

Abstract
Classic galactosemia (CG) results from profound deficiency of galactose‐1‐P uridylyltransferase (GALT). Despite early detection by newborn screening and lifelong dietary restriction of galactose, most patients grow to experience a range of long‐term complications. Recently, we developed and characterized a GALT‐null rat model of CG and demonstrated that AAV9‐hGALT, administered by tail vein injection to neonatal pups, dramatically improved plasma, liver, and brain galactose metabolites at 2 weeks posttreatment. Here we report a time‐course study of GALT restoration in rats treated as neonates with scAAV9‐hGALT and harvested at 8, 14, 30, and 60 days. Cohorts of rats in the two older groups were weaned to diets containing either 1% or 3% of calories from galactose. As expected, GALT activity in all treated animals peaked early and then diminished over time, most notably in liver, ostensibly due to dilution of the nonreplicating episomal vector as transduced cells divided. All treated rats showed dramatic metabolic rescue through 1 month, and those weaned to the lower galactose diet showed continued strong metabolic rescue into adulthood (2 months). Prepubertal growth delay and cataracts were both partially rescued by treatment. Finally, we found that UDP glucose pyrophosphorylase (UGP), which offers a metabolic bypass around missing GALT, was 3‐fold more active in brain samples from adult rats than from young pups, offering a possible explanation for the improved ability of older GALT‐null rats to metabolize galactose. Combined, these results document promising metabolic and phenotypic efficacy of neonatal GALT gene replacement in a rat model of classic galactosemia.
Keywords: AAV9, galactosemia, GALT, gene therapy, metabolite, rat, scAAV9
1. INTRODUCTION
Classic galactosemia (CG) is a potentially lethal autosomal recessive disorder that results from profound deficiency of galactose‐1‐P uridylyltransferase (GALT), the middle enzyme in the Leloir pathway of galactose metabolism. Despite early detection by newborn screening and life‐long dietary restriction of galactose, which is the current standard of care, most infants with CG grow to experience long‐term developmental and other complications. 1 , 2 The mechanisms underlying these complications remain unclear and likely differ by tissue. 3 At present, there is no known intervention that prevents or resolves the long‐term complications of CG.25. 2
Studies of patients 2 , 4 document that following exposure to breast milk or cow's milk, which contain approximately 19% and 15% of calories from galactose, respectively, 5 infants with CG demonstrate dramatically elevated levels of galactose, galactitol, and galactose‐1‐phosphate (gal‐1‐P) in blood and tissues. Upon rigorous dietary galactose restriction these metabolite levels drop quickly, at least in blood, but in most cases do not fully normalize, 1 ostensibly reflecting the effects of endogenously produced galactose. 6 , 7 Both the time frame of vulnerability, and which, if any, of the galactose metabolites that accumulate in patients cause most long‐term complications, remain unknown.
Recently, we developed and characterized a GALT‐null rat model that mimics many of the biochemical and long‐term complications of CG. 3 Time course studies of metabolites in these rats revealed two striking patterns. First, the levels and proportions of accumulated galactose, galactitol, and gal‐1‐P varied across tissues and through development. Second, over time after weaning, and despite a consistent diet and complete and continued absence of GALT activity, as the rats matured their levels of accumulated galactose metabolites dropped by 2‐fold to 10‐fold in all tissues tested. 3 Whether this drop reflected the reported diminished endogenous production of galactose with age, 6 , 7 or a hypothesized increase 3 in GALT‐independent bypass routes, 8 , 9 , 10 or other factors, was unclear.
Previously, we also applied our GALT‐null rats to test whether human GALT (hGALT), expressed from an adeno‐associated viral vector (AAV9), might suffice to rescue the metabolic abnormalities otherwise seen in nursing GALT‐null pups; it did. 11 However, whether this neonatal treatment would suffice to maintain lowered galactose metabolite levels into adulthood, and whether it would provide long‐term phenotypic benefit beyond cataracts, was untested. Here we have extended from that small pilot study to define the longer‐term impacts of GALT restoration in cohorts of GALT‐null rats followed into early adulthood.
The current study had three goals. First, we wanted to define the extent and kinetics of GALT restoration achieved in liver and brain following neonatal intravenous administration of viral vector. To be clear, we anticipated that GALT would be restored to multiple tissues as has been documented for AAV9 transduction of other transgenes in rodent hosts, 12 but here we focused on liver because it is the organ where most galactose metabolism occurs in mammals, and on brain because of its relevance to the cognitive, behavioral, and motor complications experienced by many patients. Second, we wanted to define the extent and kinetics of metabolic rescue achieved in liver, brain, and blood, and the impact of continued dietary galactose exposure on that metabolic rescue. Finally, we wanted to know whether the extent and timing of GALT restoration and metabolic rescue achieved in GALT‐null rats treated as neonates with a nonreplicating, largely nonintegrating vector would suffice to minimize or prevent two representative complications of GALT deficiency: cataracts and pre‐pubertal growth delay. To the limits of our study, the results presented here confirm the compelling possibility that early GALT gene replacement by scAAV9 may offer a powerful tool toward future intervention for CG.
2. MATERIALS AND METHODS
2.1. Rats and rat husbandry
Outbred wild‐type and GALT‐null Sprague Dawley rats were maintained in space overseen by the Division of Animal Resources at Emory. Pups were weaned at 21 to 24 days of age. Rats intended to experience a post‐wean diet of 1% calories from galactose were weaned to ad libitum LabDiet 5053 chow, plus plain water. Having confirmed that our rats consume about 5 g solid chow and drink about 10 mL of water daily per 100 g body mass, rats were weaned to LabDiet 5053 plus water containing 1% (w/v) galactose to achieve a combined post‐wean diet of about 3% calories from galactose, which is the same level seen in rat breast milk. 5
2.2. Viral stock and administration to 2‐day‐old rat neonates
The viral stock used here was generated from a plasmid (pscAAV.CBh.HA‐hGALT) assembled and confirmed by VectorBuilder (www.vectorbuilder.com) on a fee‐for‐service basis. This plasmid included a modified chicken beta‐actin promoter fused to a CMV early enhancer 13 driving expression of an HA‐tagged human GALT coding sequence that we had previously expressed successfully in yeast. 14 To enable generation of a self‐complementary viral genome the AAV 3′ ITR sequence carried a depleted terminal resolution site. 15 The scAAV9 viral stock was received from VectorBuilder at a concentration of 1.04 × 1013 vg/mL. Negative control vector (AAV9‐CMV‐GFP) and virus were prepared by the same vendor; that viral stock was received at a concentration of 3.06 × 1013 vg/mL.
For tail vein injections of scAAV9‐hGALT or AAV9‐GFP virus, we diluted the appropriate viral stock in sterile PBS for a final dose of 2.5 × 1013 vg/kg. Rat neonates were anesthetized briefly via isoflurane inhalation and the tail was wiped with betadine. The injection volume (8 μL/g body weight) was administered slowly into the dorsal tail vein using a 30‐gauge insulin syringe before gently applying pressure at the injection site until bleeding stopped. Neonates were returned to the nest with their mother for recovery and monitoring.
2.3. Euthanasia and tissue harvest
Rats to be harvested at 8 days were euthanized under deep isoflurane anesthesia and rapidly decapitated. Solid tissues to be processed for DNA isolation, enzyme assays, or metabolite analyses were removed, cut with a razor into small pieces, flash frozen on dry ice, and stored at −80°C until use. The following solid tissues were collected: whole brain and whole liver. Rats to be harvested at 14, 30, or 60 days were euthanized under deep isoflurane anesthesia, perfused with ice‐cold PBS to remove blood from solid tissues, and harvested as described previously. 3 , 11
2.4. Quantitative real‐time PCR to define AAV9 viral genome copy number
Quantitative real‐time PCR was performed using DNA isolated from samples of liver, as described previously. 11 The results are presented as vector genome copy number per diploid rat genome, or vg copy number/cell.
2.5. Immunohistochemistry of fixed liver samples
Tissues intended for immunohistochemistry (IHC) were fixed with 4% paraformaldehyde and prepared for staining as described previously. 11 Anti‐HA primary antibody (Cell Signaling Technology #3724) was used at a 1:1000 dilution in antibody diluent (Agilent Dako S0809) with incubation overnight at 4°C. The secondary antibody was Goat Anti‐Rabbit IgG (H + L), Peroxidase (Vector Labs PI‐1000‐1) used at 1:500 dilution in antibody diluent with incubation for 30 minutes at room temperature. Anti‐GALT antibody (Novus Biologicals #4C11) was also used at a 1:1000 dilution in antibody diluent with incubation overnight at 4°C. The secondary antibody was Horse Anti‐Mouse IgG (H + L), Peroxidase (Vector Labs PI‐2000‐1) used at a 1:500 dilution in antibody diluent with incubation for 30 minutes at room temperature. All slides were treated with DAB (Vector Labs SK‐4100) and counterstained with Mayer's hematoxylin (Sigma‐Aldrich MHS16) prior to re‐dehydration and mounting with Cytoseal 60 (Thermo Scientific 8310‐16).
2.6. Quantitation of IHC staining
Stained slides were scanned at 40X magnification and the fraction of stained (brown) cells was estimated as the fraction of stained pixels, scored using custom software developed in our lab that will be described in detail elsewhere (Patel et al, in preparation). Background and staining thresholds used to distinguish empty space from stained and unstained tissue were calculated from control slides, allowing for 0.1% misclassification per pixel. These thresholds were then applied to quantify the fraction of IHC‐stained pixels in each of 20 randomly placed 500 × 500‐pixel boxes on each slide, excluding boxes that contained more than half empty space or detectable artifacts such as wrinkles or folds. The average of brown pixels detected in these 20 boxes was used to define the staining fraction in each slide.
2.7. Immunofluorescent imaging of fixed brain sections
Tissues intended for immunofluorescence (IF) were fixed with 4% paraformaldehyde and prepared for staining as described previously. 11 All antibodies were diluted as listed below in antibody diluent (Agilent Dako S0809) with incubation overnight at 4°C. Anti‐GALT primary antibody (Novus Biologicals #4C11) was used at a 1:100 dilution; anti‐NeuN (Cell Signaling #24307 16 ) and anti‐GFAP (Invitrogen #PA5‐16291 17 ) were each used at a 1:1800 dilution. The secondary antibodies used were: anti‐Mouse IgG (H + L) Alexa Fluor 568 (Abcam 175 473) and anti‐Rabbit IgG (H + L) Alexa Fluor 488 (Cell Signaling #4412), each used at 1:500 dilution in antibody diluent with incubation for 1 hour at room temperature. All slides were treated with 300 μM DAPI (Invitrogen #D1306) and mounted with ProLong Diamond Antifade Mountant (Life Technologies #P36970) for 24‐hours at room temperature prior to imaging. Staining for Purkinje cells using anti‐calbindin‐E‐28K (KD‐15) antibody (Millipore Sigma, #C7354 18 ) was performed as described previously. 19
2.8. Quantitation of GALT and UGP activities
Enzyme assays for GALT were performed in up to three technical replicates from liver, brain, and RBC essentially as described previously. 11 For measurement of GALT activity from RBC, 100 μL of washed RBC was used to make lysates as described for liver and brain. To be within the linear range of the assay, lysates from GALT‐null animals were diluted 2‐fold to make the final concentration added to reactions, and from wild‐type animals were diluted 10‐fold. After incubating reactions for 30 minutes at 37°C and quenching with 450 μL ice cold HPLC grade water, reactions were incubated at 100°C for 10 minutes with gentle agitation half‐way through. Samples were then centrifuged at 16 000g for 10 minutes at room temperature, and the supernatant centrifuged through 0.22‐μm Costar Spin‐X centrifuge tube filters at 4000g for 4 minutes at 4°C. Finally, reactants and products were separated and quantified by HPLC following the same method used for liver and brain assays. Activity for RBC GALT activity is reported as pmol UDPgal formed per microgram of hemoglobin per minute.
UGP activity was measured in lysates prepared from samples of brain weighing approximately 40 mg; lysates were prepared as for GALT assays. UGP activity from each sample was measured at 25, 50, and 75 μg of total protein per 50 μL reaction, all of which were determined to be within the linear range of the assay, and each reaction was run in triplicate. The initial reaction component concentrations were: 11.2 mM MgCl2, 56 mM Tris pH 8.0, 2.1 mM dithiothreitol, 2.5 mM UTP, and 60 mM gal‐1‐P. Reactions were initiated by the addition of 30 μL of sample lysate to 20 μL of reaction mixture. Reactions were incubated at 37°C for 15 minutes, then quenched by the addition of 450 μL ice cold HPLC‐grade water. Stopped reactions were then centrifuged at 4000g for 4 minutes at 4°C through 0.22‐μm Costar Spin‐X columns. Reactants and products were separated and quantified by HPLC following the same method as for GALT enzyme assays. Activity was calculated from the conversion of gal‐1‐P to UDPgal and presented as pmol UDPgal formed per μg protein per minute.
2.9. Quantitation of galactose, galactitol, and gal‐1‐P in samples of plasma, RBC, liver, and brain
Galactose, galactitol, and gal‐1‐P from plasma, RBC, liver, and brain were quantified as described previously. 3 To be clear, gal‐1‐P is an intracellular metabolite that is not detected in plasma.
2.10. Scoring cataract intensity and growth delay
Following euthanasia and PBS perfusion (for rats ≥14 days old), eyelids were removed and for rats 30 days and older profile photographs were taken of each eye using a Nikon V1 model camera. The eyes of younger rats often demonstrated rapid clouding following eyelid removal that confounded photography.
Cataracts were scored manually using a 4‐point scale: 0 = no cataract, 1 = mild, 2 = moderate, and 3 = severe, as described previously. 3 , 11 Rat growth was analyzed using data sets generated by weighing each pup every day after birth.
2.11. Statistical analyses
To compare growth of different cohorts of pups we used separate linear mixed effects models at each age tested (2 and 22 days). For each model, the pup weight in grams was the response variable, with comparison group, litter size, and sex defined as predictor variables. The comparison group variable combined GALT genotype with treatment status (PBS vs scAAV9‐hGALT).
To evaluate the effects of scAAV9‐hGALT treatment on GALT restoration and metabolic rescue we used a nonparametric Wilcoxon Rank‐Sum test because our comparison groups were too small to confirm normal distribution of data. Specifically, we compared the GALT activity levels found in liver and brain samples from GALT‐null rats administered PBS vs scAAV9‐hGALT as neonates and harvested at 8, 14, 30, or 60 days (Figure 1). We also used Wilcoxon Rank‐Sum tests to compare the GALT activity levels found in liver and brain samples from treated rats harvested at different ages to examine the kinetics of GALT loss in both tissues (Figure 1).
FIGURE 1.
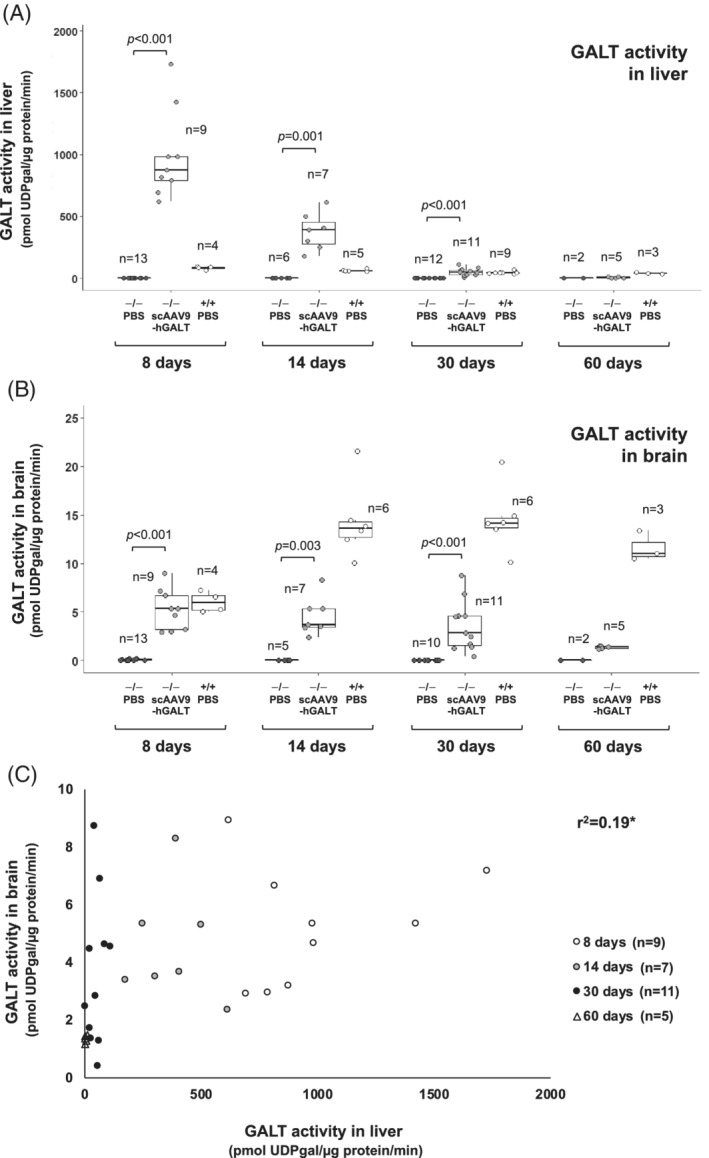
GALT activity over time in tissues from treated and control rats. Box and whisker plots show GALT activity in liver (panel A) and brain (panel B) from PBS‐treated GALT‐null rats (black symbols), scAAV9‐hGALT‐treated GALT‐null rats (gray symbols), and PBS‐treated wild‐type rats (white symbols). The top and bottom of each box represent the 75th and 25th percentiles, respectively, the middle line in each box represents the median for that set. The whiskers, if any, represent the maximum and minimum values in the data set within 1.5 interquartile range (IQR) of the upper and lower quartiles. Statistical comparisons between PBS vs scAAV9‐hGALT‐treated GALT‐null pups in each age group were performed as described in Materials and Methods; groups with n < 3 were not assessed. Comparisons of GALT activity in scAAV9‐hGALT‐treated GALT‐null rats between time points yielded the following P values: for liver 8 to 14 days (P < .001), 14 to 30 days (P < .001), 30 to 60 days (P = .002); for brain 8 to 14 days (P = .525), 14 to 30 days (P = .328), and 30 to 60 days (P = .052). Panel C shows the relationship between GALT activity detected in liver and brain samples from the same animals described in panels A and B, with 8‐day‐old rats represented by white circles, 14‐day‐old rats represented by gray circles, 30‐day‐old rats represented by black circles, and 60‐day‐old rats represented by white triangles. *r 2 as presented is confounded by the inclusion of multiple time points because restored GALT activity is lost from liver and brain at different rates
To assess the significance of metabolite levels between groups, we again used Wilcoxon Rank‐Sum tests for the pairwise comparisons indicated in Figures 2, S1, and S5; we refrained from performing statistical tests on any comparison groups with a sample size <3. We applied a similar approach to compare the fraction of IHC‐stained cells in liver sections from treated animals (Figure S4) and also to compare the brain UGP activities found in rats harvested at 8 vs 60 days (Figure 4). To be clear, we first tested whether wild‐type and GALT‐null rats of the same age showed any significant difference in brain UGP activity; they did not, and so the genotypes were combined for subsequent analyses.
FIGURE 2.
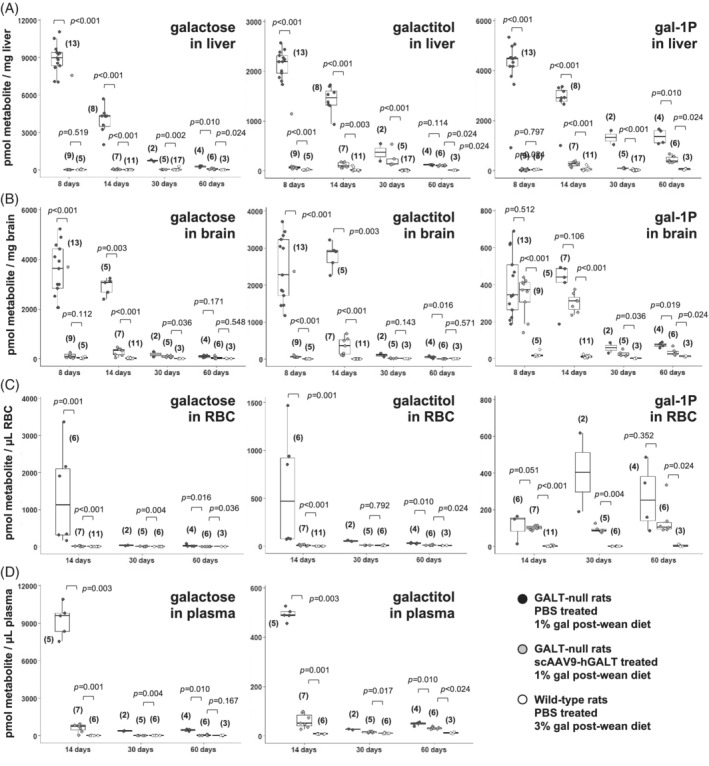
Metabolites quantified over time in tissues from treated and control rats. Box and whisker plots show galactose, galactitol, and gal‐1‐P levels measured in samples of liver (panel A), brain (panel B), red blood cells (RBC, panel C), and plasma (panel D) collected from treated and control rats of the indicated ages. The top and bottom of each box represent the 75th and 25th percentiles, respectively, the middle line in each box represents the median for that set, and the whiskers, if any, represent the maximum and minimum values in the data set within 1.5 interquartile range (IQR) of the upper and lower quartiles. In all graphs, PBS‐treated GALT‐null rats are represented by black symbols, scAAV9‐hGALT‐treated GALT‐null rats by gray symbols, and PBS‐treated wild‐type rats by white symbols
FIGURE 4.
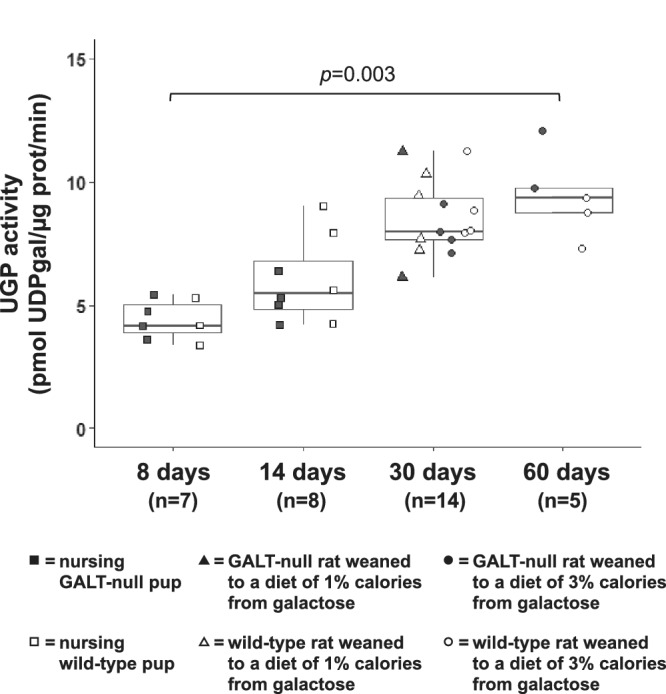
UGP activity in brain samples from GALT‐null and wild‐type rats of different ages. Box and whisker plots show UGP activity measured toward gal‐1‐P in samples of brain harvested from wild‐type (+/+, white boxes) and GALT‐null (−/−, shaded boxes) rats of the indicated ages. The top and bottom of each box represent the 75th and 25th percentiles, respectively, the middle line in each box represents the median for that set, and the whiskers, if any, represent the maximum and minimum values in the data set within 1.5 interquartile range (IQR) of the upper and lower quartiles
3. RESULTS
3.1. Kinetics of GALT restoration in liver and brain following neonatal tail vein administration of scAAV9‐hGALT
The first goal of this study was to define the kinetics of GALT restoration achieved in the livers and brains of GALT‐null rats by neonatal tail vein administration of scAAV9 virus encoding human GALT (hGALT). Specifically, we wanted to ask how quickly GALT activity could be restored in liver and brain, and how long it would persist. As described in Section 2, we administered self‐complementary AAV9 encoding an N‐terminally HA‐tagged hGALT transgene at 2.5 × 1013 vg/kg to GALT‐null rats on day 2 of life. To quantify transduction efficiency and the kinetics of GALT restoration, we euthanized cohorts of treated animals at 8, 14, 30, and 60 days and collected liver, brain, and blood for enzyme activity measurements, metabolite quantitation, and DNA analyses. Following fixation of solid tissues, we also performed staining, IHC, and IF. Wild‐type and GALT‐null rats administered either vehicle alone (sterile phosphate buffered saline, PBS), or AAV9‐GFP, were included as controls. As expected, results were essentially indistinguishable between GALT‐null rats receiving PBS vs GFP (eg, Figure S1). Of note, while the galactitol levels in 8‐day‐old wild‐type rat pups administered PBS were slightly higher than the corresponding levels seen in GFP‐treated pups, for gal‐1‐P the order was reversed, suggesting this is not a biologically meaningful difference. For the majority of subsequent experiments, we used PBS injection as a negative control.
GALT activity assays confirmed strong transgene expression in both liver and brain in all treated rats harvested on day 8, with diminishing albeit clearly detectable GALT activity persisting throughout the timeframe of the study (Figure 1A,B and Table S1). To be clear, although the level of GALT activity detected in liver at 60‐days in treated GALT‐null rats appears very close to 0 in Figure 1A due to the expanded Y‐axis, it is still well above the level detected in mock‐treated GALT‐null rats, as presented in Table S1 (6.14 ± 4.31 vs 0.05 ± 0.09 pmol UDPgal/μg prot/min, respectively).
At all time points tested, GALT activity in liver exceeded that in brain, but the magnitude of the difference changed over time. In treated GALT‐null rats the ratio of liver to brain GALT activity was >200‐fold at 8 days, about 79‐fold at 14 days, about 17‐fold at 30 days, and about 4 to 5‐fold at 60 days (Table S1). To put these ratios in context, in untreated or PBS‐treated wild‐type rats at 8 days GALT activity was almost 14‐fold higher in liver than in brain; at 14 days the difference was about 5‐fold, and at both 30 and 60 days the difference was 3‐fold to 4‐fold.
As reported previously, 11 among treated GALT‐null animals we saw a weak but positive correlation between GALT activity detected in liver and brain (Figure 1C), and also a correlation between the level of GALT activity seen in liver and the number of scAAV9 viral genomes detected per cell (Figure S2). Of note, despite finding strong GALT activity in both liver and brain in treated GALT‐null rats, we were not able to detect any GALT activity in RBC from the same animals, although we did see strong GALT activity in RBC from wild‐type rats (Table S2). 12
To quantify the fraction of individual cells expressing GALT in treated rats, we performed IHC on sections of fixed tissue from rats harvested at different ages. To confirm specificity of the IHC signal for GALT transgene product we first stained sequential 5 μm sections of tissue using antibodies against either the N‐terminal HA tag, or the GALT protein itself (see Section 2). As expected, both antibodies decorated the same cells (Figure S3); the anti‐HA antibody was selected for further use in these IHC experiments.
The fraction of HA‐positive cells in each IHC slide was estimated using custom software that calculated the fraction of pixels demonstrating above a threshold of normalized red minus blue color, as described in detail elsewhere (Patel et al, in preparation). As seen for GALT activity, the fraction of pixels staining positive in liver decreased over time from a median of about 0.34 at 14 days to above zero but <0.05 at 60 days (Figure S4A). To be clear, the fraction of GALT+ cells never went to zero, as the level of GALT activity per mg tissue also never went to zero (Table S1). Also, as expected, there was a clear positive relationship between the level of GALT activity detected in liver samples processed for enzyme activity and the fraction of HA‐positive pixels seen in fixed liver from the same animal (Figure S4B).
Finally, to test whether there might be any detectable liver or brain histopathology associated with GALT‐null status, GALT over‐expression, or scAAV9 exposure, we prepared H&E‐stained sections of fixed liver and brain from both treated and mock‐treated GALT‐null rats and wild‐type controls. Scans of slides coded to hide genotype and treatment group were reviewed by an experienced veterinary pathologist (Dr. Uriel Blas‐Machado, University of Georgia) who reported no visible pathology in either liver or brain that correlated with GALT genotype, GALT transgene expression level, or scAAV9‐exposure (data not shown).
3.2. GALT expression in brain
While GALT activity assays of brain homogenate (Figure 1B and Table S1) confirmed effective transduction of scAAV9‐hGALT in brain, these results did not address whether GALT was restored across different regions of the brain and/or to different cell types. To answer these questions, we performed double‐label IF procedures using an anti‐GALT antibody together with either anti‐NeuN, which stains neurons, 16 or anti‐GFAP, which stains astrocytes, 17 or anti‐calbindin, which stains Purkinje cells. 19 We saw evidence of GALT signal in at least three brain regions: frontal cortex, cerebellum, and hippocampus (Figure S6). We did not see GALT signal in parallel sections derived from PBS‐injected GALT‐null rats (data not shown). We also saw clear evidence of GALT staining in both neurons and Purkinje cells. While we did not see GALT‐staining of GFAP+ cells, we cannot rule out that GALT might also have been transduced into some astrocytes. We suspect the apparent preferential transduction of neurons in our rats may reflect the very early age (P2) at which the rats were injected. 20
3.3. Metabolic efficacy of scAAV9‐hGALT administered by tail vein injection to neonatal GALT‐null rats
Our second goal was to define the extent and kinetics of metabolic rescue achieved by scAAV9‐mediated GALT restoration in GALT‐null rats. Specifically, we measured galactose, galactitol, and gal‐1‐P in liver, brain, and blood samples from treated and control GALT‐null rats harvested at 8, 14, 30, and 60 days. As explained above, for the older cohorts we also tested the impact of dietary galactose exposure on metabolites by weaning some pups to a diet with 1% of calories from galactose, and other pups to a diet with 3% of calories from galactose—which is the same level found in rat breast milk. 5
Our results (Figure 2A‐D) demonstrated three important points. First, all three metabolites were present at extremely high levels in the livers and brains of PBS‐treated GALT‐null rats at 8 and 14 days, but at normal or near‐normal levels in their scAAV9‐treated counterparts. Second, metabolic rescue remained strong in the treated rats at both post‐wean time points (30 and 60 days), but in some cases rescue was not as complete as that seen in younger rats. Third, galactose and galactitol seemed the “easiest” metabolites to correct, and gal‐1‐P the most difficult, especially in brain and RBC.
To assess the impact of dietary galactose exposure on metabolites in post‐wean animals we compared the galactose, galactitol, and gal‐1‐P levels in liver, brain, and blood from treated and control GALT‐null rats weaned to diets with 1% vs 3% of calories from galactose (see Section 2). With some exceptions, diet mattered, especially in treated GALT‐null rats euthanized at 60 days. Specifically, treated GALT‐null rats weaned to the lower galactose diet showed near‐normalized metabolites at 60 days, whereas their counterparts weaned to the higher galactose diet showed much higher metabolite levels (Figures 2 and S5).
3.4. Phenotypic efficacy of neonatal GALT restoration in GALT‐null rats
Our third goal was to probe the relationship between GALT restoration, metabolic rescue, and long‐term outcomes of treated GALT‐null rats. The two phenotypes we followed were cataracts and pre‐pubertal growth delay.
As presented in Table S1, as a group the scAAV9‐hGALT‐treated GALT‐null rats harvested at ages 8 and 14 days showed notably milder cataracts than their PBS‐treated counterparts, for example, average score 1.33 (n = 18) vs 2.77 (n = 26). However, in each treated group one or more rats still showed severe cataracts. By 30 days, however, scAAV9‐hGALT‐treated GALT‐null rats showed a striking and more consistent rescue of cataracts (Table S1 and Figure 3A,B); for example, at 30 days the treated rats demonstrated an average cataract score of 0.09 with only one treated animal showing even mild cataracts, while the PBS‐treated GALT‐null rats in this age group showed an average cataract score of 1.7. By 60 days, it appeared that both virus and PBS‐treated GALT‐null rats had only mild cataracts, though the cohort sizes in this age group were very small.
FIGURE 3.
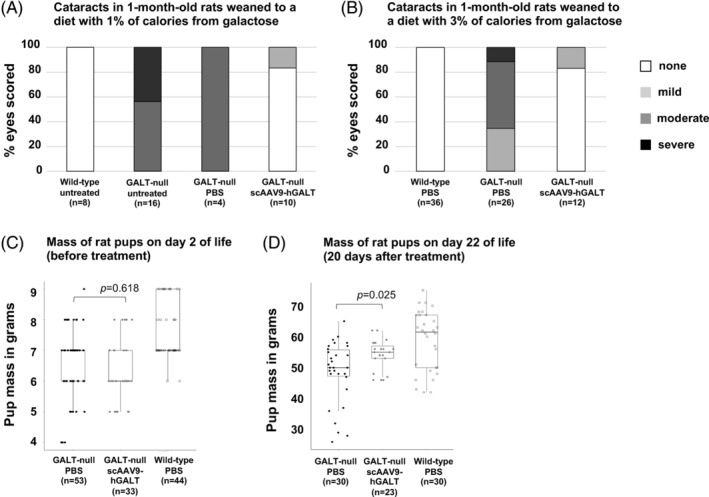
Impact of GALT restoration on cataracts and growth of GALT‐null rats. Cataracts were scored post‐mortem using a 4‐point scale (white = no cataract, light gray = mild cataract, darker gray = moderate cataract, black = severe cataract) in treated and control 30‐day‐old rats weaned either to a diet containing 1% of calories from galactose (panel A) or 3% of calories from galactose (panel B). Box and whisker plots show body mass in treated and control rat pups on day 2 (just before treatment, panel C) and day 22 (20 days after treatment, panel D)
Finally, with regard to growth, treated GALT‐null pups showed a significant (P = .025), albeit partial rescue of pre‐pubertal growth delay at day 22 of life relative to PBS‐treated GALT‐null rats (Figure 3D). As expected, prior to treatment the two groups showed body mass values that were indistinguishable (P = .618; Figure 3C).
3.5. UDP‐glucose pyrophosphorylase activity increases over time in rat brain
One of the striking observations that emerged from our prior 3 and current (Figures 2 and S5) characterization of tissue metabolites in untreated or PBS‐treated GALT‐null rats was the steady drop in galactose metabolite levels seen over time in post‐wean rats despite an unchanging diet. In an effort to explain this pattern, we quantified the level of UDP‐glucose pyrophosphorylase (UGP) activity toward gal‐1‐P in brain samples from both wild‐type and GALT‐null rats 8 to 60 days old (Figure 4). The pattern was clear; UGP activity rose steadily as the rats matured, regardless of GALT genotype and also regardless of whether the rats were weaned to a diet of close to 1% or 3% calories from galactose. Whether the trajectory would have continued beyond 60 days will be a subject of future investigation.
4. DISCUSSION
The work presented here achieved three goals. First, we demonstrated that even a moderate dose of scAAV9‐hGALT (2.5 × 1013 vg/kg) administered intravenously to neonatal GALT‐null rats was sufficient to achieve strong GALT replacement within 1 week, with partial GALT replacement persisting to young adulthood (Figure 1 and Table S1). Loss of transgene expression over time was most evident in liver, and likely reflected dilution of the nonreplicating episomal virus due to proliferation of the juvenile tissue. To be clear, while the level of GALT activity detected in the livers of treated GALT‐null rats decreased over time, even at 60 days it remained clearly above the level seen in untreated GALT‐null rats of the same age (6.14 ± 4.31 vs 0.05 ± 0.09 pmol UDPgal/μg prot/min, respectively).
Second, we demonstrated that the level of GALT replacement achieved was sufficient to provide near‐complete metabolic rescue to young adulthood, at least under conditions of partial galactose restriction. To be clear, even the more restrictive diet tested here (almost 1% of calories from galactose) included far more galactose than is consumed by most patients with classic galactosemia. 21
Finally, we demonstrated that the level of GALT restoration and metabolic rescue achieved here in treated GALT‐null rats was sufficient to minimize two representative complications of GALT deficiency: cataracts and pre‐pubertal growth delay. This is an important result, in part because while cataracts involve an isolated tissue (ocular lens) with a known mechanism of pathology (galactitol accumulation 22 ), growth delay does not. That the metabolic correction achieved here minimized a whole‐body outcome suggests that other long‐term outcomes, not tested here, may also have been modified.
4.1. Improved metabolism of galactose with age
As reported previously, 3 and confirmed here, GALT‐null rats demonstrate diminished galactose metabolite accumulation in blood and tissues over time despite an unchanging post‐wean diet. This result mirrors a similar phenomenon reported for patients, 23 made all the more notable given that patient diets tend to include more, not less nondairy galactose as the patients grow from childhood to adulthood. 23 If changing dietary galactose load is not the answer, the remaining options include diminished endogenous production of galactose, which has been reported for patients, 7 , 24 , 25 and increased capacity of bypass routes, which has been hypothesized previously, but not tested. Here we quantified UGP activity toward gal‐1‐P in samples of brain from neonates through adults and found a close to 3‐fold increase. The mechanism underlying this increase remains unclear, but the result may explain, at least in part, the improved ability of rats, and humans, to metabolize galactose as they mature despite a continued absence of GALT.
4.2. Differential rescue of metabolites in tissues, and correction of metabolites in blood despite an apparent failure to transduce RBCs
We observed two intriguing patterns related to metabolites in treated GALT‐null rats. First, at most time points and in most tissues, galactose showed the most complete rescue, followed by galactitol, followed by gal‐1‐P. One possible explanation is that while galactose and galactitol can cross the cell membrane so that transduced cells can help to metabolize the galactose and galactitol that would otherwise accumulate in their nontransduced neighbors, gal‐1‐P is strictly intracellular, and therefore cell autonomous.
The second pattern relates to metabolites in RBC and plasma. Specifically, as expected from prior studies of AAV9 12 we did not restore GALT activity in RBCs, and GALT is intracellular and therefore not found in plasma. However, all three galactose metabolites showed partial to near‐complete correction in the RBC and/or plasma of treated GALT‐null rats. We hypothesize that this result reflects the impact of GALT in transduced liver and/or other tissues, effectively “scrubbing” galactose from the blood. This is important, not because blood is a relevant tissue for long‐term outcomes in CG, but because this result confirms that GALT restored to one tissue may lessen the accumulation of galactose metabolites in another tissue. This result also raises the possibility that GALT restored to less mitotic extra‐hepatic tissues, such as skeletal or cardiac muscle, could establish a long‐term GALT repository offering broad metabolic benefit to the animal. Testing this possibility will be a focus of future experiments. Finally, another implication of finding metabolic correction in blood despite absence of GALT activity in RBC is that while measuring GALT activity in RBC samples collected from patients who receive gene therapy for GALT may not be informative, checking the galactose metabolites in both plasma and RBC from these patients should be very informative.
4.3. Limitations
This study presents a major step toward improved intervention for classic galactosemia, but it had important limitations. For example, at no point in their lives did our rats experience the level of dietary galactose restriction recommended for patients, so that the results presented here reflect more of a metabolic “worst case” rather than a “best case” scenario. We also had very limited numbers of animals in some comparison groups, ended the experiment at 60 days, and due to small cohort size and limited time frame, did not test cohorts for cognitive or motor function. Further, we did not test for GALT restoration in skeletal or cardiac tissues. Considering the well‐documented ability of AAV9 to transduce numerous extra‐hepatic tissues, 12 and the ubiquitous promoter used here, we fully expect that GALT was restored to these other tissues, and indeed, that persisting GALT in these other tissues might have contributed to the metabolic rescue observed here, especially at later time points. Finally, we administered scAAV9‐hGALT to neonatal pups at a single dose of 2.5 × 1013 vg/kg. Considering the partial rescue observed for both cataracts and pre‐pubertal growth delay, we suspect a higher dose might have achieved a stronger and potentially also a longer‐lived metabolic and phenotype impact.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Jenna Daenzer helped to design and interpret experiments, performed or oversaw the majority of biochemical and metabolic work reported, and participated in writing and editing of the manuscript. Shauna Rasmussen helped to design and interpret experiments, performed or oversaw the majority of animal work reported, and participated in writing and editing of the manuscript. Sneh Patel quantified IHC staining, performed all statistical analyses, and participated in writing and editing of the manuscript. James McKenna performed much of the IF work and participated in editing of the manuscript. Judith Fridovich‐Keil oversaw the design and interpretation of all experiments, coordinated the activities of all co‐authors, helped with some of the animal work, and did most of the writing and editing of the manuscript.
ETHICS APPROVAL STATEMENT
This manuscript does not include any human subjects work. All animal work was performed with approval of the Emory IACUC (Protocol PROTO201700095; PI: JL Fridovich‐Keil) and oversight by the Emory Division of Animal Resources.
Supporting information
Table S1 Animals used in this study. Neonates were administered either vehicle (sterile PBS) or viral stock (scAAV9‐hGALT) at 2.5 × 1013vg/kg on P2 and euthanized at the indicated age for tissue harvest (see Section 2). *Rats weaned to a diet containing 1% of calories from galactose. **Rats weaned to a diet containing 3% of calories from galactose.
Table S2 GALT activity in red blood cells.
Figure S1 Metabolites in GALT‐null and wild‐type rats administered PBS vs AAV9‐GFP. Box and whisker plots showing metabolites in tissues from 8‐day‐old to 14‐day‐old GALT‐null and wild‐type rats administered PBS vs AAV9‐GFP.
Figure S2 Liver GALT activity correlates with viral genome copy number. GALT activity in liver samples from rats injected as neonates with PBS (black circles) or scAAV9‐hGALT (gray circles) and harvested at 8, 14, 30, or 60 days.
Figure S3 Immunohistochemistry (IHC) of sequential liver sections from rat 60C stained with anti‐HA vs anti‐GALT antibody. Brown color denotes regions stained by each antibody.
Figure S4 IHC reveals the fraction of cells in liver expressing GALT. (A) Digital analysis was used to score the fraction of pixels stained brown in anti‐HA‐stained sections of liver from GALT‐null rats administered scAAV9‐hGALT on day 2 and harvested on day 14, 30, or 60. In each box and whisker plot the top and bottom of each box represent the 75th and 25th percentiles, respectively, the middle line in each box represents the median for that set, and the whiskers, if any, represent the maximum and minimum values in the data set within 1.5 interquartile range (IQR) of the upper and lower quartiles. (B) Positive correlation between the fraction of pixels stained brown in a fixed section of liver and GALT enzyme activity quantified in lysate from the same liver. Black circles represent rats harvested at 14 days; gray circles represent rats harvested at 30 days, white circles represent rats harvested at 60 days.
Figure S5 Impact of elevated post‐wean dietary galactose load on metabolites at 30 and 60 days. Box and whisker plots show galactose, galactitol, and gal‐1‐P levels measured in samples of liver (panel A), brain (panel B), red blood cells (RBC, panel C), and plasma (panel D) collected from rats weaned to diets containing either 1% or 3% of calories from galactose and harvested at 30 or 60 days. The top and bottom of each box represent the 75th and 25th percentiles, respectively, the middle line in each box represents the median for that set, and the whiskers, if any, represent the maximum and minimum values in the data set within 1.5 interquartile range (IQR) of the upper and lower quartiles. In all graphs, PBS‐treated GALT‐null rats are represented by black symbols, scAAV9‐hGALT‐treated GALT‐null rats by gray symbols, and PBS‐treated wild‐type rats by white symbols.
Figure S6 Immunofluorescent (IF) detection of GALT transgene expression in three brain regions and two cell types. Double‐label IF of fixed brain sections from a GALT‐null rat (rat 30C from Table S1) administered scAAV9‐hGALT on P2 and harvested at 30 days demonstrates GALT signal in frontal cortex, cerebellum, and hippocampus. Co‐localization is seen with neurons and Purkinje cells, but not with astrocytes.
ACKNOWLEDGMENTS
We are grateful to numerous colleagues for their contributions to this project. We specifically thank Dr. Uriel Blas‐Machado, of the University of Georgia, for reviewing our histology slides, and Dr. Lyra Griffiths, of the Emory Integrated Cores, for performing the quantitative PCR required to calculate viral genome copy number in tissue samples. We also thank all members of the Fridovich‐Keil lab for constant support, and the many professionals working in the Emory Division of Animal Resources and the Emory IACUC without whom this work could not have been conducted. This work was supported in part by grants from the National Institutes of Health R01DK107900 and R21HD092785 (both to JLFK), and in part by the Emory Integrated Genomics Core (EIGC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities.
Daenzer JMI, Rasmussen SA, Patel S, McKenna J III, Fridovich‐Keil JL. Neonatal GALT gene replacement offers metabolic and phenotypic correction through early adulthood in a rat model of classic galactosemia. J Inherit Metab Dis. 2022;45(2):203‐214. doi: 10.1002/jimd.12471
Jennifer M. I. Daenzer and Shauna A. Rasmussen contributed equally to this study.
Communicating Editor: Gerard T. Berry
Funding information National Institutes of Health, Grant/Award Numbers: R01DK107900, R21HD092785; University Research Committee, Emory University, Grant/Award Number: 00097374
REFERENCES
- 1. Welling L, Bernstein LE, Berry GT, et al. International clinical guideline for the management of classical galactosemia: diagnosis, treatment, and follow‐up. J Inherit Metab Dis. 2017;40:171‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Berry GT. Classic Galactosemia and Clinical Variant Galactosemia. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews®. Seattle, WA: University of Washington; 2021. [PubMed] [Google Scholar]
- 3. Rasmussen SA, Daenzer JMI, MacWilliams JA, et al. A galactose‐1‐phosphate uridylyltransferase‐null rat model of classic galactosemia mimics relevant patient outcomes and reveals tissue‐specific and longitudinal differences in galactose metabolism. J Inherit Metab Dis. 2020;43:518‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Quan‐Ma R, Wells HJ, Wells WW, Sherman FE, Egan TJ. Galactitol in the tissues of a galactosemic child. Am J Dis Child. 1966;112:477‐478. [DOI] [PubMed] [Google Scholar]
- 5. Jenness R. Proceedings: biosynthesis and composition of milk. J Invest Dermatol. 1974;63:109‐118. [DOI] [PubMed] [Google Scholar]
- 6. Berry GT, Nissim I, Lin ZP, Mazur AT, Gibson JB, Segal S. Endogenous synthesis of galactose in normal men and patients with hereditary galactosemia. Lancet. 1995;346:1073‐1074. [DOI] [PubMed] [Google Scholar]
- 7. Schadewaldt P, Kamalanathan L, Hammen HW, Kotzka J, Wendel U. Endogenous galactose formation in galactose‐1‐phosphate uridyltransferase deficiency. Arch Physiol Biochem. 2014;120:228‐239. [DOI] [PubMed] [Google Scholar]
- 8. Lai K, Elsas LJ. Overexpression of human UDP‐glucose pyrophosphorylase rescues galactose‐1‐phosphate uridyltransferase‐deficient yeast. Biochem Biophys Res Commun. 2000;271:392‐400. [DOI] [PubMed] [Google Scholar]
- 9. Leslie N, Yager C, Reynolds R, Segal S. UDP‐galactose pyrophosphorylase in mice with galactose‐1‐phosphate uridyltransferase deficiency. Mol Genet Metab. 2005;85:21‐27. [DOI] [PubMed] [Google Scholar]
- 10. Mehta DV, Kabir A, Bhat PJ. Expression of human inositol monophosphatase suppresses galactose toxicity in Saccharomyces cerevisiae: possible implications in galactosemia. Biochim Biophys Acta. 1999;1454:217‐226. [DOI] [PubMed] [Google Scholar]
- 11. Rasmussen SA, Daenzer JMI, Fridovich‐Keil JL. A pilot study of neonatal GALT gene replacement using AAV9 dramatically lowers galactose metabolites in blood, liver, and brain and minimizes cataracts in GALT‐null rat pups. J Inherit Metab Dis. 2021;44:272‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mattar CN, Wong AM, Hoefer K, et al. Systemic gene delivery following intravenous administration of AAV9 to fetal and neonatal mice and late‐gestation nonhuman primates. FASEB J. 2015;29:3876‐3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gray SJ, Foti SB, Schwartz JW, et al. Optimizing promoters for recombinant adeno‐associated virus‐mediated gene expression in the peripheral and central nervous system using self‐complementary vectors. Hum Gene Ther. 2011;22:1143‐1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Elsevier JP, Wells L, Quimby BB, Fridovich‐Keil JL. Heterodimer formation and activity in the human enzyme galactose‐1‐phosphate uridylyltransferase. Proc Natl Acad Sci U S A. 1996;93:7166‐7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McCarty DM, Fu H, Monahan PE, Toulson CE, Naik P, Samulski RJ. Adeno‐associated virus terminal repeat (TR) mutant generates self‐complementary vectors to overcome the rate‐limiting step to transduction in vivo. Gene Ther. 2003;10:2112‐2118. [DOI] [PubMed] [Google Scholar]
- 16. Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201‐211. [DOI] [PubMed] [Google Scholar]
- 17. Jacque CM, Vinner C, Kujas M, Raoul M, Racadot J, Baumann NA. Determination of glial fibrillary acidic protein (GFAP) in human brain tumors. J Neurol Sci. 1978;35:147‐155. [DOI] [PubMed] [Google Scholar]
- 18. Park SY, Yoo YM, Jung EM, Jeung EB. Distribution of and steroid hormone effects on calbindin‐D9k in the immature rat brain. Brain Res Bull. 2019;152:225‐235. [DOI] [PubMed] [Google Scholar]
- 19. Reith RM, Way S, McKenna J 3rd, Haines K, Gambello MJ. Loss of the tuberous sclerosis complex protein tuberin causes Purkinje cell degeneration. Neurobiol Dis. 2011;43:113‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Calcar SC, Bernstein LE, Rohr FJ, Scaman CH, Yannicelli S, Berry GT. A re‐evaluation of life‐long severe galactose restriction for the nutrition management of classic galactosemia. Mol Genet Metab. 2014;112:191‐197. [DOI] [PubMed] [Google Scholar]
- 22. Ai Y, Zheng Z, O'Brien‐Jenkins A, et al. A mouse model of galactose‐induced cataracts. Hum Mol Genet. 2000;9:1821‐1827. [DOI] [PubMed] [Google Scholar]
- 23. Waggoner DD, Buist NR, Donnell GN. Long‐term prognosis in galactosaemia: results of a survey of 350 cases. J Inherit Metab Dis. 1990;13:802‐818. [DOI] [PubMed] [Google Scholar]
- 24. Berry GT, Moate PJ, Reynolds RA, et al. The rate of de novo galactose synthesis in patients with galactose‐1‐phosphate uridyltransferase deficiency. Mol Genet Metab. 2004;81:22‐30. [DOI] [PubMed] [Google Scholar]
- 25. Schadewaldt P, Kamalanathan L, Hammen HW, Wendel U. Age dependence of endogenous galactose formation in Q188R homozygous galactosemic patients. Mol Genet Metab. 2004;81:31‐44. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Animals used in this study. Neonates were administered either vehicle (sterile PBS) or viral stock (scAAV9‐hGALT) at 2.5 × 1013vg/kg on P2 and euthanized at the indicated age for tissue harvest (see Section 2). *Rats weaned to a diet containing 1% of calories from galactose. **Rats weaned to a diet containing 3% of calories from galactose.
Table S2 GALT activity in red blood cells.
Figure S1 Metabolites in GALT‐null and wild‐type rats administered PBS vs AAV9‐GFP. Box and whisker plots showing metabolites in tissues from 8‐day‐old to 14‐day‐old GALT‐null and wild‐type rats administered PBS vs AAV9‐GFP.
Figure S2 Liver GALT activity correlates with viral genome copy number. GALT activity in liver samples from rats injected as neonates with PBS (black circles) or scAAV9‐hGALT (gray circles) and harvested at 8, 14, 30, or 60 days.
Figure S3 Immunohistochemistry (IHC) of sequential liver sections from rat 60C stained with anti‐HA vs anti‐GALT antibody. Brown color denotes regions stained by each antibody.
Figure S4 IHC reveals the fraction of cells in liver expressing GALT. (A) Digital analysis was used to score the fraction of pixels stained brown in anti‐HA‐stained sections of liver from GALT‐null rats administered scAAV9‐hGALT on day 2 and harvested on day 14, 30, or 60. In each box and whisker plot the top and bottom of each box represent the 75th and 25th percentiles, respectively, the middle line in each box represents the median for that set, and the whiskers, if any, represent the maximum and minimum values in the data set within 1.5 interquartile range (IQR) of the upper and lower quartiles. (B) Positive correlation between the fraction of pixels stained brown in a fixed section of liver and GALT enzyme activity quantified in lysate from the same liver. Black circles represent rats harvested at 14 days; gray circles represent rats harvested at 30 days, white circles represent rats harvested at 60 days.
Figure S5 Impact of elevated post‐wean dietary galactose load on metabolites at 30 and 60 days. Box and whisker plots show galactose, galactitol, and gal‐1‐P levels measured in samples of liver (panel A), brain (panel B), red blood cells (RBC, panel C), and plasma (panel D) collected from rats weaned to diets containing either 1% or 3% of calories from galactose and harvested at 30 or 60 days. The top and bottom of each box represent the 75th and 25th percentiles, respectively, the middle line in each box represents the median for that set, and the whiskers, if any, represent the maximum and minimum values in the data set within 1.5 interquartile range (IQR) of the upper and lower quartiles. In all graphs, PBS‐treated GALT‐null rats are represented by black symbols, scAAV9‐hGALT‐treated GALT‐null rats by gray symbols, and PBS‐treated wild‐type rats by white symbols.
Figure S6 Immunofluorescent (IF) detection of GALT transgene expression in three brain regions and two cell types. Double‐label IF of fixed brain sections from a GALT‐null rat (rat 30C from Table S1) administered scAAV9‐hGALT on P2 and harvested at 30 days demonstrates GALT signal in frontal cortex, cerebellum, and hippocampus. Co‐localization is seen with neurons and Purkinje cells, but not with astrocytes.