

Abstract
Sensors capable of accurate, continuous monitoring of biochemistry are crucial to the realization of personalized medicine on a large scale. Great strides have been made to enhance tissue compatibility of long-term in vivo biosensors using biomaterials strategies such as tissue-integrating hydrogels. However, the low level of oxygen in tissue presents a challenge for implanted devices, especially when the biosensing function relies on oxygen as a measurand—either as a primary analyte or as an indirect marker to transduce levels of other biomolecules. This work presents a method of fabricating inorganic-organic interpenetrating network (IPN) hydrogels to optimize the oxygen transport through injectable biosensors. Capitalizing on the synergy between the two networks, various physicochemical properties (e.g. swelling, glass transition temperature, and mechanical properties) are shown to be independently adjustable while maintaining a 250% increase in oxygen permeability relative to poly(2-hydroxyethyl methacrylate) (pHEMA) controls. Finally, these gels, when functionalized with a Pd(II) benzoporphyrin phosphor, track tissue oxygen in real time for 76 days as subcutaneous implants in a porcine model while promoting tissue ingrowth and minimizing fibrosis around the implant. These findings support IPN networks for fine-tuned design of implantable biomaterials in personalized medicine and other biomedical applications.
Keywords: silicone hydrogels, biosensors, tissue integration, biocompatible, implants
TOC Summary:
Interpenetrating network (IPN) hydrogels are fabricated with inorganic and organic networks as optical biosensors. Synergy between both polymer networks allows independent tuning of physicochemical properties while optimizing oxygen transport. IPN sensors with inverted colloidal crystal microarchitecture track tissue oxygen in real time for 76 days as subcutaneous implants in a porcine model, promoting tissue ingrowth and minimizing fibrotic encapsulation.
Graphical Abstract
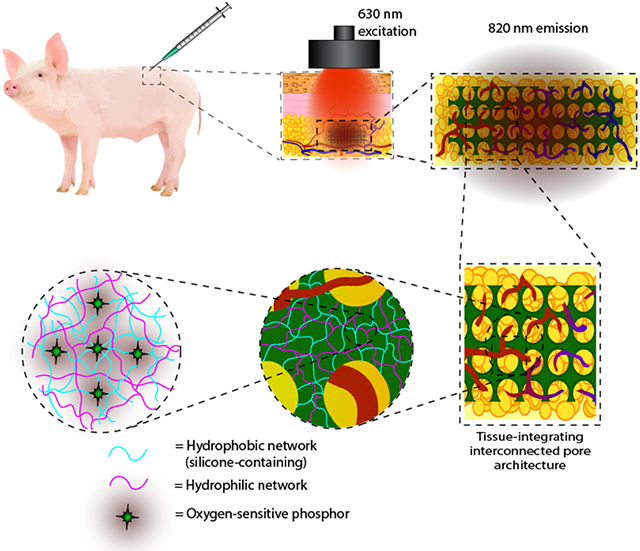
1. Introduction
Polymeric biomaterials have been used in medical devices for decades, functioning as a whole or part of the device. These act as passive, standalone materials for structural support (e.g., dental fixation devices, joint replacements, bone cements), or as active, stimuli-responsive materials to interact with, modulate, or respond to its surrounding environment (e.g., biosensors, drug delivery devices, biodegradable tissue engineering scaffolds). [1-6] Hydrogels— crosslinked polymer networks which imbibe water without dissolving—are particularly attractive in the biomaterials world due to the wide range material properties that may be achieved to mimic biological tissue. One of the first hydrogels developed was comprised of poly(2-hydroxyethyl methacrylate) (pHEMA); pHEMA-based hydrogels and their copolymers have been effectively used in contact lenses as well as in fully-implantable devices. [7] Over the past few decades, hydrogel materials and their processing methods have been refined to improve important attributes for medical device applications. In the case of contact lenses, these attributes include excellent optical clarity, high oxygen permeability, low modulus with flexural toughness, wettability/hydration, and resistance to fouling by proteins and other particulate matter. [2, 3, 8-20]
Additional features have also been added to contact lenses, such as incorporation of small molecules that are released for local therapeutic effect. For example, the Sheardown group has functionalized pHEMA hydrogels with various comonomer combinations including [3-methacryloyloxypropyl] tris trimethylsilyl siloxy silane (TRIS) and dimethylacrylamide (DMA) for the release of various drugs to the anterior eye. [12, 13, 15, 21, 22] These formulations have been shown to partition against the drug, enabling a sustained zero-order release over two weeks with only 20-30% TRIS, the agent used to control the partitioning. However, because TRIS is hydrophobic, in some cases these hydrogels only contained 30% water (equilibrium water content, EWC) and lost their optical clarity with TRIS contents above 30% due to silicone phase separation from the water, undesirable properties for optical lens purposes. [22] Despite these advances, recent studies highlight the potential for novel fabrication strategies to overcome these limitations with traditional hydrogels which impact numerous applications including contact lenses, drug delivery, tissue engineering, and wound healing. [23-26]
Emerging device concepts such as optical biosensors have very similar material property requirements to contact lenses. [27] Long-term, tissue-integrating oxygen sensors based on pHEMA have been demonstrated in rats, pigs, and humans for over two years in the body. [28-35] Additionally, we have demonstrated pHEMA hydrogels may be functionalized for detection of lactate and glucose. [36-38] Proof-of-concept performance with these pHEMA copolymer hydrogels, functionalized with oxidoreductase enzymes and palladium(II) benzoporphyrin dyes, has been shown. [36, 38] Evaluation of preclinical performance of our glucose sensors in porcine models demonstrated the ability to closely track real-time measured blood glucose values in vivo after injection; however, long-term performance (i.e. on the order of months) of this device has yet to be realized in preclinical studies. [38, 39] Loss of function in vivo has been attributed to the extreme low oxygen environment of subcutaneous tissue, because explanted devices exhibit preserved function. This is consistent with observations related to diminished sensor longevity in vivo in many electrochemical sensors, which has been related to a dynamically-changing tissue environment from the acute immune response and natural tissue healing process as well as generally high metabolic activity of skin interstitium. [40] Possible approaches to alleviate complications associated with shifting tissue oxygen levels are: 1) increase oxygen transport into the sensor; and/or 2) induce tissue integration throughout the device for more uniform substrate transport. [34]
As noted above, increasing oxygen permeability is a materials chemistry matter. Inorganic-organic polymers—such as siloxane-containing materials—have been utilized in biomedical applications because of unique properties between both classes of polymers and their potential for biomedical applications (e.g., tissue engineering). [39, 41-44] Further, interpenetrating network hydrogels (IPNs) are an emerging class of materials that have gained interest in materials science because of the ability to combine two polymer materials with different properties (e.g., miscibility) and harness a unique synergy when combined, exhibiting properties superior to those of either polymer network alone. [2, 9, 45-47] IPNs are defined as two networks containing no covalent bonds between them but are intertwined such that neither network can be separated from the other without breaking the covalent bonds of the individual polymer networks. [48] Synthesizing IPN hydrogels affords two possible polymerization routes: 1) sequential (polymerization of one network, infiltration of second network precursor) or 2) simultaneous (both networks polymerized at the same time through non-interfering mechanisms). [19] In this study, sequential polymerization is used to fabricate the IPN hydrogels, illustrated below in Figure 1 and described further in Section 4.1.2.
Figure 1.

Sequential polymerization process used in this study to fabricate the IPN gels.
IPNs of TRIS and DMA have been specifically exploited to create contact lenses with finer control over oxygen permeability and hydration in the lens material. Nicholson and Wang describe the TRIS and DMA construct morphology as isotropic, yet heterogeneous, with distinct, intertwining, hydrophobic silicone domains and hydrophilic DMA domains to allow molecular transport between each other, resulting in high oxygen transport properties in the overall gel. [2, 16] Fortuitously, the same oxygen diffusion properties are desired for the subcutaneous, fully-implantable biosensors described above. However, to our knowledge, IPNs have not been evaluated for performance in these optical sensing applications.
On the other hand, tissue integration relates to the structure of the as-formed materials. Sphere-templated hydrogels have been used as tissue integrating constructs for a variety of tissue engineering applications. [49-52] These templated hydrogels exhibit an inverted colloidal crystal (ICC) structure that uses the scaffold morphology to promote angiogenesis throughout the construct and avoid the use of anti-inflammatory agents or growth factors which increase material cost, complicate the device fabrication process, limit the drug payload, and restrict spatiotemporal control and bioactivity. [53, 54] Materials used in these studies include pHEMA, poly (N-isopropyl acrylamide) (poly(NIPAAm)), and poly(acrylic acid) among others. [20, 52, 54-57] While a variety of materials have shown cellular infiltration, most studies have implemented ICC hydrogels as standalone matrices and were not functionalized to allow real-time monitoring of tissue biochemistry. Only recently have studies have used pHEMA ICC implants to monitor tissue oxygen. [34] In principle, incorporating sensing chemistry within additional materials would enable the study of relationships between tissue ingrowth and oxygen supply to the implant. For example, zones of cellular necrosis in ICC scaffolds during cell culture experiments have been reported, believed to be due to nutrient (e.g. oxygen and glucose) transport limitations in non-optimized scaffolds. [56] Additionally, Somo, et al. investigated large pore sizes (> 100 μm) in ICC hydrogels and reported that the interconnection between the pores has a profound effect on the penetration of these blood vessels throughout a sphere templated gel. [58] In both cases, monitoring the environment throughout the sensor could further the understanding of tissue ingrowth in relation to substrate levels over time. Expanding the library of hydrogel materials used as tissue-integrating biosensors is the first step towards achieving this goal.
The aim of this study is to design and fabricate novel inorganic-organic silicone-containing IPN hydrogels functionalized with an optical biosensing assay and evaluate the relevant various physicochemical properties (water content, optical clarity, substrate transport, mechanical properties, and overall device performance) compared to similarly functionalized pHEMA-based controls. Capitalizing on the synergistic behavior of hydrophobic, inorganic TRIS alkylsiloxane with hydrophilic, organic DMA in an IPN hydrogel structure, we investigate the ability to control gel hydration and mechanical properties independently from substrate (oxygen) transport. Further, by also utilizing an ICC scaffold architecture, we investigate proof-of-concept for these IPN hydrogels as long-term, tissue-integrating implants. To our knowledge, this is the first study proposing silicone-based IPNs in fully-implantable biosensors as long-term implants. We anticipate this inorganic-organic polymer matrix system will impart better control of device properties to fine-tune sensor function, optimize dynamic range, and extend device operational lifetime for oxygen-based biosensing platforms.
2. Results and Discussion
2.1. In Vitro Characterization
2.1.1. Swelling Ratios (Equilibrium Water Content)
Hydrogel discs of four IPN formulations were tested for water content. Samples were prepared with a tetramethacrylated benzoporphyrin dye (PdBP) as well as without dye (blank); all formulations were compared to pHEMA controls, also prepared with and without dye. [36, 38] A general decrease in overall water content (EWC) is observed with increasing TRIS content (Figure 2A). This trend was expected because of the hydrophobic nature of TRIS; thus, a gel with higher TRIS content should imbibe less water than those with higher DMA content relative to TRIS.
Figure 2.

Swelling (A), optical clarity (B), and confocal microscopy images for matrix homogeneity (50:50, 60:40, 70:30, and 80:20 TRIS:DMA for C, D, E, and F, respectively). Swelling data represent triplicate averages; error bars represent 95% confidence intervals.
Considering the PdBP-functionalized gels, there is a prominent, roughly linear decrease in EWC with increasing TRIS content; all gel formulations appear to follow this trend. Among the blank gels, the EWC follows a similar trend based on the TRIS content, with the only exception being with the 60:40 and 70:30 formulations, likely due to decreased crosslinking efficiency with increasing TRIS content as TRIS is known to inhibit crosslinking efficiency in similar acrylate-based gel systems. [59, 60] As TRIS content increases and crosslinking efficiency decreases, uncrosslinked poly(TRIS-co-DMA) chains may introduce matrix inhomogeneities, allowing for water uptake profiles across formulations deviating from the prominent linear trend among dye-containing gels.
It is noteworthy to remember that because PdBP contains four methacrylate groups, it acts as a tetrafunctional crosslinker in the matrix. Based on these observations of the EWC data, the tetrafunctional nature of the PdBP must explain the deviations from the trends seen with the blank gels where the only crosslinker present is TEGDMA (difunctional). Regardless of TRIS:DMA ratio, all IPN gels were significantly more hydrated than pHEMA single network gels, taking up 5-10X more water per unit mass of matrix material. This result was fully anticipated, given the highly hydrophilic nature of DMA compared to pHEMA. Similar trends have been observed in previous studies using DMA as a wetting agent for contact lenses. [12, 18, 19, 21, 61]
2.1.2. Optical Clarity and Homogeneity
Transmission spectra show that with 70% and 80% TRIS content, the gels begin to lose optical clarity over all wavelengths tested. However, 60:40 and 50:50 TRIS:DMA gels both exhibit similar optical transparency behavior to pHEMA, particularly in the primary PdBP excitation/emission window of 630 to 820 nm measured in vivo by the optical instrumentation described in section 4.3.1 (denoted by dashed black lines, Figure 2B, Figure S1). The 50:50 gel even exhibits a higher transmittance than pHEMA at lower wavelength ranges (i.e. 400 to 580 nm) while 60:40 transmittance remains slightly lower than pHEMA. In contrast, 70:30 and 80:20 TRIS:DMA become drastically less transparent than pHEMA at these lower wavelengths.
These data suggest the IPN morphology successfully prevents the phase separation and opacity characteristically reported in the literature for single network silicone hydrogels. [18, 44] Decreased transparency in gels with higher TRIS content (70-80%) is likely due to incomplete crosslinking of the TRIS chains due to the highly branched structure of the TRIS pendant group causing steric hindrance effects, blocking access of unreacted methacrylate groups to free radicals and slowing the propagation of the free radical polymerization. Steric hindrance effects have been reported in the literature in similar free-radical initiated systems using ethylene glycol-derived crosslinkers. [62] While siloxane materials are notorious for their oxygen-permeability, this characteristic can be disadvantageous during processing due to inhibition of free radical polymerizations by oxygen acting as a free-radical scavenger. In the case of TRIS, a siloxane methacrylate, higher amounts of TRIS may have allowed enough oxygen back into the system after purging to stunt the crosslinking with TEGMDA. In fact, oxygen inhibition in similar photocurable siloxanes crosslinked with ethylene glycol-derived crosslinkers has been reported in research and the coatings and resins industry. [63] In the current system, the polymer chains containing high amounts of TRIS may not have fully crosslinked in the first matrix by the tetraethylene glycol dimethacrylate (TEGDMA) crosslinker due to either oxygen inhibition or steric hindrance effects. This could have prevented the second pDMA network from fully reinforcing the first silicone-containing network if polymer chains were not fully tethered to the first matrix, resulting in phase separation in the IPN hydrogel.
Despite some variations in transparency with higher silicone content, the pDMA second network seems to effectively prevent silicone phase separation. This conclusion derives from our previous observations of substantial TRIS phase separation in a single network monolith hydrogel with only 20 vol% TRIS. [18, 39, 44] All formulations here have higher TRIS content and increased optical clarity, indicating the IPN morphology reinforces the silicone and prevents phase separation. This is consistent with previous TRIS-containing IPN hydrogel reports for contact lenses. [16, 19] However, the loss in transparency seen here was considerable and therefore was investigated further for an underlying cause. We hypothesized that, because TRIS is hydrophobic, at the higher concentrations of TRIS (70-80% in the first network) portions of pTRIS would not be adequately reinforced by the pDMA second network. The unreinforced pTRIS would then phase separate from the rest of the gel, manifesting itself as precipitated domains throughout the matrix. For the purposes of investigating morphological inhomogeneities such as phase separation, the gels were stained with Nile Red following an established protocol. [64, 65] It is known that Nile Red selectively adheres to hydrophobic components such as silicone. Staining the gels with Nile Red was expected to allow visualization of phase separation in the gels. Confocal microscopy images suggest a higher degree of phase separation in the 80:20 TRIS:DMA, which was consistent with the loss in optical clarity indicated from the UV-Vis measurements (Figure 2B, 2F, Figure S1, Figure S2). Lighter regions of the confocal images indicate areas where more dye was sequestered within the gel. The 80:20 TRIS:DMA confocal image shows darker spherical regions dispersed throughout a lighter bulk gel material, whereas the other formulations appear much more uniform (Figure 2C-E). It is important to note that the phase separation behavior seen in the confocal images is contrary to the hypothesized mode of phase separation where the dye-stained silicone regions would be manifested as the dispersed phase, with silicone regions distributed throughout the hydrophilic DMA phase. We believe the opposite phase separation behavior occurred here in part because four times as much silicone by volume was used to fabricate the 80:20 gels than in previous silicone gels used with this sensing platform. [44] This could allow the DMA to become dispersed in microdomains throughout the bulk silicone material. Additionally, these gels were made with a substantial amount of dichloromethane (DCM) as a precursor co-solvent (35 vol% of the total precursor solution). In previous studies of silicone-containing hydrogels, phase separation was induced by using DCM as the primary solvent in the precursor solution which allowed the silicone to remain in the continuous phase; similar behavior is seen here. [64, 65]
2.1.3. Thermal and Mechanical Properties
The viscoelastic properties of the IPNs were assessed by measuring their response to an applied oscillatory force varying in frequency from 1 Hz to 100 Hz at a constant shear stress amplitude of 1 Pa and constant temperature of 25 °C and then 37 °C. Figure 3A-D show the real elastic modulus (G’) and imaginary viscous modulus (G”) results for all formulations tested within their viscoelastic region. As expected, all G’ values were found to be higher than their associated G” value, indicating a predominantly elastic response in all IPN formulations. [47] Although the difference between G’ and G” decreases with increasing TRIS concentration, G’ is still greater than G” for all frequency and temperature combinations tested. Therefore, the elastic response dominates since tan(δ) < 1 (Equation 2). For the application of in vivo implants, the gels will remain more elastic than viscous.
Figure 3.

Dynamic mechanical testing comprised of storage and loss modulus at 25 °C (A and C) and 37 °C (B and D) within the viscoelastic region. C and D: averages of measurements within the viscoelastic range (n=11 ± 95% confidence interval). E: glass transition temperatures of dried gels (averages (n=3) ± 95% confidence interval).
The G’ and G” values for the 50:50 and 60:40 TRIS: DMA formulations at both temperatures were not significantly different from each other; otherwise, there is a general increase in both G’ and G” with increasing TRIS content—increasing hydrophobicity (one-way ANOVA, Tukey-Kramer post-hoc test, α = 0.05). It is important to note the slight increase in G’ with increasing frequency for the 80:20 TRIS:DMA IPN, indicating physical mechanical hindrance of the bulky TRIS sidechains at higher frequencies. [66] Increasing temperature resulted in an overall decrease in both G’ and G”, which was expected given that elevated temperatures have been shown to transform hydrogels to a more rubbery state as a result of increased mobility in the chain backbone. [67] This is relevant here because the gels will experience temperatures of 37 °C in vivo.
The DSC results (Figure 3E, Figure S3) show no statistically significant difference compared among 50:50, 60:40, or 70:30 TRIS:DMA formulations; however, the 80:20 TRIS:DMA gel exhibited a significantly depressed Tg, dropping around 50 °C lower than the three other IPN gels (one-way ANOVA, Tukey-Kramer post-hoc test, α = 0.05). This lowering of the Tg is attributed to the high siloxane content of the 80:20 formulation, as siloxane bonds are known for their remarkable torsional flexibility and the branched TRIS pendant group structure is known for its plasticizing ability. [3, 11, 14] Similarly, all IPN formulations, excluding 80:20, had significantly higher Tg than the pHEMA standard which we attribute to the higher mobility of the hydroxyethyl pendant group compared to the DMA pendant group.
While we predicted a steady decline in Tg with increasing siloxane content, we believe we only observed a decrease in Tg with the 80:20 formulation because the siloxane linkages were located in the polymer pendant groups, not in the backbone. Comparatively, PDMS has one of the lowest Tg values among polymers; however, those siloxane linkages are in the polymer backbone. With TRIS, the polymer backbone is a vinyl backbone without siloxane bonds, which could explain the fact that depression of the Tg is not seen until the first network is 80% TRIS. Because Tg is characterized by increased segmented mobility in the backbone, we anticipate a stronger correlation between Tg and siloxane content with a siloxane backbone polymer (e.g. PDMS). Furthermore, the entire second network is pDMA; Tg values for 50% - 70% TRIS are similar to Tg values reported for pDMA which we attribute to the second network comprising of pDMA. [68]
2.1.3. Oxygen Transport: Stern-Volmer Kinetics
Compared to pHEMA standards, all IPN formulations displayed a drastic increase in oxygen permeability according to Stern-Volmer diffusion kinetics (Figure 4, Table 1), as evidenced by the increased linear slopes (KSV values). Specifically, the IPN monoliths exhibited an average of 256% increase in oxygen permeability based on KSV and a 210% increase based on kq. Among IPNs, all formulations were significantly different from pHEMA (t-test, α=0.05). Comparing IPNs among themselves, only the 80:20 formulation was found to be significantly different from 50:50 and 70:30; otherwise, all formulations showed no statistical difference (t-test, α = 0.05). These results indicate that one can maintain a significantly increased oxygen permeability (compared to pHEMA) with these IPN gels irrespective of the silicone content.
Figure 4.

Stern-Volmer oxygen diffusion plot. Data represent averages (n=3) ± 95% confidence intervals.
Table 1.
In Vitro oxygen sensor Stern-Volmer Figures of Merit
| Figure of Merit | 50:50 TRIS:DMA |
60:40 TRIS:DMA | 70:30 TRIS:DMA |
80:20 TRIS:DMA | pHEMA |
|---|---|---|---|---|---|
| Ksv (μM−1) | 0.112 ± 0.007 | 0.086 ± 0.019 | 0.084 ± 0.006 | 0.083 ± 0.002 | 0.0256 ± 0.002 |
| τ0 (μs) | 267 ± 4 | 276 ± 20 | 307 ± 11 | 307 ± 5 | 248 ± 2 |
| kq [10−4 (μs-μM)−1] | 4.18 ± 0.30 | 3.15 ± 0.82 | 2.75 ± 0.18 | 2.70 ± 0.05 | 1.03 ± 0.08 |
2.2. In Vivo Performance and Biocompatibility
As mentioned in Section 1, pHEMA hydrogel ICCs have been used as tissue-integrating oxygen sensors in rats, pigs, and humans. [28-35] While other materials have been used as ICC implants in vivo, most studies have implemented ICC hydrogels as standalone matrices without functionalizing them to allow real-time monitoring of tissue biochemistry.[69] Therefore, evaluating in vivo response of these porphyrin-functionalized gels is of greatest practical importance. Toward this aim, the following sections discuss 1) the baseline signal values at physiological oxygen over time, 2) real-time oxygen tracking behavior, and 3) tissue integration and foreign body response.
2.2.1. Oxygen Modulation Performance – Baseline Lifetime Analysis
Phosphorescence lifetime data from implants (Figure 5) reveal low average baseline values soon after implantation (Day 0) for the IPN ICC implants (31.6±0.3 μs and 10±3.8 μs for Implant 1 and 2, respectively, n=10 lifetime measurements, Figure 5C), while pHEMA implants demonstrated, on average, an over three-fold higher baseline after implantation (69.3±16.9 μs, average of n=3 implant baselines, each baseline average calculated from n=10 lifetime measurements, Figure 5D). Note that IPN Implant 3 was unable to be located transdermally on Day 0, likely due to the fresh capillary blood around and throughout the sensor, resulting in high local oxygen levels which is known to decrease phosphorescence intensity of oxygen-quenched porphyrins (section 4.2.6). [36, 38, 44, 70-73] For both IPN and pHEMA implants, the sensors were the most sensitive (based on total percent change in signal from baseline to peak, Table S1, Table S2) on Day 0, which we attribute to the microporated architecture of the ICC scaffolds (higher surface area to volume ratio) allowing analytes from the stimulated tissue of the fresh injection wound to penetrate the scaffold more rapidly after implantation. It is understood that the local tissue (i.e. immediate surrounding layers of cells) will be inflamed and experience increased perfusion in the acute phase due to mast cells mobilized in response to the initial trauma from the injection needle. Comparing Day 0 oxygen response profiles between the the IPN and pHEMA implants, the IPN implants exhibited a more stable baseline lifetime profile (before oxygen modulation) and higher sensitivity (Figure 5A & B, Table S1, Table S2). This is substantiated by the distinct response plateau around 130 μs at Day 0 (IPN) and the subsequent rapid decline to the baseline lifetime value upon resuming 100% inspired oxygen level. On the contrary, the pHEMA implants started with a higher baseline before oxygen modulation and did not reach a distinct plateau during the oxygen modulation on Day 0 (Figure 5B). We attribute this to the wound healing around the implant. As local oxygen levels decrease, porphyrins are known be more sensitive to changes in local oxygen levels. [71, 72, 74, 75]
Figure 5.

Top: Real-time oxygen sensor response profile at 100% inspired oxygen (baseline) and 15% inspired oxygen (modulation, gray region) for two representative ICC implants (60:40 TRIS:DMA IPN (A)) and pHEMA (B)). Bottom: Luminescence lifetime averages immediately before starting the oxygen modulation experiment (100% inspired oxygen, baseline) for the 60:40 TRIS:DMA IPN (C) and the pHEMA implant (D). Data represent average (n=10 data points) ± 95% confidence interval.
Figure 5 (C-D) shows baseline lifetime averages for all six ICC implants as observed of a 10-week period. Similar trends are observed for each formulation among time points. There is notable variability in the starting baseline lifetime of one IPN sensor (Implant 1) compared to the other two IPN sensors. We attribute this to possible manual aberrations in the injection of the sensors and subsequent tissue trauma leading to varying amounts of microcapillary damage around the sensor. All three pHEMA oxygen sensors exhibited baseline lifetimes trending closely with one another, even during the first two weeks of implantation. However, pHEMA was observed to be over three times less permeable to oxygen based on in vitro calculations of Ksv on pHEMA monoliths, so differences in oxygen levels around the implants during the wound healing phase may be less apparent. Overall, variations in oxygen levels around the implants are expected due to the nature of the wound healing response as it is known that immune cells consume oxygen to carry out both pro-inflammatory and pro-angiogenic immune responses as the tissue remodels or reaches a resolution stage. [74-77]
Both IPN and pHEMA sensors reveal steadily increasing baseline lifetimes over the first three weeks. For both materials, the baseline lifetimes eventually plateau around 200 μs; this occurs at different times, roughly at 20 and 28 days of implantation for the IPN and pHEMA sensors, respectively. This finding was unexpected based on in vitro results indicating IPNs are much more permeable to oxygen. Therefore, we expected the starting baseline lifetimes to be lower than those of pHEMA ICCs, even after tissue ingrowth. This behavior could be explained by the manner in which the tissue integrated throughout the sensor, as some collagen deposition was observed towards the interior of the implant (Figure 6, discussed further in section 2.2.3).
Figure 6.

Scanning Electron Microscope images of the IPN ICC surface, respectively, at 100X and 540X (A&B) and 20X (C&D) magnification. Histology images of ICC hydrogels after explantation: Haemotoxylin and Eosin stained image (C) show the presence of tissue ingrowth (pink) and cell nuclei (purple) while the Masson’s Trichrome stain (D) further differentiates the presence of loosely-woven collagen (blue) and cellular penetration (red).
2.2.2. In Vivo Oxygen Modulation: Real-Time Tracking of Tissue Oxygen Levels
It was observed that both types of ICC sensors exhibit high sensitivity and maintain good responsiveness over 2-3 months in vivo, with a notably stronger sensitivity in the first 6 days (Figure 5, Table S1). Local oxygen fluctuations are reduced over time as the tissue heals, leading to a more consistent steady-state behavior that persists for the remaining duration of implantation. We attribute this to the tissue-integrating nature of the ICC scaffold. While the porous architecture was primarily intended as a means to reduce fibrosis and promote tissue ingrowth throughout the ICC construct, the observed increase in baseline lifetime and lower modulation is likely due to the tissue returning to low-oxygen homeostasis after the initial stimulation of acute inflammation that occurs after injection. Over time, the scaffold baseline lifetime becomes more consistent; however, the sensitivity decreases as the tissue heals throughout and around the implant (Table S1, Table S2). Around one month of implantation, near the tail-end of the acute immune response phase, both pHEMA and IPN implants showed the lowest sensitivity. Between two and three months of implantation, after the acute wound healing process, the implants regained some sensitivity, indicating potential of these sensors to function long term (i.e., several months) as it is known that the first month of the wound healing response is accompanied with the most volatile tissue environmental changes from a wound healing perspective. [31, 34, 38, 70, 73, 78] While there is a reduction of sensor sensitivity based on change in signal level, the sensor response is remarkably consistent (no significant difference in percent signal change – baseline lifetime to peak lifetime – among timepoints, p < 0.05) from day 20 to 76 for both ICC pHEMA and IPN implants (Figure 5, C-D, Table S1, Table S2).
2.2.3. Morphology, Tissue Integration, and Foreign Body Response
SEM imaging of the IPN hydrogels confirm the highly-porous, interconnected structure. Figure 6A suggests an average pore size of ca. 50 μm after lyophilization; the slight decrease in pore size is attributed to preparation steps of the hydrogels for imaging. The interconnected structure is typical of ICC templating methods. This was confirmed by imaging as indicated in Figure 6B, where a second layer of pores can be seen within the first. [52]
Tissue integration through the oxygen sensor was hypothesized to enhance sensor performance by ameliorating the extent of fibrotic encapsulation caused by the foreign body response, as seen in previous ICC materials. [20, 51, 52, 57, 69] Figure 6C shows tissue ingrowth throughout the pores of the ICC implant, as indicated by the red color (Haemotoxylin and Eosin stain) interspersed throughout the implant image. Additionally, Figure 6D shows loosely-packed collagen indicated by the light teal/blue fibrous structured regions (Mason’s Trichrome stain) surrounding the ICC implant, whereas a denser, thicker, and more tightly-packed fibrotic encapsulation has been reported in previous work. [78-80] A side-by-side comparison of the foreign body response surrounding the IPN ICC sensors and IPN monolith sensors is shown in supplemental information Figure S4.
It is important to note that while the ICCs promote tissue ingrowth and the fibrotic capsule around the ICC sensors is loosely woven, negligible vascularization was observed throughout the ICC implant. This was contrary to our hypothesis that tissue ingrowth will carry with it benefits of neovascularization within the implant since other studies have shown angiogenesis throughout ICC implants.[69, 81-83] The scope of this study was to show initial preclinical proof of concept of these IPN ICC sensors as biomedical implants. Therefore, the bead template diameter was kept constant at 80 μm to directly compare to the pHEMA ICC implants which were being used as part of a larger study in the same animal. Investigation of alternate pore sizes in future studies will help refine the design of these IPN ICC structures to minimize collagen deposition within the pores and promote angiogenesis throughout the implant. Prior work from the Ratner group shows smaller pore sizes (i.e. ~35-40 μm diameter) may result in slightly different distribution of collagen and other biological components interspersed throughout the ICCs compared to what was seen here. [51, 52, 57, 58] On the contrary, other studies have shown larger pore sizes are able to promote vascular ingrowth.[81-83] Therefore, further investigation of ICC pore size for this application is needed.
While oxygen has been identified as one of the most important analytes in biological sensing, optimizing oxygen sensitivity in these implants also paves the way for detecting other analytes with refined sensitivity, such as glucose and lactate.[71] Ongoing efforts include: 1) further investigating the biocompatibility of these IPNs in additional porcine models (i.e. various pore sizes and solid vs. porous morphologies) and 2) functionalizing these IPNs with optical assays to transduce concentrations of other biomolecules with relevance to continuous or on-demand monitoring (e.g. glucose, lactate, ethanol, etc.).
3. Conclusion
We have demonstrated the ability to fabricate inorganic-organic interpenetrating network silicone hydrogels and effectively deploy them as long-term, fully-implantable optical biosensors, responding for 72 days in vivo, by capitalizing on the synergy of both networks and altering gel physicochemical properties independently of one another. To our knowledge, this is the first account of successfully fabricating sequentially-polymerized IPN gels with an inverted colloidal crystal microarchitecture. Maintaining an interconnected pore architecture with this sequentially-polymerized hydrogel matrix can enable the design of tissue-integrating implants with similar mechanical properties to pHEMA ICC implants while optimizing oxygen transport properties. This is promising for applications including, but not limited to, biosensing and tissue engineering.
4. Materials and Methods
4.1. Hydrogel Fabrication
4.1.1. Reagents and Instrumentation
For photocuring, the UV lamp (Blak Ray-B-100 SP) was purchased from UVP-LLC (Upland, CA). DMA, Nile Red, Irgacure 651, and PBS were purchased from Sigma Aldrich (St. Louis, MO). TRIS was purchased from Silar Laboratories (Wilmington, NC). Tetraethylene glycol dimethacrylate (TEGDMA) was purchased from Polysciences (Warrington, PA). The tetramethacrylated benzoporphyrin dye (PdBP) and poly(methyl methacrylate) (PMMA) beadcakes were provided by Profusa, Inc. Prior to use, DMA, TRIS, and TEGDMA were separately passed through individual inhibitor removal columns (Sigma) to remove the MEHQ inhibitor. All other reagents were used as received unless otherwise stated.
4.1.2. Monolith Synthesis
IPN hydrogels were synthesized using a sequential polymerization process illustrated in Figure 1, Section 1. In a typical synthesis, appropriate amounts of TRIS and DMA were added to Irgacure 651 (1:99 w/v% initiator:monomer) and vortexed to dissolve the photoinitiator. Next, TEGDMA was added as a crosslinker (2:98 v/v% crosslinker:monomer). The oxygen-sensitive phosphor, a tetramethacrylated benzoporphyrin dye (PdBP) was dissolved in dichloromethane (DCM) at a concentration of 10 mM; 25 μL of dye solution was added to the precursor solution and vortexed to mix. Finally, appropriate amounts of DCM and isopropanol (IPA) were added; necessary amounts of each solvent were determined based on the ratio of TRIS:DMA used. Too high of an IPA content renders the gel “tacky” and unmanageable, whereas too much DCM renders the gel too brittle and unable to be removed from the glass slide mold. A summary of TRIS:DMA ratios and corresponding cosolvent ratios are summarized in Table 2. Finally, the precursor solutions were purged with nitrogen gas for three minutes, transferred via pipette to a glass slide mold consisting of a Teflon spacer (760 μm thick) secured with clips, and photopolymerized for 10 minutes (five minutes per side).
Table 2.
Hydrogel formulations
| First Network | Second Network | Morphology | Sensing Chemistry | Co-Solvents | Monomer:Solvent Ratios (v:v) |
|---|---|---|---|---|---|
| 50:50 TRIS:DMA | pDMA homopolymer | IPN | 10 mM PdBP in DCM | 75 μL IPA; 50 μL DCM | 1:1.04 |
| 60:40 TRIS:DMA | pDMA homopolymer | IPN | 10 mM PdBP in DCM | 62.5 μL IPA; 62.5 μL DCM | 1:1.04 |
| 70:30 TRIS:DMA | pDMA homopolymer | IPN | 10 mM PdBP in DCM | 50 μL IPA; 75 μL DCM | 1:1.04 |
| 80:20 TRIS:DMA | pDMA homopolymer | IPN | 10 mM PdBP in DCM | 37.5 μL IPA; 87.5 μL DCM | 1:1.04 |
| pHEMA homopolymer | N/A | Single network | 10 mM PdBP in DMSO | 90 μL ethylene glycol; 100 μL DI water | 1:1 |
Following photocuring, the gels were removed from the mold and placed in a second precursor solution (3 mL total volume; 50:50 v/v% DMA:IPA; 98:2 v/v% DMA:TEGDMA; 1:99 w/v% Irgacure 651:DMA) and allowed to soak on a tabletop shaker overnight. The next day, the soaked gel permeated with the second DMA precursor was blotted on a KimWipe to remove excess surface DMA precursor, then photocured between two glass slides to form the second network. The resulting IPN gel was placed in phosphate buffer saline (100 mM, pH 7.4) overnight to hydrate.
4.1.3. Inverted Colloidal Crystals (ICC)
To fabricate IPNs as ICC scaffolds, a PMMA colloidal crystal “beadcake” was used as a sacrificial template similar to methods described in the literature. [20, 57, 58, 84] During fabrication of the first network, the precursor was injected directly over the beadcake and placed under the house vacuum for ~20 seconds to facilitate wicking of the precursor throughout the beadcake and eliminate any entrapped air pockets. The first network was photocured in the same manner as the monolith gels. After photocuring, the beadcake/silicone gel composite was removed from the glass slide mold by gently scraping with a razor blade and placed in the second network precursor to soak overnight, then photocured in the same manner as the monolith gels. Due to the formation of a gel “skin” layer between the interface of the beadcake and the glass slide on both sides of the gel composite, the IPN-beadcake composite was gently scraped on both surfaces with a razor blade to remove the skin layer. The “de-skinned” IPN/beadcake composite was placed in 50 mL of DCM in an Erlenmeyer flask with a rubber stopper overnight to dissolve the PMMA beads. The next day, the ICC scaffolds were removed from the DCM flask and placed in a fume hood to allow the DCM to evaporate under air. Air-dried scaffolds were placed in a vacuum chamber under house vacuum overnight to remove any trace levels of solvents after air drying before sending for ethylene oxide sterilization.
4.2. In Vitro Performance
4.2.1. Swelling Ratios
Five discs from each monolith, both with dye and one without dye (blank), were punched with a disposable biopsy punch (5 mm diameter, VWR). Each disc was blotted on a Kimwipe to remove excess water and weighed on an analytical balance (Mettler Toledo, Model XS64) to obtain the hydrated mass. The gel discs were dried under house vacuum overnight (> 24 hours) and weighed again to obtain the dried weight (Wd). The EWC was calculated from the percent difference in the two weights, according to Equation 1.
4.2.2. Optical Clarity
Five-millimeter “blank” discs of each formulation were placed in a 96-well flat bottom polystyrene well plate, and 200 μL phosphate buffer saline (Sigma, 10 mM, pH 7.4) was dispensed over each gel. An absorbance scan covering 400 to 850nm was conducted in a plate reader (Tecan Model M200) and transmittance was calculated.
4.2.3. Nile Red Staining and Confocal Laser Scanning Microscopy
Staining of the gels with Nile Red was adapted from Bailey, et al. [64] First, a 20 mg/mL solution of Nile Red (Sigma) in methanol was dissolved in a dram vial by vortexing for one minute. A 75 μL aliquot of this dye solution was added to 8 mL of DI water in a 20 mL scintillation vial and vortexed for one minute. Next, the 8 mL solution was diluted by adding it to 120 mL PBS in a beaker under constant stirring. Gel discs (5 mm diameter) were punched from the IPN gels with a biopsy punch. Each disc was placed in a separate centrifuge tube filled with 50 mL of the diluted Nile Red solution in PBS and placed on a nutating mixer at room temperature for 24 hours. After 24 hours, the Nile Red solution in PBS was discarded, and the stained gel discs were placed in fresh centrifuge tubes with 50 mL fresh PBS, allowing to nutate at room temperature for 24 more hours. The washing process was repeated three times, and the discs were stored in PBS until imaging. Confocal Laser Scanning Microscopy (CLSM) images were taken on an Olympus FV1000 confocal microscope. Excitation of the Nile Red stained gels was accomplished with a HeNe laser (543 nm); emission was collected from 650-700 nm. Optical sections were obtained at a depth of 100 μm into the sample.
4.2.4. DSC
Differential scanning calorimetry (TA Instruments, Q200) was performed on blank hydrogel monolith strips of each formulation. The blank strips were dried in a 2.5 mL microcentrifuge tube under house vacuum overnight (> 24 hours). Five to 10 mg of each dried gel strip were weighed into an aluminum TZero pan, then sealed hermetically. Thermal scans were performed from 20 – 200 °C at a ramp rate of 10 °C/min. Two heat/cool cycles were run for each sample; the first cycle served to erase any previous thermal history in the polymer while the second cycle was used to obtain the glass transition temperature (Tg). Tg values were calculated in the TA Universal Analysis software using the glass transition function. Data reported are the calculated midpoint values for each run (n = 3).
4.2.5. Rheology
Dynamic mechanical analysis to determine the viscoelastic response was performed on 3 separate hydrogel discs, 10 mm in diameter, of each formulation using a rheometer (Anton Paar, MCR 301). Excess moisture was carefully removed by blotting the sample with a Kim Wipe before analysis. A stainless steel, parallel plate geometry, 10 mm in diameter, was used for frequency tests over the range of 1-100 Hz at constant strain amplitude of 1 Pa. The temperature, 25 °C and then 37 °C, was controlled by a circulating water bath and built in Peltier heater. The elastic and viscous behaviors were compared by evaluating the loss tangent, defined in Equation 2 below.
Equation 2:
Definition of the loss tangent, a measurement of viscoelastic damping behavior
| (2) |
4.2.6. SEM
ICC gels were flash frozen in liquid nitrogen then lyophilized (Labconco, Model 7811020) overnight to preserve scaffold morphology. After lyophilizing, the gels were mounted on an aluminum SEM stub using double-sided carbon tape and sputter-coated (Ted Pella, Model Cressington 108) for one minute under vacuum to generate a ca. 2 nm layer of gold. ICC gels were imaged at 10 kV acceleration voltage under low vacuum (JEOL USA, Inc., JCM-5000 Neoscape).
4.2.7. In Vitro Oxygen Modulation (Stern-Volmer)
The Stern-Volmer oxygen diffusion experiments were carried out by immobilizing dye-loaded monolith gel strips in an acrylic sample chamber sealed with a circular gasket and secured with screws. Samples were immobilized in wells with O-rings flush with the side of the well. Oxygen modulation experiments were carried out in an incubator using a recirculating flow system similar to previously described. [36] Oxygen levels were controlled using a mass flow controller (MKS) with two gas flow line inputs: nitrogen and house air. Dissolved oxygen levels were verified using a picoammeter (Unisense, Denmark). Gas was bubbled into a 2L Erlenmeyer flask filled with 1L PBS and sealed with a rubber stopper with three openings: two for liquid influent and effluent and the other for the ceramic gas bubbling tube (VWR).
Luminescence lifetimes of the benzoporphyrin dye in these sensors depend on the local oxygen concentration around the dye, governed by oxygen flux into the sensor. The Stern-Volmer relationship illustrates the behavior of collisional quenching of dye phosphorescence by oxygen which can be related to the diffusion kinetics underlying the glucose response according to the Stern-Volmer relationship (Equation 3), where τ0 and I0 are the luminescence lifetime and intensity in the absence of oxygen, respectively, τ and I are the luminescence lifetime and intensity at a particular oxygen concentration, [O2], and KSV is the Stern-Volmer constant representing the linear relationship between the ratio τ0/τ and [O2].
To calculate the Stern-Volmer constant for each formulation, the luminescence lifetime ratios were calculated and plotted against oxygen concentrations. Data were fit with a linear curve (R2 > 0.99). The Stern-Volmer quenching constant for each sample was calculated according to Equation 3 to serve as a measurement of comparative oxygen permeability.
4.3. In Vivo Performance
4.3.1. Implantation and Oxygen Modulation
In vivo inspired oxygen modulation procedures were approved by the Texas A&M IACUC (AUP #2013-0108). A female Sinclair mini-pig was obtained from Sinclair Bio-Resources (Columbia, MO), anesthetized with an injection of Telazol® (5 mg/kg) and buprenorphine (0.01 mg/kg), sedated, and intubated. Dorsal hair was shaved and skin was cleaned and prepared for injections by brushing with dye-free ChloraPrep applicators (CareFusion, San Diego, CA). Anesthesia was maintained with 1.0-2.5% isoflurane (v/v in O2) to effect during sensor injections and oxygen modulation. Sensors were injected subcutaneously into the dorsum of the pig (42.0 kg, one year old) from an 18-gauge cannula with a reverse action plunger. The sensors were targeted for insertion at ~5 mm deep; average and standard deviation of implant depths were determined to be 5.4 ± 1.9 mm from post-experiment histological evaluation of n = 60 implants (some implants were used only as controls and for histology for separate studies, so they are not detailed here).
A custom measurement system designed in collaboration with Profusa, Inc. and described in Unruh, et al. [38, 39] was used to acquire phosphorescence lifetime data. Briefly, a red LED (4mW, 630nm, Phillips) is used to illuminate samples using a square waveform (500 μsec ON, 2500 μsec OFF) and a silicon PMT (SensL) is used to measure of emitted photons passing through a long-pass filter. Lifetime is calculated by fitting the emission decay measured after LED is turned off to an exponential model equation. In each case, a reader was placed and adhered on the skin over the injected IPN or pHEMA oxygen biosensor and allowed to collect baseline signal while the anesthesia was delivered with 100% O2 (fraction of inspired oxygen, fiO2 = 1.00). Systemic oxygen challenges were then achieved through reduction of fiO2. to approximately 0.15 (v/v balance N2) for up to 15 min. After the hypoxic period, the fiO2 was returned to 1.00.
4.3.2. Histology
Following humane euthanasia of the animal, the dorsal pelt was removed via scalpel and sensors were located by transdermal interrogation. [38] Once located in the pelt, tissue plugs containing the sensors were isolated with 8 mm biopsy punches. The tissue plugs were immediately placed in formaldehyde and sent to CVPath (Gaithersburg, MD) for histological processing. The histology slides were imaged as received using a slide scanner with a 20X objective (Mikroscan, Model S2). Images were digitally processed using standard imaging software (Aperio ImageScope, version 11.1.2.760).
Supplementary Material
Acknowledgements
The authors would like to thank several people and facilities that contributed to this work. C.W. Peak of the Inspired Nanomaterials and Tissue Engineering (iNanoTE) Laboratory at Texas A&M University is acknowledged for assistance with the rheometry measurements. Michael Frassica of the Grunlan Research Group in the Department of Biomedical Engineering at Texas A&M University is acknowledged for his guidance in the Nile Red staining of the hydrogels. Dr. Stanislav Vitha of the Microscopy and Imaging Center of Texas A&M University is acknowledged for assistance with the Confocal Laser Scanning Microscopy images. The use of the Microscopy and Imaging Center facility at Texas A&M University is acknowledged. The Olympus FV1000 confocal microscope acquisition was supported by the Office of the Vice President for Research at Texas A&M University.
This work was supported by NIH project number 1R01EB016414.
Contributor Information
Rachel M. Unruh, 5045 Emerging Technologies Building, 3120 TAMU, College Station, TX 77843, USA
Lindsey R. Bornhoeft, 5045 Emerging Technologies Building, 3120 TAMU, College Station, TX 77843, USA
Scott P. Nichols, Profusa, Inc., 5959 Horton St #450, Emeryville, CA 94608
Natalie A. Wisniewski, Medical Device Consultancy, 1219 18th Street, San Francisco, CA 94107
Michael J. McShane, 5045 Emerging Technologies Building, 3120 TAMU, College Station, TX 77843, USA
References
- [1].Nichols SP, Koh A, Storm WL, Shin JH, Schoenfisch MH, Chemical Reviews 2013, 113, 2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Nicolson PC, Vogt J, Biomaterials 2001, 22, 3273. [DOI] [PubMed] [Google Scholar]
- [3].Refojo MF, Eye & Contact Lens 1996, 22, 38. [Google Scholar]
- [4].Gaharwar AK, Peppas NA, Khademhosseini A, Biotechnology and bioengineering 2014, 111, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ratner BD, Hoffman AS, Schoen FJ, Lemons JE, Biomaterials science: an introduction to materials in medicine, Academic press, 2004. [Google Scholar]
- [6].Ratner BD, Hoffman AS, Schoen FJ, Lemons JE, Biomaterials science: an introduction to materials in medicine 2004, 1. [Google Scholar]
- [7].Wichterle O, Lim D, Nature 1960, 185, 117. [Google Scholar]
- [8].Abbasi F, Mirzadeh H, Katbab A-A, Polymer International 2001, 50, 1279. [Google Scholar]
- [9].Chekina NA, Pavlyuchenko VN, Danilichev VF, Ushakov NA, Novikov SA, Ivanchev SS, Polymers for Advanced Technologies 2006, 17, 872. [Google Scholar]
- [10].Iyer AV, Sahin A, Investigative Ophthalmology & Visual Science 2015, 56, 6096. [Google Scholar]
- [11].Lai Y-C, Journal of Applied Polymer Science 1995, 56, 317. [Google Scholar]
- [12].Lasowski F, Conant G, Sheardown H, Investigative Ophthalmology & Visual Science 2014, 55, 6044. [Google Scholar]
- [13].Lasowski F, Sheardown H, Investigative Ophthalmology & Visual Science 2015, 56, 6072. [Google Scholar]
- [14].Madkour TM, Mohamed SK, Barakat AM, Polymer 2002, 43, 533. [Google Scholar]
- [15].Van Beek M, Jones L, Sheardown H, Biomaterials 2008, 29, 780. [DOI] [PubMed] [Google Scholar]
- [16].Wang J, Li X, Journal of Applied Polymer Science 2010, 116, 2749. [Google Scholar]
- [17].Weeks A, Morrison D, Alauzun JG, Brook MA, Jones L, Sheardown H, Journal of Biomedical Materials Research Part A 2012, 100A, 1972. [DOI] [PubMed] [Google Scholar]
- [18].Zhao Z, Xie H, An S, Jiang Y, The Journal of Physical Chemistry B 2014, 118, 14640. [DOI] [PubMed] [Google Scholar]
- [19].Xu J, Yang P, Zhang L, Huo G, Journal of Applied Polymer Science 2015, 132. [Google Scholar]
- [20].Teng W, Long TJ, Zhang Q, Yao K, Shen TT, Ratner BD, Biomaterials 2014, 35, 8916. [DOI] [PubMed] [Google Scholar]
- [21].Guidi G, Korogiannaki M, Sheardown H, Eye & Contact Lens 2014, 40, 269. [DOI] [PubMed] [Google Scholar]
- [22].Lasowski F, Sheardown H, Optometry and Vision Science 2016, 93, 404. [DOI] [PubMed] [Google Scholar]
- [23].Tran N-P-D, Yang M-C, Journal of Polymer Research 2019, 26, 143. [Google Scholar]
- [24].Seitz ME, Wiseman ME, Hilker I, Loos J, Tian M, Li J, Goswami M, Litvinov VM, Curtin S, Bulters M, Polymer 2017, 118, 150. [Google Scholar]
- [25].Jacob S, Nair AB, Shah J, Sreeharsha N, Gupta S, Shinu P, Pharmaceutics 2021, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ibrahim MM, Patel PS, Wu ZJ, Chien JS, Wisniewski NA, Mohammed MM, Klitzman B, Surgery 2020, 168, 926. [DOI] [PubMed] [Google Scholar]
- [27].Cunningham DD, Stenken JA, In vivo glucose sensing, Vol. 174, John Wiley & Sons, 2009. [Google Scholar]
- [28].Chien JS, Mohammed M, Eldik H, Ibrahim MM, Martinez J, Nichols SP, Wisniewski N, Klitzman B, Scientific Reports 2017, 7, 8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kanick SC, Schneider PA, Klitzman B, Wisniewski NA, Rebrin K, Microvascular Research 2019, 124, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Montero-Baker MF, Au-Yeung KY, Wisniewski NA, Gamsey S, Morelli-Alvarez L, Mills JL, Campos M, Helton KL, Journal of Vascular Surgery 2015, 61, 1501. [DOI] [PubMed] [Google Scholar]
- [31].Nichols SP, Balaconis MK, Gant RM, Au-Yeung KY, Wisniewski NA, in Oxygen Transport to Tissue XL, (Eds: Thews O, LaManna JC, Harrison DK), Springer International Publishing, Cham: 2018, 351. [Google Scholar]
- [32].Nichols SP, Balaconis MK, Gant RM, Au-Yeung KY, Wisniewski NA, Adv Exp Med Biol 2018, 1072, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rivera KR, Pozdin V, Dieffenderfer J, Wisniewski NA, Bozkurt AY, Daniele MA, "Microsystems for Measuring Oxygen Using Tissue Integrated Phosphorescence-Based Soft Sensors", presented at Optical Sensors and Sensing Congress (ES, FTS, HISE, Sensors), San Jose, California, 2019/June/25, 2019. [Google Scholar]
- [34].Wisniewski NA, Nichols SP, Gamsey SJ, Pullins S, Au-Yeung KY, Klitzman B, Helton KL, in Oxygen Transport to Tissue XXXIX, (Eds: Halpern HJ, LaManna JC, Harrison DK, Epel B), Springer International Publishing, Cham: 2017, 377. [Google Scholar]
- [35].Wisniewski NA, Nichols SP, Gamsey SJ, Pullins S, Au-Yeung KY, Klitzman B, Helton KL, Adv Exp Med Biol 2017, 977, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Andrus L, Unruh R, Wisniewski N, McShane M, Biosensors 2015, 5, 398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Roberts JR, Park J, Helton K, Wisniewski N, McShane MJ, Journal of Diabetes Science and Technology 2012, 6, 1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Unruh RM, Roberts JR, Nichols SP, Gamsey S, Wisniewski NA, McShane MJ, Journal of Diabetes Science and Technology 2015, 9, 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Unruh RM, Dissertation, Texas A&M University; 2017. [Google Scholar]
- [40].Wilson GS, Gifford R, Biosensors and Bioelectronics 2005, 20, 2388. [DOI] [PubMed] [Google Scholar]
- [41].Munoz-Pinto DJ, Jimenez-Vergara AC, Hou Y, Hayenga HN, Rivas A, Grunlan M, Hahn MS, Tissue Eng Part A 2012, 18, 1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Frassica MT, Jones SK, Diaz-Rodriguez P, Hahn MS, Grunlan MA, Acta biomaterialia 2019, 99, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Munoz-Pinto DJ, McMahon RE, Kanzelberger MA, Jimenez-Vergara AC, Grunlan MA, Hahn MS, Journal of biomedical materials research. Part A 2010, 94, 112. [DOI] [PubMed] [Google Scholar]
- [44].Unruh RM, Weaver JL, McShane MJ, "Hydrogel matrix effects on oxygen diffusion: Controlling properties for biosensor applications", presented at 2015 IEEE SENSORS, 1-4 Nov. 2015, 2015. [Google Scholar]
- [45].Matsuda T, Nakajima T, Fukuda Y, Hong W, Sakai T, Kurokawa T, Chung U.-i., Gong JP, Macromolecules 2016, 49, 1865. [Google Scholar]
- [46].Iyer AV, Investigative ophthalmology & visual science 2015, 56, 6096. [Google Scholar]
- [47].Shi Y, Ma C, Peng L, Yu G, Advanced Functional Materials 2015, 25, 1219. [Google Scholar]
- [48].Dragan ES, Chemical Engineering Journal 2014, 243, 572. [Google Scholar]
- [49].Liu Y, Wang S, Lee JW, Kotov NA, Chemistry of Materials 2005, 17, 4918. [Google Scholar]
- [50].Zhang Y, Wang S, Eghtedari M, Motamedi M, Kotov NA, Advanced Functional Materials 2005, 15, 725. [Google Scholar]
- [51].Long TJ, Takeno M, Sprenger CC, Plymate SR, Ratner BD, Tissue Eng Part C Methods 2013, 19, 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Madden LR, Mortisen DJ, Sussman EM, Dupras SK, Fugate JA, Cuy JL, Hauch KD, Laflamme MA, Murry CE, Ratner BD, Proceedings of the National Academy of Sciences 2010, 107, 15211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lee K, Silva EA, Mooney DJ, Journal of The Royal Society Interface 2011, 8, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Marshall AJ, Ratner BD, AIChE Journal 2005, 51, 1221. [Google Scholar]
- [55].Long TJ, Sprenger CC, Plymate SR, Ratner BD, Biomaterials 2014, 35, 8164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Long TJ, Takeno M, Sprenger CC, Plymate SR, Ratner BD, Tissue Engineering Part C: Methods 2013, 19, 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ratner BD, Regenerative Biomaterials 2016, 3, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Somo SI, Akar B, Bayrak ES, Larson JC, Appel AA, Mehdizadeh H, Cinar A, Brey EM, Tissue Engineering Part C: Methods 2015, 21, 773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Corcione CE, Frigione M, Thermochimica acta 2012, 534, 21. [Google Scholar]
- [60].Lai YC, Journal of Applied Polymer Science 1995, 56, 317. [Google Scholar]
- [61].Lasowski F, Sheardown H, Optometry & Vision Science 2016, 93, 404. [DOI] [PubMed] [Google Scholar]
- [62].Loshaek S, Fox TG, Journal of the American Chemical Society 1953, 75, 3544. [Google Scholar]
- [63].Esposito Corcione C, Frigione M, Thermochimica Acta 2012, 534, 21. [Google Scholar]
- [64].Bailey BM, Fei R, Munoz-Pinto D, Hahn MS, Grunlan MA, Acta Biomaterialia 2012, 8, 4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Bailey BM, Nail LN, Grunlan MA, Acta Biomaterialia 2013, 9, 8254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Vincent JF, Structural biomaterials, Princeton University Press, 2012. [Google Scholar]
- [67].Anseth KS, Bowman CN, Brannon-Peppas L, Biomaterials 1996, 17, 1647. [DOI] [PubMed] [Google Scholar]
- [68].Haraguchi K, Farnworth R, Ohbayashi A, Takehisa T, Macromolecules 2003, 36, 5732. [Google Scholar]
- [69].Joao CFC, Vasconcelos JM, Silva JC, Borges JP, Tissue Engineering Part B: Reviews 2014, 20, 437. [DOI] [PubMed] [Google Scholar]
- [70].Guo S. a., DiPietro LA, Journal of dental research 2010, 89, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Papkovsky DB, Dmitriev RI, Chemical Society Reviews 2013, 42, 8700. [DOI] [PubMed] [Google Scholar]
- [72].Papkovsky DB, Ponomarev GV, Trettnak W, O'Leary P, Analytical Chemistry 1995, 67, 4112. [Google Scholar]
- [73].Schreml S, Szeimies R, Prantl L, Karrer S, Landthaler M, Babilas P, British Journal of Dermatology 2010, 163, 257. [DOI] [PubMed] [Google Scholar]
- [74].Guo S, DiPietro LA, Journal of Dental Research 2010, 89, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Schreml S, Szeimies RM, Prantl L, Karrer S, Landthaler M, Babilas P, British Journal of Dermatology 2010, 163, 257. [DOI] [PubMed] [Google Scholar]
- [76].Anderson JM, Rodriguez A, Chang DT, Seminars in immunology 2008, 20, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sen CK, Wound repair and regeneration : official publication of the Wound Healing Society [and] the European Tissue Repair Society 2009, 17, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Wisniewski N, Reichert M, Colloids and Surfaces B: Biointerfaces 2000, 18, 197. [DOI] [PubMed] [Google Scholar]
- [79].Wisniewski N, Klitzman B, Miller B, Reichert WM, Journal of Biomedical Materials Research 2001, 57, 513. [DOI] [PubMed] [Google Scholar]
- [80].Wisniewski N, Moussy F, Reichert W, Fresenius' journal of analytical chemistry 2000, 366, 611. [DOI] [PubMed] [Google Scholar]
- [81].Cai X, Zhang Y, Li L, Choi S-W, MacEwan MR, Yao J, Kim C, Xia Y, Wang LV, Tissue Eng Part C Methods 2013, 19, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Choi SW, Zhang Y, Macewan MR, Xia Y, Advanced healthcare materials 2013, 2, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].MARSHALL AJ, Polymer Preprints 2004, 45, 100. [Google Scholar]
- [84].Zhang Y, Wang S, Eghtedari M, Motamedi M, Kotov NA, Advanced Functional Materials 2005, 15, 725. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.