysosome contact untethering may lead to inactive Rab7-GDP which can no longer bind these effectors to modulate lysosomal dynamics. Thus, mitochondria-lysosome contacts may play an important part in mediating the simultaneous and bidirectional regulation of both mitochondrial and lysosomal network dynamics.
Roles for mitochondria-lysosome contacts in regulating organelle function
Mitochondria-lysosome contacts have also been found to play a crucial role in the maintenance of metabolite homeostasis by serving as the primary site for exchange of different ions and lipids between both organelles. Indeed, both mitochondria and lysosomes are important organelles for storing various metabolites including calcium, cholesterol and iron (Figure 3). Mitochondria-lysosome contacts were recently shown to regulate intracellular calcium dynamics by mediating the transfer of calcium from lysosomes to mitochondria [37]. The lysosomal cation channel transient receptor potential mucolipin 1 (TRPML1) serves as a calcium efflux channel mediating release of calcium to mitochondria at contact sites. Using live cell microscopy, activation of TRPML1 by agonists such as ML-SA1 was demonstrated to increase mitochondrial calcium, preferentially for mitochondria which were in contact with lysosomes. Furthermore, dominant-negative pore mutant TRPML1 led to reduced mitochondrial calcium influx in comparison to wildtype TRPML1. On the mitochondria, voltage-dependent anion channel 1 (VDAC1) located in the outer mitochondrial membrane and mitochondrial calcium uniporter (MCU) in the inner mitochondrial membrane mediated mitochondrial influx of lysosomal calcium at contact sites [37]. Thus, the storage of calcium in mitochondria and lysosomes can be acutely regulated by mitochondria-lysosome contact tethering dynamics.
Figure 3. Metabolite transfer at mitochondria-lysosome contact sites.
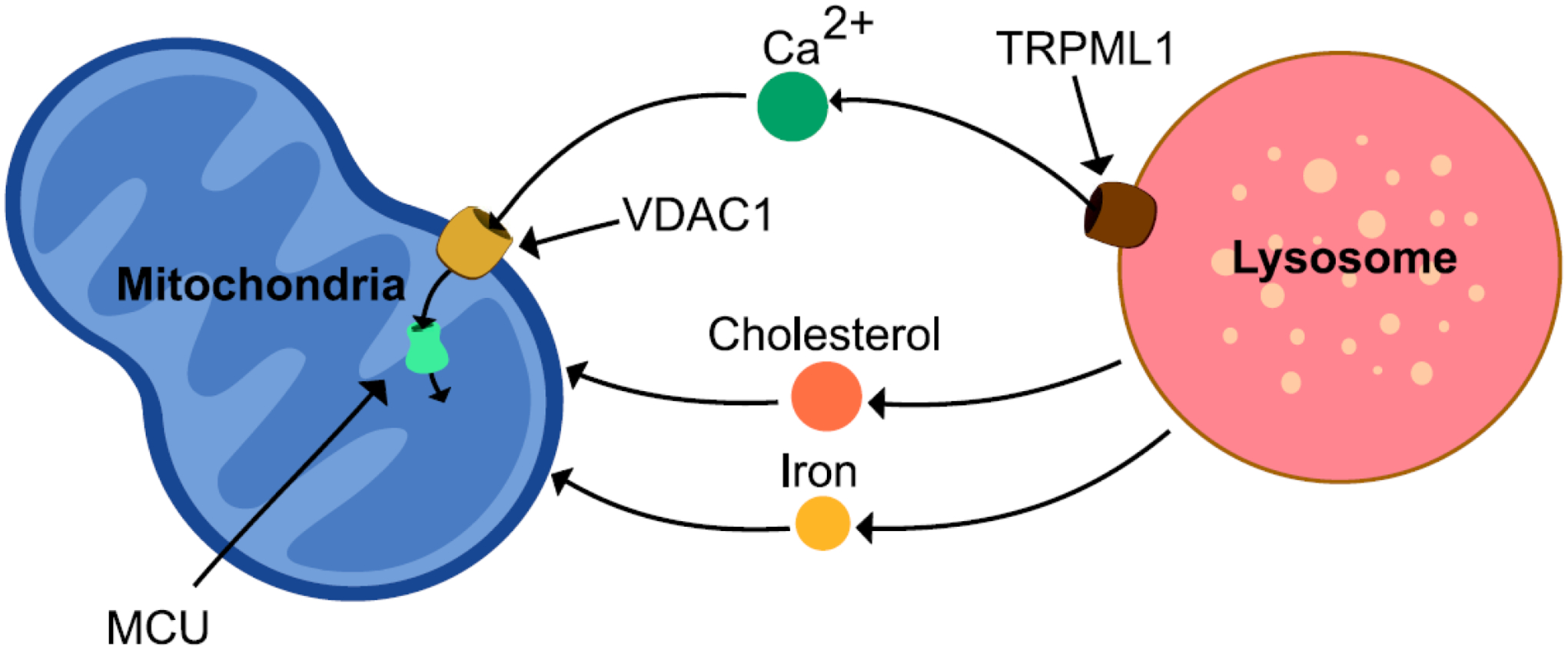
Mitochondria-lysosome contact sites can mediate the exchange of metabolites, such as calcium, cholesterol and iron. The transport of calcium from lysosomes to mitochondria occurs via the lysosomal membrane channel TRPML1 which promotes efflux of lysosomal calcium into the mitochondrial matrix via the outer and inner mitochondrial membrane channels VDAC1 and MCU at mitochondria-lysosome contacts. Cholesterol and iron have also been shown to be transferred from lysosomes to mitochondria at contact sites.
Mitochondria-lysosome contacts have also been implicated in regulating the dynamics of other metabolites. Cholesterol homeostasis is essential for maintaining membrane integrity as well as serving as a precursor for multiple classes of signaling molecules. While Niemann Pick C1 (NPC1) on the lysosomal membrane may help shuttle cholesterol from lysosomes to the ER through interactions with the ER sterol-binding protein Gramd1b [38], Niemann Pick C2 (NPC2) in lysosomes has been proposed to regulate the transfer of cholesterol from lysosomes to mitochondria at mitochondria-lysosome contact sites [39]. Interestingly, loss of NPC1 was recently found to reduce ER contacts with endocytic organelles, resulting in increased mitochondria-lysosome contacts via the sterol transfer protein StAR-related lipid transfer domain-3 (STARD3) [38]. In contrast, other studies showed that loss of NPC1 instead increased ER-lysosome contacts, and led to increased cholesterol transfer from the ER to the lysosomal membrane via VAP on the ER binding to the cholesterol carrier oxysterol binding protein (OSBP) [40]. Of note, cholesterol has also been suggested to be transported from endolysosomes to mitochondria via MLN64, a steroidogenic acute regulatory protein-related lipid transfer (START) domain-containing protein which is localized to endolysosomal membranes [41, 42], although it remains unclear whether this occurs directly at mitochondria–lysosome contact sites. In addition, transferrin receptor 2 (Trf2) on endolysosomal membranes was found to mediate transferrin uptake and delivery of iron from lysosomes to mitochondria [16].
Finally, lipid homeostasis may also be regulated by these contact sites, as the vacuole and mitochondria patch (vCLAMP) in yeast [43, 44] has been shown to regulate the transfer of phospholipids such as phosphatidylserine and phosphatidylcholine [43], and is further modulated by various proteins complexes including mitochondrial Tom40/VPS39/vacuolar Rab GTPase Ypt7 [44, 45], mitochondrial MCP1/Vps13/vacuolar Ypt35 [46, 47], and Lam6 [48]. Indeed, ORP1L on the endolysosomal membrane was recently suggested to mediate the transport of lipid PI(4)P from lysosomes to mitochondria at mammalian mitochondria-lysosome contacts [33]. Moreover, misregulation of the ceramide pathway via loss of the lysosomal enzyme β-glucocerebrosidase (GCase) which catalyzes glucosylceramide into glucose and ceramide, or increased levels of glucosylceramide, was sufficient to prolong mitochondria-lysosome contact tethering duration in neurons [7], further highlighting the crosstalk between this pathway and the homeostasis of various metabolites.
A role for RNA translation was also recently shown for neuronal contacts between mitochondria and late endosomes/lysosomes, whereby mRNAs encoding proteins for mitochondrial function were found to be translated on Rab7a vesicles via contact sites in axons of Xenopus retinal ganglion cells. Ribonucleoprotein particles (RNPs) on Rab7a vesicles which dock at mitochondria serve as hotspots for de novo protein synthesis for mRNA such as LaminB2, which are important for mitochondrial integrity and axon survival [8]. Thus, as precise RNP localization and nascent protein synthesis are required at defined neuronal sites, mitochondria-lysosome contacts may have additional specialized roles in neurons to maintain these cells’ homeostasis.
Misregulation of mitochondria-lysosome contacts in genetic models of neurodegenerative diseases
Multiple neurodegenerative diseases have been linked to the dysfunction of mitochondria and lysosomes, suggesting that disrupting the crosstalk between these two organelles may be a potential mechanistic pathway contributing to neurodegeneration. Indeed, recent studies have linked mutations associated with various neurodegenerative disorders with impaired dynamics and function of mitochondria-lysosome contact sites (Table 1).
Table 1.
Genetic mutations associated with neurodegenerative diseases that have been linked to misregulation of mitochondria-lysosome contacts.
| Neurodegenerative Disease | Description | Disease Gene | References |
|---|---|---|---|
| Charcot-Marie-Tooth Disease | Peripheral neuropathy | MFN2 (Charcot-Marie-Tooth Type 2A) | [16] |
| Parkinson’s Disease | Movement disorder | GBA (Parkinson’s disease) | [64] |
| Lysosomal Storage Disorders | Multi-systemic/neurodegeneration | TRPML1 (Mucolipidosis Type IV) | [37] |
Charcot-Marie-Tooth (CMT) disease is the most common genetic group of peripheral neuropathies, involving the degeneration of peripheral sensory and motor neurons, with autosomal dominant forms resulting in axonal degeneration classified as Charcot-Marie-Tooth disease Type 2 [49]. Mutations in the GTPase Rab7 lead to Charcot-Marie-Tooth Type 2B (CMT2B) [50], and result in defective GTP hydrolysis and constitutively bound GTP [51–53]. Consistent with a role for Rab7 GTP hydrolysis in driving mitochondria-lysosome contact untethering events [13], time-lapse confocal microscopy in mammalian cell lines revealed that the most common CMT2B disease-linked mutant Rab7 (V162M) significantly increased the percentage of lysosomes in contact with mitochondria and further prolonged mitochondria-lysosome contact duration [2]. Moreover, mutant Rab7 (V162M) prolonged inter-mitochondrial contacts and decreased mitochondrial network motility [2]. Finally, CMT2B disease-linked Rab7 mutants have also been shown to decrease axonal protein synthesis, impair mitochondrial function, and compromise axonal viability in Xenopus retinal ganglion cells due to defective axonal translation of mRNAs at neuronal mitochondria-lysosome contacts, including those encoding proteins essential for mitochondrial integrity [8]. Thus, CMT2B disease-linked mutations in Rab7 directly prolong mitochondria-lysosome contact tethering, resulting in impaired mitochondrial network dynamics and function.
In addition, various other genes mutated in Charcot-Marie-Tooth disease have also been found to regulate mitochondria-lysosome contact sites. GDAP1 regulates mitochondria-lysosome contact tethering [9], and its autosomal dominant mutations lead to CMT Type 2K [54, 55], while Mfn2, which mediates outer mitochondrial membrane fusion [56], has been suggested to modulate contact formation [16] and is linked to CMT Type 2A [57]. Of note, other Charcot-Marie-Tooth Type 2 disease genes have also been implicated in mitochondrial dynamics [49]. Inverted formin 2 (INF2), which is linked to CMT Type DIE [58], was found to regulate mitochondrial fission, potentially by mediating actin polymerization for initial mitochondrial constriction prior to Drp1-mediated constriction [26, 27]. In addition, dynamin2 (Dyn2) linked to CMT Type 2M [59] may potentially play a role in mitochondrial fission [60–62] by assisting sequential constriction events following Drp1 recruitment through its transient accumulation shortly before fission [60]. Consequently, defects in mitochondrial dynamics may be a potential factor driving peripheral neurodegeneration in CMT2 pathogenesis.
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder [63], and mutations in the lysosomal enzyme GBA1 are a genetic risk factor for PD. GBA1 encodes for the lysosomal enzyme β-glucocerebrosidase (GCase) which exhibits decreased enzymatic activity in PD patients harboring GBA1 mutations (GBA1-PD) [64]. GBA1-PD patient iPSC-derived dopaminergic neurons were recently found to demonstrate prolonged mitochondria-lysosome contact tethering due to decreased TBC1D15 (Rab7-GAP) protein levels [7]. Inhibition of GCase activity by CBE (Conduritol-B-Epoxide) in control wild-type human neurons also resulted in prolonged mitochondria-lysosome contact durations, as well as decreased TBC1D15 expression levels, suggesting that loss of GCase enzymatic activity drives these defects in GBA1-PD patient neurons [7]. Of note, PD mutations in GBA1 further led to mitochondrial dysfunction including defects in mitochondrial distribution, oxidative phosphorylation and ATP levels. However, overexpression of TBC1D15 was able to rescue mitochondria-lysosome contact dynamics as well as rescue mitochondrial dysfunction in mutant neurons from GBA1-PD patients [7], suggesting that defects in contact site dynamics may potentially drive downstream neuronal dysfunction in GBA1-PD. Interestingly, the mitochondrial kinase phosphatase and tensin homologue (PTEN)-induced kinase1 (PINK1) whose loss-of-function mutations also cause familial PD was recently found to modulate contact formation between late endosomes and mitochondria, to potentially transfer mtDNA for subsequent release in extracellular vesicles [65]. While genetic forms of PD (familial PD) represent only about 10% of PD cases, the rest are considered idiopathic or sporadic. Thus, whether misregulated mitochondria-lysosome contact tethering and function are involved in sporadic PD or other familial forms of PD remains an open question to be examined in future studies.
Lysosomal storage disorders are a group of genetic diseases resulting in impaired lysosomal degradation, leading to multisystem defects including neurodegeneration [66]. Mucolipidosis type IV (MLIV) is an autosomal disease caused by loss-of-function mutations in TRMPL1 [67, 68], an efflux channel on the lysosomal membrane recently shown to mediate calcium transfer to mitochondria via mitochondria-lysosome contact sites [37]. Interestingly, fibroblasts from MLIV patients with TRPML1 mutations showed decreased mitochondrial calcium uptake compared to control fibroblasts upon activation of lysosomal calcium release from TRMPL1. Moreover, while control fibroblasts showed preferential increase in mitochondrial calcium uptake for mitochondria in contact with lysosomes, this was not observed in MLIV patient fibroblasts. In addition, MLIV patient fibroblasts had increased mitochondria-lysosome contact formation and prolonged contact tethering dynamics [37], suggesting that loss of TRPML1 lysosomal function may contribute to mitochondrial dyshomeostasis in disease through this pathway. Furthermore, the accumulation of cholesterol and other sphingolipid species in the endocytic pathway has been linked to multiple lysosomal storage disorders, including the childhood neurodegenerative disease Niemann-Pick type C (NPC) [66]. Loss of function mutations in NPC1 are the main cause of NPC, and mammalian cell lines with loss or inhibition of NPC1 as well as NPC1 patient fibroblasts demonstrated increased percentage of lysosomes in contact with mitochondria, potentially contributing to misregulated cholesterol levels [38].
Finally, other major neurodegenerative disorders such as Alzheimer’s disease and Amyotrophic Lateral Sclerosis (ALS) have also been linked to pathogenic pathways involving mitochondria and lysosomes. This raises the question of whether disrupted crosstalk between these two organelles at mitochondria-lysosome contact sites may be among the contributors to disease pathogenesis in these disorders, and remains to be further investigated.
Future studies will help to elucidate the exact roles of mitochondrial-lysosome contacts, both in the specific disease models discussed earlier, and possibly other neurodegenerative diseases. It also remains to be tested whether impaired mitochondrial-lysosome contacts in neurons contribute to disease progression, or reflect more of a corollary of other, more detrimental pathological process. Of note, many of the disease mutations discussed earlier, as well as others, affect lysosomal or mitochondrial dynamics, and clarifying the interplay between impairments in each of these organelles and mitochondria-lysosome contact tethering and/or function remains a major goal for further research in this field. Finally, as non-cell autonomous pathways have also been shown to be important in neurodegeneration, and as mitochondria-lysosome contacts can form in multiple cell types, it is possible that defective mitochondria-lysosome contact dynamics or function in non-neuronal cells may additionally contribute to disease etiology.
Concluding remarks and future perspectives
Mitochondria-lysosome contacts have become increasingly appreciated as key players in neuronal biology and homeostasis. These contacts dynamically form in the neuronal soma, axons and dendrites. Importantly, multiple protein complexes come together to regulate the initial tethering and subsequent untethering dynamics of mitochondria-lysosome contacts. Functionally, these contacts have been found to mediate multiple cellular functions including the dynamic regulation of both mitochondrial and lysosomal network dynamics and organelle motility, as well as the regulation of organelle function including metabolite transfer between the two organelles. Finally, defects in mitochondria-lysosome contacts have been observed in different models of various neurodegenerative diseases including Charcot-Marie-Tooth disease, PD and lysosomal storage disorders, suggesting that their dysfunction may potentially contribute to neurodegeneration.
Future studies on mitochondria-lysosome contacts will be important for shedding light on the role of this pathway in neuronal function and disease (see Outstanding Questions). These include identifying additional regulators of contact site tethering and possibly, new functions that may be mediated through these contacts. Moreover, whether mitochondria-lysosome contacts play distinct roles in different neuronal subtypes, or in non-neuronal cells such as astrocytes or microglia remains to be explored. Finally, investigating the role of this pathway as an underlying mechanism in various neurological disorders linked to mitochondrial and/or lysosomal dysfunction may help shed light on its contribution to neurodegenerative disease pathogenesis.
OUTSTANDING QUESTIONS BOX.
Several proteins including Rab7 GTP hydrolysis have been found to regulate mitochondria-lysosome contacts. Are there additional regulators of contact site tethering dynamics?
Mitochondria-lysosome contacts modulate the transfer of specific ions between mitochondria and lysosomes, as well as regulate the dynamics of both organelles. Are there additional events which occur at mitochondria-lysosome contacts to further modulate mitochondrial and/or lysosomal function?
Selectively vulnerable neuronal populations degenerate in neurological disorders. Do mitochondria-lysosome contacts play distinct roles in different neuronal subtypes and/or contribute to non-cell autonomous pathways by modulating the function of non-neuronal cells such as astrocytes and microglia?
Defects in mitochondria-lysosome contact tethering have been found in several experimental models involving genetic mutations associated with neurodegenerative diseases. Do mitochondria-lysosome contacts play a causal role in the pathogenesis of these diseases, or are they a downstream consequence of dysfunction in other disease pathways? Do other neurological disorders previously linked to mitochondrial and lysosomal dysfunction also display impaired mitochondria-lysosome contact dynamics and function?
Conceivably, targeting mitochondria-lysosome contact tethering and function may represent a possible therapeutic approach for neurodegenerative diseases. Would either increasing or decreasing contact formation or tethering be beneficial for preventing disease pathogenesis? What are effective strategies for directly modulating mitochondria-lysosome contact site dynamics in patients, and would this be sufficient to rescue downstream neuronal dysfunction and degeneration?
HIGHLIGHTS.
Mitochondria-lysosome contacts form in multiple cell types including neurons, and throughout different neuronal compartments including the soma, axons and dendrites.
Mitochondria-lysosome contact untethering events are driven by Rab7 GTP hydrolysis, and may be further regulated by additional protein complexes.
Multiple aspects of mitochondrial and lysosomal network dynamics are modulated by mitochondria-lysosome contacts including mitochondrial fission and inter-mitochondrial untethering events.
Mitochondria-lysosome contacts regulate various organelle functions including the transfer of different metabolites between both organelles.
Defects in mitochondria-lysosome contacts have been observed in experimental models of genetic mutations associated with multiple neurodegenerative diseases including Charcot-Marie-Tooth disease, Parkinson’s disease and lysosomal storage disorders.
ACKNOWLEDGEMENTS
This work was funded by National Institutes of Health grants from NINDS to Y.C.W (R00NS109252) and J.C. (R00NS109252-03S1).
GLOSSARY
- Charcot-Marie-Tooth disease (CMT)
the most common group of genetic peripheral neuropathies, with autosomal dominant forms caused by axonal degeneration classified as CMT Type 2
- Fis1
mitochondrial fission 1 protein; an outer mitochondrial membrane protein which recruits TBC1D15 to the mitochondria
- GBA1
a gene encoding the lysosomal enzyme β-glucocerebrosidase (GCase) which converts glucosylceramide to glucose and ceramide. GBA1 is a genetic risk factor for PD
- Ganglioside Induced Differentiation Associated Protein 1 (GDAP1)
a gene encoding an outer mitochondrial membrane protein implicated in mitochondria-lysosome contact tethering and whose mutations cause Charcot-Marie-Tooth Type 2K
- Induced pluripotent stem cells (iPSC)
cells that can be reprogrammed from human skin or blood cells into a pluripotent state, allowing for subsequent differentiation into specific cell types such as neurons
- Inter-organelle membrane contact site
a stable contact between the membranes of two different organelles that are tethered in close apposition (<30nm) without fusion, which regulates the dynamics and/or function of either organelle
- Mitochondrial calcium uniporter (MCU)
an inner mitochondrial membrane channel which mediates calcium import into the mitochondria at mitochondria-lysosome contact sites
- Mitofusin 2
an outer mitochondrial membrane protein implicated in mitochondria-lysosome contact tethering and whose mutations cause Charcot-Marie-Tooth Type 2A
- Mucolipidosis Type IV (MLIV)
a lysosomal storage disorder involving neurodegeneration, caused by loss of function mutations in the lysosomal channel TRPML1
- Niemann-Pick Type C (NPC)
a lysosomal storage disorder involving neurodegeneration, caused by loss of function mutations in the lysosomal cholesterol transporters NPC1 and NPC2
- Parkinson’s disease (PD)
the most common neurodegenerative movement disorder. PD’s pathogenesis involves degeneration of dopaminergic neurons in the substantia nigra
- Rab7 GTP hydrolysis
the conversion of Rab7 GTPase from an active GTP-bound to inactive GDP-bound state mediated by GTPase activating proteins (GAPs). Rab7 localizes to lysosomal/late endosomal membranes to regulate their dynamics through its ability to bind Rab7 effector proteins, and is mutated in Charcot-Marie-Tooth Type 2B
- Structured illumination microscopy (SIM)
super-resolution microscopy imaging that has been used to visualize mitochondria-lysosome contacts in fixed and live cells
- TBC1D15
a GAP (GTPase activating protein) for Rab7 which is cytosolic and recruited to the outer mitochondrial membrane via Fis1
- Transient receptor potential mucolipin 1 (TRPML1)
a lysosomal calcium efflux channel which mediates calcium transfer to mitochondria at mitochondria-lysosome contact sites, whose mutations cause MLIV
- Vacuole and mitochondria patch (vCLAMP)
membrane contact site between mitochondria and vacuoles in yeast
- Voltage-dependent anion channel 1 (VDAC1)
an outer mitochondrial membrane channel which mediates calcium import into the mitochondria at mitochondria-lysosome contact sites
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests in relation to this work.
REFERENCES
- 1.Giacomello M et al. (2020) The cell biology of mitochondrial membrane dynamics. Nat Rev Mol Cell Biol 21 (4), 204–224. [DOI] [PubMed] [Google Scholar]
- 2.Wong YC et al. (2019) Lysosomal Regulation of Inter-mitochondrial Contact Fate and Motility in Charcot-Marie-Tooth Type 2. Dev Cell 50 (3), 339–354 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burte F et al. (2015) Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol 11 (1), 11–24. [DOI] [PubMed] [Google Scholar]
- 4.Spinelli JB and Haigis MC (2018) The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol 20 (7), 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bonam SR et al. (2019) Lysosomes as a therapeutic target. Nat Rev Drug Discov 18 (12), 923–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ballabio A and Bonifacino JS (2020) Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol 21 (2), 101–118. [DOI] [PubMed] [Google Scholar]
- 7.Kim S et al. (2021) Dysregulation of mitochondria-lysosome contacts by GBA1 dysfunction in dopaminergic neuronal models of Parkinson’s disease. Nat Commun 12 (1), 1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cioni JM et al. (2019) Late Endosomes Act as mRNA Translation Platforms and Sustain Mitochondria in Axons. Cell 176 (1–2), 56–72 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cantarero L et al. (2021) Mitochondria-lysosome membrane contacts are defective in GDAP1-related Charcot-Marie-Tooth disease. Hum Mol Genet 29 (22), 3589–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pickles S et al. (2018) Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr Biol 28 (4), R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sugiura A et al. (2014) A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J 33 (19), 2142–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deus CM et al. (2020) Mitochondria-Lysosome Crosstalk: From Physiology to Neurodegeneration. Trends Mol Med 26 (1), 71–88. [DOI] [PubMed] [Google Scholar]
- 13.Wong YC et al. (2018) Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 554 (7692), 382–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aston D et al. (2017) High resolution structural evidence suggests the Sarcoplasmic Reticulum forms microdomains with Acidic Stores (lysosomes) in the heart. Sci Rep 7, 40620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong YC et al. (2019) Regulation and Function of Mitochondria-Lysosome Membrane Contact Sites in Cellular Homeostasis. Trends Cell Biol 29 (6), 500–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khalil S et al. (2017) A specialized pathway for erythroid iron delivery through lysosomal trafficking of transferrin receptor 2. Blood Adv 1 (15), 1181–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Itoh K et al. (2018) A brain-enriched Drp1 isoform associates with lysosomes, late endosomes, and the plasma membrane. J Biol Chem 293 (30), 11809–11822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han Y et al. (2017) Cell-permeable organic fluorescent probes for live-cell long-term super-resolution imaging reveal lysosome-mitochondrion interactions. Nat Commun 8 (1), 1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fermie J et al. (2018) Single organelle dynamics linked to 3D structure by correlative live-cell imaging and 3D electron microscopy. Traffic 19 (5), 354–369. [DOI] [PubMed] [Google Scholar]
- 20.Valm AM et al. (2017) Applying systems-level spectral imaging and analysis to reveal the organelle interactome. Nature 546 (7656), 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Q et al. (2018) Super-Resolution Tracking of Mitochondrial Dynamics with An Iridium(III) Luminophore. Small 14 (41), e1802166. [DOI] [PubMed] [Google Scholar]
- 22.Zhang XM et al. (2005) TBC domain family, member 15 is a novel mammalian Rab GTPase-activating protein with substrate preference for Rab7. Biochem Biophys Res Commun 335 (1), 154–61. [DOI] [PubMed] [Google Scholar]
- 23.Peralta ER et al. (2010) Differential effects of TBC1D15 and mammalian Vps39 on Rab7 activation state, lysosomal morphology, and growth factor dependence. J Biol Chem 285 (22), 16814–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Onoue K et al. (2013) Fis1 acts as a mitochondrial recruitment factor for TBC1D15 that is involved in regulation of mitochondrial morphology. J Cell Sci 126 (Pt 1), 176–85. [DOI] [PubMed] [Google Scholar]
- 25.Friedman JR et al. (2011) ER tubules mark sites of mitochondrial division. Science 334 (6054), 358–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korobova F et al. (2013) An actin-dependent step in mitochondrial fission mediated by the ER-associated formin INF2. Science 339 (6118), 464–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manor U et al. (2015) A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li S et al. (2015) Transient assembly of F-actin on the outer mitochondrial membrane contributes to mitochondrial fission. J Cell Biol 208 (1), 109–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore AS et al. (2016) Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat Commun 7, 12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moore AS et al. (2021) Actin cables and comet tails organize mitochondrial networks in mitosis. Nature 591 (7851), 659–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schiavon CR et al. (2020) Actin chromobody imaging reveals sub-organellar actin dynamics. Nat Methods 17 (9), 917–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kleele T et al. (2021) Distinct fission signatures predict mitochondrial degradation or biogenesis. Nature 593 (7859), 435–439. [DOI] [PubMed] [Google Scholar]
- 33.Boutry M and Kim PK (2021) ORP1L mediated PI(4)P signaling at ER-lysosome-mitochondrion three-way contact contributes to mitochondrial division. Nat Commun 12 (1), 5354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagashima S et al. (2020) Golgi-derived PI(4)P-containing vesicles drive late steps of mitochondrial division. Science 367 (6484), 1366–1371. [DOI] [PubMed] [Google Scholar]
- 35.Guo Y et al. (2018) Visualizing Intracellular Organelle and Cytoskeletal Interactions at Nanoscale Resolution on Millisecond Timescales. Cell 175 (5), 1430–1442 e17. [DOI] [PubMed] [Google Scholar]
- 36.Langemeyer L et al. (2018) Rab GTPase Function in Endosome and Lysosome Biogenesis. Trends Cell Biol 28 (11), 957–970. [DOI] [PubMed] [Google Scholar]
- 37.Peng W et al. (2020) Mitochondria-lysosome contacts regulate mitochondrial Ca(2+) dynamics via lysosomal TRPML1. Proc Natl Acad Sci U S A 117 (32), 19266–19275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoglinger D et al. (2019) NPC1 regulates ER contacts with endocytic organelles to mediate cholesterol egress. Nat Commun 10 (1), 4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Juhl AD et al. (2021) Quantitative imaging of membrane contact sites for sterol transfer between endolysosomes and mitochondria in living cells. Sci Rep 11 (1), 8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lim CY et al. (2019) ER-lysosome contacts enable cholesterol sensing by mTORC1 and drive aberrant growth signalling in Niemann-Pick type C. Nat Cell Biol 21 (10), 1206–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang M et al. (2002) MLN64 mediates mobilization of lysosomal cholesterol to steroidogenic mitochondria. J Biol Chem 277 (36), 33300–10. [DOI] [PubMed] [Google Scholar]
- 42.Charman M et al. (2010) MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann-Pick Type C1 protein. J Lipid Res 51 (5), 1023–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Honscher C et al. (2014) Cellular metabolism regulates contact sites between vacuoles and mitochondria. Dev Cell 30 (1), 86–94. [DOI] [PubMed] [Google Scholar]
- 44.Elbaz-Alon Y et al. (2014) A dynamic interface between vacuoles and mitochondria in yeast. Dev Cell 30 (1), 95–102. [DOI] [PubMed] [Google Scholar]
- 45.Gonzalez Montoro A et al. (2018) Vps39 Interacts with Tom40 to Establish One of Two Functionally Distinct Vacuole-Mitochondria Contact Sites. Dev Cell 45 (5), 621–636 e7. [DOI] [PubMed] [Google Scholar]
- 46.Bean BDM et al. (2018) Competitive organelle-specific adaptors recruit Vps13 to membrane contact sites. J Cell Biol 217 (10), 3593–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.John Peter AT et al. (2017) Vps13-Mcp1 interact at vacuole-mitochondria interfaces and bypass ER-mitochondria contact sites. J Cell Biol 216 (10), 3219–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elbaz-Alon Y et al. (2015) Lam6 Regulates the Extent of Contacts between Organelles. Cell Rep 12 (1), 7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schiavon CR et al. (2021) Impaired Mitochondrial Mobility in Charcot-Marie-Tooth Disease. Front Cell Dev Biol 9, 624823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Houlden H et al. (2004) A novel RAB7 mutation associated with ulcero-mutilating neuropathy. Ann Neurol 56 (4), 586–90. [DOI] [PubMed] [Google Scholar]
- 51.McCray BA et al. (2010) Disease mutations in Rab7 result in unregulated nucleotide exchange and inappropriate activation. Hum Mol Genet 19 (6), 1033–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Spinosa MR et al. (2008) Functional characterization of Rab7 mutant proteins associated with Charcot-Marie-Tooth type 2B disease. J Neurosci 28 (7), 1640–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang K et al. (2013) Defective axonal transport of Rab7 GTPase results in dysregulated trophic signaling. J Neurosci 33 (17), 7451–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cassereau J et al. (2009) Mitochondrial complex I deficiency in GDAP1-related autosomal dominant Charcot-Marie-Tooth disease (CMT2K). Neurogenetics 10 (2), 145–50. [DOI] [PubMed] [Google Scholar]
- 55.Chung KW et al. (2008) A novel GDAP1 Q218E mutation in autosomal dominant Charcot-Marie-Tooth disease. J Hum Genet 53 (4), 360–364. [DOI] [PubMed] [Google Scholar]
- 56.Santel A and Fuller MT (2001) Control of mitochondrial morphology by a human mitofusin. J Cell Sci 114 (Pt 5), 867–74. [DOI] [PubMed] [Google Scholar]
- 57.Zuchner S et al. (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 36 (5), 449–51. [DOI] [PubMed] [Google Scholar]
- 58.Boyer O et al. (2011) INF2 mutations in Charcot-Marie-Tooth disease with glomerulopathy. N Engl J Med 365 (25), 2377–88. [DOI] [PubMed] [Google Scholar]
- 59.Zuchner S et al. (2005) Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nat Genet 37 (3), 289–94. [DOI] [PubMed] [Google Scholar]
- 60.Lee JE et al. (2016) Multiple dynamin family members collaborate to drive mitochondrial division. Nature 540 (7631), 139–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kamerkar SC et al. (2018) Dynamin-related protein 1 has membrane constricting and severing abilities sufficient for mitochondrial and peroxisomal fission. Nat Commun 9 (1), 5239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fonseca TB et al. (2019) Mitochondrial fission requires DRP1 but not dynamins. Nature 570 (7761), E34–E42. [DOI] [PubMed] [Google Scholar]
- 63.Poewe W et al. (2017) Parkinson disease. Nat Rev Dis Primers 3, 17013. [DOI] [PubMed] [Google Scholar]
- 64.Burbulla LF et al. (2019) A modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson’s disease. Sci Transl Med 11 (514). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rabas N et al. (2021) PINK1 drives production of mtDNA-containing extracellular vesicles to promote invasiveness. J Cell Biol 220 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Platt FM et al. (2018) Lysosomal storage diseases. Nat Rev Dis Primers 4 (1), 27. [DOI] [PubMed] [Google Scholar]
- 67.Bassi MT et al. (2000) Cloning of the gene encoding a novel integral membrane protein, mucolipidin-and identification of the two major founder mutations causing mucolipidosis type IV. Am J Hum Genet 67 (5), 1110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sun M et al. (2000) Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum Mol Genet 9 (17), 2471–8. [DOI] [PubMed] [Google Scholar]