

Summary
Understanding the organization of the cell cycle has been a longstanding goal in cell biology. We combined time-lapse microscopy, highly-multiplexed single-cell imaging of 48 core cell cycle proteins, and manifold learning to render a visualization of the human cell cycle. This data-driven approach revealed the comprehensive “structure” of the cell cycle: the continuum of molecular states that cells occupy as they transition from one cell division to the next, or as they enter or exit cell cycle arrest. Paradoxically, progression deeper into cell cycle arrest was accompanied by increases in proliferative effectors such as CDKs and cyclins, which drove cell cycle re-entry by overcoming p21 induction. The structure also revealed the molecular trajectories into senescence and the unique combination of molecular features that define this irreversibly arrested state. This approach will enable the comparison of alternative cell cycles during development, in response to environmental perturbation and in disease. A record of this paper’s Transparent Peer Review process is included in the Supplemental Information.
eTOC Summary
A complete visualization of the human cell cycle is rendered by combining highly multiplexed single-cell imaging and manifold learning. The molecular trajectories through the proliferative and arrested cell cycle are revealed and the mechanisms governing cell cycle exit and re-entry are characterized. In addition, the multivariate molecular signature that defines cellular senescence is identified.
Introduction
Our current understanding of the human cell cycle has been assembled in piecemeal fashion from decades of biochemical and genetic experiments (Nurse, 2000). Early studies identifying discrete time periods of DNA synthesis and cell division (Howard and Pelc, 1951) led to adoption of a five-phase model that separates the cell cycle into four functionally distinct proliferative phases (G1, S, G2 and M) and a single state of arrest (G0). This canonical model, which has shaped our thinking for over 70 years, provides a useful framework for mapping key molecular events that govern the progression of a typical cell through the cell cycle.
Advances in molecular biology, particularly in budding yeast, led to the identification of numerous cell cycle regulators (e.g. cyclins and cyclin-dependent kinases (CDKs)) controlling DNA replication, mitosis, and cell cycle arrest (Glotzer et al., 1991; Morgan, 2007; Murray, 1994). To accommodate these discoveries, more sophisticated models were proposed to describe the cell cycle’s overall behavior. One class of models views the cell cycle as an oscillator, or molecular clock, in which the periodic activity of CDKs and expression of E2F-driven genes drive cell proliferation (Coudreuse and Nurse, 2010; Murray and Kirschner, 1989; Orlando et al., 2008). Alternative models emphasize the sequential nature of cell cycle events that, like dominoes, must be completed in a defined order before moving on to the next molecular step in the sequence (Castor, 1980; Chao et al., 2019; Smith and Martin, 1973). Both models have provided helpful frameworks for understanding the fundamental nature of cell cycle progression.
Over the last ten years, however, a wave of single-cell studies has shown that progression through the cell cycle cannot be reduced to a single, fixed sequence of molecular events. Cells can progress at different rates through each of the proliferative phases (Araujo et al., 2016; Chao et al., 2019; Shields, 1977; Smith and Martin, 1973). Individual cells may also vary in the precise molecular paths taken through these phases (Cappell et al., 2016, 2018; Liu et al., 2020; Yang et al., 2020). Moreover, transitions into—or out of—cell cycle arrest are at least as heterogeneous as the proliferative cell cycle but more poorly understood. Cells may exit into reversible (“quiescent”) or irreversible (“senescent”) states of arrest, from different phases of the proliferative cell cycle and driven by different mechanisms (Marescal and Cheeseman, 2020; Sagot and Laporte, 2019). After exiting the cell cycle, individual cells may re-enter after variable lengths of time (Barr et al., 2017; Chung et al., 2019; Overton et al., 2014; Spencer et al., 2013; Yang et al., 2017, 2020), but the mechanisms that regulate this decision remain unclear. It also appears that certain quiescent states can become “deeper” with time, requiring a larger or longer stimulus to re-enter the cell cycle (Kwon et al., 2017; Owen et al., 1989; Wang et al., 2017), and in some cases may become irreversible (Fujimaki et al., 2019; Marthandan et al., 2014; Sousa-Victor et al., 2014). These studies collectively imply that the cell cycle is a highly plastic process in which cells may traverse different mechanistic routes en route to cell division or cell cycle arrest. Although this heterogeneity is becoming increasingly appreciated, we lack a comprehensive model that integrates single-cell heterogeneity into the overall organization of cell cycle progression.
Recent work has begun using single-cell mRNA sequencing to describe the heterogeneity in cell cycle progression (Kowalczyk et al., 2015; Moussa and Măndoiu, 2020; Schwabe et al., 2020). However, it is widely appreciated that progression through the cell cycle is primarily driven by changes in protein turnover, post-translational modifications or the subcellular localization of key effectors (Morgan, 2007) — features that cannot be captured by transcriptomics. Other studies using protein-based measurements to study cell cycle progression have focused on mapping the dynamics of individual cell cycle effectors along a one-dimensional, generalized model of the cell cycle (Gookin et al., 2017; Gut et al., 2015; Mahdessian et al., 2021). These approaches provide a rich resource for describing the dynamics of proteins during a typical cell cycle. However, because they do not measure higher-order relationships among multiple effectors (e.g. simultaneous measurements of cyclins, CDKs, and CDK inhibitors in individual cells), they cannot provide a comprehensive description of single-cell states, and thus lack the capacity to map alternative cell cycle trajectories or study how multiple cell cycle regulators interact to coordinate cell cycle fate decisions.
To look for evidence of cell cycle heterogeneity at the single-cell level and to better understand the mechanistic basis of this plasticity, we performed time-lapse imaging to record the cell cycle histories of individual human epithelial cells followed by iterative immunofluorescence of 48 core cell cycle regulators to obtain high-dimensional, protein-based cell cycle signatures of thousands of individual cells. We used manifold learning to project these cells onto two- and three-dimensional surfaces to visualize the cell cycle as a sequence of continuous molecular states, ultimately revealing the underlying “structure” of the cell cycle at the protein level. We find that the primary source of heterogeneity in the proliferative cell cycle occurs shortly after cell division, when cells diverge down two distinct paths, either immediately re-entering the cell cycle or diverting into a quiescent state driven by p21 induction. Through targeted experiments, we demonstrate that cell cycle re-entry from this quiescent state does not necessarily require a reversal of the mechanisms that induced arrest, and that cells can overcome p21 induction and re-enter the cell cycle by increasing the expression of proliferative effectors such as cyclin D. Cells that remain arrested eventually transition into an irreversibly arrested senescent state. We define a unique molecular signature for this state and provide evidence that cells can enter a similar senescent state from either G1 or G2.
Results
Visualizing the structure of the cell cycle
Starting with an asynchronous population of non-transformed human retinal pigmented epithelial cells (hTERT-RPE-1, abbreviated hereafter as RPE cells; Figure 1A) expressing a single fluorophore cell cycle reporter (PCNA-mTurquoise2 (mTq2) (Zerjatke et al., 2017), we performed time-lapse imaging to record the cell cycle histories of individual cells, including the cell cycle phase (i.e. G0/G1, S, G2 or M) and age (i.e. time elapsed since previous mitosis) of each cell (Figure 1B). Following time-lapse imaging, cells were fixed and then subjected to multiple rounds of immunofluorescence using iterative indirect immunofluorescence imaging (4i) (Gut et al., 2018) to obtain measurements of 48 core cell cycle effectors (Table S1) in a total of 8850 individual cells. From this imaging dataset we extracted 246 single-cell features including the expression and localization of each protein (i.e. in the nucleus, cytosol, perinuclear region and plasma membrane), cell morphological features (e.g. the size and shape of the nucleus and cell), and features of the microenvironment (e.g. local cell density), culminating in a multivariate cell cycle signature for each cell in the entire population.
Figure 1. Probing the structure of the cell cycle.
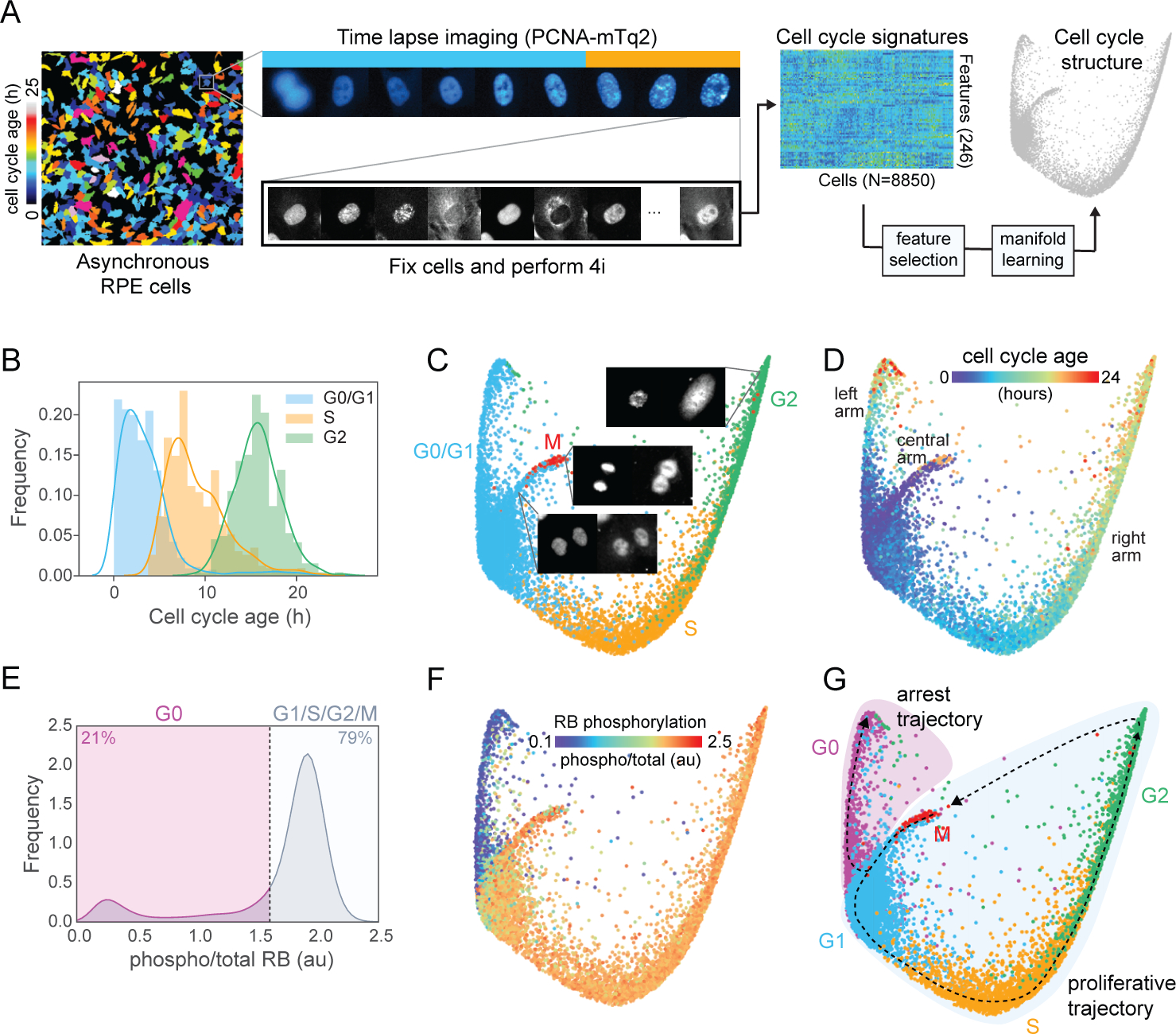
(A) Schematic of the experimental approach. (B) Distributions of cell cycle phase and age (time since mitosis) of all cells annotated by time-lapse imaging. (C-D) Cell cycle phase (C) and age (D) annotations/predictions (see Methods) of 8850 individual cells mapped onto the structure. Representative images of mitotic cells and their locations on the structure are shown in (C). (E-F) Distribution of RB phosphorylation (phospho/total nuclear intensity) in individual cells (E) and mapped on the structure (F). (G) Proliferative (G1/S/G2/M) and arrest (G0) trajectories through the five canonical phases. Scale bars = 10 μm.
Cells at similar positions in the cell cycle should possess similar cell cycle signatures (as defined by 4i), and thus be located close to one another on a lower dimensional manifold within this high-dimensional feature space. To identify and visualize this manifold, we used Potential of Heat-diffusion for Affinity-based Transition Embedding (PHATE), a nonlinear manifold learning approach that performs well for continuous and branched trajectories in high-dimensional space (Moon et al., 2019). However, if we perform PHATE using this entire 246-dimensional feature set, we obtain a two-dimensional “structure” that fails to adequately capture progression through the cell cycle (Figure S1A). We reasoned that if our feature set contains variables that differ among cells in a cell cycle-independent manner, the manifold we obtained would be convoluted by other biological processes. We therefore used a machine learning approach to enrich our feature set for variables that best predict the cell cycle ‘state’ of each cell (see Methods). We trained two random forest classifiers to predict either cell cycle phase or age (using the ground truth annotations obtained by time-lapse imaging) from the 4i signatures of individual cells (with a 95.5% accuracy for phase and an RMSE=125.8 min and R2=0.862 for age predictions) and calculated the predictive power of each feature. Features were ranked by their predictive power across both models and a minimal feature set that accurat ely predicted cell cycle state (i.e. both phase and age) was determined (Figure S1B–C, Table S2). We also used these models to infer the phase and age of cells that were not captured by time-lapse imaging (Figure 1C-D, Movies S2 and S3, Figure S1F–G). Similar inferences of phase and age were obtained using a convolutional neural network trained on the same data (Phase: 95.7% accuracy, concordance with RF model = 94.8%; Age: RMSE = 123.7 min; Figure S1H–K).
Using this minimal feature set, we obtained a continuous structure that successfully captured the progression of cells through the canonical phases of the cell cycle (G1, S, G2 and M, respectively) (2D structure in Figure 1; 3D structure in Movies S1–S4; virtual reality-compatible dataset available (see Data and materials availability)). This three-armed structure was reproducible across PHATE parameter space (Figure S2A), individual replicates (Figure S2B) and in another human epithelial cell line (human pancreatic epithelial cells, HPNE; Figure S2C). We observed that G0/G1 cells encompass most of the left and central arms, and S/G2 cells progressively reside along the rightmost arm, respectively (Figure 1C). While cell cycle progression towards mitosis is clearly represented along the right arm of the structure, mitotic cells were located at the tip of the central arm. Visual inspection of the immunofluorescence images of cells along the central arm revealed a progression through nuclear envelope reformation and cytokinesis, events consistent with the late stages of mitosis. Similarly, cells at the tip of the right arm possessed features of late G2 and early M, including DNA condensation and nuclear envelope breakdown. Mapping cell cycle age onto the structure indicated that the youngest cells lie near the tip of the central arm where newborn cells emerge out of mitosis and into G1, and that age increases as cells progress up both the left and right arms (Figure 1D).
While the PCNA-mTq2 reporter allows delineation of the proliferative phases of the cell cycle (G1/S/G2/M) (Zerjatke et al., 2017), it cannot distinguish G0 from G1 cells. The phosphorylation and inactivation of the retinoblastoma protein (RB), on the other hand, represents a major checkpoint controlling cell cycle re-entry and is often used as a molecular correlate to define the boundary between G0 (i.e. cell cycle arrest) and G1 (i.e. cell cycle commitment) (Moser et al., 2018; Pardee, 1974; Zetterberg and Larsson, 1985). In our 4i data, we observed bimodality in RB phosphorylation across the entire population (Figure 1E), and a clear delineation between these two cellular states (high vs. low RB phosphorylation) along the central arm, thus distinguishing G0 from actively cycling cells (Figure 1F, Movie S4). We use this RB phosphorylation status to identify actively proliferating versus arrested cells throughout the manuscript.
Whether we use the age of cells to infer how cells progress along the structure with time (Figure 1D), or diffusion pseudotime (DPT, Figure 2A), which measures changes in molecular state along the structure (Haghverdi et al., 2016), we infer two principal trajectories: (1) a cyclical, proliferative trajectory starting at the base of the central arm and progressing along the right arm through G1, S and G2, before looping back to the central arm during mitosis, and (2) an arrest trajectory along the left arm (Figure 1G).
Figure 2. Measuring the rate of molecular change during the cell cycle.

(A) Diffusion pseudotime (DPT) is mapped onto the cell cycle structure. (B-C) Comparisons of molecular age (pseudotime) and actual cell cycle age along the proliferative (B) and arrest (C) trajectories. Individual cells are colored by cell cycle phase. Mitotic cells are excluded.
If we compare the actual age of cells (as determined by time-lapse imaging) to their “molecular age” (as determined by DPT), there is a strong correlation along both the proliferative (r=0.895) and arrest trajectories (r=0.827) (Figure 2B–C). However, the rate of molecular change is not constant along these trajectories. Cells rapidly accumulate large molecular changes at the G1/S transition as cells initiate DNA replication, and once again in G2 after DNA replication has ceased and cells prepare for mitosis (Figure 2B). We will elaborate on the molecular changes that accompany progression along the proliferative trajectory in the next section. Along the arrest trajectory, there is a relatively constant rate of molecular change with time, except for a subpopulation of long-arrested cells that exhibit comparatively large changes in molecular state (Figure 2C). These cells possess features consistent with irreversibly arrested senescent cells and will be examined in greater detail later in this manuscript.
Mapping the mechanisms of the proliferative cell cycle (G1/S/G2/M)
To validate our approach, we used cell cycle age to temporally order actively cycling cells along the proliferative trajectory and examined well-established mechanisms known to drive cell cycle progression (Supplementary Data S1). This approach successfully ordered key molecular events that have previously been shown to regulate the G1/S transition (Figure 3A) (Hume et al., 2020). The core molecular unit regulating this decision is the RB-mediated inhibition of E2F transcription factors (Stallaert et al., 2019), which controls the expression of various S phase genes (DeGregori et al., 1995). Commitment to DNA replication is triggered by the phosphorylation and inhibition of RB by cyclin:CDK complexes. In early G1, RB is primarily phosphorylated by cyclin D:CDK4/6 (Chung et al., 2019), and we observed that cells begin their cell cycle with high cyclin D1 expression (Figure 3B). This cyclin D:CDK4/6-driven phosphorylation of RB in early G1 derepresses E2F-regulated genes important for DNA replication including expression of E2F1 itself (Figure 3C) (Narasimha et al., 2014; Sanidas et al., 2019). E2F activity also stimulates the production of cyclin E (Figure 3D), which activates CDK2 to maintain RB phosphorylation as cyclin D levels begin to decrease (Figure 3B) (Chung et al., 2019; Gookin et al., 2017). Another important event in the G1/S transition is the inactivation of APC/C complexes, which degrade and prevent the accumulation of S phase proteins during G1. The increase in cyclin E:CDK2 activity in late G1 stimulates the destruction of the Cdh1 subunit (Figure 3E), switching off APC/C and permitting S phase initiation (Cappell et al., 2016). The inactivation of APC/C also allows cyclin A to accumulate as S phase begins (Figure 3F) to replace cyclin E and maintain CDK2-dependent RB phosphorylation through to mitosis (Figure 3D).
Figure 3. Visualizing the mechanisms of the G1/S transition.
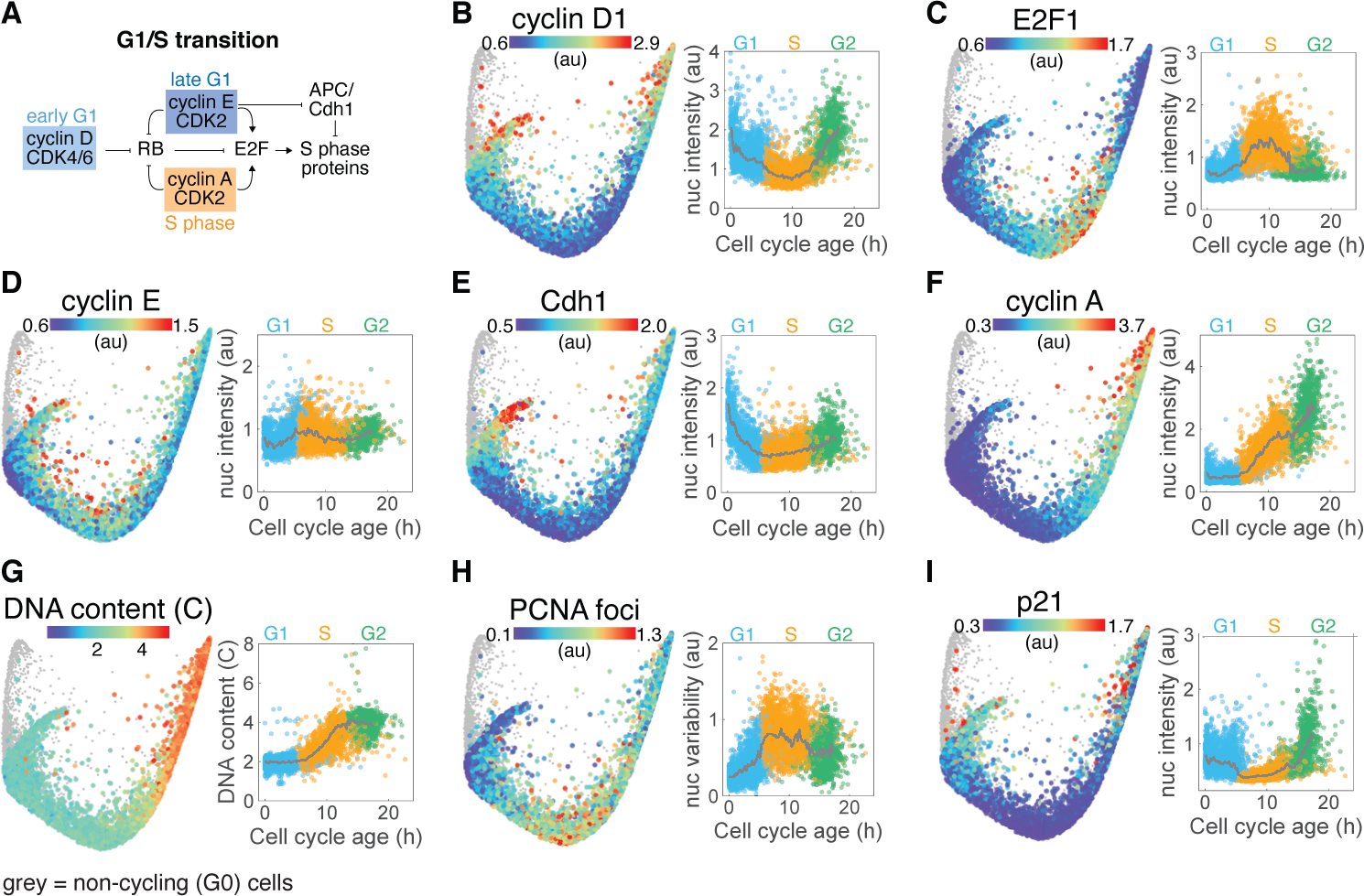
(A) Mechanistic model of the core events regulating progress through G1 and the transition to S. (B-I) Median nuclear intensities of cyclin D1 (B), E2F1 (C), cyclin E (D), Cdh1 (E), cyclin A (F) and p21 (I), DNA content (copy number) (G), and variability in nuclear PCNA intensity (H) are mapped onto cells in the proliferative trajectory (left panels) and plotted against cell cycle age (right panels). Population medians in time courses indicated by solid grey lines and individual cells are colored by cell cycle phase (G1: blue, S: orange, G2: green). Non-cycling (G0) cells (phospho/total RB < 1.6) are shown in grey on the structure and are excluded from time courses.
DNA replication was clearly visible on the structure, with DNA content doubling over the course of S phase (Figure 3G). Similarly, we localized additional S phase events important for DNA replication including the appearance of PCNA foci at replication complexes (quantified as variability in nuclear intensity) (Figure 3H) and the replication-coupled destruction of p21 (Figure 3I). Upon entry into G2, cyclin B expression increases first in the cytoplasm and then in the nucleus (Figure S3A–B) as cells move toward mitosis (Santos et al., 2012). We also observed increases in cyclin D1 (Figure 3B) and its transcription factor c-Myc (Figure S3C), as well as NF-kB activation (Figure S3D) during G2, which then remained elevated through mitosis and into the subsequent G1 phase of daughter cells, resulting in U-shape dynamics along the proliferative trajectory, as recently reported by Gookin et al. (2017). Thus, the cell cycle structure accurately recapitulated many of the key molecular events governing the progression of cells through the four proliferative cell cycle phases.
Trajectories into and out of cell cycle arrest (G0)
Next, we investigated the molecular changes that occur as cells progress along the arrest trajectory (Figure 4, Supplementary Data S2), for which comparatively less is known. We noted that all cells exited mitosis with high RB phosphorylation along a single path down the central arm and then quickly diverged along two distinct trajectories within the first 2–3 hours following mitosis: either progressing directly into G1 with sustained RB phosphorylation or exiting the cell cycle into G0 following the loss of RB phosphorylation (Figure 1F; Figure 4A; Figure S4A–B). In each daughter cell, this fate decision was found to be governed primarily by the balance between cyclin D and p21 expression (Figure 4B, Figure S4A, Figure S4C–D), as previously shown (Chen et al., 2013; Overton et al., 2014; Yang et al., 2017). We found that the decrease in the cyclin D1:p21 ratio that precedes cell cycle exit was not due to a decrease in cyclin D1, but rather by an increase in p21 in early G1 (Figure 4B–D; Figure S4A). This induction of p21 in early G1 occurred simultaneously with the loss of RB phosphorylation as cells diverted to the arrest trajectory (Figure S4A–B), consistent with the CDK2low/p21high spontaneous state of arrest first identified by Spencer et al. (2013).
Figure 4. Cell cycle exit and re-entry from arrest (G0).

(A) Divergence of cell cycle trajectories into G1 or G0 following cell division and subsequent cell cycle re-entry from G0. Age of newborn cells (<3h since mitosis) is mapped onto the structure. (B-I) The z-normalized nuclear intensity ratio of cyclin D1:p21 (B) and median nuclear expression of cyclin D1 (C), p21 (D), CDK4 (E), cyclin E (F), RB (G), E2F1 (H) and Cdt1 (I) are mapped onto G0/G1 cells. Note the dotted circle representing cell cycle re-entry in F, H and I. Non-G0/G1 cells are shown in grey. (J) List of features with the highest predictive power in a random forest regression model trained to predict arrest duration. (K) Representative single-cell traces of p21-mTq2 (green) and cyclin D1-YFP expression (blue) aligned at anaphase. The dotted line represents cell cycle re-entry as indicated by CDK2 reactivation (an increase in the cytoplasmic:nuclear ratio of DHB-mCherry, not shown). (L) p21 expression at cell cycle exit (1h post-mitosis) versus re-entry (at time of CDK2 reactivation) in individual cells. Statistical significance was determined using a Student’s paired t test. (M) Median nuclear intensities of cyclin D1 and p21 measured by time-lapse imaging at cell cycle re-entry. Individual cells are colored by their duration of arrest. N = 117 cells for panels L and M.
Further examination of the arrest trajectory revealed that cells do not exit the cell cycle into a single, static arrest state awaiting cell cycle re-entry. Instead, cells progressed further along this trajectory with time (Figure 1D) accompanied by a progressive increase in p21 expression (Figure 4D), indicating an increasing depth of arrest as has been previously suggested (Fujimaki et al., 2019; Gookin et al., 2017; Kwon et al., 2017; Wang et al., 2017). To systematically assess the molecular changes that occur as cells progress further along the arrest arm, we trained a random forest model to predict the duration of cell cycle arrest based on the 4i signature of a given cell (Figure 4J). Surprisingly, besides increased p21 expression, the most significant molecular changes that accompanied progression along the arrest arm were factors known to promote cell cycle progression, not arrest. These included increases in the expression of CDK4 (Figure 4E) and CDK2 (Data S2) as well as the G1 cyclins D1 (Figure 4C) and E (Figure 4F), and a decrease in RB expression (Figure 4G). We therefore hypothesized that this accumulation of proliferative effectors (and loss of RB) that occurs during cell cycle arrest may provide a mechanism for cell cycle re-entry—not by reversing the increase in p21 expression that originally drove cell cycle exit—but instead by overcoming it with excessive cyclin and/or CDK expression.
To test this hypothesis, we performed time-lapse imaging of RPE cells expressing fluorescent p21 (p21-mTq2) and cyclin D1 (cycD1-Venus) from their endogenous loci and a CDK2 activity sensor (DHB-mCherry) to monitor cell cycle exit and re-entry (Spencer et al., 2013). The majority of cells (86%) emerged from cell cycle arrest (as indicated by the reactivation of CDK2) in the continued presence of elevated p21, often shortly after an increase in cyclin D1 (Figure 4K, Figure S5A–B). These cells re-entered the cell cycle at equal or higher p21 than was initially required to induce cell cycle exit following cell division (Figure 4L). We also observed some instances of cell cycle re-entry that were accompanied instead by a prior reduction in p21 (Figure S5C–D). This cellular heterogeneity in effector dynamics at re-entry indicates that neither an increase in cyclin D nor a decrease in p21 alone can fully explain the timing of cell cycle re-entry in all cells. Indeed, we observed that the amount of p21 that is permissible for cell cycle re-entry is at least partially determined by cyclin D1 expression (r2= 0.587, Figure 4M), with higher cyclin D1 expression permitting re-entry at higher p21 expression suggesting that, like cell cycle exit, re-entry is also controlled by the balance between p21 and cyclin D. Altogether, these data demonstrate that a complete reversal of the p21 induction that drives cell cycle arrest is not required for cell cycle re-entry, and that increased expression of countervailing proliferative effectors such as cyclin D1 may permit cell cycle re-entry in the presence of elevated p21 expression.
Consistent with these observations, the structure revealed that cells do not simply re-enter the cell cycle by reversing back down the arrest trajectory but instead can traverse along a distinct, funnel-shaped trajectory across the inside of the structure, spanning the gap between the arrest arm and G1 (Figure 4A). Cells along this re-entry trajectory possessed increased expression of cyclin D1 (Figure 4C), a high cyclin D1:p21 ratio (Figure 4B) and high RB phosphorylation (Figure 1F, Movie S4), consistent with cell cycle re-entry. Having spent variable amounts of time in G0, these cells were relatively old compared to other G1 cells (Figure 1D, Movie S2), and exhibited characteristics of cells preparing for DNA replication including increased expression of cyclin E (Figure 4F), E2F1 (Figure 4H) and Cdt1 (Figure 4I).
Cellular senescence
Graph-based clustering resolved a subpopulation of cells at the tip of the arrest trajectory (Figure 5A) undergoing rapid molecular changes after >12 h of arrest (Figure S6A) and possessing a distinct molecular signature among arrested cells (Figure 5B). These cells had the longest durations of arrest (Figure 1D), were at least 3–4 times larger than the average arrested cell (Figure 5C), and possessed a high cytoplasm-to-DNA ratio (Figure 5D), a hallmark of irreversibly arrested senescent cells (Lanz et al., 2021; Neurohr et al., 2019). To validate this putative cluster of senescent cells and the features that define it, we combined immunofluorescence measurements with a fluorescent reporter of β-galactosidase activity (Figure S6B), an established hallmark of senescence (Hjelmeland et al., 1999). Senescent cells possessed increased abundance of CDK4, CDK6, GSK3β, and p27, as well as elevated phosphorylation of p27(Thr157), p21(Thr145) and p53(Ser6) (Figure 5E–H, Figure S6C–E). While each of these individual features were found to be significantly elevated in senescent cells, none were perfectly predictive of the senescent state on their own. Instead, senescence is cellular state characterized by coordinated changes in multiple cell cycle/signaling effectors. Our multivariate signature of cell cycle state provides a more accurate prediction of senescent cells (97.5%) than any individual feature (Figure S6F).
Figure 5. Molecular signature of cellular senescence.
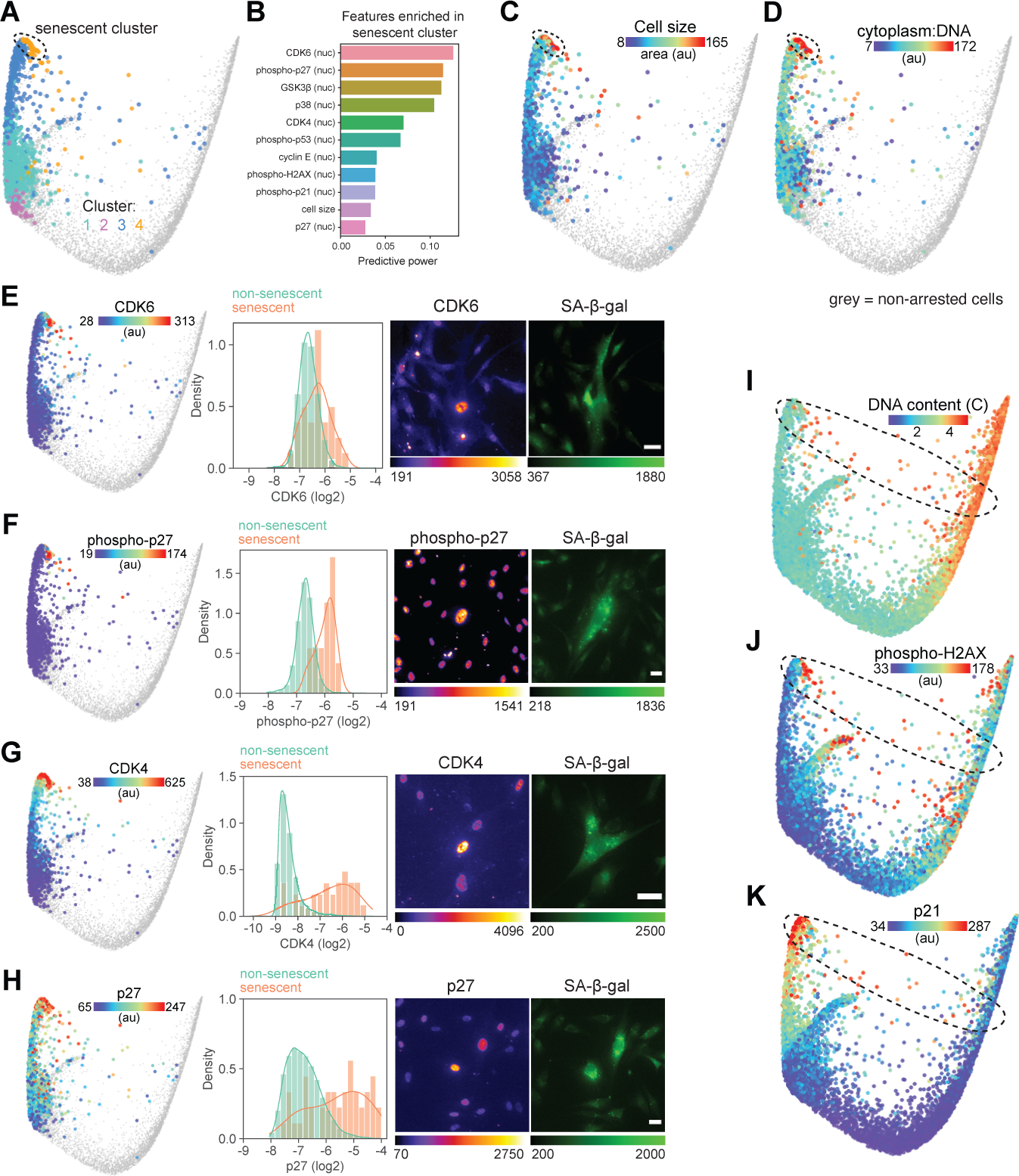
(A) Graph-based clustering of arrested cells mapped onto the cell cycle structure. Senescent cells are found in cluster 4. (B) List of features with the highest predictive power in a random forest model trained to identify senescent cells (cluster 4) from the rest of the population. (C-D) Cell area (C) and ratio of cytoplasmic area:DNA content (D) mapped onto arrested cells. (E-H) Validated features of senescent cells mapped onto the arrest trajectory of the cell cycle structure (left panels), single-cell distributions of log2-normalized feature intensity in non-senescent (β-galactosidase (β-gal)-negative, green) and senescent (β-gal-positive, orange) cells (middle panels), and representative images of feature and β-gal staining of senescent cells (right panels). All senescent features shown here (and in Figure S6) showed significant differences in feature intensity between non-senescent and senescent populations (p<0.01 using an unpaired Welch t test with unequal variance). (I-K) DNA content (I) and nuclear median intensity of phospho-H2AX (J) and p21 (K) mapped onto the structure. Scale bar = 40 μm. Non-arrested cells (phospho/total RB > 1.6) are shown in grey on cell cycle structures.
These senescent cells at the end of the arrest trajectory possessed either 2 or 4 copies of DNA (2C or 4C; Figure 5I) (and no multinucleated cells were observed), indicating that some cells must have exited the proliferative cell cycle along a second arrest trajectory that diverts after DNA replication. Indeed, we identified a population of cells residing along a path between G2 and the arrest terminus with 4 copies of DNA (Figure 5I, Movie S1), low RB phosphorylation (Figure 1D), and elevated markers of a DNA damage response including elevated phospho-H2AX (Figure 5J) and p21 (Figure 5K), consistent with cell cycle exit due to the G2/M DNA damage checkpoint (Bunz et al., 1998). Thus, cells may progress into senescence after exiting the cell cycle following cell division and remaining arrest for an extended duration or by engaging the DNA damage checkpoint after replication (Figure 6).
Figure 6. Annotated structure of the human cell cycle.

Dotted lines indicate proliferative and arrest trajectories, as well as exit and re-entry trajectories to/from the proliferative cell cycle. The molecular changes that accompany cell cycle exit/re-entry are also shown, as well as the molecular signature of cellular senescence.
Discussion
We combined time-lapse microscopy, multiplexed imaging, and manifold learning to reveal the underlying structure of the human epithelial cell cycle (Figure 6). This structure consists primarily of a cyclical, proliferative trajectory through G1/S/G2/M and a terminal arrest trajectory through G0 that diverges from the proliferative trajectory soon after mitosis. We also observe a second arrest trajectory that diverges soon after the S/G2 transition, corresponding to the G2/M DNA damage checkpoint. Both arrest trajectories converge on an irreversibly arrested, senescent state.
This structure is consistent with an emerging model of the cell cycle in which the fate of cells diverges soon after cell division (Arora et al., 2017; Min and Spencer, 2019; Overton et al., 2014; Spencer et al., 2013; Yang et al., 2020). In this model, some cells maintain a high RB phosphorylation state as they exit mitosis and proceed directly into G1, while others divert from the proliferative cell cycle to a state of transient cell cycle arrest, driven by an increase in p21 and a loss of RB phosphorylation (Figure 4A–D, Figure S4). This bifurcation in daughter cell fates was first observed by time-lapse imaging using a fluorescent reporter of CDK2 activity, revealing two distinct dynamics of CDK2 activity in cells following cell division (Spencer et al., 2013). In cells that immediately commit to cell cycle re-entry, CDK2 begins to increase soon after cell division. In cells that exit to G0, on the other hand, CDK2 activity remains low for the duration of cell cycle arrest. We observed a similar bifurcation of CDK2 activity following mitosis by time-lapse imaging and used CDK2 reactivation as an indicator of cell cycle re-entry from this spontaneous state of arrest (Figure 4K–M).
Once committed to the proliferative trajectory, we find that cells traverse a single path through DNA replication and cell division. The primary source of heterogeneity observed among cells was the bifurcation of daughter cell trajectories after mitosis (Figure 4A). Although an alternative mechanistic route through G1 has recently been observed when the canonical route is blocked (i.e. by CDK4/6 inhibition) (Liu et al., 2020), when grown under ideal culture conditions, we observed little evidence of alternative trajectories through the proliferative cell cycle. This topology was reproducibly obtained from replicate cell populations, across PHATE parameter space and in a second human epithelial cell line (Figure S2).
Progression further along the arrest trajectory is accompanied by an increase in p21 expression (Figure 4D, 4K), consistent with a “deepening” of cell cycle arrest (Fujimaki et al., 2019; Kwon et al., 2017; Wang et al., 2017). However, we also found that progression along the arrest trajectory is paradoxically accompanied by molecular changes that typically stimulate proliferation, not arrest. These changes include increased expression of multiple cyclins and CDKs (Figure 4J). This observation is consistent with a recent report that cyclin D and E increase during cell cycle arrest in MCF10A cells (Gookin et al., 2017). Using time-lapse imaging, we revealed that concomitant increases in proliferative effectors such as cyclin D1 allow cells to re-enter the cell cycle in the presence of elevated p21 (Figure 4K, Figure 4M, Figure S5A–B). Whereas p21 is the molecular entity that couples cell stress to cell cycle arrest (Arora et al., 2017; Min and Spencer, 2019), mitogenic signaling stimulates the production of cyclin D to govern cell cycle re-entry (Min et al., 2020). Like the bifurcation of daughter cells immediately following cell division, our data support a model whereby cell cycle re-entry from G0 is controlled by integrating these competing mitogenic and stress signals (Stallaert et al., 2019). While the duration of cell cycle arrest tends to increase with both p21 and cyclin D1 expression (Figure 4M), we could not identify a clear quantitative relationship that predicted the timing of cell cycle re-entry based solely on these two effectors. Instead, our data suggest that, the precise timing of cell cycle re-entry (i.e. either immediately after mitosis or after some duration in G0) is ultimately governed by the balance between the G1 cyclins (cyclin D and cyclin E) and CDK inhibitors such as p21, p27 and p16, which act respectively as “gas” and “brakes” for CDK activity to control the phosphorylation state of RB (Stallaert et al., 2019). This convergence of multiple positive and negative regulators of CDK activity allows the cell to integrate information about both its environment (e.g. mitogens, local cell density) and history (e.g. prior DNA damage, oxidative stress) to make a context-aware decision when to proliferate and when to arrest.
Cells that remain arrested for an extended duration may transition into an irreversibly arrested senescent state (Figure 6). Compared to the rest of the arrest trajectory, we observed the highest expression of many proliferative effectors in senescent cells (e.g. CDK4, CDK6, cyclin D1) (Figure 5B). These observations are consistent with a geroconversion model of senescence (Blagosklonny, 2014) in which growth factor signaling (through MAPK and mTOR pathways) continues in the presence of a strong cell cycle blockade (e.g. p21 induction). In these cells, persistent mitogenic/growth signaling drives an increase in proliferative effectors and hypertrophy (Figure 5C) in a futile attempt to overcome the cell cycle blockade. While the mechanism(s) that govern the switch between reversible and irreversible arrest remain elusive, we identified and validated a multivariate molecular signature that distinguishes senescent from quiescent cells. One interesting characteristic of cell cycle arrest in RPE cells is the decrease in total RB expression as cells progress along the arrest arm (Figure 4G). RB is the typically the point of convergence for cyclins, CDKs and their inhibitors in regulating the proliferation/quiescence decision. This loss of RB during cell cycle arrest suggests that an alternative regulatory mechanism may guard against cell cycle re-entry from these deeper states of arrest and provides a rationale for the persistence of cell cycle arrest despite high expression of cyclins and CDKs in senescent cells.
More generally, our systems-level approach enables future studies directed at comparing cell cycle progression across cell types, in different genetic backgrounds and in distinct environmental contexts. This approach may prove particularly powerful in the context of cancer cell biology, where dysregulation of cell cycle components is well appreciated, but attempts to target specific components individually have largely failed (Klein et al., 2018; Schoninger and Blain, 2020). Identifying the differences between normal and oncogenic cell cycle structures may provide novel insights into the mechanisms of tumorigenesis and lead to the development of new therapeutic targets.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeremy E. Purvis (purvisj@email.unc.edu)
Materials Availability
This study did not generate new materials.
Data and Code Availability
Single cell datasets, including data compatible for exploration in virtual reality, have been deposited at zenodo and are publicly available as of the date of publication. DOIs are listed in the key resource table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Please see a detailed list of antibodies used in Table S1 | ||
| Deposited data | ||
| Single cell imaging data | This study | http://doi.org/10.5281/zenodo.4525425. |
| Experimental models: Cell lines | ||
| Retinal pigment epithelial cells (hTERT-RPE-1) | ATCC | CRL-4000 |
| Human pancreatic epithelial cells (hTERT-HPNE) | ATCC | CRL-4023 |
| RPE-PCNA-mTurqoise2 | Laboratory of Jeremy Purvis | (Chao et al., 2019) |
| RPE-p21-mTq2/cycD1-mVenus/DHB-mCherry/H2B-mIFP | Laboratory of Jorg Mansfield | (Zerjatke et al., 2017) |
| Software and algorithms | ||
| StackReg | https://imagej.net/plugins/stackreg | (Thévenaz et al., 1998) |
| CellProfiler v3.1.8 | https://cellprofiler.org/ | (McQuin et al., 2018) |
| caret v6.0–86 | https://cran.r-project.org/web/packages/caret/vignettes/caret.html | |
| scikit-learn v0.24 | https://scikit-learn.org/stable/ | |
| Fastai | https://github.com/fastai/fastai | |
| PHATE | https://github.com/KrishnaswamyLab/PHATE | (Moon et al., 2019) |
| Leidenalg v0.8.3 | https://github.com/vtraag/leidenalg | (Traag et al., 2019) |
| scanpy v1.6 | https://github.com/theislab/scanpy | (Wolf et al., 2018) |
| singlecellVR | singlecellvr.com | (Stein et al., 2020) |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
Cell lines used in this study
Retinal pigment epithelial cells (hTERT RPE-1, ATCC, CRL-4000) were cultured in DMEM (Gibco, 11995–065) supplemented with 10% fetal bovine serum (FBS; Sigma, TMS-013-B), 2 mM L- glutamine (ThermoFisher Scientific, 25030081) and penicillin/streptomycin (P/S; ThermoFisher Scientific, 15140148). For time-lapse imaging of RPE cells, FluoroBrite™ DMEM (Gibco, A18967–01) supplemented with 10% FBS and 2 mM L- glutamine was used. Human pancreatic epithelial cells (hTERT-HPNE, ATCC, CRL-4023) were cultured in pyruvate-free DMEM (Gibco, 11965–092) supplemented with 10% FBS and P/S. All cells were cultured at 37 °C and 5% CO2. The RPE-PCNA-mTurqoise2 (RPE-PCNA-mTq2) cell line was previously described (Chao et al., 2019). The RPE-p21-mTq2/cycD1-mVenus/DHB-mCherry/H2B-mIFP cell line (Zerjatke et al., 2017) was a generous gift from Dr. Jorg Mansfield (Institute for Cancer Research, London, England) and Dr. Sabrina Spencer (University of Colorado Boulder). All cell lines were authenticated by STR profiling (ATCC) and confirmed to be mycoplasma free.
Method Details
Antibodies
High quality, previously published/validated primary antibodies were identified using BenchSci (http://app.benchsci.com) and are listed in Table S1.
Time-lapse imaging
Cells were plated in glass- bottom plates (Cellvis) coated with fibronectin (1 μg/cm2, Sigma, F1141). Fluorescence images were acquired using a Nikon Ti Eclipse inverted microscope with a Nikon Plan Apochromat Lambda 40x objective with a numerical aperture of 0.95 and an Andor Zyla 4.2 sCMOS detector. Autofocus was provided by the Nikon Perfect Focus System (PFS) and a custom enclosure (Okolabs) was used to maintain constant temperature (37°C) and atmosphere (5% CO2). For time-lapse imaging, the following filter sets were used (excitation; beam splitter; emission filter; Chroma): CFP (425–445/455/465–495nm), YFP (490–510/515/520–550nm), mCherry (540–580/585/593–668) and Cy5(590–650/660/663–738nm). Stitched 4-by-4 images were acquired every 10 min for RPE-p21-mTq2/cycD1-mVenus/DHB-mCherry/H2B-mIFP cells and every 16 min for RPE-PCNA-mTq2 cells. Uneven field illumination was corrected prior to stitching. NIS- Elements AR software was used for image acquisition and post-processing. Nuclear regions were segmented based on the H2B-mIFP or PCNA-mTq2 signal using a modified U-Net neural network (https://github.com/fastai/fastai), trained on manually annotated subsets of data. Linking of regions into tracks was computed using TrackMate (Tinevez et al., 2017). Segmentation and tracking corrections were performed manually. CDK2 activation was quantified as the ratio of background corrected cytoplasmic to nuclear intensity of the DHB-mCherry sensor (cytoplasm signal quantified as a 40th percentile in a 15-pixel ring outside the nuclear segmentation, with a 2-pixel gap between the nucleus and the ring; nuclear signal quantified as median) and p21/cyclin D1 expression was calculated as the background corrected median nuclear intensity. Cell cycle phases were annotated manually from time-lapse imaging using the appearance/disappearance of nuclear PCNA foci to mark the beginning and end of S phase, respectively, and CDK2 reactivation (cell cycle re-entry) was annotated manually as the point at which the cytoplasmic/nuclear ratio of DHB-mCherry begins to stably increase after cell division. For the time-lapse imaging that preceded 4i (see Figure 1A), approximately 25% of the total well area was imaged for a total of 24h, permitting ~27% of the total cells to be tracked.
Iterative indirect immunofluorescence imaging
Cells were plated in glass- bottom plates (Cellvis) coated with fibronectin (1 μg/cm2, Sigma, F1141), treated as required and prepared as follows. In between each step, samples were rinsed 3X times with phosphate-buffered saline (PBS) and incubations were at room temperature, unless otherwise stated. Cells were fixed with 4% paraformaldehyde (ThermoFisher Scientific, 28908) for 30 min, permeabilized with 0.1% Triton X-100 in PBS for 15 min and inspected for sample quality control following Hoechst staining in imaging buffer (IB: 700 mM N-acetyl-cysteine (Sigma, A7250) in ddH2O. Adjust to pH 7.4). Cells were rinsed 3X with ddH2O and incubated with elution buffer (EB: 0.5M L-Glycine (Sigma, 50046), 3M Urea (Sigma, U4883), 3M Guanidine chloride (ThermoFisher Scientific, 15502–016), and 70mM TCEP-HCl (Sigma, 646547) in ddH20. Adjusted to pH 2.5) 3X for 10 min on shaker to remove Hoechst stain. Sample was incubated with 4i blocking solution (sBS: 100 mM maleimide (Sigma, 129585), 100 mM NH4Cl (Sigma, A9434) AND 1% bovine serum albumin in PBS) for 1h and incubated with primary antibodies diluted as required (Table S1) in conventional blocking solution (cBS: 1% bovine serum albumin in PBS) overnight at 4°C. Samples were rinsed 3X with PBS and then incubated in secondary antibodies (Table S1) and Hoechst for 1h on shaker, then rinsed 5X with PBS and imaged in IB. Samples were imaged using the Nikon Ti Eclipse microscope described above. Stitched 8×8 images were acquired for each condition using the following filter cubes (Chroma): DAPI(383–408/425/435–485nm), GFP(450–490/495/500–550nm), Cy3(530–560/570/573–648nm), Cy5(590–650/660/663–738nm). After imaging, samples were rinsed 3X with ddH2O, antibodies were eluted and re-stained iteratively as described above. For immunofluorescence experiments that were combined with measurements of β-galactosidase activity, the CellEvent Senescence Green Detection Kit (ThermoFisher, C10850) was used according to the manufacturer’s instructions. After imaging β-galactosidase activity activity, cells were permeabilized, blocked and stained using the 4i protocol described above.
Experimental Model and Subject Details
Image processing and single cell analysis
Image registration was performed in Python (v3.7.1) using features common to multiple rounds (Hoechst or CDK2 staining) and the StackReg library (Thévenaz et al., 1998). Alignment quality was assessed manually and corrected using manually-selected fiduciary points if necessary. Segmentation and feature extraction from registered images were performed using standard modules in CellProfiler (v3.1.8) (McQuin et al., 2018). Only cells that persisted through all rounds of 4i were included in subsequent analyses.
Random forest models
Two random forest (RF) models were trained to predict cell cycle age and phase using the 4i signatures of individual cells. We used 80% of our annotated data to train the RFs and the remaining 20% was reserved as a test set. Classification accuracy was used as the error metric to train the phase model, while root mean squared error (RMSE) was used to train the age model. The phase model yielded 95.5% accuracy (95% CI: 93.5, 0.971) and a kappa of 0.925. The age model had an RMSE of 125.8 min with an R2=0.862. A third RF model was trained to predict arrest duration based on the 4i signatures of arrested cells (phospho/total RB < 1.7)(RMSE of 77.4 min and an R2=0.961). A fourth RF model was used to distinguish senescent cells (identified by graph-based clustering) from quiescent cells based on their 4i signatures with an accuracy of 97.5%. Variable importance tables were obtained from each model (Figure S1A, Table S2, Figure 4J, Figure 5B) through calculation of unconditional permutation importance. All hyperparameter tuning was performed with 10-fold cross validation optimizing model accuracy starting from default parameter values (Age: ntrees=500, mtry=246, Phase: ntrees=500, mtry=124).
We used RF models trained to predict cell cycle phase and age for feature selection to identify the minimal feature subset necessary to accurately predict cell cycle state (i.e. both phase and age) while eliminating features that vary among cells in a cell cycle-independent manner. First a combined ranking was calculated for each feature as the average of the individual rankings from the variable importance tables obtained from the age and phase models. Successive random forest models were generated for both age and phase starting with a single feature and adding additional features in order of combined ranking. Accuracy (for cell cycle phase) or error (RMSE for cell cycle age) were calculated at each iteration (Figure S1B–C). The optimal feature was defined as the top 40 features (by combined ranking).
RF models were made using R (v1.2.5001) with caret (v6.0–86) and ggplot2 (v3.3.2) packages or using Python (v3.7.1) with scikit-learn (v0.24).
Convolutional neural network
The convolutional neural network (CNN) models predicting cell cycle phase and age were trained using as an input image stacks for individual cells extracted from 4i experiments (48 fluorescent channels and 4 channels of masks representing entire cell, cell nucleus, cytoplasm and cytoplasmic ring around the nucleus respectively; 52 frames total; 100 px × 100 px) and ground truth annotations obtained from the time-lapse imaging. The Fastai (https://github.com/fastai/fastai) Python deep learning library was used for training and the initial pre-trained ResNet-50 convolutional networks were obtained directly from it. Both of the models were trained first using a low-resolution stack (50 px × 50 px) and then fine-tuned using full resolution stacks. The model predicting cell cycle phase was based on 2930 cells divided into a training set (2491 cells; 85%) and a validation set (439 cells, 15%). ResNet-50 CNN predicting phase was trained using a cross-entropy loss function. The model predicting cell cycle age was based on 2767 cells divided into a training set (2352 cells; 85%) and a validation set (415 cells, 15%). ResNet-50 CNN predicting age was trained using mean squared error loss function. Models were trained using Google Cloud VM (8 vCPUs, 52 GB memory, 1 x NVIDIA Tesla P100).
Manifold learning
Manifold learning was performed using Potential of Heat-diffusion for Affinity-based Transition Embedding (PHATE) (Moon et al., 2019) using the optimal feature set described above as input variables. PHATE was run on z-normalized variables with the following parameter sets: k-nearest neighbor (knn)=200, t=12, gamma=1.
Graph-based Clustering
To identify groups of cells with similar feature profiles, a k-nearest neighbor graph (10 neighbors) was constructed from the z-normalized optimal feature set using Euclidean distance and UMAP neighbor search. Cells were partitioned into groups using the Leiden community detection algorithm (Traag et al., 2019) with a resolution parameter of 0.30 (leidenalg v0.8.3).
Trajectory Inference
To infer the progression of cells through the cell cycle, diffusion pseudotime (Haghverdi et al., 2016) was performed using the DPT function in scanpy. A k-nearest neighbor graph (30 neighbors) was constructed from the optimal feature set (see above). Diffusion pseudotime was computed with a root cell randomly chosen from the youngest annotated cells and 10 diffusion map components. Pearson and Spearman rank correlation were computed as similarity metrics to compare pseudotime to annotated and predicted cell cycle age.
Given that DPT requires one root cell as input, we sought to evaluate its robustness to root cell choice when comparing molecular age (pseudotime) to cell cycle age. Diffusion pseudotime was computed for all potential root cells that have an annotated age of 0. The median Pearson correlation to cell cycle age for duration along the arrested trajectory was 0.8207 and proliferative trajectory was 0.8870.
Data Visualization
Data were visualized using Python (v3.7.1) and Jupyter Notebooks (v6.1.4) with matplotlib (v3.3.2), seaborn (v0.11.0) and scanpy (v1.6)(Wolf et al., 2018) libraries, as well as GraphPad Prism (v8). Scanpy was also used to prepare data for visualization in virtual reality using the singlecellVR website (singlecellvr.com) (Stein et al., 2020).
Supplementary Material
Data S1. Effector dynamics along the proliferative trajectory. Features are mapped onto the proliferative trajectory of the cell cycle structure (left panels) and plotted against cell cycle age (right panels). Population medians in time courses indicated by solid grey lines and individual cells are colored by cell cycle phase (G1: blue, S: orange, G2: green). Non-cycling (G0) cells (phospho/total RB < 1.6) are shown in grey on the structure and are excluded from time courses. Plasma membrane, PM; perinuclear, PN.
Data S2. Effector dynamics along the arrest trajectory. Features are mapped onto the arrest trajectory of the cell cycle structure (left panels) and plotted against the duration of arrest (right panels). Population medians in time courses indicated by solid grey lines. Non-arrested cells (phospho/total RB > 1.6) are shown in grey on the structure and are excluded from time courses. Plasma membrane, PM; perinuclear, PN.
Movie S1. DNA content mapped onto the 3-dimensional projection of the cell cycle structure.
Movie S2. Cell cycle age mapped onto the 3-dimensional projection of the cell cycle structure.
Movie S3. Cell cycle phase mapped onto the 3-dimensional projection of the cell cycle structure.
Movie S4. RB phosphorylation (phospho/total) mapped onto the 3-dimensional projection of the cell cycle structure.
Highlights.
Used multiplexed single-cell imaging and manifold learning to visualize the cell cycle
Measured the dynamics of 48 cell cycle proteins during proliferation and arrest
Characterized the mechanisms driving cell cycle exit and re-entry
Identified a multivariate molecular signature defining cellular senescence
Acknowledgments:
We would like to thank Sonja Mihailovic (UNC Chapel Hill), Chi Pham (UNC Chapel Hill) and Margaret Redick (Oregon State University) for assistance with cell culture and Dr. Sam Wolff (UNC Chapel Hill) for support with imaging. We would like to thank Dr. Jörg Mansfeld (ICR, London) and Dr. Sabrina Spencer (CU Boulder) for generously providing us with the RPE-p21-mTq2-cycD1-mVenus-DHB-mCherry-H2B-mIFP cell line. We would also like to thank Dr. Robert Duronio (UNC Chapel Hill) and Dr. Adam Palmer (UNC Chapel Hill) for careful reading of the manuscript. This work was supported by NIH grants R01-GM138834 (JEP), DP2-HD091800 (JEP), NSF CAREER Award 1845796 (JEP), and Chan Zuckerberg Initiative DAF grant 2020-225716 (KMK).
Footnotes
Declaration of interests:
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Araujo AR, Gelens L, Sheriff RS, and Santos SD (2016). Positive Feedback Keeps Duration of Mitosis Temporally Insulated from Upstream Cell-Cycle Events. Mol. Cell 64, 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora M, Moser J, Phadke H, Basha AA, and Spencer SL (2017). Endogenous Replication Stress in Mother Cells Leads to Quiescence of Daughter Cells. Cell Rep. 19, 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr AR, Cooper S, Heldt FS, Butera F, Stoy H, Mansfeld J, Novak B, and Bakal C (2017). DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat. Commun 8, 14728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagosklonny MV (2014). Geroconversion: irreversible step to cellular senescence. Cell Cycle 13, 3628–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, and Vogelstein B (1998). Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282, 1497–1501. [DOI] [PubMed] [Google Scholar]
- Cappell SD, Chung M, Jaimovich A, Spencer SL, and Meyer T (2016). Irreversible APC Cdh1 Inactivation Underlies the Point of No Return for Cell-Cycle Entry. Cell 166, 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappell SD, Mark KG, Garbett D, Pack LR, Rape M, and Meyer T (2018). Emi1 switches from being a substrate to an inhibitor of APC/CCdh1 to start the cell cycle. Nature 558, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castor LN (1980). A G1 rate model accounts for cell-cycle kinetics attributed to “transition probability.” Nature 287, 857–859. [DOI] [PubMed] [Google Scholar]
- Chao HX, Fakhreddin RI, Shimerov HK, Kedziora KM, Kumar RJ, Perez J, Limas JC, Grant GD, Cook JG, Gupta GP, et al. (2019). Evidence that the human cell cycle is a series of uncoupled, memoryless phases. Mol. Syst. Biol 15, e8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J-Y, Lin J-R, Tsai F-C, and Meyer T (2013). Dosage of Dyrk1a shifts cells within a p21-cyclin D1 signaling map to control the decision to enter the cell cycle. Mol. Cell 52, 87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung M, Liu C, Yang HW, Köberlin MS, Cappell SD, and Meyer T (2019). Transient Hysteresis in CDK4/6 Activity Underlies Passage of the Restriction Point in G1. Mol. Cell 76, 562–573.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudreuse D, and Nurse P (2010). Driving the cell cycle with a minimal CDK control network. Nature 468, 1074–1079. [DOI] [PubMed] [Google Scholar]
- DeGregori J, Kowalik T, and Nevins JR (1995). Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell. Biol. 15, 4215–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimaki K, Li R, Chen H, Della Croce K, Zhang HH, Xing J, Bai F, and Yao G (2019). Graded regulation of cellular quiescence depth between proliferation and senescence by a lysosomal dimmer switch. Proc. Natl. Acad. Sci. U. S. A. 116, 22624–22634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glotzer M, Murray AW, and Kirschner MW (1991). Cyclin is degraded by the ubiquitin pathway. Nature 349, 132–138. [DOI] [PubMed] [Google Scholar]
- Gookin S, Min M, Phadke H, Chung M, Moser J, Miller I, Carter D, and Spencer SL (2017). A map of protein dynamics during cell-cycle progression and cell-cycle exit. PLoS Biol. 15, e2003268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gut G, Herrmann MD, Pelkmans L (2018). Multiplexed protein maps link subcellular organization to cellular states. Science 361, eaar7042. [DOI] [PubMed] [Google Scholar]
- Gut G, Tadmor MD, Pe’er D, Pelkmans L, and Liberali P (2015). Trajectories of cell-cycle progression from fixed cell populations. Nat. Methods 12, 951–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L, Büttner M, Wolf FA, Buettner F, and Theis FJ (2016). Diffusion pseudotime robustly reconstructs lineage branching. Nat. Methods 13, 845–848. [DOI] [PubMed] [Google Scholar]
- Hjelmeland LM, Cristofolo VJ, Funk W, Rakoczy E, and Katz ML (1999). Senescence of the retinal pigment epithelium. Mol. Vis. 5, 33. [PubMed] [Google Scholar]
- Howard A, and Pelc SR (1951). Nuclear incorporation of P32 as demonstrated by autoradiographs. Exp. Cell Res. 2, 178–187. [Google Scholar]
- Hume S, Dianov GL, and Ramadan K (2020). A unified model for the G1/S cell cycle transition. Nucleic Acids Res. 48, 12483–12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein ME, Kovatcheva M, Davis LE, Tap WD, and Koff A (2018). CDK4/6 Inhibitors: The Mechanism of Action May Not Be as Simple as Once Thought. Cancer Cell 34, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalczyk MS, Tirosh I, Heckl D, Rao TN, Dixit A, Haas BJ, Schneider RK, Wagers AJ, Ebert BL, and Regev A (2015). Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 25, 1860–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon JS, Everetts NJ, Wang X, Wang W, Della Croce K, Xing J, and Yao G (2017). Controlling Depth of Cellular Quiescence by an Rb-E2 F Network Switch. Cell Rep. 20, 3223–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanz MC, Zatulovskiy E, Swaffer MP, Zhang L, Zhang S, You DS, Marinov G, McAlpine P, Elias JE, and Skotheim JM (2021). Increasing cell size remodels the proteome and promotes senescence. bioRxiv. doi: 10.1101/2021.07.29.454227. July 29, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Konagaya Y, Chung M, Daigh LH, Fan Y, Yang HW, Terai K, Matsuda M, and Meyer T (2020). Altered G1 signaling order and commitment point in cells proliferating without CDK4/6 activity. Nat. Commun. 11, 5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdessian D, Cesnik AJ, Gnann C, Danielsson F, Stenström L, Arif M, Zhang C, Le T, Johansson F, Shutten R, et al. (2021). Spatiotemporal dissection of the cell cycle with single-cell proteogenomics. Nature 590, 649–654. [DOI] [PubMed] [Google Scholar]
- Marescal O, and Cheeseman IM (2020). Cellular Mechanisms and Regulation of Quiescence. Dev. Cell 55, 259–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marthandan S, Priebe S, Hemmerich P, Klement K, and Diekmann S (2014). Long-term quiescent fibroblast cells transit into senescence. PLoS One 9, e115597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, et al. (2018). CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 16, e2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min M, and Spencer SL (2019). Spontaneously slow-cycling subpopulations of human cells originate from activation of stress-response pathways. PLoS Biol. 17, e3000178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min M, Rong Y, Tian C, and Spencer S (2020). Temporal integration of mitogen history in mother cells controls proliferation of daughter cells. Science. 368, 1261–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon KR, van Dijk D, Wang Z, Gigante S, Burkhardt DB, Chen WS, Yim K, van den Elzen A, Hirn MJ, Coifman RR, et al. (2019). Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol. 37, 1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan DO (2007). The Cell Cycle: Principles of Control. Yale J Biol Med. 80, 141–142. [Google Scholar]
- Moser J, Miller I, Carter D, and Spencer SL (2018). Control of the Restriction Point by Rb and p21. Proc. Natl. Acad. Sci. U. S. A. 115, E8219–E8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moussa M, and Măndoiu II (2020). Computational cell cycle analysis of single cell RNA-Seq data. bioRxiv. doi: 10.1101/2020.11.21.392613, Nov. 22, 2020. [DOI] [Google Scholar]
- Murray AW (1994). Cyclin-dependent kinases: regulators of the cell cycle and more. Chem. Biol. 1, 191–195. [DOI] [PubMed] [Google Scholar]
- Murray AW, and Kirschner MW (1989). Dominoes and clocks: the union of two views of the cell cycle. Science 246, 614–621. [DOI] [PubMed] [Google Scholar]
- Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, and Dowdy SF (2014). Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife 3:e02872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurohr GE, Terry RL, Lengefeld J, Bonney M, Brittingham GP, Moretto F, Miettinen TP, Vaites LP, Soares LM, Paulo JA, et al. (2019). Excessive Cell Growth Causes Cytoplasm Dilution And Contributes to Senescence. Cell 176, 1083–1097.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse P (2000). A long twentieth century of the cell cycle and beyond. Cell 100, 71–78. [DOI] [PubMed] [Google Scholar]
- Orlando DA, Lin CY, Bernard A, Wang JY, Socolar JE, Iversen ES, Hartemink AJ, and Haase SB (2008). Global control of cell-cycle transcription by coupled CDK and network oscillators. Nature 453, 944–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overton KW, Spencer SL, Noderer WL, Meyer T, and Wang CL (2014). Basal p21 controls population heterogeneity in cycling and quiescent cell cycle states. Proc. Natl. Acad. Sci. U. S. A. 111, E4386–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen TA, Soprano DR, and Soprano KJ (1989). Analysis of the growth factor requirements for stimulation of WI-38 cells after extended periods of density-dependent growth arrest. J. Cell. Physiol. 139, 424–431. [DOI] [PubMed] [Google Scholar]
- Pardee AB (1974). A restriction point for control of normal animal cell proliferation. Proc. Natl. Acad. Sci. U. S. A. 71, 1286–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagot I, and Laporte D (2019). Quiescence, an individual journey. Curr. Genet. 65, 695–699. [DOI] [PubMed] [Google Scholar]
- Sanidas I, Morris R, Fella KA, Rumde PH, Boukhali M, Tai EC, Ting DT, Lawrence MS, Haas W, and Dyson NJ (2019). A Code of Mono-phosphorylation Modulates the Function of RB. Mol. Cell 73, 985–1000.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos SDM, Wollman R, Meyer T, and Ferrell JE (2012). Spatial Positive Feedback at the Onset of Mitosis. Cell 149, 1500–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoninger SF, and Blain SW (2020). The Ongoing Search for Biomarkers of CDK4/6 Inhibitor Responsiveness in Breast Cancer. Mol. Cancer Ther. 19, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe D, Formichetti S, Junker JP, Falcke M, and Rajewsky N (2020). The transcriptome dynamics of single cells during the cell cycle. Mol. Syst. Biol. 16, e9946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields R (1977). Transition probability and the origin of variation in the cell cycle. Nature 267, 704–707. [DOI] [PubMed] [Google Scholar]
- Smith JA, and Martin L (1973). Do cells cycle? Proc. Natl. Acad. Sci. U. S. A. 70, 1263–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa-Victor P, Gutarra S, García-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardí M, Ballestar E, González S, Serrano AL, et al. (2014). Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506, 316–321. [DOI] [PubMed] [Google Scholar]
- Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, and Meyer T (2013). The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 155, 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallaert W, Kedziora KM, Chao HX, and Purvis JE (2019). Bistable switches as integrators and actuators during cell cycle progression. FEBS Lett. 593, 2805–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein DF, Chen H, Vinyard ME, and Pinello L (2020). singlecellVR: interactive visualization of single-cell data in virtual reality. bioRxiv. doi: 10.1101/2020.07.30.229534. Aug. 19, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenaz P, Ruttimann UE, and Unser M (1998). A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 7, 27–41. [DOI] [PubMed] [Google Scholar]
- Tinevez J-Y, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, and Eliceiri KW (2017). TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90. [DOI] [PubMed] [Google Scholar]
- Traag VA, Waltman L, and van Eck NJ (2019). From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Fujimaki K, Mitchell GC, Kwon JS, Della Croce K, Langsdorf C, Zhang HH, and Yao G (2017). Exit from quiescence displays a memory of cell growth and division. Nat. Commun. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HW, Chung M, Kudo T, and Meyer T (2017). Competing memories of mitogen and p53 signalling control cell-cycle entry. Nature 549, 404–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HW, Cappell SD, Jaimovich A, Liu C, Chung M, Daigh LH, Pack LR, Fan Y, Regot S, Covert M, et al. (2020). Stress-mediated exit to quiescence restricted by increasing persistence in CDK4/6 activation. Elife 9:e44571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerjatke T, Gak IA, Kirova D, Fuhrmann M, Daniel K, Gonciarz M, Müller D, Glauche I, and Mansfeld J (2017). Quantitative Cell Cycle Analysis Based on an Endogenous All-in-One Reporter for Cell Tracking and Classification. Cell Rep. 19, 1953–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterberg A, and Larsson O (1985). Kinetic analysis of regulatory events in G1 leading to proliferation or quiescence of Swiss 3T3 cells. Proc. Natl. Acad. Sci. U. S. A. 82, 5365–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Effector dynamics along the proliferative trajectory. Features are mapped onto the proliferative trajectory of the cell cycle structure (left panels) and plotted against cell cycle age (right panels). Population medians in time courses indicated by solid grey lines and individual cells are colored by cell cycle phase (G1: blue, S: orange, G2: green). Non-cycling (G0) cells (phospho/total RB < 1.6) are shown in grey on the structure and are excluded from time courses. Plasma membrane, PM; perinuclear, PN.
Data S2. Effector dynamics along the arrest trajectory. Features are mapped onto the arrest trajectory of the cell cycle structure (left panels) and plotted against the duration of arrest (right panels). Population medians in time courses indicated by solid grey lines. Non-arrested cells (phospho/total RB > 1.6) are shown in grey on the structure and are excluded from time courses. Plasma membrane, PM; perinuclear, PN.
Movie S1. DNA content mapped onto the 3-dimensional projection of the cell cycle structure.
Movie S2. Cell cycle age mapped onto the 3-dimensional projection of the cell cycle structure.
Movie S3. Cell cycle phase mapped onto the 3-dimensional projection of the cell cycle structure.
Movie S4. RB phosphorylation (phospho/total) mapped onto the 3-dimensional projection of the cell cycle structure.
Data Availability Statement
Single cell datasets, including data compatible for exploration in virtual reality, have been deposited at zenodo and are publicly available as of the date of publication. DOIs are listed in the key resource table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Please see a detailed list of antibodies used in Table S1 | ||
| Deposited data | ||
| Single cell imaging data | This study | http://doi.org/10.5281/zenodo.4525425. |
| Experimental models: Cell lines | ||
| Retinal pigment epithelial cells (hTERT-RPE-1) | ATCC | CRL-4000 |
| Human pancreatic epithelial cells (hTERT-HPNE) | ATCC | CRL-4023 |
| RPE-PCNA-mTurqoise2 | Laboratory of Jeremy Purvis | (Chao et al., 2019) |
| RPE-p21-mTq2/cycD1-mVenus/DHB-mCherry/H2B-mIFP | Laboratory of Jorg Mansfield | (Zerjatke et al., 2017) |
| Software and algorithms | ||
| StackReg | https://imagej.net/plugins/stackreg | (Thévenaz et al., 1998) |
| CellProfiler v3.1.8 | https://cellprofiler.org/ | (McQuin et al., 2018) |
| caret v6.0–86 | https://cran.r-project.org/web/packages/caret/vignettes/caret.html | |
| scikit-learn v0.24 | https://scikit-learn.org/stable/ | |
| Fastai | https://github.com/fastai/fastai | |
| PHATE | https://github.com/KrishnaswamyLab/PHATE | (Moon et al., 2019) |
| Leidenalg v0.8.3 | https://github.com/vtraag/leidenalg | (Traag et al., 2019) |
| scanpy v1.6 | https://github.com/theislab/scanpy | (Wolf et al., 2018) |
| singlecellVR | singlecellvr.com | (Stein et al., 2020) |
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.