

Abstract
Background and Aims:
Hispanics are disproportionately affected by non-alcoholic fatty liver disease, liver fibrosis, cirrhosis, and hepatocellular carcinoma. Preventive strategies and non-invasive means to identify those in this population at high risk for liver fibrosis, are urgently needed. We aimed to characterize the gut microbiome signatures and related biological functions associated with liver fibrosis in Hispanics and identify environmental and genetic factors affecting them.
Approach and Results:
Subjects of the population-based Cameron County Hispanic Cohort (n=217) were screened by vibration-controlled transient elastography (FibroScan). Among them, 144 (66.7%) had steatosis and 28 (13.0%) had liver fibrosis. The gut microbiome of subjects with liver fibrosis was enriched with immunogenic commensals (e.g. Prevotella copri, Holdemanella, Clostridiaceae 1) and depleted of Bacteroides caccae, Parabacteroides distasonis, Enterobacter and Marinifilaceae. The liver fibrosis-associated metagenome was characterized by changes in the urea cycle, L-citrulline biosynthesis and creatinine degradation pathways, and altered synthesis of B vitamins and lipoic acid. These metagenomic changes strongly correlated with the depletion of Parabacteroides distasonis and enrichment of Prevotella and Holdemanella. Liver fibrosis was also associated with depletion of bacterial pathways related to L-fucose biosynthesis. Alcohol consumption, even moderate, was associated with high Prevotella abundance. The single nucleotide polymorphisms rs3769502 and rs7573751 in the NCK2 gene positively associated with high Prevotella abundance.
Conclusion:
Hispanics with liver fibrosis display microbiome profiles and associated functional changes that may promote oxidative stress and a pro-inflammatory environment. These microbiome signatures, together with NCK2 polymorphisms, may have utility in risk modeling and disease prevention in this high-risk population.
Keywords: Metagenomics, health disparity, population study, non-alcoholic fatty liver disease, liver steatosis
Graphical Abstract

Introduction
Non-alcoholic fatty liver disease (NAFLD) affects 25% of the adult population worldwide(1) and encompasses a spectrum of disease ranging from simple steatosis to non-alcohohlic steatohepatitis (NASH). Subjects with NASH often develop liver fibrosis. Advanced liver fibrosis is the main determinant of mortality in NASH patients and a major risk factor for hepatocellular carcinoma (HCC).(1) The incidence of NASH, liver fibrosis and HCC are rising in the United States.(2, 3) The incidence of HCC in Hispanics is twice the incidence in non-Hispanic Caucasians(3) and the survival outcome is worse.(4) Hispanics in South Texas have an even higher incidence of HCC than Hispanics elsewhere in the United States,(5, 6) with a disproportionately large fraction attributed to NASH.(7)
While anti-virals have been successfully implemented for the prevention of HCC associated with viral hepatitis, no therapies for NASH have been approved yet.(8) Innovative preventive strategies and non-invasive methods to identify those with NASH at risk of fast fibrosis progression are urgently needed. The use of gut microbiome profiles to predict clinical outcome has emerged as a potential complementary strategy to the use of host factors.(9) It is well recognized that the gut microbiome contributes to the development of NASH,(10, 11) progression to advanced liver fibrosis,(12–17) and even HCC.(18–20) Most studies included heterogeneous groups of patients and studies in high-risk populations are lacking.
In this study, we aimed to identify alterations in gut microbiome associated with liver fibrosis in the high-risk population of Hispanics in South Texas. We also aimed to identify functional changes and contributing factors of these microbiome alterations. To that end, we enrolled subjects from the Cameron County Hispanic Cohort (CCHC), a large population-based Hispanic cohort in South Texas with high prevalence of obesity (51%), diabetes (28%), chronic liver injury (39%), liver steatosis (52%) and liver fibrosis (14%).(21–25)
Methods
Study participants
The study includes 217 participants from the CCHC.(25) We excluded subjects positive for hepatitis B or C virus or who had antibiotic, probiotic or proton pump inhibitor use within 30 days of stool collection. Written informed consent was obtained from each participant and the study protocol was approved by the Committees for the Protection of Human Subjects, at participating institutions. Fasting blood samples were collected and analyzed for metabolic and lipid panels. The following criteria were used as categorical or diagnostic definitions: obesity (BMI ≥30), pre-diabetes (no history of diabetic medication, plus fasting blood glucose of 100–125mg/dl or HbA1c of 5.7–6.4%), diabetes (fasting blood glucose ≥126mg/dl, HbA1c ≥6.5% or history of diabetic medication), elevated aspartate aminotransferase (AST) (>33U/L), elevated alanine aminotransferase (ALT) (>40U/L for men, >31U/L for women), heavy drinking (alcohol consumption of >20g/day for men, >10g/day for women), moderate drinking (non-zero weekly consumption that did not reach the criteria for heavy drinking), former smoking (lifetime consumption of ≥100 cigarettes plus no smoking at time of survey), current smoking (lifetime consumption of ≥100 cigarettes plus smoking at time of survey). Demographic and laboratory parameters of the study participants are described in Supporting Table S1. Dietary intake the day prior to interview was recorded using a diet questionnaire adapted from the School Physical Activity and Nutrition survey. Dietary groups were calculated from the sum of individual foods (Supporting Table S2).
Trained operators obtained controlled attenuation parameter (CAP) measurements (dB/m) for liver steatosis and liver stiffness measurements (LSM, kiloPascals, kPa) for liver fibrosis, using vibration-controlled transient elastography (FibroScan® 502 Touch or FibroScan® 530 Compact, Echosens). Presence of liver steatosis was defined as CAP ≥268 as described.(26) Significant liver fibrosis (F2-F4) was defined as LSM ≥7.1 kPa, while advanced fibrosis (F3-F4) and cirrhosis (F4) were defined as LSM ≥8.8 kPa and ≥10.4 kPa, respectively, as described.(27) LSM measurements were considered inconclusive if <10 valid measures or interquartile range-to-median ratio >0.3.
Stool DNA extraction, 16S rRNA amplicon sequencing and bioinformatic analysis
Stool samples were collected from all participants using the OMNIgene® GUT stool collection kit (DNA Genotek, Ontario, Canada). The median time interval between FibroScan and stool collection was 4.6 days. Samples were analyzed by 16S sequencing at the MD Anderson Cancer Center Microbiome Core Facility. DNA was extracted using the QIAamp Fast DNA stool mini Kit (Qiagen). The V4 region of the bacterial 16S rRNA gene was amplified by PCR (forward primer: 5’- AATGATACGGCGACCACCGAGATCTACACGCTXXXXXXXXXXXXTATGGTAATTGTGTGYCAGCMGCCGCGGTAA-3’, where XXXXXXXXXXXX is an index sequence for multiplexing libraries; reverse primer: 5’- CAAGCAGAAGACGGCATACGAGATAGTCAGCCAGCCGGACTACNVGGGTWTCTAAT-3’). Libraries were purified using Zymo I-96 columns and analyzed on the 4200 Tapestation system (Agilent). Barcoded amplicons were pooled in equal concentrations. Pooled libraries were quantified by Qubit fluorometer and the molarity was calculated based on amplicon size. Sequencing was performed using 250bp paired-end on the Illumina MiSeq platform (Read1 primer: 5’-TATGGTAATTGTGTGYCAGCMGCCGCGGTAA-3’; Read2 primer: 5’-AGTCAGCCAGCCGGACTACNVGGGTWTCTAAT-3’; and index primer: AATGATACGGCGACCACCGAGATCTACACGCT). Paired-end reads were de-multiplexed and split in QIIME. Merging of paired-end reads to create consensus sequences was done by VSEARCH v7, allowing up to a maximum of 10 mismatched. The cluster_otus command, an implementation of UPARSE algorism, was used to perform 97% related operational taxonomic units (OTU) clustering. Denoising was done by the unoise3 command. OTUs were subjected to taxonomy assignment using the Mothur with Silva database (v132). The mean of sequence reads was 90,264 (range 25,663–226,058).
Functional profiling of stool samples by shotgun metagenomic sequencing
Shotgun metagenomic sequencing was performed (CosmosID Inc., Rockville, Maryland) to a sequencing depth of 12 million reads (± 20%), on stool samples from 142 of the 217 participants, selected for having liver steatosis. DNA was isolated using the QIAGEN Dneasy PowerSoil Pro Kit (Qiagen) and quantified by Qubit (ThermoFisher). DNA libraries were prepared using the Illumina Nextera XT library preparation kit. Libraries were assessed with Qubit (ThermoFisher) and sequenced on an Illumina HiSeq platform using 150bp paired-end sequencing. The percentage of sequencing reads aligned to the human genome was determined to be minimal (mean of 0.04%) via Bowtie2 (v2.4.1),(28) using GRCh38 and major single nucleotide polymorphisms (SNPs) as the reference genome, and default Bowtie2 parameters. Initial quality control, adapter trimming and preprocessing of metagenomic sequencing reads were performed using Bbduk (https://jgi.doe.gov/data-and-tools/bbtools/). Quality-controlled reads were subjected to a translated search using Diamond against a comprehensive and non-redundant protein sequence database, UniRef 90. UniRef90 represents a clustering of all non-redundant protein sequences in UniProt, such that each sequence in a cluster aligns with 90% identity and 80% coverage of the longest sequence in the cluster. The mapping of metagenomic reads to gene sequences were weighted by mapping quality, coverage and gene sequence length to estimate community wide weighted gene family abundances as described.(29) Gene families were annotated to MetaCyc reactions (Metabolic Enzymes) to reconstruct and quantify MetaCyc metabolic pathways as described.(29) Abundance values were normalized using Total-sum scaling normalization to produce “Copies per million”.
Genome-wide association studies (GWAS) and PCR genotyping
GWAS was performed on 139 of the 217 participants with whole genome-imputed SNP genotypes. Genome-wide genotyping was performed using Illumina Multi-Ethnic Genotyping Array (MEGA) with 2.7 million SNPs, optimized for the Hispanic population. After stringent pre-imputation quality control measures including SNP/subject-wise genotyping missing rate, Hardy–Weinberg equilibrium, heterozygosity rate, sample duplication and sex inconsistency, we imputed the GWAS data to the TOPMed whole genome sequencing reference panel using the Michigan Imputation Server.(30) The R package GENESIS(31, 32) was used to perform GWAS of 9.3 million SNPs with minor allele frequency ≥3% and imputation score (R2)>0.4. The GENESIS analysis pipeline explicitly models population structure, relatedness between individuals and ancestry admixture. GWAS was performed with dichotomized Prevotella (sum of all genus subgroups) as the trait. Dichotomization of Prevotella was based on the observed bimodal peaks (Supporting Fig. S1A). Gene annotation was performed with SNPnexus v4.(33) The likelihood of having regulatory functions was predicted using RegulomeDB v2.0.(34) PhenoScanner v2(35) was used to access previous reports of associated gene expression, in expression quantitative trait loci (eQTL) studies (p<1×10−5).
SNPs rs3769502 and rs7573751 were also measured by PCR genotyping in the study participants for which genome-wide genotyping data were not available, using predesigned TaqMan™ SNP Human Genotyping Assays (ThermoFisher), the SsoAdvanced™ Universal Probes Supermix (BioRad) and the Applied Biosystems ViiA7 Real-Time PCR System (ThermoFisher). Results were analyzed using the QuantStudio™ Real-Time PCR Software v1.3 (ThermoFisher).
Statistical analyses
Alpha-diversity was estimated using QIIME, from a randomly rarefied dataset of 25,000 reads/sample with 10 iterations. Unless specified otherwise, the remaining statistical analyses were performed in R (version 4.0.0; R Foundation for Statistical Computing, Vienna, Austria). Principal coordinates analysis (PCoA) was performed using the “cmdscale” function and the weighted UniFrac distances of the OTU tables. Beta dispersion and permutational multivariate analysis of variance (PERMANOVA) tests, using weighted UniFrac distances, were performed with the Vegan package. Partitioning Around Medoids clustering based on weighed UniFrac distances was performed using the “pam” function in the cluster package. The “fviz_nbclust” function in the factoextra package suggested four clusters. Kruskal-Wallis test was performed to identify bacterial taxa with significantly different abundance across clusters. Redundancy analysis (RDA) was performed based on weighted UniFrac distances or bacterial abundance, using the “capscale” function in the Vegan package. Analysis of variance (ANOVA)-like permutation and marginal tests were performed using the “anova.cca” function in the Vegan package, to determine the significance of the model and of each explanatory variable. Fitting variables onto RDA plots were performed using the “envfit” function in the Vegan package, using linear constraints. Differences in bacterial abundance were assessed using the linear discriminant analysis (LDA) effect size (LefSe) tool,(36) with p<0.05 and log10 LDA score >2 considered significant. Taxa with ≥0.1% abundance in at least 25% of samples were included. Additional differential abundance analysis of taxa was performed with ANCOM v2.1(37), where an FDR significance threshold of 0.2 was used for calculation of W statistics. W statistics greater than or equal to the 60th percentile of the W distribution were considered significant. To determine associations between bacterial taxa and liver fibrosis, subjects were divided into low and high-abundance groups. Using the “glm.fit” function, logistic regression was performed for each threshold, to obtain odds ratios (ORs) adjusted for age, gender, BMI, diabetes and alcohol intake (g/day) (AOR) and 95% confidence intervals (CIs). Pairwise correlations were performed using Spearman’s correlation, or with Spearman’s partial correlation adjusting for LSM, using the “pcor.test” function in the ppcor package. Correlation p-values were adjusted for multiple testing by the Benjamini-Hochberg method. Differences in abundance of MetaCyc pathways and reactions were assessed by ANCOM v2.1. MetaCyc pathways and reactions with ≥0.01% abundance in at least 10% of samples were included. For performance analysis, logistic regression models were fit using the “glm” function: fitted probabilities were used for graphing receiver operating characteristic (ROC) curves and computing the area under the ROC curve (AUC) using the pROC and ROCR packages. For analysis of selected SNPs in the full dataset, we considered p<0.05 as significant for all statistical tests (Chi-squared test, Kruskal-Wallis and logistic regression).
Results
Microbiome profiles identify subject clusters that differ by liver fibrosis
We collected stool from 217 randomly selected subjects from the CCHC (Supporting Table S1). Among them, 118 (54.6%) were obese and 75 (34.7%) were diabetic. FibroScan screening showed that 144 (66.7%) had liver steatosis and 28 (13.0%) had significant liver fibrosis (kPa ≥7.1), including 14 (6.5%) with advanced fibrosis (kPa ≥8.8) or cirrhosis (kPa ≥10.4). Using 16S sequencing, we identified 6 phyla, 11 classes, 13 orders, 26 families, 68 genera and 100 species with ≥0.1% relative abundance in at least 25% of the samples. Firmicutes was the dominant phyla (mean abundance of 53.4%), followed by Bacteroidetes (35.6%), Proteobacteria (4.7%) and Actinobacteria (4.2%). Within the Firmicutes phyla, the most abundant families were Lachnospiraceae (23.6%) and Ruminococcaceae (20.3%). Within the Bacteroidetes phyla, the most abundant families were Bacteroidaceae (17.2%) and Prevotellaceae (14.9%) (Supporting Table S3).
Relationships between microbiome features and demographic or clinical parameters were revealed by Partitioning Around Medoids clustering, resulting in four groups. PCoA of the weighted UniFrac distances revealed that clusters A (34 subjects), C (83 subjects) and D (71 subjects) were three separate clusters, while cluster B (29 subjects) was an intermediate group between clusters A and C (Fig. 1A). Families and genera with significant differences in abundance across clusters are shown in Fig. 1B,C. Clusters did not differ by country of birth, age of arrival in Texas, number of years spent in Texas, age or BMI, but significant differences in other parameters were observed (Supporting Table S4). Cluster A had the highest percentage of males (52.9% vs 24.6%; OR=3.45 [95% CI=1.63–7.32], p<0.001), the highest percentage of drinkers (64.7% vs 27.3%; OR=4.88 [95% CI=2.25–10.58], p<0.001), and the highest waist-to-hip ratio (1.0 vs 0.9, p=0.031). Subjects in cluster A also displayed the highest mean FibroScan LSM (6.7kPa vs 5.1kPa, p=0.020) and were more likely to have significant liver fibrosis (23.5% vs 11.0%; OR=2.48 [95% CI=0.99–6.21], p=0.053). Cluster B had the lowest levels of HbA1c (5.9% vs 6.7%, p=0.039) and subjects with diabetes (17.2% vs 37.4%; OR=0.35 [95% CI=0.13–0.95], p=0.040). Interestingly, this cluster had the highest percentage of subjects that had lost over 10lbs in the last 6 months (20.7% vs 8.6%; OR=2.77 [95% CI=0.99=7.80], p=0.053). Cluster C had the highest levels of HbA1c (6.9% vs 6.4%, p=0.029) and fasting blood glucose (126.8md/dL vs 109.8md/dL, p=0.016). Lastly, cluster D had the lowest waist-to-hip ratios (0.9 vs 1.0, p=0.019). Overall, these results suggested that specific gut microbiome components were significantly associated with liver fibrosis.
Fig. 1.

Sample clustering and taxa contribution to differences between clusters. (A) PCoA plot based on weighted UniFrac distances. Samples were grouped by PAM clustering. Heatmap of (B) families and (C) genera (log10 abundance) with significantly different abundance across clusters, as determined by Kruskal-Wallis test. Taxa are grouped depending on which cluster has the highest median abundance. Within each group, taxa are ordered by descending mean abundance. Color key represents row Z-scores.
Microbiome signatures associated with liver fibrosis
Alpha diversity by observed OTUs or Shannon index, was not significantly different between healthy subjects and subjects with chronic liver diseases or co-morbidities (Fig. 2A,B). However, RDA indicated that liver fibrosis severity significantly contributed to variation in microbiome profiles (0.96% of variance, p=0.042) (Fig. 2C). RDA also indicated that liver fibrosis severity correlated positively with abundance of the Prevotellaceae family and Prevotella 9 genus, and negatively with the Marinifilaceae family and Butyricimonas (Fig. 2D,E).
Fig. 2.
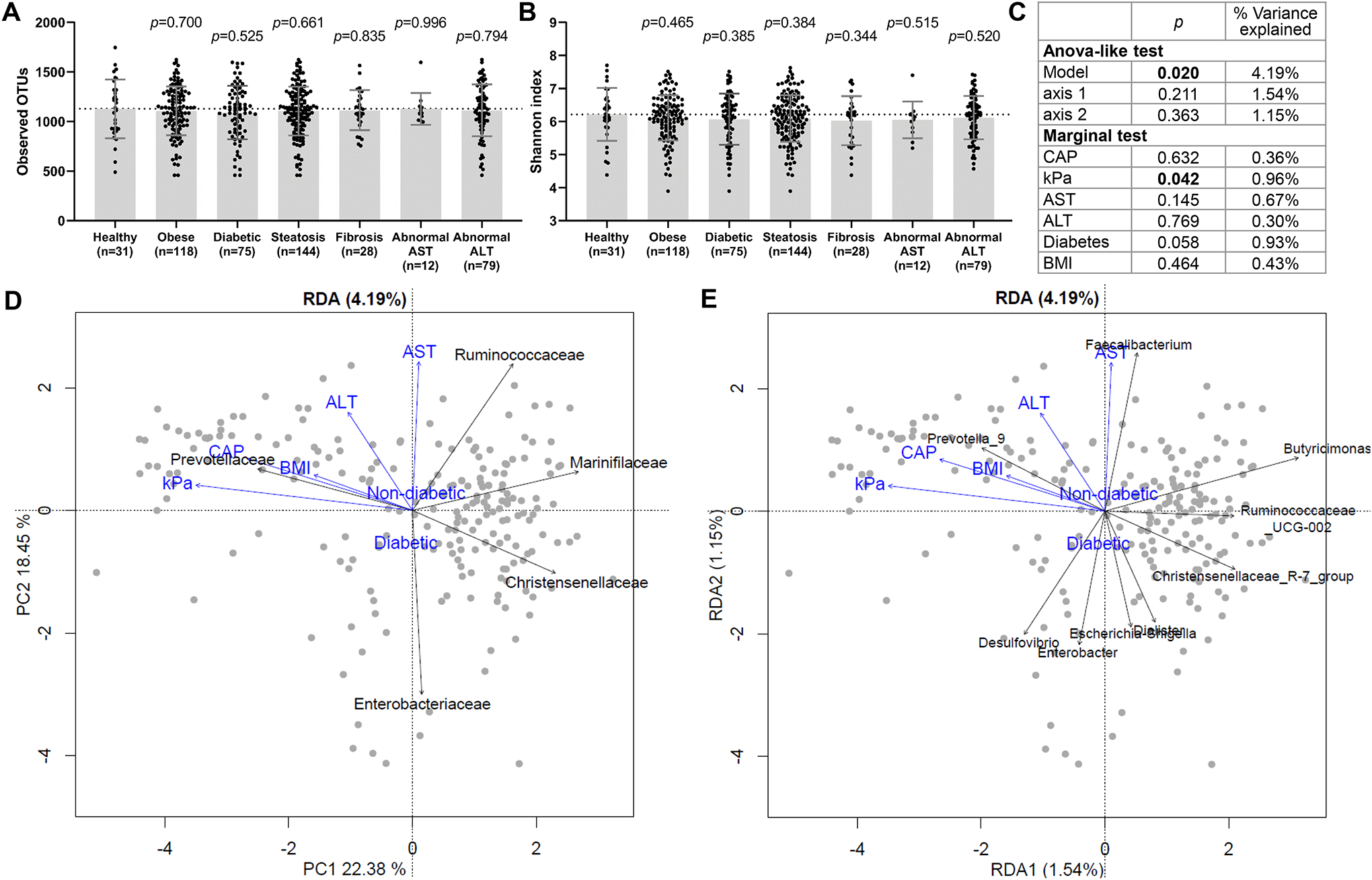
Alpha and beta diversity of stool samples in CCHC subjects. Number of (A) observed OTUs and (B) Shannon index scores, in healthy CCHC subjects versus those with disease. Bars and error bars represent mean and standard deviation. Significance compared to the healthy group was determined by unpaired t-test. (C) Weighted UniFrac distance-based RDA was performed to evaluate the relationship between selected clinical parameters and stool microbiome. ANOVA-like significance test of the model, axes and explanatory variables (clinical parameters) are shown. Triplots of the weighted Unifrac distance-based RDA with explanatory variables shown in blue and the abundance of (D) families and (E) genera shown in black. Only taxa significant by envfit permutation test (p<0.05) are shown.
To identify bacteria enriched or depleted in subjects with liver fibrosis, LEfSe analysis was performed on taxa from phylum to species levels. Twenty significant taxa were identified as shown within their phylogenetic structure in LEfSe cladograms (Fig. 3A). Of these, 18 taxa were also considered significant by ANCOM analysis (FDR of <0.2). Relative abundance and magnitude of change with liver fibrosis for these 18 taxa are illustrated in Fig. 3B,C. The strength of the associations between these 18 bacterial taxa and liver fibrosis were further determined by logistic regression analysis, adjusting for age, gender, diabetes status, BMI and alcohol intake (Fig. 3D). The strongest abundance changes and positive associations with liver fibrosis were observed for Prevotella (fold change (FC)=147; AOR=4.95 [95% CI=1.36–17.99], p=0.015), Prevotella 9 in particular with all reads assigned to Prevotella copri (FC=61.5; AOR=3.49 [95% CI=1.10–11.14], p=0.035) and for Holdemanella (FC=97.5; AOR=3.49 [95% CI=1.35–9.00], p=0.010). Liver fibrosis was also associated with enrichment of the Clostridiaceae 1 family (FC=3.63; AOR=2.83 [95% CI=1.10–7.26], p=0.031), of which almost all reads were assigned to Clostridium sensu stricto 1 (FC=3.63; AOR=2.81 [95% CI=1.09–7.22], p=0.032). Strong depletion and negative associations with liver fibrosis were observed for Bacteroides caccae (FC=-10.9; AOR=0.36 [95% CI=0.15–0.89], p=0.027), Enterobacter, Enterobacter cloacae in particular (FC=-4.7; AOR=0.34 [95% CI=0.14–0.86], p=0.022), and Parabacteroides distasonis (FC=-1.7; AOR=0.18 [95% CI=0.05–0.68], p=0.011).
Fig. 3.

Bacterial taxa with altered abundance in subjects with liver fibrosis. (A) Cladogram showing taxa with significantly different bacterial abundance between subjects with and without liver fibrosis, as assessed by the linear discriminant analysis (LDA) effect size (LEfSe) algorithm. (B) Volcano plots of ANCOM analysis showing all bacterial taxa with ≥0.1% abundance in at least 25% of samples, with significant taxa labelled as in panel (A). Significance was determined using FDR <0.2 and W statistic above the 60th percentile). The x-axis represents effect size, based on the centered log ratio (CLR)-transformed mean difference in abundance between subjects with and without liver fibrosis. Labels sharing a dot indicate taxa at different taxonomic levels, where all reads from the higher level are assigned to the taxa at the lower level. (C) Median relative abundance of significant taxa (as determined by ANCOM) in subjects with and without liver fibrosis. (D) Forest plot of significant taxa from panels (B-C), which also showed a significant association with liver fibrosis after adjusting for covariates. Adjusted odds ratios (AORs) for liver fibrosis are shown for high bacterial abundance, after adjusting for age, gender, diabetes status, BMI and alcohol intake (g/day). *Prevotella abundance is merged from all genus subgroups. Classifications at the family (f_), genus (g_), and species (s_) levels are shown.
Association between diet and liver fibrosis-associated microbiomes
To assess the contribution of diet to the liver fibrosis-associated microbiome signature, RDA was performed with the intake of dietary groups as explanatory variables and the abundances of taxa significantly associated with liver fibrosis or advanced liver fibrosis as response variables. The overall model explained 4.50% of the variation in taxa abundances (p=0.006) (Fig. 4A,B). A significant association was observed with alcohol intake (1.53% variance explained, permutation test p=0.002). PCoA confirmed that moderate/heavy drinkers exhibited a shift in microbiome composition (Fig. 4C). Total animal protein intake additionally contributed 1.04% variance (p=0.031). Only Prevotellaceae, Prevotella 9/Prevotella copri and Mollicutes were significantly associated with the RDA. Alcohol intake positively correlated with Prevotellaceae and Prevotella 9/Prevotella copri abundance, as confirmed by Spearman’s correlation (rs=0.27, p<0.001 and rs=0.26, p<0.001). The correlation remained significant after exclusion of heavy drinkers (rs=0.29, p<0.001 and rs=0.27, p<0.001).
Fig. 4.
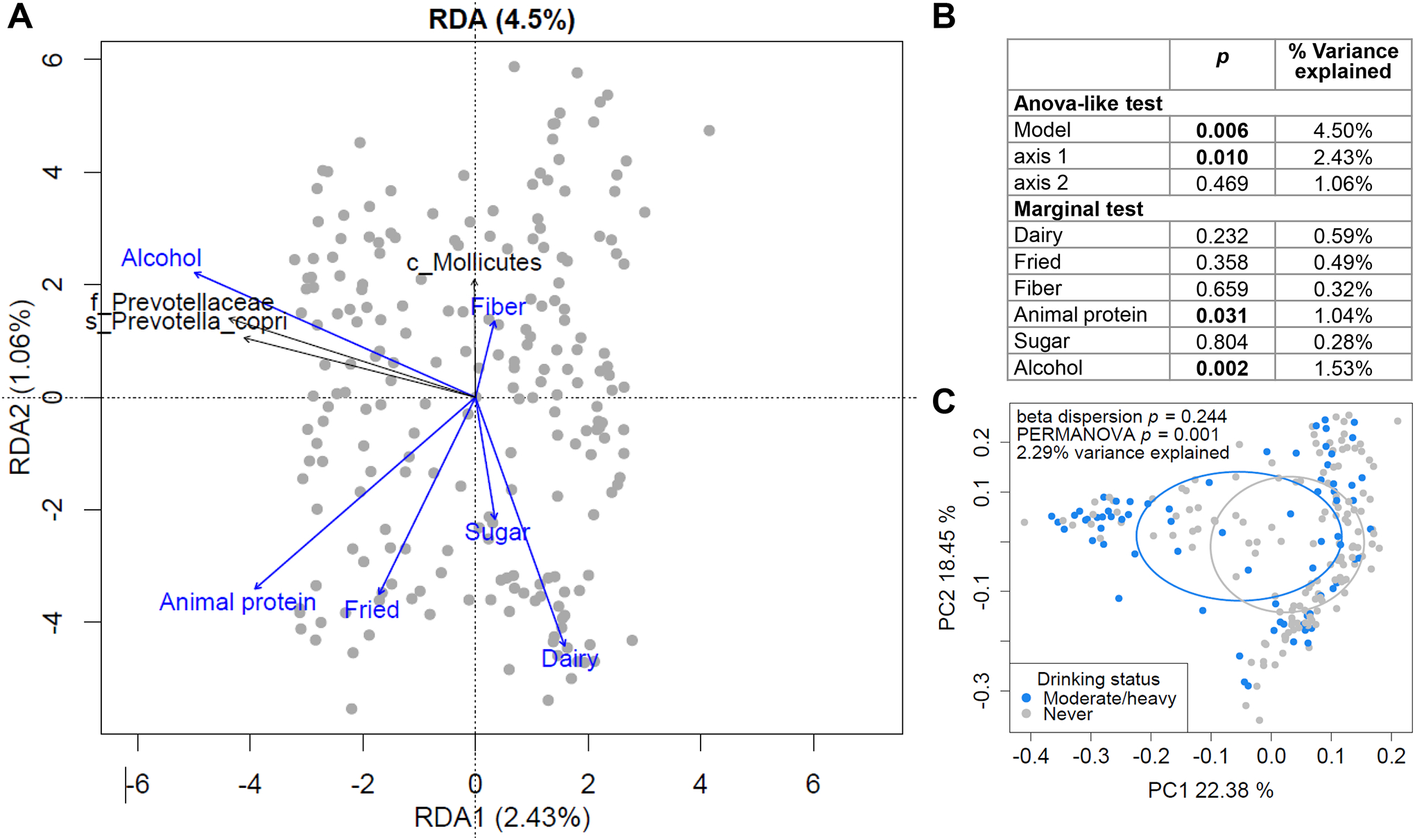
Dietary factors affecting liver fibrosis-associated microbiome profiles. (A-B) RDA to evaluate the relationship between dietary intake and the abundance profiles of taxa significantly associated with liver fibrosis. (A) Triplots of redundancy analysis. Explanatory variables (dietary groups) are shown in blue. Abundance of liver fibrosis-associated bacteria are shown in black as response variables and overlaid using the envfit function in the R vegan package on the linear constraints. Only taxa significant by envfit permutation test (p<0.05) are shown. Classifications at the class (c_), family (f_) and species (s_) levels are shown. (B) ANOVA-like significance test of the RDA model, axes and explanatory variables. (C) PCoA plot of overall gut microbiome profiles, based on weighted UniFrac distances, with samples grouped by drinking status.
Contribution of genetics to high Prevotella abundance
We next sought to determine whether genetics contributed to the microbiome changes associated with liver fibrosis in this population. GWAS was performed with high Prevotella abundance as a dichotomized trait, due to its strong association with liver fibrosis. Among the 217 CCHC subjects, 139 had genome-wide genotyping data available and PCR genotyping was performed on the remaining subjects for selected SNPs. No SNP displayed genome-wide significance by GWAS (p<5×10−8), likely due to the small sample size. However, 20 SNPs were significant at the threshold of p<1×10−5 (Supporting Fig. S1B and Supporting Table S5), including SNPs in a locus in the NCK Adaptor Protein 2 (NCK2) gene on chromosome 2 (Fig. 5A). The SNPs in this locus with the lowest p-values were rs3769502 (p=2.75×10−6) and rs7573751 (p=5.61×10−6), which displayed moderate linkage disequilibrium (0.8>r2≥0.6). rs7573751 has a RegulomeDB rank of 3a, indicating a fair likelihood of affecting transcription factor binding. The A allele for both SNPs is significantly associated with decreased gene expression of NCK2 in whole blood, based on multiple eQTL studies in the PhenoScanner database. We confirmed that the A allele for both SNPs was significantly associated with high Prevotella abundance in the 217 CCHC subjects (median abundance of 0.05%, 1.8% and 10.2% for rs3769502 GG, GA and AA genotypes, respectively, Kruskal-Wallis p=0.007; and median abundance of 0.05%, 2.3% and 7.6% for rs7573751 GG, GA and AA genotypes, respectively, Kruskal-Wallis p=0.005) (Fig. 5B,C,E,F). Logistic regression analysis further confirmed that the GA/AA and AA genotypes were significantly associated with high Prevotella abundance, even after adjusting for age, gender and presence of liver fibrosis (rs3769502-GA/AA: AOR=2.43 [95% CI=1.35–4.36], p=0.003; rs3769502-AA: AOR=2.70 [95% CI=1.08–6.74], p=0.033; rs7573751-GA/AA: AOR=2.39 [95% CI=1.34–4.24], p=0.003; rs7573751-AA: AOR=3.44 [95% CI=1.25–9.47], p=0.017) (Fig. 5D,G). Genotype frequencies for rs3769502 in CCHC were not significantly different from subjects of Mexican Ancestry in Los Angeles (MXL) nor among Europeans (EUR) from the 1000 Genomes Project. In contrast, rs7573751 GA and AA genotype frequencies in CCHC were similar to MXL (41.9% and 43.8% for GA; 9.8% and 12.5% for AA), but higher than in EUR (34.8% GA and 5.2% AA, p=0.005) (Fig. 5A).
Fig. 5.
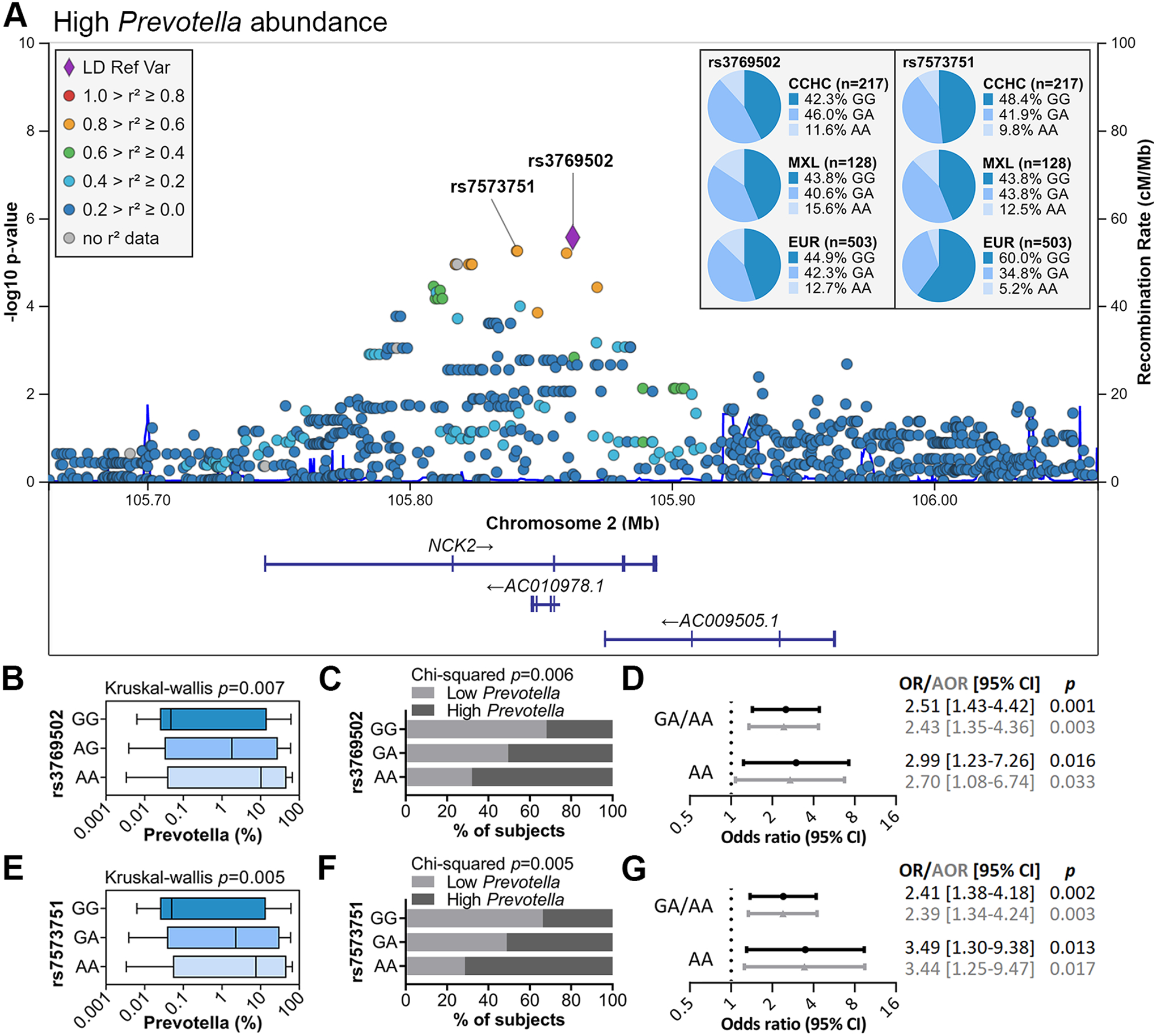
Host genetics associated with high Prevotella abundance. (A) Regional association plot for the GWAS of high Prevotella abundance. Linkage disequilibrium (LD) was based on the admixed American population from 1000 Genomes. Genotype frequencies for rs3769502 and rs7573751 are shown [MXL: subjects with Mexican ancestry in Los Angeles, California; EUR: Europeans, from 1000 Genomes]. (B,E) Prevotella abundance by rs3769502 and rs7573751 genotypes. Bars represent the median and interquartile range; error bars show the minimum and maximum abundances. (C,F) % of subjects with high and low Prevotella abundance by rs3769502 and rs7573751 genotypes. (D,G) Forest plots showing the association between rs3769502 and rs7573751 GA/AA and AA genotypes and high Prevotella abundance. OR: odds ratio; AOR: OR adjusted for age, gender and presence of liver fibrosis.
Metagenomic changes in liver fibrosis
Finally, to identify microbiome functional changes associated with liver fibrosis, shotgun metagenomic sequencing was performed on a subset of 142 CCHC study participants, selected for having liver steatosis. Among them, 27 (19.0%) had liver fibrosis. Ten MetaCyc pathways and six MetaCyc reactions were significantly depleted in subjects with liver fibrosis, as determined by ANCOM (Fig. 6A–B). Spearman’s correlation analyses between pathways and enzymes were performed to visualize co-abundant groups of functions (Fig. 6C). Correlation analysis adjusting for FibroScan LSM, was also performed between pathways/enzymes and liver fibrosis-associated bacterial taxa (Fig. 6D). Subjects with liver fibrosis had depletion of two pathways involved in de novo guanosine diphosphate (GDP)-L-fucose synthesis, namely GDP-mannose-derived O-antigen building blocks biosynthesis and colanic acid building blocks biosynthesis. Other depleted pathways included glucose and glucose-1-phosphate degradation, which correlated with Enterobacter abundance (rs=0.36, p<0.001), urea cycle and L-citrulline biosynthesis, which strongly correlated with Parabacteroides distasonis abundance (rs=0.40, p<0.001; rs=0.41, p<0.001). Depletion of malonyl-[acyl-carrier protein] O-methyltransferase, involved in vitamin B7 synthesis, and lipoyl synthase, involved in the synthesis of lipoic acid, also correlated with Parabacteroides distasonis (rs=0.34, p<0.001; rs=0.32, p=0.001). Conversely, there was enrichment of pyridoxal 5’-phosphate synthase (glutamine hydrolyzing), involved in vitamin B6 biosynthesis, and of 1,4-dihydroxy-6-naphthoate biosynthesis II pathway, involved in vitamin K2 biosynthesis, which both correlated with the abundance of Prevotella (rs=0.62, p<0.001; rs=0.31, p=0.002). Other enriched functions included creatinine degradation I, correlating with Holdemanella abundance (rs=0.31, p<0.001); D-alanine--poly(phosphoribitol) ligase, involved in incorporation of D-alanine into teichoic acid, and two pathways involved in formaldehyde-related processes: formaldehyde oxidation I and formaldehyde assimilation II (RuMP Cycle).
Fig. 6.

MetaCyc pathways and enzymes significantly altered in stool metagenome of subjects with liver fibrosis. (A-B) Significant differences in gene abundance of MetaCyc pathways (A) and enzymes (B) were identified between subjects with and without liver fibrosis. Significance was determined by ANCOM analysis (FDR <0.2). Mean abundances for the differentially abundant pathways and enzymes performing the reactions are shown. (C) Spearman’s correlation matrix between all pathways and enzymes from (A-B). P-values were corrected for multiple tests by the Benjamini-Hochberg method. +: p<0.05 and Spearman’s correlation coefficient |rs|≥0.3. (D) Partial Spearman’s correlation matrix of MetaCyc pathways and enzymes against bacterial taxa with significantly altered abundance in liver fibrosis, as determined by ANCOM analysis. Correlation was adjusted for FibroScan liver stiffness measurement (kPa). P-values were corrected for multiple tests by the Benjamini-Hochberg method. Columns represent pathways and enzymes; rows represent taxa. Only pathways, enzymes and taxa with significant positive correlation (p<0.05 and rs≥0.3, indicated with +) are shown. Bacterial classifications at the family (f_), genus (g_), and species (s_) levels are shown.
The combination of 4 MetaCyc pathways and 6 reactions altered in liver fibrosis detected with strong performance the presence of liver fibrosis in CCHC subjects (AUC=0.87 [95% CI=0.79–0.95]) (Supporting Fig. S2).
Discussion
In this study, we aimed to determine the association between the gut microbiome and chronic liver diseases disproportionately affecting Hispanics in South Texas. The study was performed on subjects from the CCHC, a population-based cohort of Hispanics in South Texas recruited from households and with a high prevalence of obesity, diabetes, NAFLD, liver fibrosis and HCC. FibroScan was used to screen the CCHC subjects for liver steatosis and fibrosis. The high homogeneity of the study cohort in terms of ethnicity and geographic location minimizes the confounding effects of these factors on the microbiome.
Liver fibrosis was most strongly associated with clustering of individual gut microbiome profiles. Cluster A, characterized by high abundance of Prevotellaceae, Prevotella 9/Prevotella copri and Holdemanella, had a particularly high degree of liver fibrosis. This was further supported by redundancy analysis, where liver fibrosis severity contributed significantly to the variation in microbiome profiles, while the degree of liver steatosis, AST and ALT levels, diabetes and BMI did not. Cluster B appeared to represent an intermediate group of samples with a transitional microbiome profile. As 20% of the subjects in cluster B had lost over 10 lbs of body weight in the 6 months prior to sample collection, weight loss may be a contributor to enterotype switching.
Fibrosis was positively associated with the abundance of Prevotellaceae, Prevotella copri, Holdemanella, Holdemanella biformis, Clostridiaceae 1 family and Clostridium sensu stricto 1, after adjusting for age, gender, diabetes status, BMI and alcohol intake. Prevotella is an immunogenic commensal linked to multiple inflammatory disorders, including metabolic disease, inflammatory bowel disease and even diseases outside of the gut, driving inflammation through activation of toll like receptor 2 and promotion of a systemic T helper 17 cell-mediated immune response.(38) Two recent studies reported a positive association between Prevotella abundance and liver fibrosis severity, including in children with NAFLD.(10, 13) Holdemanella biformis is a member of the Erysipelotricheae family, which also appear to be immunogenic commensals.(39) They have been associated with colorectal cancer, inflammation-related gastrointestinal diseases and host lipid metabolism.(40) Lastly, Clostridium_sensu_stricto_1 carries genes related to production of trimethylamine (TMA)(41), which is further metabolized by the host into trimethylamine N-oxide (TMAO), which may promote NAFLD(42).
Parabacteroides distasonis, Bacteroides caccae and Enterobacter cloacae were negatively associated with liver fibrosis. The Bacteroides genus has been frequently studied together with Prevotella due to their shared role as drivers of enterotype and their inverse correlation in abundance. We reported that toll-like receptor 4 activation by bacteria-derived lipopolysaccharide and subsequent pro-inflammatory signaling contributes to the development of liver fibrosis and HCC.(43) While members of Bacteroidetes are gram-negative and therefore major producers of lipopolysaccharide, Bacteroides species produce an immunoinhibitory form of the molecule due to an underacylated lipid A structure,(44) and may therefore have a suppressive effect on inflammation and liver fibrosis progression. Parabacteroides is a relatively newly described genus, with conflicting reports on whether it is pro- or anti-inflammatory in multiple disease settings.(45)
Alcohol and total animal protein intake significantly contributed to the variation in liver fibrosis-associated bacteria. Alcohol consumption was positively associated with Prevotellaceae and Prevotella 9/Prevotella copri, even after exclusion of heavy drinkers, indicating that even lower levels of alcohol intake can cause perturbations to the gut microbiome and contribute to NAFLD-related liver fibrosis. An increase in Prevotella abundance was observed in mice after chronic ethanol feeding(46) and in the oral microbiome of alcohol drinkers.(47) Furthermore, Prevotella species have the potential to accumulate high levels of acetaldehyde, a carcinogenic metabolite of ethanol oxidation.(48)
Beside environmental factors, host genetics can contribute to the variation in gut microbiome composition across individuals.(49) We identified a locus in the NCK2 gene, where the minor alleles for rs3769502 and rs7573751 SNPs were associated with high Prevotella abundance. Both SNPs are associated with decreased gene expression of NCK2 in whole blood. NCK2 is an adaptor protein modulating T cell receptor signaling and activity,(50) and may therefore affect adaptive immunity in the gut and/or elsewhere. The minor allele frequency for rs7573751 was significantly higher in the CCHC cohort compared to Europeans, suggesting that this variant may contribute to increased liver fibrosis susceptibility in Hispanics. The top SNPs were not found in previous microbiome-GWAS studies. Poor replication of GWAS studies on microbiome data, with inherent complexity and high dimensionality, has been recognized and discussed elsewhere.(49) Furthermore, a previous assessment concluded that <1% of participants in National Institutes of Health-funded GWAS studies were of Hispanic ancestry.(51) This disparity has also been observed in the relatively new field of microbiome GWAS studies.(49)
Finally, shotgun metagenomic sequencing showed that the microbiome signature associated with liver fibrosis was associated with depletion of bacterial pathways related to GDP-L-fucose biosynthesis. L-fucose is highly abundant in the mammalian gut and has been proposed to mediate host-commensal symbiosis.(52, 53) Certain bacteria are able to directly synthesize L-fucose de novo,(52) which becomes available for metabolism by other taxa, influencing the overall microbiome composition, bacterial virulence gene expression and metabolite production, providing a possible mechanism for affecting host health.(52, 53) We also observed metagenomic changes suggesting reduced synthesis of lipoic acid, and dysregulated synthesis of several B and K vitamins. The antioxidant, anti-inflammatory and hepatoprotective properties of lipoic acid and B vitamins and their derivatives have been demonstrated in rodent models of hepatotoxicity and NASH.(54–56) Furthermore, depletion of L-citrulline biosynthesis and urea cycle pathways suggest a potential for ammonia to accumulate, which would further contribute to oxidative stress, a key inducer of progression from simple steatosis to NASH.(57) Depletion of Parabacteroides distasonis correlated with depletion of metagenomic functions related to urea cycle, lipoic acid and vitamin B7 biosynthesis, while enrichment of Prevotella correlated with functions related to vitamin B6 and K2 biosynthesis. Enrichment of Holdemanella correlated with enrichment of the creatinine degradation I pathway, while depletion of Enterobacter correlated with depletion of the glucose and glucose-I-phosphate degradation pathway. Overall, our data suggest that a combination of dysregulated antioxidant biosynthesis and increased oxidative stress in subjects with liver steatosis, may contribute to the development of liver fibrosis. Studies demonstrating that the observed changes in the metagenome translate into changes in the metatranscriptome and circulating metabolite levels would be highly valuable.
In conclusion, we identified changes in the gut microbiome associated with liver fibrosis in Hispanics of South Texas, which may potentially mediate the development of a pro-inflammatory environment that contributes to liver fibrosis progression. Prospective cohort studies are needed to evaluate the utility of these taxonomic and functional changes in modeling risk of liver fibrosis progression in this population, and in developing novel preventive strategies.
Supplementary Material
Acknowledgments:
The authors would like to thank Rocío Uribe and her team who recruited and interviewed the CCHC participants, Marcela Morris BS and Hugo Soriano for CCHC laboratory and data support, and Norma Pérez-Olazarán BBA and Christina Villarreal BA for administrative support. We would also like to thank Dr. Tina Chang and Miriam Ortega of the MD Anderson Cancer Center Microbiome Core Facility, for their help with stool specimen processing, sequencing and analysis. Lastly, we would like to thank Valley Baptist Medical Center in Brownsville, Texas for the space used for the CCHC Center for Clinical and Translational Science Clinical Research, and the community of Brownsville and all cohort participants who so willingly participated in this study.
Grant support:
This study was funded by the MD Anderson Cancer Center SPORE in Hepatocellular Carcinoma Grant P50 CA217674 from the National Cancer Institute (NCI), and the Center for Clinical and Translational Sciences, National Institutes of Health Clinical and Translational Award grant no. UL1 TR000371 from the National Center for Advancing Translational Sciences. The MD Anderson Microbiome Core Facility is supported by the MD Anderson Cancer Center Support Grant (CCSG) P30 CA016672 from NCI.
Abbreviations:
- ALT
alanine aminotransferase
- ANCOM
Analysis of Compositions of Microbiomes
- ANOVA
analysis of variance
- AOR
adjusted odds ratio
- AST
aspartate aminotransferase
- ATPase
adenosine triphosphatase
- AUC
area under the ROC curve
- BMI
body mass index
- CAP
controlled attenuation parameter
- CCHC
Cameron County Hispanic Cohort
- CI
confidence interval
- CLR
centered log ratio
- CTP
cytidine triphosphate
- eQTL
expression quantitative trait loci
- EUR
Europeans
- FC
fold change
- GDP
guanosine diphosphate
- GTP
guanosine triphosphate
- GWAS
genome-wide association study
- HCC
hepatocellular carcinoma
- kPa
kiloPascals
- LDA
linear discriminant analysis
- LefSe
linear discriminant analysis effect size
- LSM
liver stiffness measurement
- MXL
Mexican ancestry in Los Angeles
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- NCK2
NCK Adaptor Protein 2
- NTPase
nucleoside triphosphatase
- OR
odds ratio
- OTU
operational taxonomic unit
- PCoA
principal coordinates analysis
- PCR
polymerase chain reaction
- PERMANOVA
permutational multivariate analysis of variance
- RDA
redundancy analysis
- ROC
receiver operating characteristic
- SNP
single nucleotide polymorphism
Footnotes
Disclosures: The authors have declared that no conflict of interest exists.
Data Transparency Statement: The 16S rRNA and shotgun metagenomic sequencing data have been deposited into the Sequence Read Archive (SRA) of the National Center for Biotechnology Information (NCBI) with BioProject accession number PRJNA734860. They will be made publicly available upon final publication.
References
- 1.Younossi ZM, Marchesini G, Pinto-Cortez H, Petta S. Epidemiology of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: Implications for Liver Transplantation. Transplantation. 2019;103:22–27. [DOI] [PubMed] [Google Scholar]
- 2.Estes C, Anstee QM, Arias-Loste MT, Bantel H, Bellentani S, Caballeria J et al. Modeling NAFLD disease burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the period 2016–2030. J Hepatol. 2018;69:896–904. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 4.Rich NE, Oji S, Mufti AR, Browning JD, Parikh ND, Odewole M et al. Racial and Ethnic Disparities in Nonalcoholic Fatty Liver Disease Prevalence, Severity, and Outcomes in the United States: A Systematic Review and Meta-analysis. Clin Gastroenterol Hepatol. 2018;16:198–210 e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramirez AG, Munoz E, Holden AE, Adeigbe RT, Suarez L. Incidence of hepatocellular carcinoma in Texas Latinos, 1995–2010: an update. PLoS One. 2014;9:e99365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ha J, Chaudhri A, Avirineni A, Pan JJ. Burden of hepatocellular carcinoma among hispanics in South Texas: a systematic review. Biomark Res. 2017;5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Venepalli NK, Modayil MV, Berg SA, Nair TD, Parepally M, Rajaram P et al. Features of hepatocellular carcinoma in Hispanics differ from African Americans and non-Hispanic Whites. World J Hepatol. 2017;9:391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Petroni ML, Brodosi L, Bugianesi E, Marchesini G. Management of non-alcoholic fatty liver disease. BMJ. 2021;372:m4747. [DOI] [PubMed] [Google Scholar]
- 9.Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D et al. Environment dominates over host genetics in shaping human gut microbiota. Nature. 2018;555:210–215. [DOI] [PubMed] [Google Scholar]
- 10.Schwimmer JB, Johnson JS, Angeles JE, Behling C, Belt PH, Borecki I et al. Microbiome Signatures Associated With Steatohepatitis and Moderate to Severe Fibrosis in Children With Nonalcoholic Fatty Liver Disease. Gastroenterology. 2019;157:1109–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Puri P, Sanyal AJ. The Intestinal Microbiome in Nonalcoholic Fatty Liver Disease. Clin Liver Dis. 2018;22:121–132. [DOI] [PubMed] [Google Scholar]
- 12.Adams LA, Wang Z, Liddle C, Melton PE, Ariff A, Chandraratna H et al. Bile acids associate with specific gut microbiota, low-level alcohol consumption and liver fibrosis in patients with non-alcoholic fatty liver disease. Liver Int. 2020. [DOI] [PubMed] [Google Scholar]
- 13.Dong TS, Katzka W, Lagishetty V, Luu K, Hauer M, Pisegna J et al. A Microbial Signature Identifies Advanced Fibrosis in Patients with Chronic Liver Disease Mainly Due to NAFLD. Sci Rep. 2020;10:2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loomba R, Seguritan V, Li W, Long T, Klitgord N, Bhatt A et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017;25:1054–1062 e1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oh TG, Kim SM, Caussy C, Fu T, Guo J, Bassirian S et al. A Universal Gut-Microbiome-Derived Signature Predicts Cirrhosis. Cell Metab. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharpton SR, Schnabl B, Knight R, Loomba R. Current Concepts, Opportunities, and Challenges of Gut Microbiome-Based Personalized Medicine in Nonalcoholic Fatty Liver Disease. Cell Metab. 2021;33:21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caussy C, Tripathi A, Humphrey G, Bassirian S, Singh S, Faulkner C et al. A gut microbiome signature for cirrhosis due to nonalcoholic fatty liver disease. Nat Commun. 2019;10:1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ponziani FR, Bhoori S, Castelli C, Putignani L, Rivoltini L, Del Chierico F et al. Hepatocellular Carcinoma Is Associated With Gut Microbiota Profile and Inflammation in Nonalcoholic Fatty Liver Disease. Hepatology. 2019;69:107–120. [DOI] [PubMed] [Google Scholar]
- 19.Sydor S, Best J, Messerschmidt I, Manka P, Vilchez-Vargas R, Brodesser S et al. Altered Microbiota Diversity and Bile Acid Signaling in Cirrhotic and Noncirrhotic NASH-HCC. Clin Transl Gastroenterol. 2020;11:e00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watt GP, Lee M, Pan JJ, Fallon MB, Loomba R, Beretta L et al. High Prevalence of Hepatic Fibrosis, Measured by Elastography, in a Population-Based Study of Mexican Americans. Clin Gastroenterol Hepatol. 2019;17:968–975 e965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiao J, Watt GP, Lee M, Rahbar MH, Vatcheva KP, Pan JJ et al. Cirrhosis and Advanced Fibrosis in Hispanics in Texas: The Dominant Contribution of Central Obesity. PLoS One. 2016;11:e0150978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan JJ, Fisher-Hoch SP, Chen C, Feldstein AE, McCormick JB, Rahbar MH et al. Burden of nonalcoholic fatty liver disease and advanced fibrosis in a Texas Hispanic community cohort. World J Hepatol. 2015;7:1586–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan JJ, Qu HQ, Rentfro A, McCormick JB, Fisher-Hoch SP, Fallon MB. Prevalence of metabolic syndrome and risks of abnormal serum alanine aminotransferase in Hispanics: a population-based study. PLoS One. 2011;6:e21515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fisher-Hoch SP, Rentfro AR, Salinas JJ, Perez A, Brown HS, Reininger BM et al. Socioeconomic status and prevalence of obesity and diabetes in a Mexican American community, Cameron County, Texas, 2004–2007. Prev Chronic Dis. 2010;7:A53. [PMC free article] [PubMed] [Google Scholar]
- 26.Karlas T, Petroff D, Sasso M, Fan JG, Mi YQ, de Ledinghen V et al. Individual patient data meta-analysis of controlled attenuation parameter (CAP) technology for assessing steatosis. J Hepatol. 2017;66:1022–1030. [DOI] [PubMed] [Google Scholar]
- 27.Wong VW, Vergniol J, Wong GL, Foucher J, Chan HL, Le Bail B et al. Diagnosis of fibrosis and cirrhosis using liver stiffness measurement in nonalcoholic fatty liver disease. Hepatology. 2010;51:454–462. [DOI] [PubMed] [Google Scholar]
- 28.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods. 2018;15:962–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48:1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Conomos MP, Miller MB, Thornton TA. Robust inference of population structure for ancestry prediction and correction of stratification in the presence of relatedness. Genet Epidemiol. 2015;39:276–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conomos MP, Reiner AP, Weir BS, Thornton TA. Model-free Estimation of Recent Genetic Relatedness. Am J Hum Genet. 2016;98:127–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oscanoa J, Sivapalan L, Gadaleta E, Dayem Ullah AZ, Lemoine NR, Chelala C. SNPnexus: a web server for functional annotation of human genome sequence variation (2020 update). Nucleic Acids Res. 2020;48:W185–W192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong S, Boyle AP. Predicting functional variants in enhancer and promoter elements using RegulomeDB. Hum Mutat. 2019;40:1292–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kamat MA, Blackshaw JA, Young R, Surendran P, Burgess S, Danesh J et al. PhenoScanner V2: an expanded tool for searching human genotype-phenotype associations. Bioinformatics. 2019;35:4851–4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaul A, Mandal S, Davidov O, Peddada SD. Analysis of Microbiome Data in the Presence of Excess Zeros. Front Microbiol. 2017;8:2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larsen JM. The immune response to Prevotella bacteria in chronic inflammatory disease. Immunology. 2017;151:363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L et al. Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell. 2014;158:1000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaakoush NO. Insights into the Role of Erysipelotrichaceae in the Human Host. Front Cell Infect Microbiol. 2015;5:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rath S, Rud T, Karch A, Pieper DH, Vital M. Pathogenic functions of host microbiota. Microbiome. 2018;6:174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tan X, Liu Y, Long J, Chen S, Liao G, Wu S et al. Trimethylamine N-Oxide Aggravates Liver Steatosis through Modulation of Bile Acid Metabolism and Inhibition of Farnesoid X Receptor Signaling in Nonalcoholic Fatty Liver Disease. Mol Nutr Food Res. 2019;63:e1900257. [DOI] [PubMed] [Google Scholar]
- 43.Nguyen J, Jiao J, Smoot K, Watt GP, Zhao C, Song X et al. Toll-like receptor 4: a target for chemoprevention of hepatocellular carcinoma in obesity and steatohepatitis. Oncotarget. 2018;9:29495–29507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.d’Hennezel E, Abubucker S, Murphy LO, Cullen TW. Total Lipopolysaccharide from the Human Gut Microbiome Silences Toll-Like Receptor Signaling. mSystems. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ezeji JC, Sarikonda DK, Hopperton A, Erkkila HL, Cohen DE, Martinez SP et al. Parabacteroides distasonis: intriguing aerotolerant gut anaerobe with emerging antimicrobial resistance and pathogenic and probiotic roles in human health. Gut Microbes. 2021;13:1922241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bluemel S, Wang L, Kuelbs C, Moncera K, Torralba M, Singh H et al. Intestinal and hepatic microbiota changes associated with chronic ethanol administration in mice. Gut Microbes. 2020;11:265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fan X, Peters BA, Jacobs EJ, Gapstur SM, Purdue MP, Freedman ND et al. Drinking alcohol is associated with variation in the human oral microbiome in a large study of American adults. Microbiome. 2018;6:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tsuruya A, Kuwahara A, Saito Y, Yamaguchi H, Tenma N, Inai M et al. Major Anaerobic Bacteria Responsible for the Production of Carcinogenic Acetaldehyde from Ethanol in the Colon and Rectum. Alcohol Alcohol. 2016;51:395–401. [DOI] [PubMed] [Google Scholar]
- 49.Awany D, Allali I, Dalvie S, Hemmings S, Mwaikono KS, Thomford NE et al. Host and Microbiome Genome-Wide Association Studies: Current State and Challenges. Front Genet. 2018;9:637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borroto A, Abia D, Alarcon B. Crammed signaling motifs in the T-cell receptor. Immunol Lett. 2014;161:113–117. [DOI] [PubMed] [Google Scholar]
- 51.Peprah E, Xu H, Tekola-Ayele F, Royal CD. Genome-wide association studies in Africans and African Americans: expanding the framework of the genomics of human traits and disease. Public Health Genomics. 2015;18:40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pickard JM, Chervonsky AV. Intestinal fucose as a mediator of host-microbe symbiosis. J Immunol. 2015;194:5588–5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pickard JM, Maurice CF, Kinnebrew MA, Abt MC, Schenten D, Golovkina TV et al. Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness. Nature. 2014;514:638–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bashandy SAE, Ebaid H, Abdelmottaleb Moussa SA, Alhazza IM, Hassan I, Alaamer A et al. Potential effects of the combination of nicotinamide, vitamin B2 and vitamin C on oxidative-mediated hepatotoxicity induced by thioacetamide. Lipids Health Dis. 2018;17:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gariani K, Menzies KJ, Ryu D, Wegner CJ, Wang X, Ropelle ER et al. Eliciting the mitochondrial unfolded protein response by nicotinamide adenine dinucleotide repletion reverses fatty liver disease in mice. Hepatology. 2016;63:1190–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sadek KM, Saleh EA, Nasr SM. Molecular hepatoprotective effects of lipoic acid against carbon tetrachloride-induced liver fibrosis in rats: Hepatoprotection at molecular level. Hum Exp Toxicol. 2018;37:142–154. [DOI] [PubMed] [Google Scholar]
- 57.Borrelli A, Bonelli P, Tuccillo FM, Goldfine ID, Evans JL, Buonaguro FM et al. Role of gut microbiota and oxidative stress in the progression of non-alcoholic fatty liver disease to hepatocarcinoma: Current and innovative therapeutic approaches. Redox Biol. 2018;15:467–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.