

Abstract
Translational control shapes the proteome in normal and pathophysiological conditions. Current high-throughput approaches reveal large differences in mRNA-specific translation activity but cannot identify the causative mRNA features. We developed direct analysis of ribosome targeting (DART) and used it to dissect regulatory elements within 5′ untranslated regions that confer thousand-fold differences in ribosome recruitment in biochemically accessible cell lysates. Using DART, we determined a functional role for most alternative 5′ UTR isoforms expressed in yeast, revealed a general mode of increased translation via direct binding to a core translation factor, and identified numerous translational control elements including C-rich silencers that are sufficient to repress translation both in vitro and in vivo. DART enables systematic assessment of the translational regulatory potential of 5′ UTR variants, whether native or disease-associated, and will facilitate engineering of mRNAs for optimized protein production in various systems.
Graphical Abstract
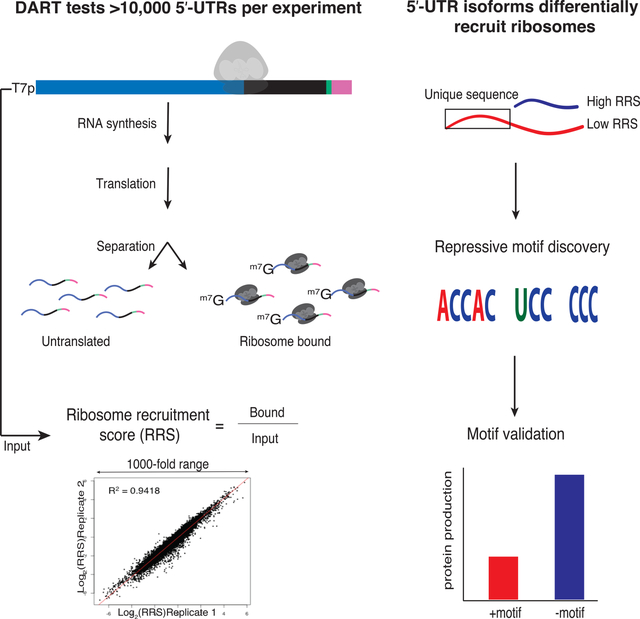
Niederer et al. develop a method to quantify translation initiation on thousands of 5′ UTRs in parallel. They uncover sequence-specific motifs that control translation by varied mechanisms and establish a platform for systematic interrogation of 5′ UTR variants that can be used to engineer mRNAs for optimized protein output.
Introduction
Translation initiation is an important step in eukaryotic gene expression whose dysregulation is linked to heritable human diseases and cancer. Systematic characterization has shown that mRNA-specific translational activity varies by orders of magnitude under normal growth conditions and is extensively regulated in response to a wide range of physiological signals (Kristensen et al., 2013; Lahtvee et al., 2017). Despite growing interest in the mRNA features responsible for this widespread translational control, current high-throughput methods, which rely on quantification of the average number of translating ribosomes per transcript, have revealed only partial correlations (R = 0.3 – 0.8) that leave many differences in translation activity unexplained (Ingolia et al., 2009; Li et al., 2019; Weinberg et al., 2016). In contrast, in-depth genetic and biochemical analysis has revealed detailed regulatory mechanisms for certain mRNAs (Dever et al., 1992; Muckenthaler et al., 1998).
Genome-wide association studies (GWAS) show diverse disease phenotypes associated with non-coding single-nucleotide polymorphisms (SNPs) found within 5′ UTRs. However, current efforts to identify causal variants, which are potential targets for new therapies, are largely restricted to non-synonymous changes within protein coding sequences. A richer understanding of mRNA sequence-specific translation mechanisms is needed to predict which 5′ UTR variants are likely to be damaging by dysregulating protein levels. We developed DART technology as a bridge between high-throughput, but mechanistically difficult to parse, in vivo approaches and mechanistically precise, but low-throughput, in vitro translation reconstitution assays. In DART, thousands of synthetic mRNAs initiate translation in vitro. Differential abundance analysis of ribosome-bound and input mRNA reveals differences in translation activity. By massively parallel testing of defined sequences, both endogenous and mutated, our approach moves beyond correlation analysis to enable rapid, hypothesis-driven dissection of the causative role of specific 5′ UTR elements to determine their mechanisms of action.
Here, we demonstrate the broad power of DART to illuminate the mechanisms underlying translational control. Overall, we tested 4354 full-length alternative mRNA isoforms from 2064 yeast genes revealing widespread translational control by alternative 5′ UTRs. We also exploited the throughput of the DART approach to systematically interrogate the effects of RNA secondary structure, start codon context and protein binding motifs on translation initiation. Our results establish a broad stimulatory role for eIF4G1 binding sequences and also demonstrate highly context-dependent control by inhibitory 5′ UTR structures. This illustrates the potential of DART to illuminate the function of putative RNA regulatory elements identified by other high-throughput approaches. We further leveraged the alternative 5′ UTR isoform comparisons to discover hundreds of previously uncharacterized translational enhancers and silencers, short sequence elements that are sufficient to promote or repress ribosome recruitment. We validated several C-rich silencers as sufficient to repress translation both in vitro and in vivo. Together, our results reveal thousands of previously unidentified functional elements within 5′ UTRs that substantially affect translation. This study establishes DART as a powerful new high-throughput method that can be broadly applied to both discover and interrogate regulatory features within 5′ UTRs.
Results
Development of DART for quantitative comparison of translation initiation
5′ UTRs directly contact the translation initiation machinery and can strongly influence translation activity (Fig. 1a), but the features that distinguish efficiently translated mRNAs are largely unknown. We developed direct analysis of ribosome targeting (DART) as a high-throughput method to enable rapid determination of the role of individual regulatory elements within 5′ UTRs. We designed a library of DNA oligos, based on deep sequencing of yeast mRNA isoforms (Pelechano et al., 2014) (Methods), that included 12,000 sequences corresponding to the full-length 5′ UTR sequences from 4,252 genes. This pool design focuses on identifying functional elements within endogenous 5′-UTRs in contrast to complementary studies that interrogate random sequences (Dvir et al., 2013; Sample et al., 2019). Our library covers 68% of genes expressed during exponential growth and includes alternative mRNA isoforms for many genes. A common (5′) T7 promoter and a (3′) priming site for reverse transcription flanked each unique mRNA sequence. The 5′ UTR sequence was extended to include at least 24 nucleotides of endogenous coding sequence to preserve potentially important downstream sequences that are protected by initiating ribosomes (Archer et al., 2016). Each oligo also included a unique 10nt barcode to distinguish closely related sequences (Fig. 1b).
Fig. 1. DART quantifies 5′ UTR-mediated translational control over a thousand-fold range.

(A) Different endogenous yeast 5′ UTRs added to a common luciferase coding sequence and 3′ UTR result in large differences in translational output per mRNA in cell lysates. Red and blue bars represent 5′ UTR isoforms from a single gene (Rojas-Duran and Gilbert, 2012). (B) Schematic of DNA pool design. All sequences begin with the T7 promoter followed by ≤122nts of 5′ UTR sequence, then a minimum of 24nts of coding sequence, followed by a 10nt identifier barcode, and finally an RT handle for library preparation. Endogenous 5′ UTR sequences were derived from RNA-seq data(Pelechano et al., 2014). (C) DART sequencing overview. (D) DART reproducibly measures ribosome recruitment over a thousand-fold range. Comparison of 3 DART replicates. See also Supplementary Fig. 1.
Next, we used these designed pools to generate dsDNA templates for in vitro transcription followed by enzymatic mRNA capping to produce the pooled RNA substrates for in vitro translation initiation. RNAs were enzymatically biotinylated at their 3′ ends to facilitate quantitative recovery. RNA pools were incubated in yeast translation extracts (Gilbert et al., 2007; Hodgman and Jewett, 2013; Iizuka et al., 1994) for 30 min, a time point that maximizes quantitative differences in ribosome recruitment between 5′ UTRs while yielding enough initiated mRNA for robust library preparation. Cycloheximide was included in the translation reaction to freeze recruited ribosomes at initiation codons and prevent ribosome run-off during ultracentrifugation to separate 80S ribosome-mRNA complexes from untranslated mRNAs. Ribosome-bound mRNA was isolated and sequenced, and the relative abundance of each sequence was compared to its abundance in the input pool to determine a ribosome recruitment score (RRS) for each 5′ UTR (Fig. 1c). DART revealed up to 1,000-fold differences in ribosome recruitment that were highly reproducible (R2 = 0.94–0.95) among three independent replicates (Fig. 1d, Supplementary Table 1). We selected six 5′ UTRs for low throughput validation and observed good agreement with DART measurements (Supplementary Fig. 1a). Thus, we established a robust method to determine the translation initiation activity of defined 5′ UTR sequences.
A caveat to the 80S/input calculation used to assess translation initiation activity is that sequences that destabilize 5′ UTRs in extracts could also lead to a reduction in 80S-bound RNA compared to the input pool. We therefore tested an alternative method of calculating ribosome recruitment scores by sequencing the untranslated mRNA from the top of the gradient in addition to the 80S fraction, RRS′ = 80S/(80S+mRNP). These two metrics, RRS and RRS′, produced correlated but not identical results (R = 0.61 p < 2.2e−16, Supplementary Fig. 1b). Notably, each of the main conclusions reported below is consistent between calculation methods. Because changes in translation initiation can indirectly affect mRNA levels (Chan et al., 2018; LaGrandeur and Parker, 1999; Miller et al., 2011; Muhlrad and Parker, 1999; Schwartz and Parker, 1999; Sun et al., 2012), the 80S/input calculation (RRS) is preferred (see Supplemental Discussion).
Systematic testing of presumed inhibitory 5′ UTR features highlights context effects
Stable RNA secondary structures are thought to impede scanning of the 5′ UTR and thereby depress protein synthesis. Low-throughput assays have shown inhibitory effects of certain engineered stable stem-loop (SL) structures, which is partially consistent with global correlations between predicted 5′ UTR folding energy and translation activity (Kozak, 1980; Weinberg et al., 2016). However, the observed correlations are modest and SL structures can have different effects depending on their position within the 5′ UTR (Li et al., 2017). We exploited the throughput of the DART approach to systematically interrogate the effects of RNA secondary structure on translation initiation (Fig. 2a). We designed SLs of varying stabilities spanning a physiologically relevant range (−5 to −31 kcal/mole) and inserted them into 20 natively unstructured 5′ UTRs (Fig. 2a). The complete set of 5′ UTR-SL constructs included 6 different stems, positioned every 3 nts across each 5′ UTR and extending 14 nts into the CDS, for a total of 1891 variants (Supplementary Table 2). All constructs were folded in silico to verify that a single structure was predicted for every sequence context.
Fig. 2. Inhibitory RNA features have highly context-dependent effects on initiation.

(A) Design of stem loop constructs. Two sets of stem loops were designed to span a range of folding energies with the second set scrambling the stem sequences of the first. Stems were inserted into 20 endogenous unstructured 5′ UTRs and positioned every 3nts. (B) Engineered stem loops repress ribosome recruitment scores (RRS). All stems repress recruitment relative to sequences without an inserted stem (black) with the strongest stem (red) showing the greatest level of repression, followed by the intermediate stem (blue) and the weakest stem (green). P value calculated using Fisher’s Test comparing sequences with introduced stems to those without. (C) Individual 5′ UTRs show distinct effects of SL insertion. Relative recruitment for the YER003C 5′ UTR with the −12kcal/mol stem inserted. In this case, the stem is most repressive near the cap and at the start codon. (D) An example 5′ UTR from YDR135C showing reduced ribosome recruitment upon mutation of preferred AUGi context nucleotides (−3A, +4G and +5C). Identity of the mutated nucleotide is indicated by color. (E) Mutations in AUGi context nucleotides cause variable effects on ribosome recruitment with changes of the −3A being generally the most deleterious. Density plot showing the probability distribution of the relative RRS at each position. See also Supplementary Fig. 2.
These well-defined SL structures inhibited ribosome recruitment to varying degrees depending on the site of insertion. Stronger inhibition was observed with increasing stem stability and SLs with different sequences but the same folding energy inhibited to a similar extent consistent with a structure-based mechanism (Fig. 2b, Supplementary Fig. 2a). Similar trends were seen using the alternative method of calculating ribosome recruitment scores, RRS′ (Supplementary Fig. 4a). Globally, the weaker SLs (−5 to −12 kcal/mole) inhibited ribosome recruitment most strongly at the cap-proximal position (Fig. 2c and Supplementary Fig. 2b). The most stable SLs (−31 kcal/mole) strongly depressed translation, particularly from central positions (Supplementary Fig. 2b). Against these global trends, individual 5′ UTRs showed large position-dependent differences in the extent of translation inhibition due to SL insertion (Supplementary Fig. 2b,c). These results highlight the pitfall of inferring general translation mechanisms from characterizing relatively few transcripts. Future DART experiments will elucidate how the single-stranded sequences that flank 5′ UTR structures potentiate or attenuate translational repression. Such context effects may contribute to mRNA-specific sensitivity to helicase activity for translation initiation in vitro and in vivo (Gupta et al., 2018). We extended our systematic characterization of 5′ UTR features to examine differences in translation caused by sequences flanking the start codon (AUGi). Current models of initiation, informed by structural studies of initiation complexes, invoke start codon context as an important feature in promoting translation. Consistent with this, upstream AUGs with optimal context are generally more inhibitory to downstream protein synthesis than those in a suboptimal context (Johnstone et al., 2016). However, AUGi context is only weakly predictive of ribosome occupancy in cells, with many efficiently translated mRNAs having poor context and vice versa, making the extent of translational enhancement by start codon context unclear (Zur and Tuller, 2013).
We used DART to interrogate the contribution of the nucleotides surrounding AUGi by altering the identity of each of three nucleotides that have previously been implicated in affecting translation (Nakagawa et al., 2008) (Fig 2d). We selected 50 native 5′ UTRs with the preferred ANNAUGGC context and mutated −3A, +4G and +4C to all other nucleotides to test a total of 415 AUGi context variants (Supplementary Table 3. Overall, changes at the −3 position were more deleterious than the other positions tested (Fig 2e). Although many mutations reduced ribosome recruitment (Fig 2d), the magnitude differed among 5′ UTRs and even included cases where changes away from the preferred context improved recruitment (Fig 2e, Supplementary Fig. 2d). For example, YDR157W showed increased ribosome recruitment when any of the preferred nucleotides were mutated (Supplementary Fig. 2d), perhaps due to disruption of structures that are predicted to occlude the start codon in the wild type sequence. Similar to the generally modest effects of mutating AUGi context nucleotides, native sequences with preferred vs. sub-optimal AUGi context showed only subtle differences in the distributions of RRS values (Supplementary Figure 2e). Overall these data support that features other than AUGi context are the primary drivers of mRNA-specific differences in translation activity in yeast. We therefore sought to identify other sequence features that contribute to the observed range of ribosome recruitment activity.
A broad stimulatory role for initiation factor binding motifs in 5′ UTRs
We hypothesized that 5′ UTR elements that bind preferentially to eukaryotic initiation factors (eIFs) may function as translational enhancers (Gilbert, 2010). Consistent with this model, diverse viruses rely on high-affinity interactions between their 5′ UTRs and cellular initiation factors and/or ribosomes for efficient translation (Filbin and Kieft, 2009). The eIF4G subunit of the cap binding complex is a prime candidate to mediate the activity of translational enhancer sequences. eIF4G contains three RNA binding domains that directly interact with mRNA and are essential for yeast growth (Berset et al., 2003; Park et al., 2011) although specific functional interactions between eIF4G and cellular mRNAs had not been characterized. We previously used a high-throughput approach, RNA Bind-n-Seq (RBNS) (Lambert et al., 2014), to interrogate the RNA binding specificity of eIF4G1. Using a library of random 20mer RNA to test ~87,380 distinct RNA 7mer motifs for binding to different concentrations of recombinant eIF4G1 we uncovered a preference for RNA sequences containing oligo-uridine (U). Consistently, we found that inserting oligo(U) into an unstructured RNA increased binding to eIF4G1 by 20-fold (Zinshteyn et al., 2017).
Here, we sought to comprehensively test the impact of endogenous oligo(U) motifs on translation initiation. Hundreds of native yeast 5′ UTRs contain oligo(U) sequences, which are evolutionarily conserved among budding yeast species and enriched in genes with regulatory roles (Zinshteyn et al., 2017). We synthesized a pool of capped mRNAs consisting of all yeast 5′ UTR sequences ≤94 nt long that contain U≥7 (168 in total, Supplementary Table 4 together with their start codons and some coding sequence. The pool included matched controls for each 5′ UTR in which the oligo(U) motif was replaced with a CA repeat of equal length (Fig. 3a). We performed DART on these native 5′ UTR sequences and compared their activity with and without their endogenous oligo(U) motifs. In all 3 replicates, wild type oligo(U)-containing mRNAs were recruited more efficiently to the ribosomal fraction than their matched mutant counterparts (Fig. 3b, Supplementary Fig. 4c). Of the 5′ UTR sequences whose ribosome recruitment differed significantly (p < 0.05 by 2-tailed paired t-test) between the wild type and mutant variants, all were recruited better with the oligo(U) motif present (Fig. 3c). These results are consistent with and substantially extend previous low-throughput experiments that showed endogenous 5′ UTR oligo(U) motifs stimulated translation of full-length mRNA in yeast lysates (Zinshteyn et al., 2017). Our results reveal a broad stimulatory role for eIF4G binding motifs within yeast 5′ UTRs and illustrate the power of DART to assess the function of putative RNA regulatory elements identified by other high-throughput approaches.
Fig. 3. eIF4G binding motifs promote translation initiation.

(A) Schematic of eIF4G1 binding to designed 5′ UTR pool including both WT oligo(U) motifs (blue) and U->CA mutants (red). (B) Violin plots of ribosome recruitment score distributions for 5′ UTRs containing U≥7, and the matched controls, for 3 independent replicates. P values calculated using Fisher’s Test. (C) Ranked changes in ribosome recruitment scores between oligo(U) 5′ UTRs and matched U->CA mutants. Dots indicate the mean of all replicates that met read coverage thresholds. Blue lines indicate the minimum and maximum value across the replicates. 5′ UTRs with a statistically significant (Bonferroni-corrected two-tailed t-test p < 0.05) difference between WT and mutant variants are colored orange.
Production of alternative 5′ UTR isoforms generally affects translation activity
Many eukaryotic genes express multiple mRNA isoforms that differ in their 5′ UTRs, which complicates the analysis of ribosome profiling data to identify translational control elements. Because ribosome-protected footprints within coding sequences cannot be correctly assigned to translation initiation by specific 5′ UTRs, the mRNA-specific translational efficiency inferred from ribosome profiling data may not accurately reflect the actual translation activity of any mRNA isoform (Fig. 4a). We therefore used DART to directly compare the activity of expressed alternative mRNA isoforms whose 5′ UTRs differ by at least 10 nucleotides (Supplementary Table 5).
Fig. 4. DART identifies translational silencers and enhancers within alternative 5′ UTRs.

(A) 5′ UTR isoforms confound the interpretation of averaged ribosome densities observed in coding sequences by ribosome profiling. (B) Most alternative 5′ UTR isoforms tested affect ribosome recruitment (RRS). 2653 soform pairs differed significantly by >2-fold (purple), 145 differed significantly, but by <2-fold (magenta), and 1332 did not confer significant differences in RRS (blue). P values from t test Bonferroni corrected. (C) Example genes with alternative 5′ UTR isoforms differing in RRS. The long (5′ extended) isoforms of YDL201W, YDR337W, YBR138C and YLR440C contain putative silencer elements and the long isoforms of YGL095C, YLR091W, YDR063W and YGR196C contain putative enhancers. (D) Violin plots of RRS values from isoform pairs showing differential ribosome recruitment (p < 0.05). The additional 5′ sequences included in longer isoforms were used as inputs to identify enriched motifs using DREME. Top two silencing (above) and enhancing (below) motifs are shown. (E) ACCAC motifs identified by DART analysis are sufficient to repress translation of full-length mRNAs in vitro (left) and in vivo (right). Translation activity in vivo is normalized to Fluc mRNA levels and total protein. See also Supplementary Fig. 3 and 4.
We analyzed RNA-seq data from wild type yeast (Methods) and identified 4354 alternative mRNA isoforms expressed from 2064 genes that we tested for isoform-specific translation activity by DART. Notably, we identified 1639 isoform pairs with reproducible differences in ribosome recruitment (p < 0.05, Bonferroni corrected two tailed t test) of which 843 differed by more than 3-fold (Supplementary Table 5). These results significantly extend previous evidence that alternative 5′ UTRs differ in ribosome association in vivo (Arribere and Gilbert, 2013; Floor and Doudna, 2016; Wang et al., 2016). DART establishes the direct contribution of differences in 5′ UTR sequences to differences in translation initiation and eliminates potentially confounding effects of co-occurring alternative 3′-UTR sequences.
C rich sequences silence translation
We leveraged these mRNA isoform comparisons to identify previously undetected translational control elements within 5′ UTRs. Enhancer and silencer elements were operationally defined as sequences present in a longer 5′ UTR variant that showed a higher RRS than a shorter alternative 5′ UTR of the same gene (enhancer) or lower RRS than the corresponding shorter alternative 5′ UTR (silencer). We identified 658 enhancer regions and 541 silencer regions by the criteria that their inclusion reproducibly changed RRS more than 2-fold (p < 0.001, n = 3 replicates) (Supplementary Table 5). Of these, 72 enhancers and 67 silencers changed RRS by more than 10-fold (Fig. 4c). Remarkably, sequence differences of only 10 nucleotides were sufficient to significantly alter translation initiation in many cases (Supplementary Table 5). Thus, DART reveals relatively short RNA sequences (10–13 nucleotides) that are sufficient to significantly alter ribosome recruitment when present near the 5′ end of the mRNA. Overall, we found that 68% of genes with alternative 5′ UTRs showed significant differences in translation initiation between isoforms.
To illuminate the mechanisms of translational control, we looked for over-represented motifs within enhancers and silencers using DREME (Bailey et al., 2015). Long 5′ UTR isoforms with higher ribosome recruitment activity were enriched for AU-rich sequences, which are distinct from the previously identified eIF4G-binding U7 motif. Intriguingly, the silencer regions were enriched in CCH motifs. We observed similar C-rich motifs in the bottom 10% of all 5′ UTRs (Fig. 4d and Supplementary Fig. 2f), which suggests a common mechanism of translational repression.
We verified the repressive effects of C-rich silencer motifs on the translational output of full-length mRNAs using luciferase reporters. For selected genes containing the motif of interest in the longer 5′ UTR isoform, we introduced mutations to disrupt the putative silencer in the long isoform, thereby testing its necessity for translation repression. In parallel, we added the motif to the short isoform, thereby testing its sufficiency. In each case, disruption of the motif from the longer isoform resulted in an increase in protein production whereas addition of the motif reduced luciferase activity from the short isoform (Fig. 4e and Supplementary Figure 3a,b). We observed similar effects with these constructs expressed in vivo (Fig. 4e and Supplementary Figure 3a,b) with somewhat smaller differences in translation between 5′ UTRs in vivo than in vitro, which is consistent with previous studies (Rojas-Duran and Gilbert, 2012). Together, these results validate the C-rich sequence motifs uncovered by DART analysis as l translational control elements that repress ribosome recruitment in vitro and protein production in cells.
Discussion
Here we present direct analysis of ribosome targeting (DART) as a new approach for high-throughput functional testing of endogenous and engineered 5′ UTR variants. We applied DART to more than 8,000 endogenous 5′ UTRs from the model eukaryote S. cerevisiae and identified C-rich motifs as translational silencers present within hundreds of 5′ UTRs. We also used DART to systematically probe the effect of oligo uridine motifs that were previously identified as preferred binding sequences for the translation initiation factor eIF4G1 (Zinshteyn et al., 2017) and thereby established a general stimulatory role for this 5′ UTR element in endogenous mRNAs. Our results illustrate the power of the DART method to uncover the regulatory elements underlying mRNA-specific differences in protein output, which we anticipate will be broadly applicable to the study of translation initiation including by human mRNAs.
We identified 5′ UTR sequences that increased or decreased ribosome recruitment by more than 1,000-fold in cell lysates. Specific sequences, such as C-rich motifs, that affected ribosome recruitment as measured by DART similarly affected protein synthesis from luciferase reporters in cells (e.g. Fig. 4d,e and Supplementary Fig. 3a,b). However, the magnitude of 5′ UTR differences was larger in vitro, as has been noted previously (Rojas-Duran and Gilbert, 2012). There are several possible explanations for this quantitative discrepancy. First, translation is simply more efficient in cells, likely due to dilution of translation factors in lysate. Some of the larger range of ribosome recruitment activities observed in lysates may reflect expansion of the low end of 5′ UTR activity in translation initiation. Competition among 5′ UTRs for limiting initiation factors may also contribute to the large range of RRS values in pooled DART assays but does not explain differences between in vitro and in vivo for luciferase reporter mRNAs tested singly.
Translation-dependent changes to mRNA turnover are another likely contributor to quantitative differences between in vitro (pre-steady state) and in vivo (steady state) assays. The apparent blunting of 5′ UTR-dependent translation differences, as observed in cells by normalizing measured protein levels to steady-state mRNA levels, is the expected result if poorly initiated mRNAs are more rapidly degraded in vivo. Consistently, reporter mRNAs with defects in translation initiation have been found to be destabilized in yeast cells (Chan et al., 2018; Miller et al., 2011; Sun et al., 2012). Furthermore, global anti-correlations between “TE” (average number of ribosomes per mRNA at steady-state) and mRNA half-life suggest a broad correspondence between decreased ribosome recruitment and increased mRNA decay (Arribere and Gilbert, 2013; Chan et al., 2018; Kristensen et al., 2013; Lahtvee et al., 2017; Miller et al., 2011; Muhlrad and Parker, 1999). Future work that combines DART with direct assessment of mRNA half-lives in intact as well as ribosome-depleted lysates may facilitate dissection of the interconnected effects of 5′ UTR sequences on translation initiation and mRNA decay.
Quantitative analysis of translation initiation in vitro has begun to reveal distinct initiation mechanisms used by different mRNAs (Gupta et al., 2018; Mitchell et al., 2010). DART increases the throughput of such experiments a thousand fold, which will be important for dissecting 5′ UTR-specific differences in the effects of features such as initiation site context and inhibitory 5′ UTR structures. The broad, but weak, correlations previously observed between predicted 5′ UTR folding energy and translation activity (Li et al., 2017; Weinberg et al., 2016) can now be understood as the probable outcome of highly context-dependent effects of RNA structures. Sequence context effects may similarly account for the failure of RNA folding to predict more than 36% of the observed variance in translational repression following inactivation of the mRNA helicase, Ded1 (Sen et al., 2015). It will be interesting to extend previous in vitro characterization of Ded1-dependent mRNAs to a much broader sample of 5′ UTRs (Gupta et al., 2018). Overall, our results demonstrate that it will be important to test many mRNA sequences to build accurate mechanistic models of initiation. The capacity of DART to quantify translation initiation on thousands of defined 5′ UTR variants in biochemically accessible cell lysates will be broadly useful to elucidate fundamental translational mechanisms.
An important application for DART technology is in predicting cellular protein production from alternative mRNA isoforms observed by RNA sequencing. Here we used DART to quantify the translational activity of 4,130 alternative 5′ UTR isoforms in budding yeast, revealing functional differences between >60% of isoform pairs. Many eukaryotic genes express multiple mRNA isoforms due to alternative transcription start site (TSS) selection, and the process of TSS selection is itself highly regulated (Carninci et al., 2006; Cheng et al., 2018; Kimura et al., 2006; Wang et al., 2008; Yamashita et al., 2011). Yeast transcript isoform analysis across different environmental conditions identified more than 1200 genes with regulated alternative 5′ UTRs (Waern and Snyder, 2013) and alternative TSS selection contributes more to mammalian mRNA isoform diversity than alternative splicing in some tissues (Pal et al., 2011). Tumor-specific changes in promoter usage have also been observed in dozens of different cancer types (Demircioglu et al., 2019). DART will be broadly useful to determine how sequence differences between alternative 5′ UTRs affect protein production, which is critical for predicting the functional output of normal or pathological transcriptional regulation that changes the type of mRNA produced as well as the number of mRNA molecules.
A related application of DART will allow rapid functional screening of 5′ UTR SNPs that have been linked to diverse disease phenotypes in genome-wide association studies (GWAS). Current efforts to identify the causal variants, which are potential targets for new therapies, are largely restricted to non-synonymous changes within protein coding sequences. The DART method will enable systematic identification of 5′ UTR variants that are likely to be damaging by dysregulating protein levels. Finally, DART will be useful to optimize 5′ UTRs for protein production in a variety of settings, including in therapeutic mRNAs.
STAR Methods
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Wendy Gilbert (wendy.gilbert@yale.edu)
Materials availability
No materials were generated during this study.
Data and code availability
All data have been deposited at GEO and accession numbers are available in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| Nano-Glo® Luciferase Assay System | Promega | Cat# N1110 |
| Bright-Glo™ Luciferase Assay System | Promega | Cat# E2610 |
| Slide-A-Lyzer™ Dialysis Cassettes, 3.5K MWCO | Thermo | Cat# 66330 |
| Deposited data | ||
| Oligo(U) DART | This study | GSE98788 |
| Yeast TL DART | This study | GSE182290 |
| Experimental models: Organisms/strains | ||
| Yeast strain (MATa trp1Δ leu2–3,112 ura3–52 gcn2Δ::hisG PGAL1-myc-UBR1::TRP1::ubr1, pRS316 <URA3>) | Zinshteyn, B et al. 2017 | YWG1245 |
| Oligonucleotides | ||
| GCTAATACGACTCACTATAGGG | This study | RN7 |
| CCTTGGCACCCGAGAATTCCA | This study | RN8 |
| GCCTTGGCACCCGAGAATTCC | This study | oWG915 |
| /5Phos/NNN NNN NNN NGA TCG TCG GAC TGT AGA ACT CTG AAC GTG /3SpC3/ | This study | oWG920 |
| AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA | This study | RP1 |
| CAAGCAGAAGACGGCATACGAGATN6GTGACTGGAGTTCCTTGGCACCCGAGAATTCCA | This study | OBC |
| Software and algorithms | ||
| STAR | Dobin et al., 2013 | |
| FastUniq | Xu et al., 2012 | |
| Cufflinks | Trapnell et al., 2012 | |
Experimental model and subject details
The yeast strain YWG1245 (MATa trp1Δ leu2–3,112 ura3–52 gcn2Δ::hisG PGAL1-myc-UBR1::TRP1::ubr1, pRS316 <URA3>) used in this assay was cultured in liquid YPAD (1% yeast extract, 2% peptone, 2% glucose, 0.01% adenine hemisulfate) at 30°C with constant shaking.
Method details
5′ UTR Pool Design and Synthesis
Each sequence in the pool consisted of ≤122 nucleotides of 5′ UTR sequence followed by at least 24 nucleotides of coding sequence followed by a randomized 10 nucleotide unique identifier barcode and an adaptor sequence used for priming reverse transcription and Illumina sequencing (Fig 1a). For 5′ UTR annotations, 5′ UTR abundances were calculated from deep RNA sequencing of wild type yeast (Pelechano et al., 2014; Pelechano et al., 2013). Each 5′ UTR was merged with its most abundant neighbor within 10 nts in either direction such that no two 5′ UTRs were within 10 nts of each other. We then imposed the following requirements for inclusion in the pool: (1) 5′ UTRs must be expressed within 25% of the mode abundance for a given 5′ UTR, and (2) 5′ UTRs must make up at least 5% of the total abundance for that ORF, unless the mode was <5% of the total, in which case we used the mode. Upstream AUGs within 761 (6.3% of all) 5′ UTR sequences were mutated to AGT such that the first AUG encountered by a scanning pre-initiation complex moving 5′ to 3′ would be the annotated AUGi. We included a parallel set of constructs testing the ribosome recruitment activity of each uAUG individually, in which case the downstream (canonical) AUG was mutated to AGT. In total, we measured recruitment to 1334 uAUGs. Designed oligos were purchased as a pool (Twist Bioscience) and PCR amplified with RN7 and RN8 (Supplementary Table 6). RNAs were produced by runoff T7 transcription from gel-purified DNA template. RNAs were 5′ capped using the Vaccinia Capping System (NEB M2080S) and 3′ biotinylated using the Pierce RNA 3′ end biotinylation kit (Thermo Scientific 20160).
Translation Assays
Yeast translation extracts were made as described (Rojas-Duran and Gilbert, 2012) from YWG1245 (MATa trp1Δ leu2–3,112 ura3–52 gcn2Δ::hisG PGAL1-myc-UBR1::TRP1::ubr1, pRS316 <URA3>) cultured in liquid YPAD (1% yeast extract, 2% peptone, 2% glucose, 0.01% adenine hemisulfate). Cultures were grown to mid-log phase, harvested and then washed two times in mannitol buffer (30mM HEPES pH 7.4, 100mM KOAc, 2mM Mg(OAc)2, 2mM DTT, 0.1mM PMSF, 8.5% mannitol). Cells were then resuspended in 1.5x volume of mannitol buffer and subsequently lysed by bead beating with 5x pellet volume of glass beads. Six 1 minute rounds of bead beating were performed with intermittent one minute cooling periods on ice. Beads were pelleted and the supernatant transferred to a fresh tube. Cell debris was pelleted by a 30 minute spin at 16,000 rcf. Supernatant was removed and dialyzed using Slide-A-lyzer, 3–12 mL, 3,500 MWCO (Pierce #66110) for four hours (30mM HEPES pH 7.4, 100mM KOAc, 2mM Mg(OAc)2, 2mM DTT), changing the buffer once. Approximately 40 pmoles of mRNA were added per 2 mL in vitro translation reaction (22 mM HEPES pH 7.4, 120 mM KOGln, 3 mM MgOGln, 3.75 mM ATP, 0.5 mM GTP, 25 mM creatine phosphate, 1.6mM DTT, 400U RNaseIn, 2mM PMSF, 650 mg creatine phosphokinase, 1X cOmplete EDTA-free protease inhibitor) containing 50% yeast extract. Reactions were incubated with 0.5mg/mL cycloheximide at 26°C for 30 minutes in a shaking thermomixer.
To measure translation activity in vivo YWG1245 was transformed with pGAL-5′ UTR-Fluc plasmids and grown to log phase in media containing 2% raffinose. 5′ UTR sequences (Supplementary Table 6) were generated by gBlock (Twist Bioscience) mRNA expression was induced by addition of galactose to a final concentration of 2%. Whole-cell lysates were prepared by vortexing with glass beads in 1× PBS with protease inhibitors (2 mM phenylmethanesulfonyl fluoride, and 1× protease inhibitor cocktail (Roche). Luciferase activity was measured using the Luciferase Bright-Glo Assay System (Promega) on a Berthold Centro XS Luminometer. Luciferase values were normalized to both FLUC mRNA levels and total protein. Fluc mRNA levels were determined by Northern blot on total cellular RNA isolated from whole-cell lysates by hot phenol extraction and normalized to U1 RNA levels. Fluc mRNA levels were determined by Northern blot on total cellular RNA isolated from whole-cell lysates by hot phenol extraction and normalized to U1 RNA levels. Probes made using radiolabeled PCR products from the following primer pairs: 5′-AAGGCGTGTTTGCTGACGTTTC-3′, 5′-CACCCGTTCCTACCAAGACC-3’ (U1); 5′-TGGGCGCGTTATTTATCGGAGTTGC-3′, 5′-GAGCCCATATCCTTGCCTGATACC-3′ (Fluc).
80S isolation
Translation reactions were loaded onto 10%−50% sucrose gradients in polysome lysis buffer (20 mM HEPES-KOH pH 7.4, 2 mM magnesium acetate, 0.1 M potassium acetate, 0.1 mg/mL cycloheximide, 1% TritonX-100) and centrifuged at 27,000 × g in a Beckman SW28 rotor for 3 hours. Gradients were fractionated from the top down using a Biocomp Gradient Station (Biocomp Instruments) with continual monitoring of absorbance at 254 nm. Fractions corresponding to the 80S peak were pooled and RNA extracted using phenol/chloroform followed by isopropanol precipitation.
Library preparation
RNA pellets from the input pool, total lysate, and gradient fractions were resuspended in binding buffer (0.5M NaCl, 20mM Tris-HCl pH 7.5, 1mM EDTA) and biotinylated 5′ UTRs were recovered using Hydrophilic Streptavidin Beads (NEB S1421S). Isolated RNA was reverse transcribed using the barcoded primer OWG921 and Superscript III (Invitrogen 18080093). Gel-purified cDNA products were ligated to the adapter OWG920 (Supplementary Table 6) using T4 RNA ligase 1 (NEB M0437M). cDNA cleanup was performed using 10 μl MyOne Silane beads (Thermo Scientific 37002D) per sample. Libraries were then PCR amplified with primers RP1 and OBC (Supplementary Table 6) and sequenced on a HiSeq 2500.
Read processing
Reads were first demultiplexed by their library-level barcodes. PCR duplicates were collapsed using FastUniq (Xu et al., 2012). Output sequences were trimmed of the 5′ decamer barcode using FastX trimmer. Reads were uniquely mapped to the sequences within the synthetic pool using STAR (Dobin et al., 2013) with the command line options < --outSAMstrandField intronMotif --alignIntronMin 200 >. Any reads that failed to align were excluded from further analysis. FPKMs were quantified using cufflinks (Trapnell et al., 2012).
Supplementary Material
Supplemental Table 2: Ribosome recruitment scores for stem loop variants; related to Fig. 2
Supplemental Table 3: Ribosome recruitment scores for context variants; related to Fig. 2
Supplemental Table 4: Ribosome recruitment scores for oligo(U) variants; related to Fig. 3
Supplemental Table 1: RNA sequences, FPKM values, Ribosome recruitment scores; related to Fig. 1
Supplemental Table 5: Ribosome recruitment scores for 5′ isoforms; related to Fig. 4
Supplemental Table 6: 5′ UTR sequences tested with motif abolished or added; related to Fig. 4
Highlights.
DART illuminates thousand-fold differences in 5′ UTR-specific translation activity
SNPs and alternative 5′ UTR isoforms affect ribosome recruitment significantly
5′ UTR motifs bind initiation factors directly, broadly stimulating translation
Sequence specific silencer motifs repress translation in vitro and in vivo
Acknowledgements
We thank members of the Gilbert lab for advice and feedback on the manuscript. This work was supported by NIH R01GM132358 to W.V.G. as well as NIH 1K99GM135533 and ACS 133477-PF-19-092-01-RMC to R.O.N.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Archer SK, Shirokikh NE, Beilharz TH, and Preiss T (2016). Dynamics of ribosome scanning and recycling revealed by translation complex profiling. Nature 535, 570–574. [DOI] [PubMed] [Google Scholar]
- Arribere JA, and Gilbert WV (2013). Roles for transcript leaders in translation and mRNA decay revealed by transcript leader sequencing. Genome Res 23, 977–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Johnson J, Grant CE, and Noble WS (2015). The MEME Suite. Nucleic Acids Res 43, W39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berset C, Zurbriggen A, Djafarzadeh S, Altmann M, and Trachsel H (2003). RNA-binding activity of translation initiation factor eIF4G1 from Saccharomyces cerevisiae. RNA 9, 871–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CA, Taylor MS, Engstrom PG, Frith MC, et al. (2006). Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet 38, 626–635. [DOI] [PubMed] [Google Scholar]
- Chan LY, Mugler CF, Heinrich S, Vallotton P, and Weis K (2018). Non-invasive measurement of mRNA decay reveals translation initiation as the major determinant of mRNA stability. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z, Otto GM, Powers EN, Keskin A, Mertins P, Carr SA, Jovanovic M, and Brar GA (2018). Pervasive, Coordinated Protein-Level Changes Driven by Transcript Isoform Switching during Meiosis. Cell 172, 910–923 e916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demircioglu D, Cukuroglu E, Kindermans M, Nandi T, Calabrese C, Fonseca NA, Kahles A, Lehmann KV, Stegle O, Brazma A, et al. (2019). A Pan-cancer Transcriptome Analysis Reveals Pervasive Regulation through Alternative Promoters. Cell 178, 1465–1477 e1417. [DOI] [PubMed] [Google Scholar]
- Dever TE, Feng L, Wek RC, Cigan AM, Donahue TF, and Hinnebusch AG (1992). Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 68, 585–596. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvir S, Velten L, Sharon E, Zeevi D, Carey LB, Weinberger A, and Segal E (2013). Deciphering the rules by which 5’-UTR sequences affect protein expression in yeast. Proc Natl Acad Sci U S A 110, E2792–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filbin ME, and Kieft JS (2009). Toward a structural understanding of IRES RNA function. Curr Opin Struct Biol 19, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floor SN, and Doudna JA (2016). Tunable protein synthesis by transcript isoforms in human cells. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert WV (2010). Alternative ways to think about cellular internal ribosome entry. J Biol Chem 285, 29033–29038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert WV, Zhou K, Butler TK, and Doudna JA (2007). Cap-independent translation is required for starvation-induced differentiation in yeast. Science 317, 1224–1227. [DOI] [PubMed] [Google Scholar]
- Gupta N, Lorsch JR, and Hinnebusch AG (2018). Yeast Ded1 promotes 48S translation pre-initiation complex assembly in an mRNA-specific and eIF4F-dependent manner. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgman CE, and Jewett MC (2013). Optimized extract preparation methods and reaction conditions for improved yeast cell-free protein synthesis. Biotechnol Bioeng 110, 2643–2654. [DOI] [PubMed] [Google Scholar]
- Iizuka N, Najita L, Franzusoff A, and Sarnow P (1994). Cap-dependent and cap-independent translation by internal initiation of mRNAs in cell extracts prepared from Saccharomyces cerevisiae. Mol Cell Biol 14, 7322–7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, and Weissman JS (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone TG, Bazzini AA, and Giraldez AJ (2016). Upstream ORFs are prevalent translational repressors in vertebrates. EMBO J 35, 706–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Wakamatsu A, Suzuki Y, Ota T, Nishikawa T, Yamashita R, Yamamoto J, Sekine M, Tsuritani K, Wakaguri H, et al. (2006). Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes. Genome Res 16, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M (1980). Influence of mRNA secondary structure on binding and migration of 40S ribosomal subunits. Cell 19, 79–90. [DOI] [PubMed] [Google Scholar]
- Kristensen AR, Gsponer J, and Foster LJ (2013). Protein synthesis rate is the predominant regulator of protein expression during differentiation. Mol Syst Biol 9, 689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaGrandeur T, and Parker R (1999). The cis acting sequences responsible for the differential decay of the unstable MFA2 and stable PGK1 transcripts in yeast include the context of the translational start codon. RNA 5, 420–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahtvee PJ, Sanchez BJ, Smialowska A, Kasvandik S, Elsemman IE, Gatto F, and Nielsen J (2017). Absolute Quantification of Protein and mRNA Abundances Demonstrate Variability in Gene-Specific Translation Efficiency in Yeast. Cell Syst 4, 495–504 e495. [DOI] [PubMed] [Google Scholar]
- Lambert N, Robertson A, Jangi M, McGeary S, Sharp PA, and Burge CB (2014). RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol Cell 54, 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JJ, Chew GL, and Biggin MD (2017). Quantitating translational control: mRNA abundance-dependent and independent contributions and the mRNA sequences that specify them. Nucleic Acids Res 45, 11821–11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JJ, Chew GL, and Biggin MD (2019). Quantitative principles of cis-translational control by general mRNA sequence features in eukaryotes. Genome Biol 20, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C, Schwalb B, Maier K, Schulz D, Dumcke S, Zacher B, Mayer A, Sydow J, Marcinowski L, Dolken L, et al. (2011). Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol Syst Biol 7, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SF, Walker SE, Algire MA, Park EH, Hinnebusch AG, and Lorsch JR (2010). The 5’−7-methylguanosine cap on eukaryotic mRNAs serves both to stimulate canonical translation initiation and to block an alternative pathway. Mol Cell 39, 950–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muckenthaler M, Gray NK, and Hentze MW (1998). IRP-1 binding to ferritin mRNA prevents the recruitment of the small ribosomal subunit by the cap-binding complex eIF4F. Mol Cell 2, 383–388. [DOI] [PubMed] [Google Scholar]
- Muhlrad D, and Parker R (1999). Recognition of yeast mRNAs as “nonsense containing” leads to both inhibition of mRNA translation and mRNA degradation: implications for the control of mRNA decapping. Mol Biol Cell 10, 3971–3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa S, Niimura Y, Gojobori T, Tanaka H, and Miura K (2008). Diversity of preferred nucleotide sequences around the translation initiation codon in eukaryote genomes. Nucleic Acids Res 36, 861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal S, Gupta R, Kim H, Wickramasinghe P, Baubet V, Showe LC, Dahmane N, and Davuluri RV (2011). Alternative transcription exceeds alternative splicing in generating the transcriptome diversity of cerebellar development. Genome Res 21, 1260–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EH, Zhang F, Warringer J, Sunnerhagen P, and Hinnebusch AG (2011). Depletion of eIF4G from yeast cells narrows the range of translational efficiencies genome-wide. BMC Genomics 12, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Wei W, Jakob P, and Steinmetz LM (2014). Genome-wide identification of transcript start and end sites by transcript isoform sequencing. Nat Protoc 9, 1740–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Wei W, and Steinmetz LM (2013). Extensive transcriptional heterogeneity revealed by isoform profiling. Nature 497, 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas-Duran MF, and Gilbert WV (2012). Alternative transcription start site selection leads to large differences in translation activity in yeast. RNA 18, 2299–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sample PJ, Wang B, Reid DW, Presnyak V, McFadyen IJ, Morris DR, and Seelig G (2019). Human 5’ UTR design and variant effect prediction from a massively parallel translation assay. Nat Biotechnol 37, 803–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DC, and Parker R (1999). Mutations in translation initiation factors lead to increased rates of deadenylation and decapping of mRNAs in Saccharomyces cerevisiae. Mol Cell Biol 19, 5247–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen ND, Zhou F, Ingolia NT, and Hinnebusch AG (2015). Genome-wide analysis of translational efficiency reveals distinct but overlapping functions of yeast DEAD-box RNA helicases Ded1 and eIF4A. Genome Res 25, 1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Schwalb B, Schulz D, Pirkl N, Etzold S, Lariviere L, Maier KC, Seizl M, Tresch A, and Cramer P (2012). Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res 22, 1350–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waern K, and Snyder M (2013). Extensive transcript diversity and novel upstream open reading frame regulation in yeast. G3 (Bethesda) 3, 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, and Burge CB (2008). Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Hou J, Quedenau C, and Chen W (2016). Pervasive isoform-specific translational regulation via alternative transcription start sites in mammals. Mol Syst Biol 12, 875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg DE, Shah P, Eichhorn SW, Hussmann JA, Plotkin JB, and Bartel DP (2016). Improved Ribosome-Footprint and mRNA Measurements Provide Insights into Dynamics and Regulation of Yeast Translation. Cell Rep 14, 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Luo X, Qian J, Pang X, Song J, Qian G, Chen J, and Chen S (2012). FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One 7, e52249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita R, Sathira NP, Kanai A, Tanimoto K, Arauchi T, Tanaka Y, Hashimoto S, Sugano S, Nakai K, and Suzuki Y (2011). Genome-wide characterization of transcriptional start sites in humans by integrative transcriptome analysis. Genome Res 21, 775–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinshteyn B, Rojas-Duran MF, and Gilbert WV (2017). Translation initiation factor eIF4G1 preferentially binds yeast transcript leaders containing conserved oligo-uridine motifs. RNA 23, 1365–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zur H, and Tuller T (2013). New universal rules of eukaryotic translation initiation fidelity. PLoS Comput Biol 9, e1003136. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 2: Ribosome recruitment scores for stem loop variants; related to Fig. 2
Supplemental Table 3: Ribosome recruitment scores for context variants; related to Fig. 2
Supplemental Table 4: Ribosome recruitment scores for oligo(U) variants; related to Fig. 3
Supplemental Table 1: RNA sequences, FPKM values, Ribosome recruitment scores; related to Fig. 1
Supplemental Table 5: Ribosome recruitment scores for 5′ isoforms; related to Fig. 4
Supplemental Table 6: 5′ UTR sequences tested with motif abolished or added; related to Fig. 4
Data Availability Statement
All data have been deposited at GEO and accession numbers are available in the key resources table. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical commercial assays | ||
| Nano-Glo® Luciferase Assay System | Promega | Cat# N1110 |
| Bright-Glo™ Luciferase Assay System | Promega | Cat# E2610 |
| Slide-A-Lyzer™ Dialysis Cassettes, 3.5K MWCO | Thermo | Cat# 66330 |
| Deposited data | ||
| Oligo(U) DART | This study | GSE98788 |
| Yeast TL DART | This study | GSE182290 |
| Experimental models: Organisms/strains | ||
| Yeast strain (MATa trp1Δ leu2–3,112 ura3–52 gcn2Δ::hisG PGAL1-myc-UBR1::TRP1::ubr1, pRS316 <URA3>) | Zinshteyn, B et al. 2017 | YWG1245 |
| Oligonucleotides | ||
| GCTAATACGACTCACTATAGGG | This study | RN7 |
| CCTTGGCACCCGAGAATTCCA | This study | RN8 |
| GCCTTGGCACCCGAGAATTCC | This study | oWG915 |
| /5Phos/NNN NNN NNN NGA TCG TCG GAC TGT AGA ACT CTG AAC GTG /3SpC3/ | This study | oWG920 |
| AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGA | This study | RP1 |
| CAAGCAGAAGACGGCATACGAGATN6GTGACTGGAGTTCCTTGGCACCCGAGAATTCCA | This study | OBC |
| Software and algorithms | ||
| STAR | Dobin et al., 2013 | |
| FastUniq | Xu et al., 2012 | |
| Cufflinks | Trapnell et al., 2012 | |