

Abstract
Microbial biosensors have diverse applications in metabolic engineering and medicine. Specific and accurate quantification of chemical concentrations allows for adaptive regulation of enzymatic pathways and temporally-precise expression of diagnostic reporters. Although biosensors should differentiate structurally similar ligands with distinct biological functions, such specific sensors are rarely found in nature and challenging to create. Using E. coli Nissle 1917, a generally regarded as safe microbe, we characterized two biosensor systems that promiscuously recognize aromatic amino acids or neurochemicals. To improve the sensors’ selectivity and sensitivity, we applied rational protein engineering by identifying and mutagenizing amino acid residues and successfully demonstrated the ligand-specific biosensors for phenylalanine, tyrosine, phenylethylamine, and tyramine. Additionally, our approach revealed insights into the uncharacterized structure of the FeaR regulator, including critical residues in ligand binding. These results lay the groundwork for developing kinetically-adaptive microbes for diverse applications.
Keywords: Aromatic amino acid, Aromatic neurochemical, Directed evolution, Probiotic, Protein engineering, Specific sensors
eTOC blurb
This work represents a considerable achievement in protein engineering and includes the ligand-specific sensors for aromatic amino acids such as phenylalanine and tyrosine as well as neurochemicals such as phenylethylamine and tyramine, which are structurally similar and have been implicated in a variety of medical conditions. It lays the groundwork for developing medically-relevant probiotic and wearable sensors with high specificity, which is critical to differentiate metabolites with divergent functions and create smart probiotics and cell-free biosensors for accurate diagnostics.
Graphical Abstract
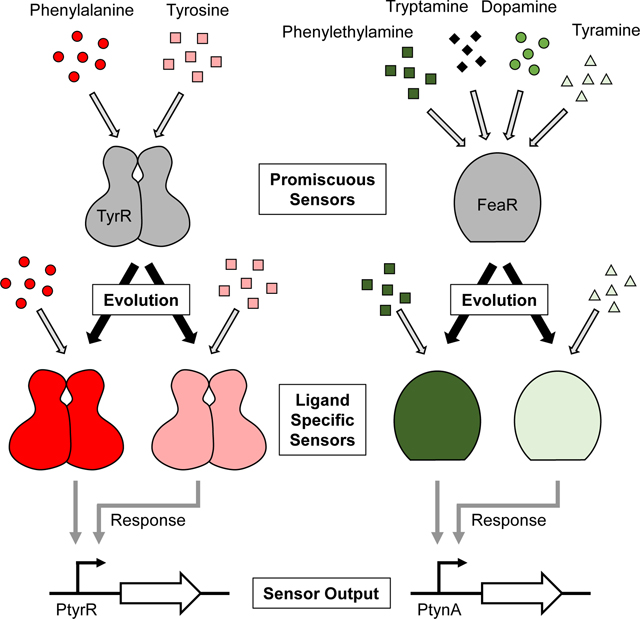
INTRODUCTION
Microbial biosensors can be utilized to kinetically regulate and quantify the products of metabolic pathways, diagnose diseases in vivo in probiotics, and analyze ex vivo samples through wearable, paper-based, and cell-free systems. Synthetic biological sensors can be created using several protein design approaches(Jones et al., 2017). However, sensors can also be obtained more efficiently by mining sensors that naturally exist in microbes(DeLorenzo et al., 2017; Hoynes-O’Connor et al., 2017). These sensors can often be directly transferred into probiotic organisms or purified for cell-free sensor applications. However, most natural sensors recognize multiple structurally similar, but functionally diverse, metabolites often found in close proximity. To accurately correlate the concentration of a chemical with biological outcomes, sensors need to differentiate between different chemicals and recognize the relevant chemical with the precise sensitivity.
Specific sensing is critical for many microbial sensor applications. A variety of protein engineering methods have been demonstrated for optimizing the selectivity and sensitivity of protein and RNA sensors. These approaches include directed evolution(Ellefson et al., 2018; Higgins et al., 2017; Meyer et al., 2019; Stainbrook and Tyo, 2019), rational design(Belsare et al., 2017), and computational de novo design(Taylor et al., 2016). Although each method has advantages, rational engineering can uniquely be performed using both basic knowledge of the protein structure and small library sizes, allowing for rapid construction and screening of libraries. In addition, when the structure of the protein is unknown, conserved or essential residues can also be identified through computational simulations or by aligning the sequence of the sensor with other proteins in the same family(Zvelebil et al., 1987). Despite advances in protein engineering, creating ligand-specific sense-and-respond systems remains challenging due to the need to couple subtle protein conformational changes with differential protein-DNA interactions and gene expression control(Glasgow et al., 2019; Peveler et al., 2016; Taylor et al., 2016), especially when the target ligands are structurally similar.
The aromatic amino acids phenylalanine (Phe) and tyrosine (tyr) are common microbial metabolic engineering products and precursors derived from the same pathway(Rodriguez et al., 2014). In addition, chronically elevated levels of structurally similar phenylalanine (Phe) and tyrosine (Tyr) are associated with the distinct disorders phenylketonuria and type 2 tyrosinemia, respectively (Table S1) (Camp et al., 2014; Cerone et al., 2002; Scriver, 2007). Sensors for both metabolites have been generated in different E. coli strains(Lin et al., 2018; Mahr et al., 2016). However, the sensors are based on the wild-type version of the multi-ligand responsive TyrR transcription factor from E. coli and have limited selectivity and low dynamic ranges. Similarly, the structurally similar amines phenylethylamine (PEA), tyramine (Tyra), and tryptamine (Trypta) are all commonly found in food and in the gut(Bhattarai et al., 2018; Chen et al., 2019; Marcobal et al., 2012; Shalaby, 1996), but contribute to distinct biological outcomes (Table S1). For example, extreme levels of PEA have been associated with a variety of psychological disorders(Irsfeld et al., 2013; Myojin et al., 1989), the presence of Tyra leads to catecholamine release and an increase in blood pressure(Bianchetti et al., 1982; Rafehi et al., 2019), and the presence of Trypta causes serotonin release and the stimulation of gastrointestinal motility(Takaki et al., 1985). Additionally, the presence of PEA, Tyra, and Trypta in food are indicators of microbial contamination(Marcobal et al., 2012), and eating foods with high levels of Tyra can lead to poisoning(Marcobal et al., 2012). Currently, there are no biosensors with high ligand specificity for these chemicals.
In this work, we generate sensors for the aromatic metabolites Phe, Tyr, PEA, and Trypta using Escherichia coli Nissle 1917 (EcN) as a host. To generate sensors for these aromatic metabolites, we identify and engineer two sensor systems: the TyrR (Phe and Tyr) sensor and the TynA-FeaR (aromatic amine) sensor system. We engineer the ligand selectivity of the TyrR and TynA-FeaR sensor systems and tune their sensitivity by rationally selecting and individually mutating amino acids in TyrR, TynA, and FeaR. This method of rational protein engineering quickly generates multiple small libraries that can be efficiently screened, making it an attractive approach for specificity engineering. Altogether, we generate the sensors specific for Phe, Tyr, PEA, or Tyra. In engineering FeaR, we also provide insights into the uncharacterized structure of FeaR and identify residues important for ligand binding. This work provides sensors with diverse applications in microbial biosensing as well as a generalizable approach to modulating the specificity of ligand-protein binding while maintaining protein-DNA interactions and thus downstream gene expression control.
Results
Developing specific phenylalanine and tyrosine sensors
Phe and Tyr are structurally similar metabolites utilized throughout the body for many processes as additives in food and animal feed and as precursors for many chemicals and pharmaceuticals (Figure 1A) (Liu et al., 2019). Notably, two distinct disorders (phenylketonuria and tyrosinemia) cause a buildup of dietary Phe or Tyr(Mitchell et al., 1990; Scriver, 2007), necessitating sensors that can differentiate between the two amino acids. Microbial TyrR regulates its target promoters in both a Phe- and Tyr-dependent manner (Figures 1B and S1A). In the absence of either amino acid, TyrR forms homodimers that bind to high-affinity recognition sites, or strong boxes(Pittard et al., 2005). When bound to Phe, the dimers interact with RNA polymerase to influence transcription initiation. When bound to Tyr, TyrR forms homohexamers that bind to both the strong box and the adjacent low affinity weak box and interact with RNA polymerase. When the binding sites are located upstream of the −35 site of the promoter, these interactions increase expression of the downstream gene. When the binding sites are overlapping or downstream of the −35 site, TyrR instead represses expression(Pittard et al., 2005). Given the complexity of DNA-TyrR binding and ligand-TyrR binding modes, we decided to take a stepwise approach to develop ligand-specific sensors with the hypothesis that targeted engineering of TyrR would enable its differential ligand binding without affecting DNA binding interactions.
Figure 1: Phenylalanine- and tyrosine-dependent regulation of the PtyrP promoter by TyrR is tyrosine-dominant.

A Structures of the two TyrR ligands phenylalanine (Phe) and tyrosine (Tyr). Structural differences between ligands are shown in red. B Schematic of how TyrR regulates the PtyrP promoter in a Phe- and Tyr-dependent manner. C Transfer curves for PtyrP with plasmid-based overexpression of wild-type TyrR in response to Phe, Tyr, and the specified concentrations of both Phe and Tyr. Points represent experimental data while lines represent the fitted curves (Table S2). Values and error bars are the average and standard deviation of biological triplicate, respectively. See also Figure S1.
We first characterized six E. coli-native promoters using genomic expression or plasmid-based overexpression of TyrR to identify the best promoter in EcN (Figures S1B and S1C). We selected PtyrP as our sensor as it displayed an activating response to Phe and a repressing response to Tyr, allowing for clear differentiation between the presence of each amino acid, especially with overexpression of TyrR (Figures 1B and 1C). The PtyrP promoter has a strong box upstream of the −35 site and a weak box overlapping the −35 site, allowing TyrR to induce expression in the presence of Phe and repress expression in the presence of Tyr. In addition, PtyrP showed the highest foldinduction in the presence of Phe (4-fold) compared to all tested promoters and strong repression in the presence of Tyr (59-fold). Notably, high expression levels of TyrR are required for Tyr-dependent repression of PtyrP, as shown by the lack of repression when only controlled by genomic TyrR expression (Figure S1B). This lack of response gives PtyrP a false appearance of Phe-specificity when paired with low TyrR expression levels. However, we found that TyrR regulated the promoter in a Tyr-dominant manner. In the presence of Tyr and overexpression of TyrR, TyrR repressed PtyrP independent of the presence of Phe (Figure 1C).
The E274Q TyrR mutation was previously shown to prevent TyrR from forming hexamers, which occurs when TyrR is bound to Tyr(Kwok et al., 1995). Since hexamerization is required for Tyr-mediated repression of PtyrP, this mutation should render TyrR nonresponsive to Tyr but maintain Phe-dependent induction. When we inserted the mutation into the plasmid-expressed TyrR, Tyr-dependent repression of PtyrP was mitigated and the Tyr-dominating response of the transcription factor was eliminated (Figure 2A). In addition, the mutant had an improved fold-induction in response to Phe (16-fold). This is the highest dynamic range demonstrated by a Phe sensor to date.
Figure 2: Development of phenylalanine- and tyrosine-specific sensors.

A Response of the PtyrP promoter to 1 mM phenylalanine (Phe), 1 mM tyrosine (Tyr), and both 1mM Phe and 1 mM Tyr with plasmid-based overexpression of wild-type (WT), E274Q mutant, and R10F mutant TyrR. B Structure of the Phe binding pocket in the N-terminal regions of the TyrR homodimer. TyrR monomers are in white and magenta. Selected amino acids with known roles in Phe-dependent modulation of RNA polymerase are shown in elemental coloring: carbon atoms are shown in green, nitrogen in blue, oxygen in red, and sulfur in yellow. The structure was obtained from RCSB Protein Data Bank (Reference ID 2JHE) and the image was generated using PyMOL. C Modulation of the Phe-induction threshold for the PtyrP promoter by saturation mutagenesis of the TyrR’s Phe binding pocket. Two TyrR mutants, E274Q+T14V and E274Q+D103S, were obtained with increased KA values (2-fold and 3-fold higher than the E274Q mutant, respectively). D-G Transfer curves for PtyrP with plasmid-based overexpression of E274Q (D), E274Q+T14V (E), E274Q+D103S (F), and R10F (G) TyrR variants in response to Phe, Tyr, and the specified concentrations of both Phe and Tyr. Points represent experimental data while lines represent the fitted curves (Table S2). The responses to Tyr (D-F) or Phe (G) were not fit due to a lack of response. Values and error bars are the average and standard deviation of biological triplicate, respectively. AA, amino acid. See also Figures S2–S5.
We next tuned the Phe-sensitivity of the E274Q mutant to create sensors that better align with variable physiological concentrations and improve the utility of the sensors for therapeutic and diagnostic applications. We selected eight amino acid positions with known roles in TyrR-mediated promoter induction (Figure 2B)(Verger et al., 2007; Yang et al., 1996). Each amino acid has been shown to be important, but potentially nonessential, for Phe binding. When we obtained the transfer curve of the parent E274Q sensor, half-maximal induction was observed at 0.04 mM Phe (Figures 2C and 2D), lower than the Phe concentrations in human intestines, serum, and urine (Table S1, 0.05–0.3 mM)(Adibi and Mercer, 1973; Kouchiwa et al., 2012; Lin et al., 2018). Thus, we performed saturation mutagenesis on each amino acid individually and screened the small targeted libraries for a reduced sensitivity to Phe (Figure S2). Three variants displayed reproducibly lower sensitivities: E274Q+T14V, E274Q+D103H, and E274Q+D103S with KA values of 0.08, 0.11, and 0.13 mM, respectively (Figures 2C and S3). The E274Q+T14V and E274Q+D103S variants were selected for further characterization as they displayed the widest range of sensitivities and maintained the highest fold-inductions. Both mutants displayed consistent sensitivities to Phe in the presence and absence of Tyr and had no apparent response to Tyr alone (Figures 2E and 2F). When we combined both mutations in E274Q TyrR, the response to Phe was eliminated. These Phe-specific sensors have the potential to be applied to kinetically diagnose and treat disorders that cause Phe-dysregulation without interference from intestinal Tyr.
We also engineered TyrR for Tyr-specificity. Because TyrR binds to Phe and Tyr using two separate binding pockets, the libraries designed to identify less sensitive Phe sensors were used to identify variants completely devoid of Phe-activity, while maintaining Tyr-activity (Figure S4). The most selective mutant, R10F, demonstrated 15-fold repression in the presence of Tyr independent of the presence of Phe (Figure 2A). The positively charged R10 residue is positioned near the region where the ligand’s carboxyl group could stabilize Phe binding. However, conversion to a hydrophobic F10 may instead prevent this stabilization. In addition, no significant response to Phe alone was observed. When we characterized the transfer curve of the mutant, we identified KA of 0.05 mM in response to Tyr (Figure 2G). In addition, we demonstrated how rational saturation mutagenesis can effectively generate functionally diverse, specific sensors from one promiscuous transcription factor. As expected for all chemical sensors, one caveat of these Phe- and Tyr-specific sensors is that they cannot differentiate elevated concentrations of the two ligands in rich medium, which has saturating concentrations of the two amino acids (Figure S5) (Sezonov et al., 2007).
Engineering ligand-specific aromatic amine sensors
The body harbors a variety of aromatic amines with diverse neurological functions. Many of these metabolites, including dopamine (DA), PEA, Tyra, and Trypta, can be found in the intestines(Bhattarai et al., 2018; Chen et al., 2019). Many commensal microbes can convert these amines into aldehydes through monoamine oxidases (Figure 3A)(Zeng and Spiro, 2013). These aldehydes can be further catabolized to carboxylic acids by aldehyde dehydrogenases(Zeng and Spiro, 2013). In some E. coli strains, expression of the monoamine oxidase TynA and aldehyde dehydrogenase FeaB are regulated by the AraC-type FeaR transcription factor, which was shown to induce expression of TynA and FeaB in the presence of multiple aldehydes formed from the corresponding amines(Zeng and Spiro, 2013). We found this tynA-feaB-feaR gene cluster to be present in select commensal E. coli strains, but not in EcN (Figure S6). This promiscuous sensing system provides an opportunity to sense a variety of important aromatic amines using engineered microbes. However, since each amine has a different function, it is essential that the microbe can differentiate the metabolites.
Figure 3: Development and characterization of sensors for aromatic amines.

A Structures of the four TynA substrates phenylethylamine (PEA), tyramine (Tyra), dopamine (DA), and tryptamine (Trypta) as well as the corresponding aldehyde ligands for FeaR. Structural differences between ligands are shown in green. B Schematic of aromatic amine sensing using the TynA-FeaR system. The TynA enzyme converts periplasmic amines to aldehydes, which are imported into the cytoplasm. In the presence of the aldehydes, FeaR induces expression from the PtynA promoter. C Validation that FeaR regulates PtynA in response to aldehydes and not amines. DA induces PtynA expression in the presence, but not absence of TynA. D Transfer curves of PtynA after 24 h of induction with DA, PEA, Tyra, and Trypta. Points represent experimental data while lines represent the fitted curves (Table S2). Values and error bars are the average and standard deviation of biological triplicate, respectively. See also Figures S6 and S7.
To develop sensors for aromatic amines, we first characterized the TynA-FeaR system in EcN. We constructed a sensor plasmid, which consisted of constitutively expressed TynA and FeaR from E. coli MG1655, and a reporter plasmid, which consisted of GFP under the control of PtynA (Figure 3B). We next confirmed that the system induces expression of PtynA in response to aromatic aldehydes, not amines (Figure 3C). In the presence of TynA, the addition of 0.5 mM DA resulted in a 410-fold increase in GFP expression. In contrast, the induction was eliminated when TynA was inactivated. We next characterized the sensitivity and selectivity to different ligands. The system displayed the strongest response to the addition of PEA or Tyra, inducing expression of the reporter by more than 1500-fold with the addition of 1.25 mM of each amine (Figures 3D and S7). The addition of DA resulted in the next highest response, with approximately 900-fold induction. Trypta had the weakest response, inducing expression by approximately 280-fold. The system showed a similar sensitivity to each ligand, with half-maximal induction occurring at 0.07–0.09 mM (Table S2).
Due to their superior induction and roles in numerous processes, we selected PEA and Tyra as targets for engineering selectivity in the TynA-FeaR system. This system presents the interesting opportunity to optimize ligand-selectivity using two different knobs of control. Either TynA or FeaR can be engineered for selectivity. However, the protein structure and mode of ligand-binding of FeaR have not been elucidated. In contrast, the structure of TynA has been characterized. Thus, we first focused on improving the amine selectivity of TynA. Several residues in TynA have been identified as catalytically essential, including D413 and Y496 (Figures 4A and S8)(Saysell et al., 2002; Wilmot et al., 1997). To optimize the selectivity of TynA to PEA and Tyra without fully eliminating enzymatic activity, we elected to individually mutate the amino acids adjacent to these two essential residues, including positions 411–412, 414–416, 493–495, and 497–498.
Figure 4: Identification of phenylethylamine- and tyramine-selective sensors through TynA engineering.

A Structure of the TynA catalytic domain. Unmutagenized amino acids with known catalytic roles are in red. Amino acids selected for mutagenesis are in blue. The structure was obtained from RCSB Protein Data Bank (Reference ID 6EZZ) and the image was generated using PyMOL. B GFP fluorescence from PtynA when induced for 24 h with 0 mM and 1 mM dopamine (DA), phenylethylamine (PEA), tyramine (Tyra), and tryptamine (Trypta). PtynA was regulated by wild-type TynA (left), G494S TynA (middle), or G415H TynA (right) and either wild-type or A81T (*) FeaR. C and D Transfer curves of PtynA after 24 h of induction with DA, PEA, Tyra, and Trypta. PtynA was regulated by the G494S TynA mutant with the A81T FeaR mutant (C) or the G415H TynA mutant with wild-type FeaR (D). Points represent experimental data while lines represent the fitted curves (Table S2). The responses of G494S* to DA, Tyr, and Trypta and the responses of G415H to DA and Trypta were not fit due to a lack of response. Values and error bars are the average and standard deviation of biological triplicate, respectively. See also Figures S8–S12.
To quickly screen variants from the libraries for a response to each ligand, we developed a growth-based assay by incorporating the carbenicillin resistance gene, bla, and the sucrose counterselection gene, sacB, onto the reporter plasmid under the control of PtynA (Figure S9). When the constitutively expressed TynA produces an aldehyde, both Bla and SacB are expressed. To isolate variants with improved selectivity to a specific amine, rounds of positive selection (with the amine of interest and carbenicillin) and negative selection (with all other amines and sucrose) can be performed. In positive selection, cells that fail to respond to the amine of interest will be killed by carbenicillin. In negative selection, cells that remain promiscuous and respond to one of the undesired amines will be killed by sucrose. After multiple rounds of these dual selections, the activities of variants were validated by fluorescence measurements.
We successfully identified two PEA-specific variants (Figure 4B). Both variants possessed a G494S mutation in TynA. However, one variant displayed a stronger fluorescence output in response to PEA. Upon further investigation, we identified an A81T mutation in the FeaR protein of the stronger sensor. Under the pressure of carbenicillin in our dual-selection system, a cell bearing the weak PEA-specific G494S TynA mutant additionally acquired the A81T FeaR mutation to improve bla expression and cell survival. This double mutant sensor (G494S*) displayed 140-fold induction in response to PEA with an insignificant response to other tested amines (Figures 4C and S10A), while the single mutant sensor (G494S) displayed 50-fold induction by PEA (Figure S11).
We also identified two variants with improved selectivity to Tyra (Figure S12). The best sensor, G415H, induced expression of the reporter by 200-fold and 79-fold in response to Tyra and PEA, respectively, with minimal response to DA and Trypta (Figures 4B, 4D, and S10B). The G494S and G494S* variants are the PEA-specific biosensors and the G415H variant is the Tyra-selective biosensor. In addition, this work further demonstrates how the ligand specificity of a protein can be quickly and effectively optimized through targeted mutagenesis of catalytically adjacent residues.
Characterization and engineering of FeaR
Intrigued by the appearance of the A81T FeaR mutation in the G494S* sensor, we next sought to understand the importance of the 81st residue for ligand binding. A protein motif search indicated that the position 12–185 of FeaR is a ligand-binding domain of the AraC-like transcription factor, and the position 218–298 is its DNA-binding domain with a helix-turn-helix motif at positions 258–298. Since no experimentally derived structures of FeaR exist, we applied computational simulations based on comparative modeling to predict the structure. The algorithm utilized the moderate homology of FeaR to CuxR, another AraC-like transcription factor with a previously solved structure, to comparatively predict the structure of FeaR(Schaper et al., 2017). Visualization of the predicted structure revealed that A81 is part of a solvent-accessible beta-barrel, and the alanine sidechain is oriented towards the interior of the barrel (Figure S13). Based on this information, we hypothesized that the beta-barrel is likely the aromatic aldehyde ligand binding site. In agreement with this theory, our ligand docking simulations using wild-type FeaR found the beta-barrel to be the most thermodynamically favorable ligand-binding site within the protein, with A81 oriented in close proximity to the bound ligand (Figure 5A).
Figure 5: Engineering FeaR to identify improved phenylethylamine-specific sensors.

A Computationally predicted ligand-binding region of FeaR. Key residues are shown with (magenta) and without (green) phenylethylamine-aldehyde (PEA-aldehyde; cyan) complexed. The structure was predicted using the Robetta web server. Ligand docking was modelled using the Rosetta Ligand Docking Server hosted by ROSIE. The image was generated using PyMOL. B Fluorescence from PtynA when induced for 24 h with 0.25 mM dopamine (DA), phenylethylamine (PEA), tyramine (Tyra), and tryptamine (Trypta). PtynA was regulated by wild-type TynA and 20 different FeaR variants with the specified amino acid in the 81st position. Residues for each group are listed from left to right in order of increasing size. Non-zero values and error bars are the average and standard deviation of biological triplicate, respectively. Variants with significant expression in response to PEA were determined by unpaired t-tests (P < 0.05). C Transfer curve of PtynA after 24 h of induction with DA, PEA, Tyra, and Trypta. PtynA was regulated by wild-type TynA and the A81L FeaR mutant. Points represent experimental data while the line represents the fitted curve (Table S2). The responses to DA, Tyr, and Trypta were not fit due to a lack of response. Values and error bars are the average and standard deviation of biological triplicate, respectively. D Reorganization of Figure 6B with respect to the size and hydropathy index of the 81st residue (Table S3). The blue area represents characteristic values that permit binding of Tyra. See also Figures S13–S18.
To further elucidate the role of the A81T mutation in ligand selectivity, we inserted the A81T mutation into the FeaR-TynA sensor plasmid with wild-type TynA. The mutation had an insignificant impact on the maximum response of the sensor to PEA and Tyra, but significantly reduced the responses to DA and Trypta (Figure S14). However, when we inserted the same mutation into the sensor plasmids with the Tyra-selective S414M and G415H TynA variants, both sensors became PEA-selective (Figure S14). The fluorescence output of each variant in response to PEA was also improved by the A81T mutation. We hypothesize that the improved PEA-dependent output and PEA-selectivity created by the A81T mutation may be a result of an altered sensitivity profile, where A81T FeaR is more sensitive to PEA-aldehyde and less sensitive to the remaining aldehydes. While the S414M and G415H TynA mutations weaken the catalytic activity of the enzyme, unequally reducing the amount of each aldehyde produced (as shown by the lower fluorescence output of the two variants relative to wild-type TynA; Figure S12), the A81T FeaR mutation may improve the sensitivity, and thereby selectivity, of FeaR to PEA-aldehyde. However, the high catalytic activity of wild-type TynA towards Tyra may be sufficient to overcome the elevated barrier of activation required by A81T FeaR to induce Tyra-dependent expression. Indeed, the A81T FeaR mutant demonstrated a lower KA value than wild-type FeaR when paired with both wild-type and G494S TynA (Figure S15). The A81S FeaR mutant paired with G494S TynA showed a very similar transfer function to that of A81T FeaR/G494S TynA, possibly due to the structural similarity of the side chains (Figures S15B and S16).
Noting the PEA selectivity given by the A81T mutation and the smallest size of the PEA-aldehyde (no OH group), followed by that of Tyra- (1 OH), DA- (2 OHs), and Trypta- (additional 5-membered ring) aldehyde (Figure 3A), we hypothesized that small residues, including alanine as in wild-type FeaR, would cause ligand promiscuity, with ligand size-dependent response as shown in Figure 3D. Slightly larger residues such as threonine would have improved selectivity towards the smallest PEA-aldehyde, but much larger or charged residues may fully eliminate ligand binding. We also hypothesized that the hydropathy index of the 81st residue may also play a role in the sensor’s performance due to differences in ligand polarities caused by the different number of hydroxyl groups. Notably, amines are positively charged in physiological pH, but the true ligands are uncharged aldehydes.
To test this prediction, we mutated A81 and paired each mutant with the promiscuous wild-type TynA (Figure 5B). Consistent with our predictions, A81L, A81P, A81I, and A81N were identified as PEA-specific variants, with 580-fold induction for the A81L sensor (Figure 5C). The A81L and A81P variants also demonstrated improved sensitivities to PEA relative to the G494S* PEA-specific sensor, with half maximal expression at 0.14 and 0.05 mM PEA, respectively (Figures 5C, S17, and S18 and Table S2). As predicted, a clear pattern between the identity of residue 81 and ligand selectivity was observed (Figure 5D and Table S3). FeaR recognized Tyra-aldehyde at a range of size and hydropathy index values (Figure 5D; blue area). The ranges with Tyra-dependent activity were non-discrete (as shown by the shape of the blue area). This suggests that the size and polarity of the 81st residue collaboratively dictate ligand selectivity. Small deviations outside of this space allowed FeaR to maintain PEA-aldehyde activity, but not Tyra activity. Similarly, the ranges that allowed binding of FeaR to the larger DA-aldehyde and Trypta-aldehyde ligands were smaller. Large deviations in either residue size or hydropathy index caused a complete loss of activity in response to all ligands.
We performed structural simulations of the wild-type (promiscuous), A81T (Tyra- and PEA-responsive), A81L (PEA-specific), and A81H (non-functional) variants to understand how the mutations may alter the structure and thus ligand binding of FeaR. Residues of increasing size (A<T<L) protruded further into and occupied more space in the ligand binding pocket, which is consistent with the observed size effect. All three mutations also shifted the positions of several side chains within the beta-barrel relative to the wild-type structure (Figure S19), contributing to the contrasting ligand selectivity. Of particular interest are the differences in the three residues forming the top of the pocket, M83, L108, and W110, as each has severe differences in rotation angles. However, no consistent trends exist between the size of the 81st residue and the orientation of these positions.
Given their significant rotations, we hypothesized that residues M83, L108, and W110 may also play important roles in ligand binding, potentially by making contacts with alternative functional groups in the ligand. We hypothesized that mutagenizing these residues may reveal a sensor for DA, Tyra, or Trypta. We individually mutated each residue and successfully identified multiple ligand-specific sensors for PEA or Tyra (Figures 6A–6C and S20). Randomizing the amino acids in positions 108 and 110 was only able to generate Tyra-specific variants, while randomizing the amino acid in position 83 was able to generate both Tyra- and PEA-specific variants. Each amino acid position demonstrated unique size and hydropathy index profiles for ligand specificity. We further characterized these ligand-specific sensors to assess their ligand sensitivity (Figures 6D, 6E and S21). The M83Y FeaR variant displayed the most sensitive response of all PEA-specific sensors, with half-maximal expression at 0.012 mM PEA. The best Tyra-specific variant, M83N, recognized its ligand with a comparable sensitivity.
Figure 6: Engineering additional FeaR amino acids to identify amine-specific sensors.

A-C Fluorescence from PtynA when induced for 24 h with 0.25 mM dopamine (DA), phenylethylamine (PEA), tyramine (Tyra), and tryptamine (Trypta). PtynA was regulated by wild-type TynA and 20 different FeaR variants with the specified amino acid in the (A) 83rd, (B) 108th, or (C) 110th position. Residues for each group are listed with their size and hydropathy index (Table S3). Values are the average of biological triplicate. To obtain the relative fluorescence, fluorescence values for each variant were normalized to the fluorescence of the wild-type sensor (see STAR Methods, Equation 3). D and E Transfer curves of PtynA after 24 h of induction with DA, PEA, Tyra, and Trypta. The expression of PtynA was regulated by wild-type TynA with (D) M83Y or (E) M83N FeaR variants. Points represent experimental data while lines represent the fitted curves (see STAR Methods and Table S2). Values and error bars are the average and standard deviation of biological triplicate, respectively. See also Figures S19–S22.
To further confirm the specificity of the best performing sensors, we next tested the response of each to the four amines and the four respective carboxylic acids, 3,4-dihydroxyphenylacetic acid (DOPAC), phenylacetic acid (PAA), 4-hydroxyphenylacetic acid (HPAA), and indole-3-acetic acid (IAA) in minimal medium with casamino acids and LB medium (Figure S22). No sensors significantly responded to the carboxylic acids in either medium, confirming the specificity of FeaR to only aldehydes. All mutant sensors remained specific to PEA or Tyra in minimal medium with casamino acids. However, all sensors displayed no output in LB medium, suggesting that the system may be regulated by catabolite repression.
Together, this work provides insights into the structure and activity of FeaR. In addition, by engineering FeaR, we provide the best performing PEA- and Tyra-specific sensors for future applications. Future work could include random approaches of protein engineering such as error-prone PCR-based directed evolution to develop additional ligand-specific sensors for the larger DA and Trypta amines. In addition, further exploration of the relevant regulatory pathways is required to uncouple the activity of the sensors with resource availability.
DISCUSSION
The ligand-specific sensors developed here have the potential for diverse applications, including (i) monitoring food quality, (ii) diagnosing or treating metabolic, digestive, and neurological disorders in probiotics or ex vivo wearable, paper-based and cell-free systems, and (iii) dynamically regulating enzymatic pathways for microbial metabolic engineering. The high degree of ligand specificity shown by the engineered sensors allows them to effectively differentiate between diverse structurally similar aromatic metabolites. We demonstrate an efficient and effective method of rational protein engineering by individually performing saturation mutagenesis on logically selected amino acid residues. We show the generalizability of this method by applying it to three protein systems, including both enzymes and transcription factors. The simplicity of the approach and small library sizes make it an attractive first step in sensor engineering. Although this protein engineering method requires basic structural knowledge of the protein of interest, the scope of the required information is less than fully computation-based engineering approaches. However, we also show how protein simulations can be used to identify important residues from uncharacterized proteins.
Protein engineering for specific ligand-protein interactions has been extensively performed(Meyer et al., 2019), especially for facilitating chemical drug screening. However, coupling ligand-protein binding with an output response has remained challenging(Peveler et al., 2016). Current approaches include designing proteins that fluoresce upon ligand binding(Dou et al., 2018) or employing ligand-binding transcription factors (TFs)(Taylor et al., 2016). While the former can be used to develop sensitive sensors, the latter can generate sensors as well as controllers for downstream functions such as gene expression. Because this TF-based system requires the maintenance or engineering of DNA-protein interaction in addition to engineering ligand-protein binding, developing ligand-specific sensors using this system has been challenging, especially when the target ligands are structurally similar. To address this challenging issue, we demonstrated two approaches: optimizing ligand-TF binding specificity and sensitivity by leveraging differential multimerization patterns of TyrR without affecting DNA-TF binding interactions (Figures 1–2) and employing the dual-knob modulation of the specificity and sensitivity of substrate-enzyme and ligand-TF interactions while maintaining DNA-TF binding interactions (Figures 3–6).
The feaR-tynA system represents a unique way to develop sensors using two different control knobs. As shown in Figures 4 and S14–S16, Tyra-selective sensors (S414M or G415H TynA) can be transformed into PEA-specific sensors by introducing A81T FeaR; sensor sensitivity (G494S TynA) can be improved by A81T or A81S FeaR mutations; and PEA-Tyra selective sensors (A81T or A81S FeaR) can become PEA-specific sensors when combined with G494S TynA. One caveat is that TynA and FeaR mutagenesis often led to reduced output signals, sometimes making a PEA-specific sensor (FeaR-A81P) not responsive to any ligand due to lowered aldehyde concentrations (e.g., TynA-G494S + FeaR-A81P). The output of the sensors could be further improved by optimization of the ribosome binding site of the reporter or mutagenesis of the PtynA reporter promoter. Furthermore, for engineered TynA variants, increasing the expression of the enzyme through optimization of its promoter or ribosome binding site may improve signal output by increasing the amine to aldehyde conversion rate. Notably, the intermediate aldehydes are rarely found in natural environments, eliminating the possibility of extracellular aldehydes contributing to sensor outputs. However, until the sensor activity is uncoupled from catabolite repression, applications may be limited to environments lacking a preferred carbon or nitrogen source(Zeng and Spiro).
Altogether, we generated the specific microbial sensors for Phe, Tyr, PEA, and Tyra. This work provides ligand-specific sensors that can be applied to create probiotics for diverse applications. In addition, the generalizable protein engineering techniques demonstrated here can be used to quickly and effectively engineer enzymes and transcription factors for ligand specificity and sensitivity. Although we specifically developed sensors with potential applications primarily in medicine and food quality, enzymes and sensors can be similarly engineered to produce fuels, pharmaceuticals, and commodity chemicals in response to the levels of metabolites or the products in a dynamic way. This work represents a considerable achievement in the challenging goal of engineering ligand-specific sense-and-respond systems for medically-relevant chemicals through coupling protein conformational changes caused by ligand binding with DNA interactions.
STAR METHODS
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact, Tae Seok Moon (tsmoon@wustl.edu).
Materials availability
Plasmids generated in this paper are available upon request from the Lead Contact. This study did not generate additional new unique reagents.
Data and code availability
The data that support the findings of this study are provided in the main text, supplementary information, or Data S1.
This paper does not report original code.
Method details
Plasmids, strains, and reagents
All plasmids, strains, and genetic parts used in this study are summarized in Tables S4, S5, and S6, respectively. All plasmids were assembled in E. coli DH10B using the Gibson Assembly or Golden Gate Assembly methods. EcN was transformed with the purified and sequence-verified plasmids for testing. The EcN strain used in this work lacks its native plasmids (obtained from DSMZ). The tynA, feaR, and tyrR genes and PtynA, PtyrP, PtyrR, Pmtr, PtyrB, ParoF, and ParoP promoters were obtained from E. coli MG1655 genomic DNA. pHJY23 containing the inactivated tynA gene (Figure 3C) was constructed by removing the internal region (973 – 2230th bases) of tynA from plasmid pHJY22 by digesting with EaeI and ligating the linear product. The pHJY028 PtynA reporter plasmid was optimized by randomizing the ribosome binding site for GFP and assaying variants for a response to DA.
Plasmid DNA was isolated using the PureLink Quick Plasmid Miniprep Kit (Invitrogen, Walthem, MA, USA), and PCR products were extracted from electrophoresis gels using the Zymoclean Gel DNA Recovery Kit (ZYMO research, Irvine, CA, USA). Enzymes were purchased from New England Biolabs (Ipswich, MA, USA). Chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) or Gold Biotechnology (Olivette, MO, USA). All sequencing was performed by Genewiz (South Plainfield, NJ, USA). Primers were purchased from Integrated DNA Technologies (Coralville, IA, USA).
Aromatic amino acid sensing assay
Everything but (Eb) medium was prepared by supplementing M9 minimal medium with 1 mM MgSO4, 100 μM CaCl2, 0.4% w/v glucose, and all non-aromatic amino acids (0.8 mM alanine, 5 mM arginine, 0.4 mM asparagine, 0.4 mM aspartate, 0.1 mM cysteine, 0.6 mM glutamate, 0.6 mM glutamine, 0.8 mM glycine, 0.2 mM histidine, 0.4 mM isoleucine, 0.8 mM leucine, 0.4 mM lysine, 0.2 mM methionine, 0.4 mM proline, 10 mM serine, 0.4 mM threonine, and 0.6 mM valine). Single colonies of EcN containing the relevant sensor plasmids were transferred to 5 mL Eb medium in 14 mL round bottom tubes and incubated overnight at 250 rpm and 37oC. Experimental cultures were prepared by diluting overnight cultures 200x into 0.6 mL fresh Eb medium supplemented with the respective ligands (Phe and Tyr) in 2 mL 96-deep well plates (Eppendorf, Hamburg, Germany). Cultures were grown for 8 h at 37oC and 250 rpm before sampled for fluorimetry or flow cytometry analysis. All medium was supplemented with the relevant antibiotics for plasmid maintenance (34 μg/ml chloramphenicol and 100 μg/ml spectinomycin).
Aromatic amine sensing assays
For fluorescence-based quantification, single colonies were transferred to 5 mL LB medium (VWR, Radnor, PA, USA) in 14 mL round bottom tubes and incubated overnight at 250 rpm and 37oC. Unless otherwise specified, experimental cultures were prepared by diluting overnight cultures 100x into 0.6 mL M9 minimal medium supplemented with 1 mM MgSO4, 100 μM CaCl2, and 2% w/v glycerol as well as the respective ligands (DA, PEA, Tyra, and Trypta) in 2 mL 96-deep well plates. Cultures were grown for 24 h at 37oC and 250 rpm. After 5 h and 24 h of incubation, samples were obtained from the cultures for flow cytometry analysis.
For growth-based library screening, bla (encoding β-lactamase) and sacB (encoding levansucrase) were incorporated downstream of gfp in the pHJY028 reporter plasmid (Table S4). For optimization, ribosome binding site libraries were designed for bla and sacB using the RBS Calculator (Table S6)(Salis et al., 2009). The optimization resulted in reporter pCX008. To test sensor variants using the pCX008 reporter, single colonies were transferred to 0.6 mL LB medium in 2 mL 96-deep well plates and incubated overnight at 250 rpm and 37oC. The overnight cultures were diluted 50x into 0.6 mL M9 minimal medium supplemented with 1 mM MgSO4, 100 μM CaCl2, and 2% w/v glycerol in 2 mL 96-well deep well plates. For positive selection, cultures were supplemented with 300 μg/mL carbenicillin. Cultures were then incubated for 2 h at 250 rpm and 37oC. After the 2 h incubation, 2 μL of cells were plated onto M9 agar plates supplemented with 2% glycerol and either 300 μg/mL carbenicillin and 1 mM of the amine of interest (positive selection) or 5% (w/v) sucrose and 1 mM of the non-desired amines (negative selection). Plates were incubated overnight at 37oC.
Fluorimetry
200 μL culture samples were collected and transferred to 96-well assay microplates (clear, flat bottom black, Greiner Bio-One). The fluorescence and culture absorbance (Abs) were measured using a Tecan microplate reader (Infinite M200 Pro) as previously described(Lee et al., 2016). The fluorescence of GFP was measured with an excitation at 483 nm and emission at 530 nm. The Abs of the samples was measured at 600 nm. The measured fluorescence was normalized by dividing by the Abs and subtracting the same ratio obtained from non-fluorescent wild-type cells (Equation 1).
| (1) |
Flow cytometry
Flow cytometry was performed as previously described(Shopera et al., 2017). Culture samples were collected and diluted to a final OD600 of ~0.005–0.01 in 200 μL filtered phosphate-buffered saline supplemented with 2 mg/ml kanamycin in 96-well assay microplates (U-bottom, REF-353910 from BD Biosciences, San Jose, CA, USA). The fluorescence of the samples was measured using a Millipore Guava EasyCyte High Throughput flow cytometer with a 488 nm excitation laser and 512/18 nm emission filter.
Cytometry data was gated by forward and side scatter. FlowJo (TreeStar Inc.) was used to obtain the arithmetic mean of the fluorescence distribution. The averages of the arithmetic means were calculated from three biological replicates. The average fluorescence of the non-fluorescent wild-type cell was subtracted from each sample to obtain the final fluorescence (au) values (Equation 2). To obtain the relative fluorescence in Figures 6A–6C, fluorescence values for each variant were normalized to the fluorescence of the wild-type sensor using Equation 3.
| (2) |
| (3) |
Hill equation fitting
The Hill equation (Equations 4 and 5) was used to fit lines to the fluorescence data. The model was fit to the experimentally collected data by minimizing the root mean square error (RMSE; Equation 6). Fitted values are listed in Table S2.
For repressible constructs(DeLorenzo et al., 2018):
| (4) |
For inducible constructs(DeLorenzo et al., 2017):
| (5) |
where
F = Calculated fluorescence
Fmax = Maximum fluorescence
Fmin = Minimum fluorescence
KA = Half maximal constant
n = Hill coefficient
[L] = Ligand concentration
| (6) |
where
F = Calculated fluorescence
Fexp = Actual experimental fluorescence
N = Number of data points
FeaR protein modeling
FeaR structural motifs were annotated using the MOTIF Search web tool (genome.jp). Structural predictions were made using the comparative modeling function of the Robetta web server with the wild-type or mutant FeaR amino acid sequences as inputs(Song et al., 2013). Predicted structures were used as the basis for ligand docking simulations. Three-dimensional conformers of the aldehydes of DA, PEA, Tyra, and Trypta were generated using Chem3D 16.0 (PerkinElmer, Waltham, MA). Ligand conformer and protein structure .pdb files were uploaded to the Rosetta Ligand Docking Server hosted by ROSIE, with the ligand of interest initially centered at the coordinates of the 81st residue’s side chain(Combs et al., 2013; Lyskov et al., 2013). All protein structures were visualized using PyMOL 2.4.1 (Schrödinger, Inc., New York, NY). Values for amino acid size and hydropathy index were obtained from literature for the analysis in Figure 5D and are shown in Table S3(Kyte and Doolittle, 1982; Perkins, 1986).
Quantification and statistical analysis
All statistical details of experiments, including significance criteria and sample size can be found in the figure legends. No sample size calculations were performed during the design of experiments. No samples were excluded.
Supplementary Material
Data S1. Source Data
Table S2: Fitted Hill equation parameters for the specified figures. The Hill equation, described in the STAR Methods, was fit to each set of fluorescence (au) values by minimizing the root mean square error (RMSE). Fmax and Fmin represent the fitted maximum and minimum fluorescence (au) values, respectively. The half-maximal constant (KA) is in mM of the respective ligand. The fitted Hill coefficient (n) and RMSE are also reported. See Excel file.
Table S4: Plasmids used in this work. See Excel file.
Table S6: Genetic parts used in this work. See Excel file.
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| E. coli DH10B | Invitrogen | EC0113 |
| E. coli Nissle 1917 (plasmid free) | DSMZ | DSM 16700 |
| Chemicals, peptides, and recombinant proteins | ||
| Agar | Sigma | A1296 |
| L-Alanine | Sigma | 05129 |
| Anhydrotetracycline HCl | Sigma | 37919 |
| L-Arginine monohydrochloride | Sigma | A6969 |
| L-Asparagine | Sigma | A4159 |
| L-Aspartic acid | Sigma | A9256 |
| Bacto casamino acids | ThermoFisher | 223050 |
| CaCl2 2H2O | Sigma | C5080 |
| carbenicillin disodium salt | Sigma | C1389 |
| Chloramphenicol | Gold Biotechnology | C-105 |
| L-Cysteine hydrochloride monohydrate | Sigma | C6852 |
| Deoxynucleotide Set (dNTPs) | G-Biosciences | 786-460 |
| 3,4-Dihydroxyphenylacetic acid | Sigma | 850217 |
| 1,4-Dithiothreitol (DTT) | Sigma | 3483-12-3 |
| Dopamine hydrochloride | Sigma | H8502 |
| DpnI | New England BioLabs | R0176 |
| D-(+)-Glucose | Sigma | G8270 |
| L-Glutamine | Sigma | G3126 |
| L-Glutamic acid | Sigma | G1251 |
| Glycerol | Fisher Scientific | BP229-1 |
| Glycine | Sigma | 410225 |
| Histidine | Sigma | H8000 |
| 4-Hydroxyphenylacetic acid | Sigma | H50004 |
| Indole-3-acetic acid | Sigma | I5148 |
| L-Isoleucine | Sigma | I7403 |
| Kanamycin monosulfate | Gold Biotechnology | K-120 |
| KCl | Sigma | P5405 |
| KH2PO4 | Sigma | P9791 |
| LB broth, Miller | VWR | J106 |
| L-Leucine | Sigma | 61819 |
| L-Lysine | Sigma | L5501 |
| M9 minimal salts, 5X | Sigma | M6030 |
| L-Methionine | Sigma | M9625 |
| MgCl2 | Sigma | 7786-30-3 |
| MgSO4 7H2O | Sigma | 63138 |
| Na2HPO4 | Sigma | S5136 |
| NaCl | Sigma | S7653 |
| NAD+ | Sigma | 53-84-9 |
| PEG-8000 | Sigma | 1546605 |
| Phenylacetic acid | Sigma | P16621 |
| L-Phenylalanine | Sigma | P5482 |
| Phenylethylamine hydrochloride | Sigma | P6513 |
| Phusion high-fidelity DNA polymerase | New England BioLabs | M0530 |
| L-Proline | Sigma | P5607 |
| SapI | New England BioLabs | R0569 |
| L-Serine | Sigma | S4311 |
| Spectinomycin dihydrochloride pentahydrate | Gold Biotechnology | S-140 |
| T4 DNA ligase | New England BioLabs | M0202 |
| T5 exonuclease | New England BioLabs | M0663 |
| Taq DNA Ligase | New England BioLabs | M0208 |
| L-Threonine | Sigma | T8441 |
| Tris base | Sigma | 77-86-1 |
| Tryptamine | Sigma | 193747 |
| L-Tryptophan | Sigma | T0254 |
| Tyramine hydrochloride | Sigma | T2879 |
| L-Tyrosine | Sigma | T8566 |
| L-Valine | Sigma | V0513 |
| Recombinant DNA | ||
| See Tables S4 and S6 | ||
| Software and algorithms | ||
| Chem3D 16.0 | PerkinElmer | N/A |
| MOTIF Search | Genome.jp | N/A |
| Rosetta Ligand Docking Server | N/A | |
| Robetta web server | Song et al., 2013 | N/A |
| Other | ||
| 14 mL round bottom tubes | Fisher Scientific | 14-959-11B |
| 96-deep well plates | Fisher Scientific | E951032808 |
| 96-well black assay microplates | Fisher Scientific | 07-000-088 |
| 96-well clear round bottom assay microplates | Corning | 353910 |
| PureLink Quick Plasmid Miniprep Kit | Invitrogen | K210011 |
| Zymoclean Gel DNA Recovery Kit | ZYMO Research | D4008 |
Highlights.
Created specific sensors for structurally similar amino acids and neurochemicals
Developed a protein engineering method requiring only a basic structural understanding
Provided insights into the otherwise uncharacterized structure of a transcription factor
Acknowledgments
We thank Prof. Brian Pfleger for the gift of the pMP11 and pgRNAcm plasmids. This work was supported by the National Institutes of Health (R01 AT009741 to T.S.M.), the Office of Naval Research (N00014-17-1-2611 and N00014-19-1-2357 to T.S.M.), the United States Department of Agriculture (2020-33522-32319 to T.S.M.), the National Science Foundation (CBET-1350498 to T.S.M.), and the National Science Foundation Graduate Research Fellowship Program (DGE-1745038 to M.B.A.).
Footnotes
Declaration of Interests
The authors have filed a provisional application with the US Patent and Trademark Office on this work.
Inclusion and diversity statement
One of the authors is a female scientist.
A record of this paper’s Transparent Peer Review process is included in the Supplemental Information.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adibi SA, and Mercer DW (1973). Protein digestion in human intestine as reflected in luminal, mucosal, and plasma amino acid concentrations after meals. J Clin Invest 52, 1586–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsare KD, Andorfer MC, Cardenas FS, Chael JR, Park HJ, and Lewis JC (2017). A Simple Combinatorial Codon Mutagenesis Method for Targeted Protein Engineering. ACS Synth Biol 6, 416–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattarai Y, Williams BB, Battaglioli EJ, Whitaker WR, Till L, Grover M, Linden DR, Akiba Y, Kandimalla KK, Zachos NC, et al. (2018). Gut Microbiota-Produced Tryptamine Activates an Epithelial G-Protein-Coupled Receptor to Increase Colonic Secretion. Cell Host Microbe 23, 775–785 e775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchetti MG, Minder I, Beretta-Piccoli C, Meier A, and Weidmann P (1982). Effects of tyramine on blood pressure and plasma catecholamines in normal and hypertensive subjects. Klin Wochenschr 60, 465–470. [DOI] [PubMed] [Google Scholar]
- Camp KM, Parisi MA, Acosta PB, Berry GT, Bilder DA, Blau N, Bodamer OA, Brosco JP, Brown CS, Burlina AB, et al. (2014). Phenylketonuria Scientific Review Conference: state of the science and future research needs. Mol Genet Metab 112, 87–122. [DOI] [PubMed] [Google Scholar]
- Cerone R, Fantasia AR, Castellano E, Moresco L, Schiaffino MC, and Gatti R (2002). Pregnancy and tyrosinaemia type II. J Inherit Metab Dis 25, 317–318. [DOI] [PubMed] [Google Scholar]
- Chen H, Nwe PK, Yang Y, Rosen CE, Bielecka AA, Kuchroo M, Cline GW, Kruse AC, Ring AM, Crawford JM, et al. (2019). A Forward Chemical Genetic Screen Reveals Gut Microbiota Metabolites That Modulate Host Physiology. Cell 177, 1217–1231 e1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs SA, Deluca SL, Deluca SH, Lemmon GH, Nannemann DP, Nguyen ED, Willis JR, Sheehan JH, and Meiler J (2013). Small-molecule ligand docking into comparative models with Rosetta. Nat Protoc 8, 1277–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLorenzo DM, Henson WR, and Moon TS (2017). Development of Chemical and Metabolite Sensors for Rhodococcus opacus PD630. ACS Synth Biol 6, 1973–1978. [DOI] [PubMed] [Google Scholar]
- DeLorenzo DM, Rottinghaus AG, Henson WR, and Moon TS (2018). Molecular Toolkit for Gene Expression Control and Genome Modification in Rhodococcus opacus PD630. ACS Synth Biol 7, 727–738. [DOI] [PubMed] [Google Scholar]
- Dou J, Vorobieva AA, Sheffler W, Doyle LA, Park H, Bick MJ, Mao B, Foight GW, Lee MY, Gagnon LA, et al. (2018). De novo design of a fluorescence-activating β-barrel. Nature 561, 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellefson JW, Ledbetter MP, and Ellington AD (2018). Directed evolution of a synthetic phylogeny of programmable Trp repressors. Nat Chem Biol 14, 361–367. [DOI] [PubMed] [Google Scholar]
- Glasgow AA, Huang YM, Mandell DJ, Thompson M, Ritterson R, Loshbaugh AL, Pellegrino J, Krivacic C, Pache RA, Barlow KA, et al. (2019). Computational design of a modular protein sense-response system. Science 366, 1024–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins SA, Ouonkap SVY, and Savage DF (2017). Rapid and Programmable Protein Mutagenesis Using Plasmid Recombineering. ACS Synth Biol 6, 1825–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoynes-O’Connor A, Shopera T, Hinman K, Creamer JP, and Moon TS (2017). Enabling complex genetic circuits to respond to extrinsic environmental signals. Biotechnol Bioeng 114, 1626–1631. [DOI] [PubMed] [Google Scholar]
- Irsfeld M, Spadafore M, and Pruss BM (2013). beta-phenylethylamine, a small molecule with a large impact. Webmedcentral 4. [PMC free article] [PubMed] [Google Scholar]
- Jones AM, Atkinson JT, and Silberg JJ (2017). PERMutation Using Transposase Engineering (PERMUTE): A Simple Approach for Constructing Circularly Permuted Protein Libraries. Methods Mol Biol 1498, 295–308. [DOI] [PubMed] [Google Scholar]
- Kouchiwa T, Wada K, Uchiyama M, Kasezawa N, Niisato M, Murakami H, Fukuyama K, and Yokogoshi H (2012). Age-related changes in serum amino acids concentrations in healthy individuals. Clin Chem Lab Med 50, 861–870. [DOI] [PubMed] [Google Scholar]
- Kwok T, Yang J, Pittard AJ, Wilson TJ, and Davidson BE (1995). Analysis of an Escherichia coli mutant TyrR protein with impaired capacity for tyrosine-mediated repression, but still able to activate at sigma 70 promoters. Mol Microbiol 17, 471–481. [DOI] [PubMed] [Google Scholar]
- Kyte J, and Doolittle RF (1982). A simple method for displaying the hydropathic character of a protein. J Mol Biol 157, 105–132. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Hoynes-O’Connor A, Leong MC, and Moon TS (2016). Programmable control of bacterial gene expression with the combined CRISPR and antisense RNA system. Nucleic Acids Res 44, 2462–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, and Bork P (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res 47, W256–W259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Jair YC, Chou YC, Chen PS, and Yeh YC (2018). Transcription factor-based biosensor for detection of phenylalanine and tyrosine in urine for diagnosis of phenylketonuria. Anal Chim Acta 1041, 108–113. [DOI] [PubMed] [Google Scholar]
- Liu X, Niu H, Li Q, and Gu P (2019). Metabolic engineering for the production of l-phenylalanine in Escherichia coli. 3 Biotech 9, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyskov S, Chou FC, Conchuir SO, Der BS, Drew K, Kuroda D, Xu J, Weitzner BD, Renfrew PD, Sripakdeevong P, et al. (2013). Serverification of molecular modeling applications: the Rosetta Online Server that Includes Everyone (ROSIE). PLoS One 8, e63906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahr R, von Boeselager RF, Wiechert J, and Frunzke J (2016). Screening of an Escherichia coli promoter library for a phenylalanine biosensor. Appl Microbiol Biotechnol 100, 6739–6753. [DOI] [PubMed] [Google Scholar]
- Marcobal A, De las Rivas B, Landete JM, Tabera L, and Munoz R (2012). Tyramine and phenylethylamine biosynthesis by food bacteria. Crit Rev Food Sci Nutr 52, 448–467. [DOI] [PubMed] [Google Scholar]
- Meyer AJ, Segall-Shapiro TH, Glassey E, Zhang J, and Voigt CA (2019). Escherichia coli “Marionette” strains with 12 highly optimized small-molecule sensors. Nat Chem Biol 15, 196–204. [DOI] [PubMed] [Google Scholar]
- Mitchell G, Larochelle J, Lambert M, Michaud J, Grenier A, Ogier H, Gauthier M, Lacroix J, Vanasse M, Larbrisseau A, et al. (1990). Neurologic crises in hereditary tyrosinemia. N Engl J Med 322, 432–437. [DOI] [PubMed] [Google Scholar]
- Myojin T, Taga C, and Tsuji M (1989). Concentrations of beta-phenylethylamine in plasma and plateletes of schizophrenics. Jpn J Psychiatry Neurol 43, 171–176. [DOI] [PubMed] [Google Scholar]
- Perkins SJ (1986). Protein volumes and hydration effects. The calculations of partial specific volumes, neutron scattering matchpoints and 280-nm absorption coefficients for proteins and glycoproteins from amino acid sequences. Eur J Biochem 157, 169–180. [DOI] [PubMed] [Google Scholar]
- Peveler WJ, Yazdani M, and Rotello VM (2016). Selectivity and Specificity: Pros and Cons in Sensing. ACS Sens 1, 1282–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittard J, Camakaris H, and Yang J (2005). The TyrR regulon. Mol Microbiol 55, 16–26. [DOI] [PubMed] [Google Scholar]
- Rafehi M, Faltraco F, Matthaei J, Prukop T, Jensen O, Grytzmann A, Blome FG, Berger RG, Krings U, Vormfelde SV, et al. (2019). Highly Variable Pharmacokinetics of Tyramine in Humans and Polymorphisms in OCT1, CYP2D6, and MAO-A. Front Pharmacol 10, 1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Martinez JA, Flores N, Escalante A, Gosset G, and Bolivar F (2014). Engineering Escherichia coli to overproduce aromatic amino acids and derived compounds. Microb Cell Fact 13, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salis HM, Mirsky EA, and Voigt CA (2009). Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol 27, 946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saysell CG, Tambyrajah WS, Murray JM, Wilmot CM, Phillips SE, McPherson MJ, and Knowles PF (2002). Probing the catalytic mechanism of Escherichia coli amine oxidase using mutational variants and a reversible inhibitor as a substrate analogue. Biochem J 365, 809–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- chaper S, Steinchen W, Krol E, Altegoer F, Skotnicka D, Sogaard-Andersen L, Bange G, and Becker A (2017). AraC-like transcriptional activator CuxR binds c-di-GMP by a PilZ-like mechanism to regulate extracellular polysaccharide production. Proc Natl Acad Sci U S A 114, E4822–E4831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scriver CR (2007). The PAH gene, phenylketonuria, and a paradigm shift. Hum Mutat 28, 831–845. [DOI] [PubMed] [Google Scholar]
- Sezonov G, Joseleau-Petit D, and D’Ari R (2007). Escherichia coli physiology in Luria-Bertani broth. J Bacteriol 189, 8746–8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalaby AR (1996). Significance of biogenic amines to food safety and human health. Food Research International 29, 675–690. [Google Scholar]
- Shopera T, Henson WR, and Moon TS (2017). Dynamics of sequestration-based gene regulatory cascades. Nucleic Acids Res 45, 7515–7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, DiMaio F, Wang RY, Kim D, Miles C, Brunette T, Thompson J, and Baker D (2013). High-resolution comparative modeling with RosettaCM. Structure 21, 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stainbrook SC, and Tyo KEJ (2019). Model-guided mechanism discovery and parameter selection for directed evolution. Appl Microbiol Biotechnol 103, 9697–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaki M, Mawe GM, Barasch JM, Gershon MD, and Gershon MD (1985). Physiological responses of guinea-pig myenteric neurons secondary to the release of endogenous serotonin by tryptamine. Neuroscience 16, 223–240. [DOI] [PubMed] [Google Scholar]
- Taylor ND, Garruss AS, Moretti R, Chan S, Arbing MA, Cascio D, Rogers JK, Isaacs FJ, Kosuri S, Baker D, et al. (2016). Engineering an allosteric transcription factor to respond to new ligands. Nat Methods 13, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verger D, Carr PD, Kwok T, and Ollis DL (2007). Crystal structure of the N-terminal domain of the TyrR transcription factor responsible for gene regulation of aromatic amino acid biosynthesis and transport in Escherichia coli K12. J Mol Biol 367, 102–112. [DOI] [PubMed] [Google Scholar]
- Wilmot CM, Murray JM, Alton G, Parsons MR, Convery MA, Blakeley V, Corner AS, Palcic MM, Knowles PF, McPherson MJ, et al. (1997). Catalytic mechanism of the quinoenzyme amine oxidase from Escherichia coli: exploring the reductive half-reaction. Biochemistry 36, 1608–1620. [DOI] [PubMed] [Google Scholar]
- Yang J, Camakaris H, and Pittard AJ (1996). Further genetic analysis of the activation function of the TyrR regulatory protein of Escherichia coli. J Bacteriol 178, 1120–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J, and Spiro S (2013). Finely tuned regulation of the aromatic amine degradation pathway in Escherichia coli. J Bacteriol 195, 5141–5150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zvelebil MJ, Barton GJ, Taylor WR, and Sternberg MJ (1987). Prediction of protein secondary structure and active sites using the alignment of homologous sequences. J Mol Biol 195, 957–961. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Source Data
Table S2: Fitted Hill equation parameters for the specified figures. The Hill equation, described in the STAR Methods, was fit to each set of fluorescence (au) values by minimizing the root mean square error (RMSE). Fmax and Fmin represent the fitted maximum and minimum fluorescence (au) values, respectively. The half-maximal constant (KA) is in mM of the respective ligand. The fitted Hill coefficient (n) and RMSE are also reported. See Excel file.
Table S4: Plasmids used in this work. See Excel file.
Table S6: Genetic parts used in this work. See Excel file.
Data Availability Statement
The data that support the findings of this study are provided in the main text, supplementary information, or Data S1.
This paper does not report original code.