

Abstract
Pancreatic β cells secrete insulin in response to glucose, a process that is regulated at multiple levels, including a network of input signals from other organ systems. Impaired islet function contributes the pathogenesis of type 2 diabetes mellitus (T2DM) and targeting inter-organ communications, such as GLP-1 signalling, to enhance β cell function has been proven to be a successful therapeutic strategy in the last decade. In this review, we will discuss recent advances in inter-organ communication from the metabolic, immune, and neural system to pancreatic islets, their biological implication in normal pancreas endocrine function, as well as their role in the (mal)adaptive responses of islet to nutrition-induced stress.
Keywords: Pancreatic β cells, type 2 diabetes mellitus, GLP-1, inter-organ crosstalk
Graphical Abstract
In this review, we discussed inter-organ signaling that are beneficial or detrimental to islet function and survival. Multiple distal metabolic organs, such as liver, adipose, intestine, bone, and skeletal muscle, modulate islet function through a variety of circulating factors. Multiple components in the pancreatic microenvironment, such as sympathetic/parasympathetic nerves, infiltrating immune cells, and exocrine compartment, also crosstalk with islet in both normal physiological and pathological settings.
Introduction
Already a global epidemic, the incidence of T2DM is expected to rapidly escalate in the coming decades. In 2018, 34.2 million people (or 10.5% of the population) within the United States were diabetic; by the year 2030, the diabetic population in the United States is expected to increase by 35% [1]. Accordingly, diabetes has been classified as a pandemic disease. Diabetes underpins a multitude of severely debilitating pathologies in patients, including blindness, kidney failure, stroke, cancer, heart disease, depression, neuropathy, loss of limbs, infertility, and death. Because of these complications, millions of people rely daily upon medications to regulate their fundamental metabolic processes.
β cell dysfunction and failure are the driving force of diabetes pathogenesis, and is characterized by defective insulin secretion, ER stress, eventual β cell loss and disease progression[2–5]. A number of stress factors, including excessive nutrients (glucose and/or lipids) systemic inflammation and insulin resistance, contribute to β cell dysfunction[6, 7]. Inside β cells, stress induces proinflammatory responses[8], ER stress[9], oxidative stress[10, 11], mitochondrial dysfunction[12], and dedifferentiation[13, 14], causing reduced insulin synthesis and secretion, eventually resulting in β cell failure[15, 16]. Though many therapeutic approaches are deployed to combat hyperglycemia, few (if any) treatments directly target β cell pathogenesis. Thus, long-term control of disease progression remains a persistent challenge.
The islet functional loss in the T2DM is a combinatory result of genetics and complex whole body metabolic dysregulation. Compared to the well-characterized intra-islet signalling and cell-autonomous regulation of the pancreatic endocrine cells, the inter-organ communication that regulates islet function, and how it is disrupted in obesity and T2DM, has just started to be elucidated in the past decades. There is accumulating evidence suggesting that secreted factors deriving from distant organs are crucial regulators of islet homeostasis, dysfunction, and regeneration. In this review, we focus on the metabolic tissues that communicate with islets through direct contact or circulating factors (Figure 1), and discuss the implication of tissue crosstalk in diabetes pathogenesis as well as the potential for therapeutic development.
Figure 1. Inter-organ crosstalk regulating pancreatic islet function.
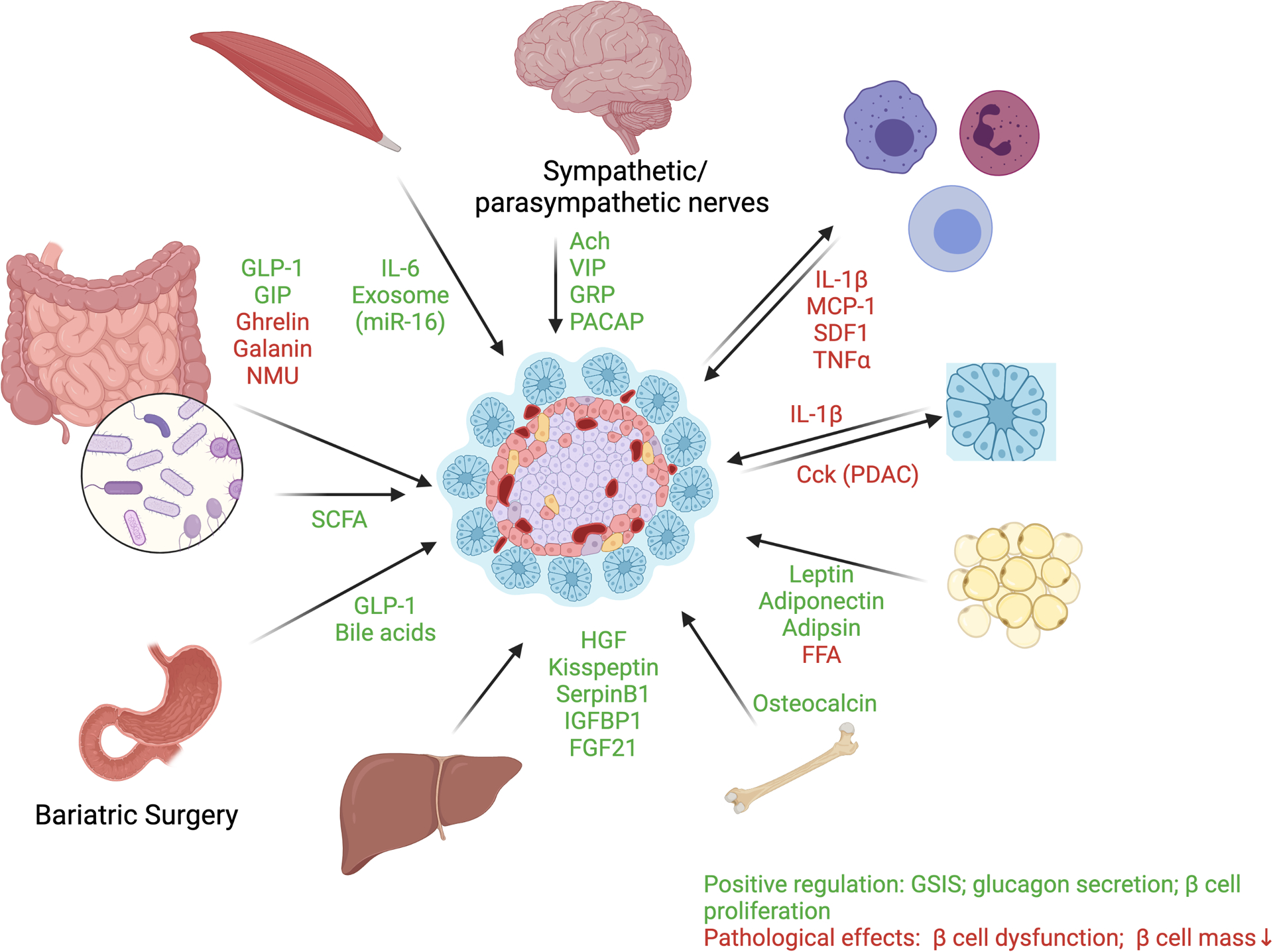
Multiple distal metabolic organs, such as liver, adipose, intestine, bone, and skeletal muscle, modulate islet function through a variety of circulating factors. The specific mediators for the inter-organ crosstalk include intestine-released incretin/decretin (GLP-1, GIP, Ghrelin, Galanin, and NMU) and metabolites from microbiome such as SCFA; adipose secreted leptin, adiponectin, adipsin, and free fatty acids (FFA); liver-secreted HGF, Kisspeptin, SerpinB1, IGFBP1 and FGF21; skeletal muscle secreted IL-6 and miR-16; and bone secreted Osteocalcin. Multiple components in the pancreatic microenvironment, such as sympathetic/parasympathetic nerves, infiltrating immune cells, and exocrine compartment, also crosstalk with islet in both normal physiological and pathological settings, such as pro-inflammatory cytokines (IL-1β, etc) from immune cells and neuropeptides (VIP, etc.) from sympathetic/parasympathetic nerves. The inter-organ signaling converge at islet to promote or impair glucose stimulated insulin response in both homeostatic and dysfunctional setting. Red: signaling molecules which cause islet dysfunction. Green: signaling molecules which enhance islet function. Created with Biorender.com.
Communication from the gastrointestinal system to pancreatic islets
Incretins and decretins
The concept of gut secreting hormones can be traced back to early 20th century, when it was recognized that simple nutrients trigger the release of secretory factors from gut mucosa to regulate blood glucose[17]. The incretin concept was later confirmed by administrating the same dose of oral or intravenous glucose. Oral glucose lead to a higher level of circulating insulin[18]. The best characterized incretins are glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP). GLP-1 is a cleaved product from pre-proglucagon, secreted from intestinal L cells (and from alpha cells), and released into circulation[19]. It binds to GLP-1 receptor (GLP-1R), and directly and indirectly, suppress alpha cell glucagon secretion while increasing insulin secretion in a glucose dependent manner[20]. Re-expression of pancreatic GLP-1R in the background of whole body GLP-1R knockout mice restored the glucose tolerance, suggesting a direct role for GLP-1 signalling in pancreatic islets[21]. Interestingly, mice with β cell specific deletion of GLP-1R remain glucose tolerant with oral glucose, but become glucose intolerant with intra-peritoneal glucose injection[22]. These results suggest that the mechanisms of GLP-1 on β cell insulin secretion likely include paracrine signalling by GLP-1 from alpha cells, or inputs from neural systems. Besides acute insulin secretion, GLP-1 also induce β cell proliferation and up-regulate insulin biosynthesis while reducing ER stress[23]. Therefore, it is likely that the chronic benefit of GLP-1 activation in improving islet function is a combinatory result of enhancing insulin secretion and preserving the β cell mass. Alongside β cells, GLP-1 also supresses glucagon secretion from alpha cells[24] while concurrently promoting somatostatin secretion from delta cells[25]. Whether these actions are mediated directly by GLP-1R on alpha/delta cells, or through indirect crosstalk between β cells and alpha/delta cells, remains to be determined.
The clinical success of GLP-1R agonists (reviewed in [19]) also suggests the therapeutic potential in other incretins such as GIP. While the administration of synthetic GIP failed to enhance the GSIS in human T2DM patients[26], recent progress in the development of GLP-1;GIP dual agonist has resulted in encouraging clinical data[27]. Future mechanistic studies will be needed to address whether the superior glycemia benefits are mediated directly by islet endocrine cells.
Contrary to incretins, decretins are secreted by intestine to dampen insulin secretion from β cells[20]. Typically induced by fasting, decretins suppress insulin secretion to protect against hypoglycaemia. The decretin peptide family includes ghrelin[28], galanin[29] and neuromedin U (NMU) [30], all of which suppress insulin secretion in ex vivo and in vivo conditions. Ghrelin is produced in stomach epithelium and islet epsilon cells. Pharmacological inhibition of the ghrelin O-acyltransferase (GOAT), the enzyme required for ghrelin activation, improved GSIS in animal models[31]. Galanin is expressed in neurons and intestine acting to inhibit insulin secretion through G02 proteins in the β cells[29]. NMU is additionally found in Drosophila, the human stomach and duodenum, and is able to inhibit GSIS in human islets[30]. Given their significance in physiological regulation of insulin secretion, decretins could be valuable targets in future T2DM therapeutic development.
Bariatric surgery
Initially developed to treat morbid obesity, bariatric surgery procedures quickly attracted the attention in diabetes researchers, as clinical studies consistently revealed that the procedure strongly improves glucose metabolism (see review from Douros JD et. al. [32]). Although the beneficial effects of bariatric surgery come from a complex combination of weight loss, insulin sensitivity and insulin secretion, there is substantial evidence that the islet function is significantly improved directly and indirectly by bariatric surgery. Here we summarize the clinical and preclinical evidence of signalling changes that contribute to islet function improvement after bariatric surgery.
Bariatric surgeries, such as Roux-en-Y gasctric bypass (RYGB), vertical sleeve gastrectomy (VSG), adjustable gastric band (AGC) and biliopancreatic diversion (BPD), changes the anatomy of gastrointestinal tract, achieve long term weight loss and importantly, significantly improve glucose control [33–36]. The effects of bariatric surgeries on T2DM is striking, as half of the patients with diabetes are able to achieve a nondiabetic HbA1C without diabetic medication[37]. Remarkably, RYGB, VSG, and BPD dramatically improve glucose control immediate after surgery[38, 39], suggesting mechanisms independent of weight loss. Distinct mechanisms such as caloric restriction, improvement of insulin resistance, changed postprandial glycemia, and increased insulin secretion, may all contribute to the superior glucose control after bariatric surgery[32].
The improvement of islet function in T2DM patient after bariatric surgery is supported by consistent findings that first phase insulin release is quickly restored in T2DM patients underwent RYGB or BPD[40–43]. This is consistent with the observation that patients after RYGB or VSG have a significant increase of GLP-1 release after meals[44], which is likely caused by the rapid passage of nutrient into intestine[45–47]. As GLP-1 signalling is critical for enhancing insulin secretion, it is natural to speculate that the increased L cell secretion of GLP-1 accounts for the improvement of glucose metabolism. However, a series of studies consistently showed that although GLP-1R blockade by exendin-(9–39) impair the insulin secretion after meal, the changes are no different from that before surgery, arguing against the hypothesis that increased GLP-1 is responsible for the enhanced insulin secretion after surgery[48–52]. Consistent with the clinical observation, mice with whole body deletion of GLP-1R demonstrate similar benefits in glucose tolerances after VSG or RYGB surgeries[53–55].
Besides GLP-1, other incretins, circulating hormones and metabolites are also altered and could potentially contribute to the beneficial effects of bariatric surgeries[32]. For example, mice with whole body deletion of bile acid receptor FXR failed to show weight reduction or glucose improvement after VSG, strongly suggesting that circulating bile acids are signalling mediators[56]. However, it should be noted that total knock-out of FXR are leaner and have better glucose tolerance before surgery[56]. Importantly, it is shown recently that the intestinal bile acids and lipid absorption is reduced in VSG and contribute to the metabolic benefits[57]. However, the exact molecular modulators for the improvement of insulin secretion on islets after bariatric surgeries remains to be identified.
Intestinal microbiota
The roles of intestinal microbiota in health and disease have become an emerging area of interest. It is clear that in both T1DM and T2DM, the intestinal microbiota is significantly altered[58, 59]. Mechanistically, the alteration of bacterial composition leads to changes of metabolites and vitamin levels, many of which have profound influence on islet function. An example is short-chain fatty acids (SCFAs). Produced by distal gut from fermentation of non-digestive carbohydrates[60], circulating SCFAs are anti-inflammatory mediators in both immune components and β cells[61–64]. SCFAs, such as sodium acetate and sodium propionate, act through the receptor FFAR2 to potentiate GSIS and prevent cytokine-induced apoptosis in both mouse and human islets [65]. Besides altering β cell function, SCFAs also induce production of CRAMP, an anti-microbial peptide in β cells, which induces an increase of regulatory macrophages, regulatory dendritic cells, and Treg cells in the pancreatic microenvironment[63]. The complex interorgan communication among islets, immune environment, and intestinal microbiota, and the underlying signalling and mechanisms, are still largely a black box and remains to be explored.
Communication from the adipose tissue to pancreatic islets
Adipose tissue actively produces adipokines, in response to nutrient and other systemic signals, to influence whole body metabolism. As the most extensively characterized adipokine, leptin is secreted from adipose and regulates both food intake and energy expenditure. Global mutations in both leptin and the leptin receptor result in mild to severe impairment to islet insulin secretion[66]. At least part of the effect of leptin action on islet is direct via activation of the KATP and NMDA channels[67–70]. These indirect actions of leptin, possibly through neural innervation, remain to be characterized.
Another key adipokine, adiponectin, is thought to act on the regeneration of β cells. In the β cell ablation model, adiponectin stimulates islet regeneration through HNF4α and PPARα mediated anti-lipotoxic effects [71, 72]. In gestational diabetes mellitus, adiponectin also promotes β cell proliferation through lactogen expression[73]. Therefore, it is likely the downstream mediators of adiponectin in β cells are context dependent.
Recently, a novel adipokine, adipsin, was identified an essential factor for islet insulin secretion and glucose tolerance[74]. Genetic deletion of adipsin results in insulinopenia[74]. Conversely, administration of adipsin in diabetic mice increases insulin secretion and reverses hyperglycaemia[74, 75]. Adipsin generates C3a and increases ATP, OxPhos, and Ca2+ levels in β cells[75]. Mechanistically, this acts through phosphatase Dusp26 to increase insulin secretion and promote β cell survival in murine and human islets[75]. These data suggest that adipsin and its downstream pathways are possible targets for T2DM therapeutics.
Communication from skeletal muscle to pancreatic islets
It is widely known that, exercise can prevent or mitigate metabolic diseases such as obesity, T2DM, and cardiovascular diseases[76]. Similarly, PPARδ agonists (aka: exercise mimetics) also powerful mitigators of metabolic disease, by promoting oxidative metabolism, lowering blood glucose, enhancing muscle performance and stimulating weight loss[76, 77]. Besides its role in consuming carbohydrates and lipids, skeletal muscle produces multiple endocrine signals, known as myokines, which regulate distal metabolic organs including pancreatic islets. As one of the best characterized myokines, IL-6 is secreted from contracting muscle and release into circulation[78]. As a pleiotropic factor, IL-6 can trigger nutrient availability and improve insulin sensitivity[79, 80]. Besides its direct effect on β cells, IL-6 also act on enteroendocrine cells (L-cells) and islet alpha cells to increase prohormone convertase 1/3, proglucagon, and GLP-1 expression, enhancing the GSIS function of islet[81]. Importantly, exercise increases the IL-6 expression in skeletal muscle[81], providing a molecular explanation for the exercise benefit.
Besides myokines, skeletal muscle also secretes exosome-like vesicles, which contains key microRNAs that may target islet function. In insulin resistant muscle, excess palmitate induces release of exosome-like vesicles containing overexpressed miR-16[82]. miR-16 regulates the expression of Ptch1 and other developmental genes, which may also enable the expansion of islet mass[82]. The molecular underpinnings of skeletal muscle derived exosomes and miRNAs in human islets and its therapeutic values remains an important emerging field.
Communication from liver to pancreatic islets
As a major organ for energy storage, liver oversees glucose and lipid homeostasis and can therefore contribute to islet health or dysfunction by changing the circulating metabolite composition and levels. In addition, other liver-derived hormones and growth factors, such as IGF-1 can influence islet function and mass.
Kisspeptin (encoded by KISS1) is widely expressed in many tissues and initially characterized as a tumour suppressor[83]. However, recent studies revealed novel roles of kisspeptin and its receptor, KISS1R, in endocrine regulation, from puberty and fertility to islet function[83]. Importantly, the liver production of kisspeptin is regulated by glucagon[84]. Kisspeptin is able to suppress GSIS from β cells[84]. Importantly, this crosstalk is defective in T2DM models, as hyperglucagonemia enhances kisspeptin production to ultimately reduce insulin secretion[84].
FGF21 is a critical metabolic regulator, which is secreted from liver upon fasting or ketogenic diet[85]. An early study demonstrated that FGF21 is able to improve β cell function and survival by activating Akt signalling[86]. However, the benefit of FGF21 on islet function may be a result of multiple parallel mechanisms, such as activation of insulin-like growth factor proteins (IGFBPs)[87]. When activated by FGF21[88], IGFBPs improve insulin sensitivity, protect against atherosclerosis, and promote β cell regeneration in multiple animal models[89, 90].
Another regenerative factor from liver is hepatic growth factor (HGF). HGF acts on its receptor c-Met, to promote regeneration in multiple tissues including islets[91]. While c-Met deletion does not disrupt β cell development or normal physiology, it does impair the regenerative potential of β cells in low dose streptozotocin or partial pancreatectomy-induced β cell loss[92].
In addition to injury-induced β cell regeneration, it is well documented that obesity and/or insulin resistance induce β cells proliferation[93]. Insulin resistance induce widespread changes in the liver secretome, which act on islets to promote β cell proliferation in an attempt to restore glucose homeostasis[94]. More recently, a search for potential β cell expansion factors in liver insulin receptor knock-out mice discovered serpinB1, a circulating factor that inhibits elastase to facilitate β cell regeneration[95]. Small molecule compound sivelestat, which targets elastase, also resulted in similar regenerating capacity in cultured and transplanted islets[95]. Together, these studies revealed the potential therapeutic value of targeting serpinB1 and elastase. SerpinB1 as a hepatocyte-secretory protease inhibitor regulating b-cell proliferation in humans, mice, and zebrafish. SerpinB1 acts by modulating canonical growth and survival signalling pathways.
Communication from nervous system to pancreatic islets
Rodent pancreatic islets are innervated by both parasympathetic nervous system and sympathetic nervous system[96]. In humans, 3D imaging of the pancreatic tissue also showed dense innervation similar to rodents[97]. In hyperglycaemia, parasympathetic nerves release acetylcholine which activate muscarinic receptor signalling (m3AchR) in β cells to potentiate insulin secretion[98]. Other parasympathetic nerves also release peptides, including pituitary adenylate cycle-activating peptide (PACAP), vasointestinal polypeptide (VIP) and gastrin-related peptide (GRP), also influence insulin secretion from β cells[96]. It is known that the insulin secretion in response to feeding can happen before the increase of blood glucose, which is termed as the cephalic-phase insulin release[99]. The cephalic phase of insulin secretion is mediated by vagal cholinergic signals and contribute to glucose tolerance[99]. Besides the regulation of cephalic insulin release, neural control is also likely to contribute to postprandial insulin secretion, though the specific regulator remains to be defined[100].
The signals from central nervous system also involve in glucagon secretion[101]. In contrast to the hyperglycaemia-induced activation of parasympathetic nervous system, hypoglycaemia induce activation of sympathetic nervous system induces glucagon secretion from islet alpha cells[97]. The glucagon response to hypoglycaemia is impaired when islets are denervated, supporting the causality between sympathetic activity and alpha cell function[97]. The postprandial glucagon secretion, which influence the nutrient induced insulin secretion, is also partly regulated by the parasympathetic input [102].
Though the neural input is considered as a significant contributor to islet function in both rodents and human, the innervation patterns are strikingly different between species[103]. In mouse islets display a dense innervation of parasympathetic nerves in the islet core, while sympathetic nerves are enriched in the periphery[103]. In contrast, in human islets parasympathetic nerves typically avoid the islet core, and sympathetic nerves projected to the intra-islet blood vessels[104]. Thus, for humans, the alpha cells, (rather than the cholinergic nerves), are the main source of acetylcholine that regulate β cell function[104]. This type of structural divergence suggests that despite a dominance of similarities, variation in the regulation of islet function by neural inputs between rodents and humans is to be expected and that therapeutic outcomes in one species may not always extend to the other.
Communication from innate immune system to pancreatic islets
Chronic stress and inflammation within the islet has an indispensable role in T2DM pathogenesis[7]. Typically mediated by the innate immune system, the resulting elevated circulation of paracrine proinflammatory signals has long been considered as a principal cause for islet dysfunction and loss of β cell mass. In T2DM, obesity and overnutrition create an increased demand on insulin production. This chronic provocation in turn induces conjoined inflammatory responses in multiple tissues, including adipose, liver, and pancreatic islets[105]. This pathologic cascade results in an increased level of lipids, gut derived antigens (such as lipopolysaccharide), and damage associated molecular proteins (DAMP)[106]. As expected, an increase of macrophage population is observed in adipose and islets[105, 107]. Due to proximity these local macrophages tend to be more inflammatory, demonstrated by an increase level of TNFα and chemokines, and align with traditional “M1 macrophages”[107–109]. Enlargement of the adipose depot, linked to high fat diets, results in insulin resistance which triggers proinflammatory macrophages to increase cytokine production [105, 110]. The islet macrophages are considered as a “sensor” of the inflammatory signals, which effectively become the paracrine cytokines that drive β cells stress and eventually, β cell failure [108, 109, 111]. In this pathologic state, proinflammatory cytokines such as IL-1β and chemokines such as MCP1 are produced by β cells under stress from high free fatty acids and hyperglycaemia[112]. The chemokines are known to increase infiltration of monocyte-derived macrophages into the islets[4]. Conversely, inhibition of M1 macrophage accumulation protects β cells from palmitate acid induced β cell loss[4]. Insulin resistance and the stress induced by high glucose activates NLRP3-dependent inflammasome activation in islet residential macrophages, leading to IL-1β cleavage by the proinflammatory caspase-1 [113]. The resulting high level of IL-1β represses β cell lineage gene expression, induces β cell dysfunction, and ultimately β cell death[114–116]. Therefore, the IL-1β signalling is a key pathway responsible for the vicious cycle of the crosstalk between β cell and macrophages in the obesity settings.
It should also be noted that the action of IL-1β on islets partly depends on its concentration. While the high level of IL-1β clearly is detrimental for islet function, low level of IL-1β is known to, paradoxically, increase the insulin secretion[117]. It has been shown that nutrient uptake induced release by low level IL-1β will actually enhance GSIS[117]. Therefore, the differential mechanisms of IL-1β in regulating both normal physiological responses and pathological inflammatory response will be an interesting topic for future study and a potential area to exploit. In addition, other chemotaxis signals, such as fractalkine/CX3CR1 and SDF1/CXCR4, are also known to increase GSIS and β cell proliferation/regeneration[118–120]. Therefore, the inflammatory pathways mediated by islet macrophages may also promote islet function in the context of obesity and nutrient stress.
Another outstanding question remains the origin of islet macrophages. In recent years the lineage tracing studies suggest that the residential islet macrophages are mainly from yolk-sac whereas the recruited macrophages in obesity are from bone marrow (Ly6c+CD11b+)[121]. The classic view of the origin of islet macrophages is that the increased number of islet residential macrophages are mainly recruited from circulating monocytes[121]. This was challenged by a recent study which characterized two distinct populations, the peri-islet macrophages (F4/80hiCd11c−) and the intra-islet macrophage (F4/80loCd11chi)[109]. In obese mice, the increase of residential macrophages appears to be of intra-islet origin [109]. Further studies using genetic tracing models will be needed to solve this discrepancy.
In brief, obesity induces a complex pathologic cascade principally comprised of islet residential and infiltrating immune populations, especially macrophages. These changes clearly correlate and contribute to islet dysfunction, and diabetes progression.
Communication from bone to pancreatic islet
Bone is now considered as an endocrine organ that is able to crosstalk with other metabolic organs. In mice lacking the protein tyrosine phosphatase OST-PTP in osteoblasts treated with high fat diet (HFD), β cell proliferation and insulin secretion are enhanced, and glucose metabolism is improved, compared to wild type HFD-treated mice[122]. Mechanistically, OST-PTP negatively regulates Osteocalcin[122]. Mice with osteocalcin deletion, on the other hand, have defects in β cell proliferation and are glucose intolerant[122]. The β cell protective effect of Osteocalcin is mediated by β cell specific expression of its receptor Gprc6a[123]. Other than Osteocalcin, Neuropeptide Y (NPY) from osteoblast is also shown to promote insulin gene expression in β cells[124]. Together, these data support the model that distant crosstalk between osteoblasts and β cells modulate the function of β cells.
Exocrine-endocrine crosstalk
While the crosstalk between distant organs and islets are essential for islet function and pathology, it should not be ignored that islets are embedded in the exocrine pancreas (and ducts) and the interactions between endocrine and exocrine pancreas are critical not only for islet function but for exocrine pathology as well. This is best demonstrated in the case of pancreatogenic diabetes, or type 3c diabetes (T3cDM), a distinct class of diabetes secondary to pancreatic exocrine disease[125]. It is well known that chronic pancreatitis[126, 127] and cystic fibrosis[128] are associated with islet dysfunction and diabetes, possibly due to injury, inflammation and fibrosis in the acini. Elevated proinflammatory cytokines, such as IL-1β, and inflammatory infiltration are likely the causes of β cell dysfunction in both pancreatitis[129] and cystic fibrosis[130].
The exocrine-endocrine crosstalk in the pancreatic ductal adenocarcinoma (PDAC) is complex and intriguing. Diabetes frequency is elevated in PDAC patients[131]. 74% of the PDAC patients with diabetes are diagnosed within 24 months before PDAC diagnosis[132], suggesting a potential link between new-onset diabetes and PDAC. Most importantly, a large portion of PDAC patients with new onset diabetes have resolved their diabetes after surgery, strongly suggesting that a casual relation between PDAC and T3cDM[131]. A number of studies have revealed potential diabetogenic mediators of T3cDM in PDAC include Connexin 26[133], S-100A8 N-terminal peptide[134], vanin-1[135], MMP9[135], Adrenomedullin[136], and exosomes[137]. Inside the islets, it has been shown that β cells are dedifferentiated in PDAC patients[138]. The direct signalling cues that mediate this crosstalk, however, remain undetermined.
Besides the mounting evidence of exocrine-endocrine crosstalk in causing islet dysfunction, it should also be noted that the dysfunctional islets could also drive the carcinogenesis. Recently, Chuang et. al. elegantly showed that obesity leads to aberrant expression of cholecystokinin expression in islets and subsequently drives the PDAC development in leptin knock-out, Kras-driven mouse pancreatic cancer model[139]. Whether a similar mechanism exists in human PDAC remain to be determined.
Conclusions and Perspectives
In the past decades, significant advances have been made in our understanding of how signals from other organs regulate islet function in homeostasis and dysfunction. It is now clear that the islet is an integrative hub for circulating signals from a variety of metabolically related tissues (Figure 1). Besides sensing glucose and nutrients, circulating factors from other distal organs influence the baseline of islet function. The dysregulation of these inter-organ communications is key to pancreatic islet pathogenesis. However, a number of outstanding questions remain: how does the regulation of islet physiology differ in humans vs. rodents? What are the roles of exosomes and microbiome-derived metabolites in regulating β cell function? What are the chronic transcriptional and epigenetic responses of β cells to individual signaling? Further investigations in these areas may provide insights into developing next-generation diabetes therapies.
Acknowledgments
Due to the limits on space, we were not able to include all important papers in the field. We apologize to those whose work was not cited directly. We thank C. Brondos for administrative help.
Funding
Work in R.M.E’s laboratory is supported by grants from the NIH (HL088093, HL105278, R01DK120480, R01DK057978, P42ES010337), the Glenn Foundation for Medical Research, the Leona M. and Harry B. Helmsley Charitable Trust (#2017PG-MED001), Ipsen/Biomeasure, California Institute for Regenerative Medicine, The Ellison Medical Foundation. R.M.E is a NOMIS Foundation Distinguished Scientist and Scholar at the Salk Institute. Work in Z.W.’s laboratory is supported by NIH (K01DK120808), Mayo Clinic Center for Biomedical Discovery, ASU-Mayo Collaborative Fund, Roubos Family Fund in Research, and Integrated Islet Distribution Program. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations:
- GLP-1
glucagon-like peptide-1
- GIP
glucose-dependent insulinotropic peptide
- GLP-1R
GLP-1 receptor
- NMU
neuromedin U
- RYGB
Roux-en-Y gasctric bypass
- VSG
vertical sleeve gastrectomy
- AGC
adjustable gastric band
- BPD
biliopancreatic diversion
- SCFAs
short-chain fatty acids
- IGFBPs
insulin-like growth factor proteins
- DAMP
damage associated molecular proteins
- HFD
high fat diet
- PACAP
pituitary adenylate cycle-activating peptide
- VIP
vasointestinal polypeptide
- GRP
gastrin-related peptide
- DAMP
damage associated molecular proteins
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- 1.National Diabetes Statistics Report. in https://www.cdc.gov/diabetes/pdfs/data/statistics/national-diabetes-statistics-report.pdf.
- 2.Halban PA, Polonsky KS, Bowden DW, Hawkins MA, Ling C, Mather KJ, Powers AC, Rhodes CJ, Sussel L & Weir GC (2014) beta-cell failure in type 2 diabetes: postulated mechanisms and prospects for prevention and treatment, Diabetes care. 37, 1751–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashcroft FM & Rorsman P (2012) Diabetes mellitus and the beta cell: the last ten years, Cell. 148, 1160–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donath MY, Dalmas E, Sauter NS & Boni-Schnetzler M (2013) Inflammation in obesity and diabetes: islet dysfunction and therapeutic opportunity, Cell metabolism. 17, 860–72. [DOI] [PubMed] [Google Scholar]
- 5.Weyer C, Bogardus C, Mott DM & Pratley RE (1999) The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus, The Journal of clinical investigation. 104, 787–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prentki M, Peyot ML, Masiello P & Madiraju SRM (2020) Nutrient-Induced Metabolic Stress, Adaptation, Detoxification, and Toxicity in the Pancreatic beta-Cell, Diabetes. 69, 279–290. [DOI] [PubMed] [Google Scholar]
- 7.Donath MY & Shoelson SE (2011) Type 2 diabetes as an inflammatory disease, Nature reviews. 11, 98–107. [DOI] [PubMed] [Google Scholar]
- 8.Donath MY, Boni-Schnetzler M, Ellingsgaard H & Ehses JA (2009) Islet inflammation impairs the pancreatic beta-cell in type 2 diabetes, Physiology (Bethesda, Md. 24, 325–31. [DOI] [PubMed] [Google Scholar]
- 9.Ghosh R, Colon-Negron K & Papa FR (2019) Endoplasmic reticulum stress, degeneration of pancreatic islet beta-cells, and therapeutic modulation of the unfolded protein response in diabetes, Mol Metab. 27S, S60–S68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J & Wang H (2017) Oxidative Stress in Pancreatic Beta Cell Regeneration, Oxid Med Cell Longev. 2017, 1930261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee J, Moulik M, Fang Z, Saha P, Zou F, Xu Y, Nelson DL, Ma K, Moore DD & Yechoor VK (2013) Bmal1 and beta-cell clock are required for adaptation to circadian disruption, and their loss of function leads to oxidative stress-induced beta-cell failure in mice, Molecular and cellular biology. 33, 2327–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haythorne E, Rohm M, van de Bunt M, Brereton MF, Tarasov AI, Blacker TS, Sachse G, Silva Dos Santos M, Terron Exposito R, Davis S, Baba O, Fischer R, Duchen MR, Rorsman P, MacRae JI & Ashcroft FM (2019) Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic beta-cells, Nature communications. 10, 2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Talchai C, Xuan S, Lin HV, Sussel L & Accili D (2012) Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure, Cell. 150, 1223–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo S, Dai C, Guo M, Taylor B, Harmon JS, Sander M, Robertson RP, Powers AC & Stein R (2013) Inactivation of specific beta cell transcription factors in type 2 diabetes, The Journal of clinical investigation. 123, 3305–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hudish LI, Reusch JE & Sussel L (2019) beta Cell dysfunction during progression of metabolic syndrome to type 2 diabetes, The Journal of clinical investigation. 129, 4001–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campbell JE & Newgard CB (2021) Mechanisms controlling pancreatic islet cell function in insulin secretion, Nature reviews. 22, 142–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bayliss WM & Starling EH (1902) On the causation of the so-called ‘peripheral reflex secretion’ of the pancreas. (Preliminary communication.), Proceedings of the Royal Society of London. 69, 352–353. [Google Scholar]
- 18.McIntyre N, Holdsworth CD & Turner DS (1964) New Interpretation of Oral Glucose Tolerance, Lancet. 2, 20–1. [DOI] [PubMed] [Google Scholar]
- 19.Drucker DJ, Habener JF & Holst JJ (2017) Discovery, characterization, and clinical development of the glucagon-like peptides, The Journal of clinical investigation. 127, 4217–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hussain MA, Akalestou E & Song WJ (2016) Inter-organ communication and regulation of beta cell function, Diabetologia. 59, 659–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamont BJ, Li Y, Kwan E, Brown TJ, Gaisano H & Drucker DJ (2012) Pancreatic GLP-1 receptor activation is sufficient for incretin control of glucose metabolism in mice, The Journal of clinical investigation. 122, 388–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith EP, An Z, Wagner C, Lewis AG, Cohen EB, Li B, Mahbod P, Sandoval D, Perez-Tilve D, Tamarina N, Philipson LH, Stoffers DA, Seeley RJ & D’Alessio DA (2014) The role of beta cell glucagon-like peptide-1 signaling in glucose regulation and response to diabetes drugs, Cell metabolism. 19, 1050–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yusta B, Baggio LL, Estall JL, Koehler JA, Holland DP, Li H, Pipeleers D, Ling Z & Drucker DJ (2006) GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress, Cell metabolism. 4, 391–406. [DOI] [PubMed] [Google Scholar]
- 24.Drucker DJ (2018) Mechanisms of Action and Therapeutic Application of Glucagon-like Peptide-1, Cell metabolism. 27, 740–756. [DOI] [PubMed] [Google Scholar]
- 25.de Heer J, Rasmussen C, Coy DH & Holst JJ (2008) Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas, Diabetologia. 51, 2263–70. [DOI] [PubMed] [Google Scholar]
- 26.Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R & Creutzfeldt W (1993) Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus, The Journal of clinical investigation. 91, 301–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baggio LL & Drucker DJ (2021) Glucagon-like peptide-1 receptor co-agonists for treating metabolic disease, Mol Metab. 46, 101090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prado CL, Pugh-Bernard AE, Elghazi L, Sosa-Pineda B & Sussel L (2004) Ghrelin cells replace insulin-producing beta cells in two mouse models of pancreas development, Proceedings of the National Academy of Sciences of the United States of America. 101, 2924–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang G, Wang Y, Park S, Bajpayee NS, Vi D, Nagaoka Y, Birnbaumer L & Jiang M (2012) Go2 G protein mediates galanin inhibitory effects on insulin release from pancreatic beta cells, Proceedings of the National Academy of Sciences of the United States of America. 109, 2636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alfa RW, Park S, Skelly KR, Poffenberger G, Jain N, Gu X, Kockel L, Wang J, Liu Y, Powers AC & Kim SK (2015) Suppression of insulin production and secretion by a decretin hormone, Cell metabolism. 21, 323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barnett BP, Hwang Y, Taylor MS, Kirchner H, Pfluger PT, Bernard V, Lin YY, Bowers EM, Mukherjee C, Song WJ, Longo PA, Leahy DJ, Hussain MA, Tschop MH, Boeke JD & Cole PA (2010) Glucose and weight control in mice with a designed ghrelin O-acyltransferase inhibitor, Science (New York, NY. 330, 1689–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Douros JD, Tong J & D’Alessio DA (2019) The Effects of Bariatric Surgery on Islet Function, Insulin Secretion, and Glucose Control, Endocrine reviews. 40, 1394–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schauer PR, Bhatt DL, Kirwan JP, Wolski K, Brethauer SA, Navaneethan SD, Aminian A, Pothier CE, Kim ES, Nissen SE, Kashyap SR & Investigators, S. (2014) Bariatric surgery versus intensive medical therapy for diabetes--3-year outcomes, The New England journal of medicine. 370, 2002–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rubino F, Nathan DM, Eckel RH, Schauer PR, Alberti KG, Zimmet PZ, Del Prato S, Ji L, Sadikot SM, Herman WH, Amiel SA, Kaplan LM, Taroncher-Oldenburg G, Cummings DE & Delegates of the 2nd Diabetes Surgery, S. (2017) Metabolic Surgery in the Treatment Algorithm for Type 2 Diabetes: a Joint Statement by International Diabetes Organizations, Obes Surg. 27, 2–21. [DOI] [PubMed] [Google Scholar]
- 35.Ikramuddin S, Korner J, Lee WJ, Connett JE, Inabnet WB, Billington CJ, Thomas AJ, Leslie DB, Chong K, Jeffery RW, Ahmed L, Vella A, Chuang LM, Bessler M, Sarr MG, Swain JM, Laqua P, Jensen MD & Bantle JP (2013) Roux-en-Y gastric bypass vs intensive medical management for the control of type 2 diabetes, hypertension, and hyperlipidemia: the Diabetes Surgery Study randomized clinical trial, JAMA. 309, 2240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mingrone G, Panunzi S, De Gaetano A, Guidone C, Iaconelli A, Leccesi L, Nanni G, Pomp A, Castagneto M, Ghirlanda G & Rubino F (2012) Bariatric surgery versus conventional medical therapy for type 2 diabetes, The New England journal of medicine. 366, 1577–85. [DOI] [PubMed] [Google Scholar]
- 37.Buchwald H, Avidor Y, Braunwald E, Jensen MD, Pories W, Fahrbach K & Schoelles K (2004) Bariatric surgery: a systematic review and meta-analysis, JAMA. 292, 1724–37. [DOI] [PubMed] [Google Scholar]
- 38.Pories WJ, Swanson MS, MacDonald KG, Long SB, Morris PG, Brown BM, Barakat HA, deRamon RA, Israel G, Dolezal JM & et al. (1995) Who would have thought it? An operation proves to be the most effective therapy for adult-onset diabetes mellitus, Annals of surgery. 222, 339–50; discussion 350–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schauer PR, Burguera B, Ikramuddin S, Cottam D, Gourash W, Hamad G, Eid GM, Mattar S, Ramanathan R, Barinas-Mitchel E, Rao RH, Kuller L & Kelley D (2003) Effect of laparoscopic Roux-en Y gastric bypass on type 2 diabetes mellitus, Annals of surgery. 238, 467–84; discussion 84–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salinari S, Bertuzzi A, Guidone C, Previti E, Rubino F & Mingrone G (2013) Insulin sensitivity and secretion changes after gastric bypass in normotolerant and diabetic obese subjects, Annals of surgery. 257, 462–8. [DOI] [PubMed] [Google Scholar]
- 41.Lin E, Liang Z, Frediani J, Davis SS Jr., Sweeney JF, Ziegler TR, Phillips LS & Gletsu-Miller N (2010) Improvement in ss-cell function in patients with normal and hyperglycemia following Roux-en-Y gastric bypass surgery, American journal of physiology. 299, E706–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reed MA, Pories WJ, Chapman W, Pender J, Bowden R, Barakat H, Gavin TP, Green T, Tapscott E, Zheng D, Shankley N, Yieh L, Polidori D, Piccoli SP, Brenner-Gati L & Dohm GL (2011) Roux-en-Y gastric bypass corrects hyperinsulinemia implications for the remission of type 2 diabetes, The Journal of clinical endocrinology and metabolism. 96, 2525–31. [DOI] [PubMed] [Google Scholar]
- 43.Martinussen C, Bojsen-Moller KN, Dirksen C, Jacobsen SH, Jorgensen NB, Kristiansen VB, Holst JJ & Madsbad S (2015) Immediate enhancement of first-phase insulin secretion and unchanged glucose effectiveness in patients with type 2 diabetes after Roux-en-Y gastric bypass, American journal of physiology. 308, E535–44. [DOI] [PubMed] [Google Scholar]
- 44.Dirksen C, Bojsen-Moller KN, Jorgensen NB, Jacobsen SH, Kristiansen VB, Naver LS, Hansen DL, Worm D, Holst JJ & Madsbad S (2013) Exaggerated release and preserved insulinotropic action of glucagon-like peptide-1 underlie insulin hypersecretion in glucose-tolerant individuals after Roux-en-Y gastric bypass, Diabetologia. 56, 2679–87. [DOI] [PubMed] [Google Scholar]
- 45.Basso N, Soricelli E, Castagneto-Gissey L, Casella G, Albanese D, Fava F, Donati C, Tuohy K, Angelini G, La Neve F, Severino A, Kamvissi-Lorenz V, Birkenfeld AL, Bornstein S, Manco M & Mingrone G (2016) Insulin Resistance, Microbiota, and Fat Distribution Changes by a New Model of Vertical Sleeve Gastrectomy in Obese Rats, Diabetes. 65, 2990–3001. [DOI] [PubMed] [Google Scholar]
- 46.Chambers AP, Jessen L, Ryan KK, Sisley S, Wilson-Perez HE, Stefater MA, Gaitonde SG, Sorrell JE, Toure M, Berger J, D’Alessio DA, Woods SC, Seeley RJ & Sandoval DA (2011) Weight-independent changes in blood glucose homeostasis after gastric bypass or vertical sleeve gastrectomy in rats, Gastroenterology. 141, 950–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chambers AP, Stefater MA, Wilson-Perez HE, Jessen L, Sisley S, Ryan KK, Gaitonde S, Sorrell JE, Toure M, Berger J, D’Alessio DA, Sandoval DA, Seeley RJ & Woods SC (2011) Similar effects of roux-en-Y gastric bypass and vertical sleeve gastrectomy on glucose regulation in rats, Physiology & behavior. 105, 120–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jimenez A, Mari A, Casamitjana R, Lacy A, Ferrannini E & Vidal J (2014) GLP-1 and glucose tolerance after sleeve gastrectomy in morbidly obese subjects with type 2 diabetes, Diabetes. 63, 3372–7. [DOI] [PubMed] [Google Scholar]
- 49.Jimenez A, Casamitjana R, Viaplana-Masclans J, Lacy A & Vidal J (2013) GLP-1 action and glucose tolerance in subjects with remission of type 2 diabetes after gastric bypass surgery, Diabetes care. 36, 2062–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jorgensen NB, Dirksen C, Bojsen-Moller KN, Jacobsen SH, Worm D, Hansen DL, Kristiansen VB, Naver L, Madsbad S & Holst JJ (2013) Exaggerated glucagon-like peptide 1 response is important for improved beta-cell function and glucose tolerance after Roux-en-Y gastric bypass in patients with type 2 diabetes, Diabetes. 62, 3044–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shah M, Law JH, Micheletto F, Sathananthan M, Dalla Man C, Cobelli C, Rizza RA, Camilleri M, Zinsmeister AR & Vella A (2014) Contribution of endogenous glucagon-like peptide 1 to glucose metabolism after Roux-en-Y gastric bypass, Diabetes. 63, 483–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vetter ML, Wadden TA, Teff KL, Khan ZF, Carvajal R, Ritter S, Moore RH, Chittams JL, Iagnocco A, Murayama K, Korus G, Williams NN & Rickels MR (2015) GLP-1 plays a limited role in improved glycemia shortly after Roux-en-Y gastric bypass: a comparison with intensive lifestyle modification, Diabetes. 64, 434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wilson-Perez HE, Chambers AP, Ryan KK, Li B, Sandoval DA, Stoffers D, Drucker DJ, Perez-Tilve D & Seeley RJ (2013) Vertical sleeve gastrectomy is effective in two genetic mouse models of glucagon-like Peptide 1 receptor deficiency, Diabetes. 62, 2380–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Douros JD, Lewis AG, Smith EP, Niu J, Capozzi M, Wittmann A, Campbell J, Tong J, Wagner C, Mahbod P, Seeley R & D’Alessio DA (2018) Enhanced Glucose Control Following Vertical Sleeve Gastrectomy Does Not Require a beta-Cell Glucagon-Like Peptide 1 Receptor, Diabetes. 67, 1504–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mokadem M, Zechner JF, Margolskee RF, Drucker DJ & Aguirre V (2014) Effects of Roux-en-Y gastric bypass on energy and glucose homeostasis are preserved in two mouse models of functional glucagon-like peptide-1 deficiency, Mol Metab. 3, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ryan KK, Tremaroli V, Clemmensen C, Kovatcheva-Datchary P, Myronovych A, Karns R, Wilson-Perez HE, Sandoval DA, Kohli R, Backhed F & Seeley RJ (2014) FXR is a molecular target for the effects of vertical sleeve gastrectomy, Nature. 509, 183–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ding L, Zhang E, Yang Q, Jin L, Sousa KM, Dong B, Wang Y, Tu J, Ma X, Tian J, Zhang H, Fang Z, Guan A, Zhang Y, Wang Z, Moore DD, Yang L & Huang W (2021) Vertical sleeve gastrectomy confers metabolic improvements by reducing intestinal bile acids and lipid absorption in mice, Proceedings of the National Academy of Sciences of the United States of America. 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Utzschneider KM, Kratz M, Damman CJ & Hullar M (2016) Mechanisms Linking the Gut Microbiome and Glucose Metabolism, The Journal of clinical endocrinology and metabolism. 101, 1445–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Siljander H, Honkanen J & Knip M (2019) Microbiome and type 1 diabetes, EBioMedicine. 46, 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morrison DJ & Preston T (2016) Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism, Gut Microbes. 7, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Asarat M, Apostolopoulos V, Vasiljevic T & Donkor O (2016) Short-Chain Fatty Acids Regulate Cytokines and Th17/Treg Cells in Human Peripheral Blood Mononuclear Cells in vitro, Immunol Invest. 45, 205–22. [DOI] [PubMed] [Google Scholar]
- 62.Nastasi C, Fredholm S, Willerslev-Olsen A, Hansen M, Bonefeld CM, Geisler C, Andersen MH, Odum N & Woetmann A (2017) Butyrate and propionate inhibit antigen-specific CD8(+) T cell activation by suppressing IL-12 production by antigen-presenting cells, Scientific reports. 7, 14516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun J, Furio L, Mecheri R, van der Does AM, Lundeberg E, Saveanu L, Chen Y, van Endert P, Agerberth B & Diana J (2015) Pancreatic beta-Cells Limit Autoimmune Diabetes via an Immunoregulatory Antimicrobial Peptide Expressed under the Influence of the Gut Microbiota, Immunity. 43, 304–17. [DOI] [PubMed] [Google Scholar]
- 64.Miani M, Le Naour J, Waeckel-Enee E, Verma SC, Straube M, Emond P, Ryffel B, van Endert P, Sokol H & Diana J (2018) Gut Microbiota-Stimulated Innate Lymphoid Cells Support beta-Defensin 14 Expression in Pancreatic Endocrine Cells, Preventing Autoimmune Diabetes, Cell metabolism. 28, 557–572 e6. [DOI] [PubMed] [Google Scholar]
- 65.Pingitore A, Gonzalez-Abuin N, Ruz-Maldonado I, Huang GC, Frost G & Persaud SJ (2019) Short chain fatty acids stimulate insulin secretion and reduce apoptosis in mouse and human islets in vitro: Role of free fatty acid receptor 2, Diabetes, obesity & metabolism. 21, 330–339. [DOI] [PubMed] [Google Scholar]
- 66.Kleinert M, Clemmensen C, Hofmann SM, Moore MC, Renner S, Woods SC, Huypens P, Beckers J, de Angelis MH, Schurmann A, Bakhti M, Klingenspor M, Heiman M, Cherrington AD, Ristow M, Lickert H, Wolf E, Havel PJ, Muller TD & Tschop MH (2018) Animal models of obesity and diabetes mellitus, Nature reviews. 14, 140–162. [DOI] [PubMed] [Google Scholar]
- 67.Kulkarni RN, Wang ZL, Wang RM, Hurley JD, Smith DM, Ghatei MA, Withers DJ, Gardiner JV, Bailey CJ & Bloom SR (1997) Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice, The Journal of clinical investigation. 100, 2729–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen PC, Kryukova YN & Shyng SL (2013) Leptin regulates KATP channel trafficking in pancreatic beta-cells by a signaling mechanism involving AMP-activated protein kinase (AMPK) and cAMP-dependent protein kinase (PKA), The Journal of biological chemistry. 288, 34098–34109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wu Y, Shyng SL & Chen PC (2015) Concerted Trafficking Regulation of Kv2.1 and KATP Channels by Leptin in Pancreatic beta-Cells, The Journal of biological chemistry. 290, 29676–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu Y, Fortin DA, Cochrane VA, Chen PC & Shyng SL (2017) NMDA receptors mediate leptin signaling and regulate potassium channel trafficking in pancreatic beta-cells, The Journal of biological chemistry. 292, 15512–15524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ye R, Holland WL, Gordillo R, Wang M, Wang QA, Shao M, Morley TS, Gupta RK, Stahl A & Scherer PE (2014) Adiponectin is essential for lipid homeostasis and survival under insulin deficiency and promotes beta-cell regeneration, eLife. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ye R, Wang M, Wang QA & Scherer PE (2015) Adiponectin-mediated antilipotoxic effects in regenerating pancreatic islets, Endocrinology. 156, 2019–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Qiao L, Saget S, Lu C, Hay WW Jr., Karsenty G & Shao J (2021) Adiponectin Promotes Maternal beta-Cell Expansion Through Placental Lactogen Expression, Diabetes. 70, 132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lo JC, Ljubicic S, Leibiger B, Kern M, Leibiger IB, Moede T, Kelly ME, Chatterjee Bhowmick D, Murano I, Cohen P, Banks AS, Khandekar MJ, Dietrich A, Flier JS, Cinti S, Bluher M, Danial NN, Berggren PO & Spiegelman BM (2014) Adipsin is an adipokine that improves beta cell function in diabetes, Cell. 158, 41–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gomez-Banoy N, Guseh JS, Li G, Rubio-Navarro A, Chen T, Poirier B, Putzel G, Rosselot C, Pabon MA, Camporez JP, Bhambhani V, Hwang SJ, Yao C, Perry RJ, Mukherjee S, Larson MG, Levy D, Dow LE, Shulman GI, Dephoure N, Garcia-Ocana A, Hao M, Spiegelman BM, Ho JE & Lo JC (2019) Adipsin preserves beta cells in diabetic mice and associates with protection from type 2 diabetes in humans, Nature medicine. 25, 1739–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fan W & Evans RM (2017) Exercise Mimetics: Impact on Health and Performance, Cell metabolism. 25, 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E, Mihaylova MM, Nelson MC, Zou Y, Juguilon H, Kang H, Shaw RJ & Evans RM (2008) AMPK and PPARdelta agonists are exercise mimetics, Cell. 134, 405–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pedersen BK & Febbraio MA (2008) Muscle as an endocrine organ: focus on muscle-derived interleukin-6, Physiological reviews. 88, 1379–406. [DOI] [PubMed] [Google Scholar]
- 79.Febbraio MA, Steensberg A, Keller C, Starkie RL, Nielsen HB, Krustrup P, Ott P, Secher NH & Pedersen BK (2003) Glucose ingestion attenuates interleukin-6 release from contracting skeletal muscle in humans, The Journal of physiology. 549, 607–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, Ramm G, Prelovsek O, Hohnen-Behrens C, Watt MJ, James DE, Kemp BE, Pedersen BK & Febbraio MA (2006) Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase, Diabetes. 55, 2688–97. [DOI] [PubMed] [Google Scholar]
- 81.Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, Eppler E, Bouzakri K, Wueest S, Muller YD, Hansen AM, Reinecke M, Konrad D, Gassmann M, Reimann F, Halban PA, Gromada J, Drucker DJ, Gribble FM, Ehses JA & Donath MY (2011) Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells, Nature medicine. 17, 1481–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jalabert A, Vial G, Guay C, Wiklander OP, Nordin JZ, Aswad H, Forterre A, Meugnier E, Pesenti S, Regazzi R, Danty-Berger E, Ducreux S, Vidal H, El-Andaloussi S, Rieusset J & Rome S (2016) Exosome-like vesicles released from lipid-induced insulin-resistant muscles modulate gene expression and proliferation of beta recipient cells in mice, Diabetologia. 59, 1049–58. [DOI] [PubMed] [Google Scholar]
- 83.Hussain MA, Song WJ & Wolfe A (2015) There is Kisspeptin - And Then There is Kisspeptin, Trends in endocrinology and metabolism: TEM. 26, 564–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Song WJ, Mondal P, Wolfe A, Alonso LC, Stamateris R, Ong BW, Lim OC, Yang KS, Radovick S, Novaira HJ, Farber EA, Farber CR, Turner SD & Hussain MA (2014) Glucagon regulates hepatic kisspeptin to impair insulin secretion, Cell metabolism. 19, 667–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kliewer SA & Mangelsdorf DJ (2019) A Dozen Years of Discovery: Insights into the Physiology and Pharmacology of FGF21, Cell metabolism. 29, 246–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wente W, Efanov AM, Brenner M, Kharitonenkov A, Koster A, Sandusky GE, Sewing S, Treinies I, Zitzer H & Gromada J (2006) Fibroblast growth factor-21 improves pancreatic beta-cell function and survival by activation of extracellular signal-regulated kinase 1/2 and Akt signaling pathways, Diabetes. 55, 2470–8. [DOI] [PubMed] [Google Scholar]
- 87.Shirakawa J, De Jesus DF & Kulkarni RN (2017) Exploring inter-organ crosstalk to uncover mechanisms that regulate beta-cell function and mass, European journal of clinical nutrition. 71, 896–903. [DOI] [PubMed] [Google Scholar]
- 88.Wang X, Wei W, Krzeszinski JY, Wang Y & Wan Y (2015) A Liver-Bone Endocrine Relay by IGFBP1 Promotes Osteoclastogenesis and Mediates FGF21-Induced Bone Resorption, Cell metabolism. 22, 811–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rajwani A, Ezzat V, Smith J, Yuldasheva NY, Duncan ER, Gage M, Cubbon RM, Kahn MB, Imrie H, Abbas A, Viswambharan H, Aziz A, Sukumar P, Vidal-Puig A, Sethi JK, Xuan S, Shah AM, Grant PJ, Porter KE, Kearney MT & Wheatcroft SB (2012) Increasing circulating IGFBP1 levels improves insulin sensitivity, promotes nitric oxide production, lowers blood pressure, and protects against atherosclerosis, Diabetes. 61, 915–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lu J, Liu KC, Schulz N, Karampelias C, Charbord J, Hilding A, Rautio L, Bertolino P, Ostenson CG, Brismar K & Andersson O (2016) IGFBP1 increases beta-cell regeneration by promoting alpha- to beta-cell transdifferentiation, The EMBO journal. 35, 2026–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mellado-Gil J, Rosa TC, Demirci C, Gonzalez-Pertusa JA, Velazquez-Garcia S, Ernst S, Valle S, Vasavada RC, Stewart AF, Alonso LC & Garcia-Ocana A (2011) Disruption of hepatocyte growth factor/c-Met signaling enhances pancreatic beta-cell death and accelerates the onset of diabetes, Diabetes. 60, 525–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Alvarez-Perez JC, Ernst S, Demirci C, Casinelli GP, Mellado-Gil JM, Rausell-Palamos F, Vasavada RC & Garcia-Ocana A (2014) Hepatocyte growth factor/c-Met signaling is required for beta-cell regeneration, Diabetes. 63, 216–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Heit JJ, Karnik SK & Kim SK (2006) Intrinsic regulators of pancreatic beta-cell proliferation, Annual review of cell and developmental biology. 22, 311–38. [DOI] [PubMed] [Google Scholar]
- 94.El Ouaamari A, Kawamori D, Dirice E, Liew CW, Shadrach JL, Hu J, Katsuta H, Hollister-Lock J, Qian WJ, Wagers AJ & Kulkarni RN (2013) Liver-derived systemic factors drive beta cell hyperplasia in insulin-resistant states, Cell reports. 3, 401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.El Ouaamari A, Dirice E, Gedeon N, Hu J, Zhou JY, Shirakawa J, Hou L, Goodman J, Karampelias C, Qiang G, Boucher J, Martinez R, Gritsenko MA, De Jesus DF, Kahraman S, Bhatt S, Smith RD, Beer HD, Jungtrakoon P, Gong Y, Goldfine AB, Liew CW, Doria A, Andersson O, Qian WJ, Remold-O’Donnell E & Kulkarni RN (2016) SerpinB1 Promotes Pancreatic beta Cell Proliferation, Cell metabolism. 23, 194–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Thorens B (2014) Neural regulation of pancreatic islet cell mass and function, Diabetes, obesity & metabolism. 16 Suppl 1, 87–95. [DOI] [PubMed] [Google Scholar]
- 97.Tang SC, Baeyens L, Shen CN, Peng SJ, Chien HJ, Scheel DW, Chamberlain CE & German MS (2018) Human pancreatic neuro-insular network in health and fatty infiltration, Diabetologia. 61, 168–181. [DOI] [PubMed] [Google Scholar]
- 98.Gautam D, Han SJ, Duttaroy A, Mears D, Hamdan FF, Li JH, Cui Y, Jeon J & Wess J (2007) Role of the M3 muscarinic acetylcholine receptor in beta-cell function and glucose homeostasis, Diabetes, obesity & metabolism. 9 Suppl 2, 158–69. [DOI] [PubMed] [Google Scholar]
- 99.Teff KL (2011) How neural mediation of anticipatory and compensatory insulin release helps us tolerate food, Physiology & behavior. 103, 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.D’Alessio DA, Kieffer TJ, Taborsky GJ Jr. & Havel PJ (2001) Activation of the parasympathetic nervous system is necessary for normal meal-induced insulin secretion in rhesus macaques, The Journal of clinical endocrinology and metabolism. 86, 1253–9. [DOI] [PubMed] [Google Scholar]
- 101.Osundiji MA & Evans ML (2013) Brain control of insulin and glucagon secretion, Endocrinology and metabolism clinics of North America. 42, 1–14. [DOI] [PubMed] [Google Scholar]
- 102.Capozzi ME, Svendsen B, Encisco SE, Lewandowski SL, Martin MD, Lin H, Jaffe JL, Coch RW, Haldeman JM, MacDonald PE, Merrins MJ, D’Alessio DA & Campbell JE (2019) beta Cell tone is defined by proglucagon peptides through cAMP signaling, JCI Insight. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rodriguez-Diaz R, Abdulreda MH, Formoso AL, Gans I, Ricordi C, Berggren PO & Caicedo A (2011) Innervation patterns of autonomic axons in the human endocrine pancreas, Cell metabolism. 14, 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rodriguez-Diaz R, Dando R, Jacques-Silva MC, Fachado A, Molina J, Abdulreda MH, Ricordi C, Roper SD, Berggren PO & Caicedo A (2011) Alpha cells secrete acetylcholine as a non-neuronal paracrine signal priming beta cell function in humans, Nature medicine. 17, 888–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Reilly SM & Saltiel AR (2017) Adapting to obesity with adipose tissue inflammation, Nature reviews. 13, 633–643. [DOI] [PubMed] [Google Scholar]
- 106.Wisse BE (2004) The inflammatory syndrome: the role of adipose tissue cytokines in metabolic disorders linked to obesity, J Am Soc Nephrol. 15, 2792–800. [DOI] [PubMed] [Google Scholar]
- 107.Ying W, Fu W, Lee YS & Olefsky JM (2020) The role of macrophages in obesity-associated islet inflammation and beta-cell abnormalities, Nature reviews. 16, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Calderon B, Carrero JA, Ferris ST, Sojka DK, Moore L, Epelman S, Murphy KM, Yokoyama WM, Randolph GJ & Unanue ER (2015) The pancreas anatomy conditions the origin and properties of resident macrophages, The Journal of experimental medicine. 212, 1497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ying W, Lee YS, Dong Y, Seidman JS, Yang M, Isaac R, Seo JB, Yang BH, Wollam J, Riopel M, McNelis J, Glass CK, Olefsky JM & Fu W (2019) Expansion of Islet-Resident Macrophages Leads to Inflammation Affecting beta Cell Proliferation and Function in Obesity, Cell metabolism. 29, 457–474 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Olefsky JM & Glass CK (2010) Macrophages, inflammation, and insulin resistance, Annu Rev Physiol. 72, 219–46. [DOI] [PubMed] [Google Scholar]
- 111.Ferris ST, Zakharov PN, Wan X, Calderon B, Artyomov MN, Unanue ER & Carrero JA (2017) The islet-resident macrophage is in an inflammatory state and senses microbial products in blood, The Journal of experimental medicine. 214, 2369–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA & Donath MY (2002) Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets, The Journal of clinical investigation. 110, 851–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Westwell-Roper C, Nackiewicz D, Dan M & Ehses JA (2014) Toll-like receptors and NLRP3 as central regulators of pancreatic islet inflammation in type 2 diabetes, Immunology and cell biology. 92, 314–23. [DOI] [PubMed] [Google Scholar]
- 114.Wei Z, Yoshihara E, He N, Hah N, Fan W, Pinto AFM, Huddy T, Wang Y, Ross B, Estepa G, Dai Y, Ding N, Sherman MH, Fang S, Zhao X, Liddle C, Atkins AR, Yu RT, Downes M & Evans RM (2018) Vitamin D Switches BAF Complexes to Protect beta Cells, Cell. 173, 1135–1149 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Blum B, Roose AN, Barrandon O, Maehr R, Arvanites AC, Davidow LS, Davis JC, Peterson QP, Rubin LL & Melton DA (2014) Reversal of beta cell de-differentiation by a small molecule inhibitor of the TGFbeta pathway, eLife. 3, e02809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Herder C, Dalmas E, Boni-Schnetzler M & Donath MY (2015) The IL-1 Pathway in Type 2 Diabetes and Cardiovascular Complications, Trends in endocrinology and metabolism: TEM. 26, 551–63. [DOI] [PubMed] [Google Scholar]
- 117.Dror E, Dalmas E, Meier DT, Wueest S, Thevenet J, Thienel C, Timper K, Nordmann TM, Traub S, Schulze F, Item F, Vallois D, Pattou F, Kerr-Conte J, Lavallard V, Berney T, Thorens B, Konrad D, Boni-Schnetzler M & Donath MY (2017) Postprandial macrophage-derived IL-1beta stimulates insulin, and both synergistically promote glucose disposal and inflammation, Nature immunology. 18, 283–292. [DOI] [PubMed] [Google Scholar]
- 118.Kayali AG, Lopez AD, Hao E, Hinton A, Hayek A & King CC (2012) The SDF-1alpha/CXCR4 axis is required for proliferation and maturation of human fetal pancreatic endocrine progenitor cells, PloS one. 7, e38721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lee YS, Morinaga H, Kim JJ, Lagakos W, Taylor S, Keshwani M, Perkins G, Dong H, Kayali AG, Sweet IR & Olefsky J (2013) The fractalkine/CX3CR1 system regulates beta cell function and insulin secretion, Cell. 153, 413–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yano T, Liu Z, Donovan J, Thomas MK & Habener JF (2007) Stromal cell derived factor-1 (SDF-1)/CXCL12 attenuates diabetes in mice and promotes pancreatic beta-cell survival by activation of the prosurvival kinase Akt, Diabetes. 56, 2946–57. [DOI] [PubMed] [Google Scholar]
- 121.Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, Ogata F, Yagi N, Ohto U, Kimoto M, Miyake K, Tobe K, Arai H, Kadowaki T & Nagai R (2012) Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation, Cell metabolism. 15, 518–33. [DOI] [PubMed] [Google Scholar]
- 122.Lee NK, Sowa H, Hinoi E, Ferron M, Ahn JD, Confavreux C, Dacquin R, Mee PJ, McKee MD, Jung DY, Zhang Z, Kim JK, Mauvais-Jarvis F, Ducy P & Karsenty G (2007) Endocrine regulation of energy metabolism by the skeleton, Cell. 130, 456–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wei J, Hanna T, Suda N, Karsenty G & Ducy P (2014) Osteocalcin promotes beta-cell proliferation during development and adulthood through Gprc6a, Diabetes. 63, 1021–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lee NJ, Nguyen AD, Enriquez RF, Luzuriaga J, Bensellam M, Laybutt R, Baldock PA & Herzog H (2015) NPY signalling in early osteoblasts controls glucose homeostasis, Mol Metab. 4, 164–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Andersen DK, Korc M, Petersen GM, Eibl G, Li D, Rickels MR, Chari ST & Abbruzzese JL (2017) Diabetes, Pancreatogenic Diabetes, and Pancreatic Cancer, Diabetes. 66, 1103–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wakasugi H, Funakoshi A & Iguchi H (1998) Clinical assessment of pancreatic diabetes caused by chronic pancreatitis, J Gastroenterol. 33, 254–9. [DOI] [PubMed] [Google Scholar]
- 127.Malka D, Hammel P, Sauvanet A, Rufat P, O’Toole D, Bardet P, Belghiti J, Bernades P, Ruszniewski P & Levy P (2000) Risk factors for diabetes mellitus in chronic pancreatitis, Gastroenterology. 119, 1324–32. [DOI] [PubMed] [Google Scholar]
- 128.Moran A, Dunitz J, Nathan B, Saeed A, Holme B & Thomas W (2009) Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality, Diabetes care. 32, 1626–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sun J, Ni Q, Xie J, Xu M, Zhang J, Kuang J, Wang Y, Ning G & Wang Q (2019) beta-Cell Dedifferentiation in Patients With T2D With Adequate Glucose Control and Nondiabetic Chronic Pancreatitis, The Journal of clinical endocrinology and metabolism. 104, 83–94. [DOI] [PubMed] [Google Scholar]
- 130.Hull RL, Gibson RL, McNamara S, Deutsch GH, Fligner CL, Frevert CW, Ramsey BW & Sanda S (2018) Islet Interleukin-1beta Immunoreactivity Is an Early Feature of Cystic Fibrosis That May Contribute to beta-Cell Failure, Diabetes care. 41, 823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pannala R, Leirness JB, Bamlet WR, Basu A, Petersen GM & Chari ST (2008) Prevalence and clinical profile of pancreatic cancer-associated diabetes mellitus, Gastroenterology. 134, 981–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pelaez-Luna M, Takahashi N, Fletcher JG & Chari ST (2007) Resectability of presymptomatic pancreatic cancer and its relationship to onset of diabetes: a retrospective review of CT scans and fasting glucose values prior to diagnosis, The American journal of gastroenterology. 102, 2157–63. [DOI] [PubMed] [Google Scholar]
- 133.Pfeffer F, Koczan D, Adam U, Benz S, von Dobschuetz E, Prall F, Nizze H, Thiesen HJ, Hopt UT & Lobler M (2004) Expression of connexin26 in islets of Langerhans is associated with impaired glucose tolerance in patients with pancreatic adenocarcinoma, Pancreas. 29, 284–90. [DOI] [PubMed] [Google Scholar]
- 134.Basso D, Greco E, Fogar P, Pucci P, Flagiello A, Baldo G, Giunco S, Valerio A, Navaglia F, Zambon CF, Falda A, Pedrazzoli S & Plebani M (2006) Pancreatic cancer-derived S-100A8 N-terminal peptide: a diabetes cause?, Clin Chim Acta. 372, 120–8. [DOI] [PubMed] [Google Scholar]
- 135.Huang H, Dong X, Kang MX, Xu B, Chen Y, Zhang B, Chen J, Xie QP & Wu YL (2010) Novel blood biomarkers of pancreatic cancer-associated diabetes mellitus identified by peripheral blood-based gene expression profiles, The American journal of gastroenterology. 105, 1661–9. [DOI] [PubMed] [Google Scholar]
- 136.Sah RP, Nagpal SJ, Mukhopadhyay D & Chari ST (2013) New insights into pancreatic cancer-induced paraneoplastic diabetes, Nat Rev Gastroenterol Hepatol. 10, 423–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Javeed N, Sagar G, Dutta SK, Smyrk TC, Lau JS, Bhattacharya S, Truty M, Petersen GM, Kaufman RJ, Chari ST & Mukhopadhyay D (2015) Pancreatic Cancer-Derived Exosomes Cause Paraneoplastic beta-cell Dysfunction, Clin Cancer Res. 21, 1722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang Y, Ni Q, Sun J, Xu M, Xie J, Zhang J, Fang Y, Ning G & Wang Q (2020) Paraneoplastic beta Cell Dedifferentiation in Nondiabetic Patients with Pancreatic Cancer, The Journal of clinical endocrinology and metabolism. 105. [DOI] [PubMed] [Google Scholar]
- 139.Chung KM, Singh J, Lawres L, Dorans KJ, Garcia C, Burkhardt DB, Robbins R, Bhutkar A, Cardone R, Zhao X, Babic A, Vayrynen SA, Dias Costa A, Nowak JA, Chang DT, Dunne RF, Hezel AF, Koong AC, Wilhelm JJ, Bellin MD, Nylander V, Gloyn AL, McCarthy MI, Kibbey RG, Krishnaswamy S, Wolpin BM, Jacks T, Fuchs CS & Muzumdar MD (2020) Endocrine-Exocrine Signaling Drives Obesity-Associated Pancreatic Ductal Adenocarcinoma, Cell. 181, 832–847 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]