

Abstract
Sex differences in cellular and systems biology have been evolutionarily selected for to optimize reproductive success in all species with little (sperm) and big (ova) gamete producers. They are evident from the time of fertilization and accrue throughout development through genetic, epigenetic, and circulating sex hormone dependent mechanisms. Among other effects, they significantly impact on chromatin organization, metabolism, cell cycle regulation, immunity, longevity, and cancer risk and survival. Sex differences in cancer should be expected and accounted for in basic, translational, and clinical oncology research.
Keywords: Sex differences, cancer, DNA repair, X chromosome, Tumor Suppressor, Immunity
Introduction
Significant sex differences in cancer incidence and outcome exist around the world and across genetic ancestries[1–4]. Unfortunately, what they mean for cancer risk, treatment response, and survival are under-explored. Here we briefly review potential mechanisms underlying sex differences in cancer with the hope that discussions like these can prompt greater accounting for sex in laboratory and clinical cancer research. This accounting would be the most rigorous approach to the biology of sex differences in cancer, it would fulfill the NIH mandate, and it would likely improve outcomes for all patients as it would add a key feature to the personalization of therapeutic regimens and minimize the potential harm done to patients when sex as a biological variable is not adequately addressed in preclinical and early phase clinical investigation[5].
Sex differences are not the same as sexual dimorphism (Figure 1A). Sexual dimorphism refers to dichotomous features like ovaries versus testes. The way in which sex differences are distinct from sexual dimorphism is well-illustrated by considering human height. While most males and females in the population are between approximately 5 and 6 feet tall, at the normal extremes of short and tall are exclusively females and males, respectively[6]. Moreover, there is a significant difference between mean male and female heights. So, even though the sexes have substantially over-lapping heights, males and females occupy opposing poles in the spectrum of human height and differ in their population distributions between those poles. These observations suggest that sexual differentiation (Figure 1B), which involves genetic, epigenetic, and gonadal hormone actions, exerts male and female biased effects on height. These divergent developmental pathways have been evolutionarily selected to ensure reproductive success (Figure 1C). Defining the mechanisms by which sexual differentiation similarly affects cancer, and determining how cancer is also affected by gender, warrants sustained effort until the clinical ramifications of these differences are more completely accounted for and understood.
Figure 1: The Nature of Sex Differences.
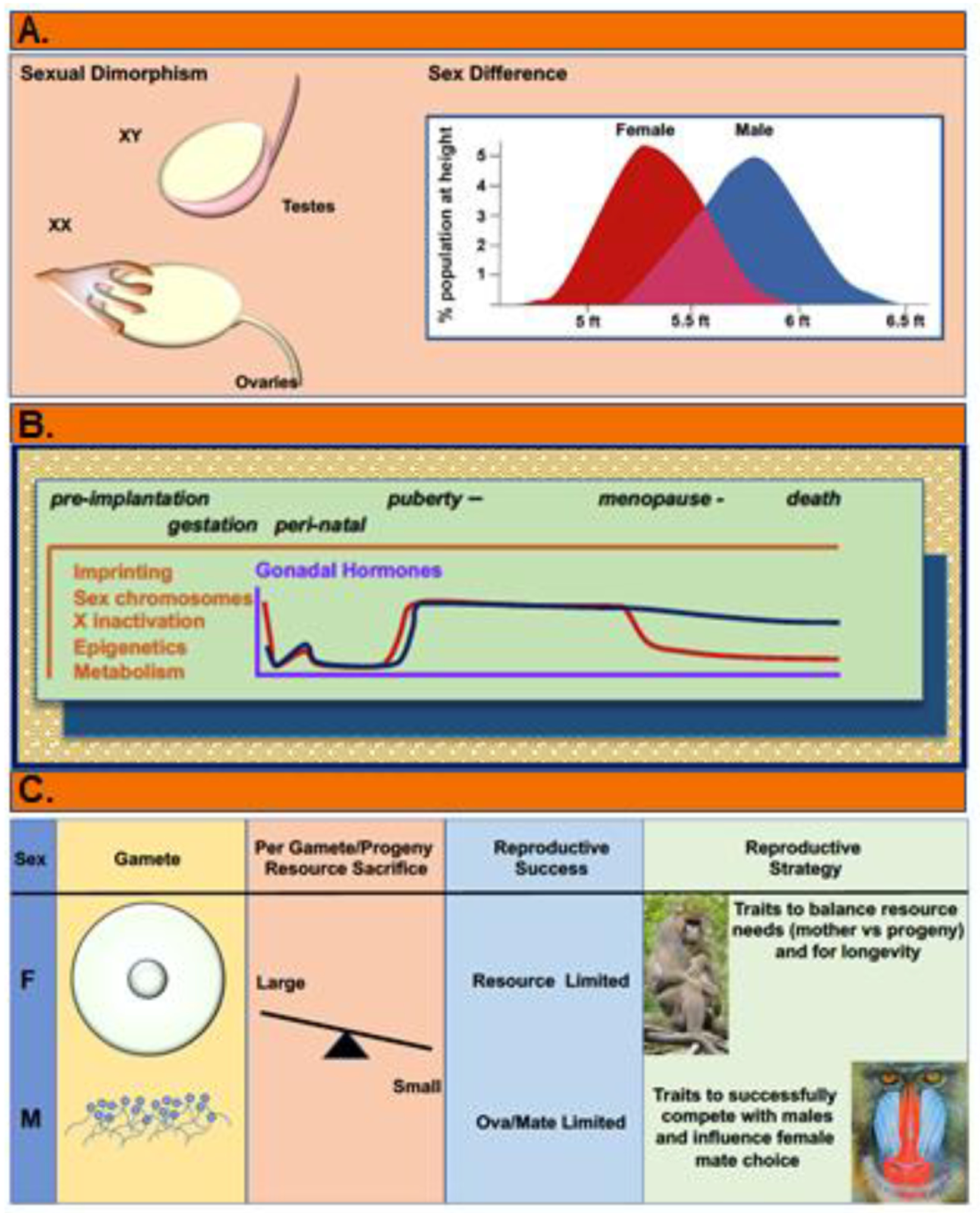
(A) Sexual dimorphism refers to dichotomous differences such as ovaries versus testes. Most differences between males and females are not dichotomous. Rather they are more akin to sex differences in height in which the extremes of human heights are distinctly female vs male but the bulk of the population have heights between these poles, and substantially overlap. This is the nature of sex differences in cancer. Thus, we should expect that sex differences in cancer will lie along a spectrum and that optimal personalized cancer treatment will sex-adapted, not sex-specific. (B) Sex differences accrue and change in magnitude across the lifespan as a consequence of genetic and gonadal hormone influences on epigenetics, gene expression and metabolism at the cellular and systems levels. Key to cancer risks and response to treatment are the resultant differences in growth regulation and metabolism that are extant from the time of fertilization at the cellular level, and the systems level differences in metabolism, immunity, mutational burden and aging. (C) Sexual selection is the evolutionary process by which sex specific reproductive biology have been optimized. The fulcrum for sexual selection across species involves the abundance of big (ova) and little (sperm) gametes, and the per progeny resource sacrifice made by the gamete producers. Per progeny resource sacrifice is a parameter that is measured from the resources required for gamete production, for gestation of fetuses and nurturing of newborns, as well as differing requirements for longevity (tissue maintenance, disease protection). Per progeny resource sacrifice is large and limiting for female reproductive success whereas it is quite small for males and has little impact on their reproductive success. For males, reproductive success is limited by their access to females. These alternate pressures drive the independent selection of female and male traits to optimize their reproductive success.
The numbers of described sex differences in male and female biology is vast. Based on evolutionary and developmental biology, significant sex differences in cancer biology should be expected. But like height, sex-related cancer phenotypes are not likely to be dichotomous and we should not expect that one treatment will be effective for men and another for women. Instead, what we should expect is that sex differences research will support appropriate clinical trial design and more completely informed treatment recommendations based on patient-specific data. It should also lead to greater biological fidelity in basic and translational sciences. Still, not all pathways and mechanisms will exhibit sex differences and not all sex differences will impact on cancer rates and survival. The goal of this piece is to provide some focus on key mechanisms by which sex is likely to impact on cancer risk. To this end, we consider sex differences in DNA repair, the X chromosome and tumor suppressor function, and immunity.
The current paradigm for the most cutting-edge systemic cancer treatments is the deployment of advanced therapeutics that are specifically indicated for genetic events involving their targets such as mutation, copy number alteration, or an aberrant level of expression[7]. This approach is fundamentally grounded in a univariate paradigm in which mutation or level of expression involving a single gene or protein directly qualifies a patient for treatment with a drug that targets the wildtype or mutant protein product. But that approach can only work if the gene or protein target has the same therapeutic value regardless of all the other factors that make individual patients different from each other. Many of these precision therapeutics target pathways such as, cell cycle regulation, metabolism, epigenetics, angiogenesis, and immunity, each of which has been explicitly demonstrated to exhibit significant sex differences from the cellular to the systems levels[8]. Moreover, similar expression levels of multiple genes have differing correlations with survival in male versus female patients[9, 10]. Overall, recognition that important sex-biased and sex-neutral mechanisms operate at all levels of biology, in both the little (sperm) and the big (ova) gamete producers of all sexually-reproducing species, is critical to meaningfully move biomedical sciences forward[11].
DNA damage and Repair
As DNA alterations are fundamental to carcinogenesis, we first consider whether sex differences in DNA damage and repair might contribute to the male bias in cancer rates. To this end, we examine cancer rates in populations exposed to ionizing radiation or affected by germline defects in DNA repair and consider the regulation of DNA repair by sex hormones (Figure 2).
Figure 2:

The normal biology of sex differences directly impacts on cancer incidence. Many elements of cancer biology exhibit substantial sex differences. These differences frequently correlate with sex differences in cancer incidence, response to treatment, and survival. Among these are: 1) the basal sex differences in ROS regulation and DNA repair, 2) X chromosome effects on expression of mutant alleles, and regulation of the p53 network, 3) the profound sex differences in immunity, 4) Multiple other mechanisms that affect stem cell biology, tissue maintenance and longevity, cellular and systemic metabolism, particularly those elements that affect drug pharmacokinetics.
Population level considerations
Cancer rates following the intentional or accidental exposure of large populations to ionizing radiation have been closely followed. Overall, sex differences in cancer risk are evident after exposure to ionizing radiation, with much of that risk biased towards males. When sex-specific cancers are excluded, solid tumors occur more frequently in male survivors of the Hiroshima and Nagasaki atomic bombings (93.7 and 86.9 per 104 person-years, respectively) compared to female survivors (63.7 and 48.8, respectively)[12]. The rates of hematological malignancies are similarly biased towards males exhibiting an excess absolute rate (EAR) ratio of 0.66 (female:male)[13]. Moreover, pediatric ductal thyroid carcinoma (DTC) following the accidental radiation exposures at Chernobyl and Fukushima have been well-studied. The rates of DTC in unexposed control populations from Belarus and Tokyo were 19% and 11% male, respectively[14]. Ingestion of I131-contaminated water and food following the accidents, resulted in an increase in DTC diagnosis in males to 36% in both populations. No significant increase in thyroid cancer has been reported following the Three-Mile Island meltdown[15].
Studying secondary cancers after primary cancer treatment provides another approach to examining possible sex differences in the cancer risk following irradiation. Based on data obtained through the Surveillance, Epidemiology, and End Results Program (SEER), the lifetime Excess Absolute Risk (EAR: cancer risk/10,000 person-years) of secondary cancers is slightly higher in females and includes a large number of breast cancer cases[16]. The rates of secondary cancers in males whose initial diagnosis and treatment was at less than 60 years of age is consistently greater than that in females. Importantly for laboratory-based investigations, male mice and rats develop more cancer than their female littermates in multiple carcinogenesis models that involve mutagen exposure [17–20]. Thus, they are appropriate models for investigations into the basis for this effect of sex.
Sex differences in cancer rates in those affected by germline mutations affecting DNA repair are also informative regarding the relationship between DNA alteration and cancer. Lynch syndrome, the most common form of inherited colorectal cancer risk, arises from germline mutations in the DNA mismatch repair genes MLH1 or MSH2. Individuals with Lynch Syndrome carry a substantially increased risk for colorectal, and less frequently, several other cancers. The risk is significantly lower for affected females compared to males across the lifespan[21, 22].
Germline mutations in TP53 in those with Li-Fraumeni Syndrome (LFS) increase the risk for multiple cancers that vary as a function of age, location, and sex. In children, girls suffer more from adrenocortical carcinoma, while choroid plexus carcinoma and sarcomas affect more boys[23]. In adults, breast cancer is overall the most frequent diagnosis in LFS. Brain tumors, which are frequent in both males and females with LFS, occur, with an overall increased risk in males.
Together, population level data thus far suggest that the relation between DNA damage and cancer may be steeper for males compared to females. An important follow up question is whether there are sex differences in the accumulation of DNA damage as a function of age. Genome instability is a “hallmark” feature of aging[24, 25] and consequently has been extensively studied with careful attention to sex differences. Sex differences with age in chromosomal mosaicism[26] and mutational burden in nuclear DNA[27] is significantly biased towards greater mutational burden in males. Mitochondrial heteroplasmy is an indicator of mitochondrial DNA mutation and positively correlates with breast[28] and prostate[29] cancer. The frequency of mitochondrial DNA mutation in the population has been well-established using data from commercial geneology sequencing services. In an analysis of nearly one million individuals of European ancestry females exhibited lower heteroplasmy[30]. It is likely that higher ROS levels in male mitochondria contributes to sex differences in mitochondrial DNA alteration[31].
Sex hormone regulation of DNA repair
Higher mutational burden and a steeper relation between exposure to ionizing radiation and cancer in males, raises questions about whether sex differences in DNA repair and/or the cellular response to DNA damage might be an underlying cause for male bias in incidence and mortality rates in cancer. In this regard, studies of the DNA damage response (DDR) in prostate and breast cancer have provided important insights into reciprocal regulation of DDR genes by androgen (AR) and estrogen receptor-α (ERα), and of AR and ERα transcriptional activity by components of the DDR.
Both AR- and ERα-mediated transcription is associated with a low level of DNA damage suggesting that androgen and estrogen regulation of DDR may be a homeostatic mechanism for preserving genome integrity[32, 33]. Androgen effects on DDR gene expression have been well-studied in prostate cancer cell lines, patient-derived xenografts, and patient specimens[34, 35]. Both positive and negative regulation of multiple DDR genes by androgens/AR exhibited significant variability across cell lines and patient specimens. Overall, AR appears to stimulate DDR gene expression, and concordantly, either androgen deprivation or AR inhibition is associated with decreased DDR gene expression[36–38] and enhanced response to PARP inhibition[39].
Estrogen receptors ligation also regulates DNA repair. In ER-positive breast cancer cell lines, estrogen treatment reduced the extent of double strand breaks following etoposide treatment, which correlated with enhanced plasmid end-joining and increased complex formation between ERα, CBP, and BRCA1[40]. While no effects of estrogen treatment on DDR gene expression were detected, ERα has been shown to increase DNA repair by inducing the expression of the catalytic subunit of DNA-PK[41]. DDR proteins also regulate AR and ERα functions. DNA-PK[41, 42], PARP[43, 44], and MDC1[45, 46], all enhance AR- and ERα- mediated transcription. Finally, sex differences in the cellular response to DNA damage may also underlie disparate cancer rates and survival. Following irradiation of patient and murine cell models, female cells have been shown to more readily undergo senescence[47].
Tumor suppressor effects of the X chromosome
While the accumulation of DNA alterations with age is inevitable, cell-intrinsic tumor suppressor responses to those DNA alterations are the safeguards by which normal tissue function is preserved over time, and the frontline protection from cancer. Age-dependent increases in cancer rise more steeply in males compared to females[48, 49], and here we consider sex differences in tumor suppressor functions as a contributing factor. We focus on the role of the X-chromosome in tumor suppression (Figure 2).
X chromosome mutation buffering effects
The X chromosome encodes a number of known tumor suppressor genes, epigenetic regulators, and interactors with the p53 pathway[50, 51]. As in X-linked disorders, the presence of two X chromosomes can buffer against X allele mutation in an individual with more than one X chromosome. Buffering could occur by two mechanisms. The first involves the approximately 15% of X alleles that escape X-inactivation. Many of these comprise the pseudo-autosomal genes with accompanying Y paralogs. However, several essential tumor suppressors are within the non-pseudo-autosomal component of the X-chromosome. These alleles are not genetically or functionally equilibrated through X inactivation and thus, single allelic mutation results in complete versus heterozygous alteration of function in males versus females, respectively. These genes, KDM6A, ATRX, KDM5C, CNKSR2, DDX3X, and MAGEC3[52] have important epigenetic and tumor suppressor functions. Mutated alleles are more frequently detected in male cancers and may account for a portion of the excess male cancer cases.
Mutations in KDM6A appear to account for much of the sex bias in patients with bladder cancer (M:F cases = 3–5:1[53]. Experiments in which chromosomal and gonadal sex effects are distinguishable by using the four-core genotypes murine model of sexual differentiation, indicate that both sex chromosomes complement and gonadal hormones both influence the sex disparity in bladder cancer. The sex chromosome effect was dependent upon catalytically active Kdm6a and involved downstream effectors, Cdkn1a and Perp [54]. Analysis of patient gene expression data confirmed the sex bias in KDM6A expression and its prognostic significance and suggested a role for DDX3X in sex disparity. Similar sex biased KDM6A tumor suppressor functions are indicated in multiple cancers. As an exception, KDM6A and its Y paralog, UTY, appear to have similar tumor suppressor functions in pancreatic cancer [55]. As UTY has little to no catalytic activity, these data suggest that tumor suppression can, in some instances, be independent of demethylase activity. Instead, it likely involves the roles of these proteins in COMPASS complex function. ATRX mutation exhibits a similar pattern of male-biased mutation frequency[52].
The second mechanism of buffering is X chromosome inactivation itself. Random and mosaic X chromosome inactivation provides a mechanism for preserved tissue function even if mutated genes are expressed in some component cells. This mechanism could provide a measure of protection against tumor suppressor loss, though a growth advantage in a cell with deficient tumor suppressor function could lead to skewed X chromosome activity and abrogation of a protective buffering effect. In cancer, there is reduced expression of mutated alleles indicating that X chromosome inactivation can buffer against expression of mutated alleles and protect against cancer[56]. Components of an X-chromosome - p53 pathway network are prominent among the unexpressed mutated X alleles and may account for the biased occurrence of p53 mutations in male cancers (Text box 1).
Text Box 1. X interactions with p53.
Critical interactions between the X chromosome and the p53 pathway are evident in developmental and cancer biology. Complete germline loss of p53 function in mice results in selective demise of female fetuses because of a sex difference in neural tube defects. Female fetuses develop lethal rostral neural tube defects while male fetuses develop viable caudal neural tube defects[108]. Male and female differences in neural tube defects arise because of aberrant X chromosome inactivation and loss of gene dosage compensation[109], due to failed expression of X-inactivation specific (Xist). Xist is a long non-coding RNA that is required for X chromosome inactivation and is a direct transcriptional target of p53.
Sex differences in p53 effects have also been observed in a Drosophila aging model in which overexpression of wildtype p53 extended the lives of male flies while shortening the lives of female flies[110]. These observations are relevant to sex differences in p53 in cancer, including the male bias in the frequency of p53 mutations in cancer[56], sex differences in cell cycle regulation following loss of p53[111, 112], and sex- and mutation- specific gains of aberrant and oncogenic transcriptional activity in missense mutations within the DNA binding domain of p53[113].
Multiple X-encoded alleles have been identified as regulators of p53 functions. Mutations in these p53 interactors exhibit significant and informative patterns of expression in male and female cancers[50, 56]. Overall expression of mutated X alleles is repressed in female cancers compared to males. Prominent among the non-expressed mutations were those in p53 interactors FLNA, MED12, HUWE1, and ATRX. However, expression of mutated p53-X interactors is common in both male and female cancers indicating their importance in p53 tumor suppressor functions. Sex bias in the expression of X-encoded p53 interactors was further demonstrated in healthy tissues. KDM6A, DDX3X and UBA1, are more highly expressed in healthy female tissues as compared to male tissues. These observations suggest that the X chromosome has tumor suppressive functions and that much of that protection is mediated through general buffering against expression of mutated alleles and more specifically through regulation of p53 functions.
Sex chromosome loss in cancer
Given the tumor suppressor functions of the X chromosome, loss of a single X chromosome is a common event in cancer. Significant loss of an entire X chromosome has been reported in neuroblastoma[57], myelodysplastic diseases[58] and leukemia[59], medulloblastoma[60], and other solid tumors[61]. With age, there is frequent loss of the Y chromosome in male cells and this too is associated with cancer, including lung cancer[62] and leukemia[59], glioblastoma[63], and renal tumors[64].
Together, these considerations emphasize the important and distinct tumor suppressor functions of sex chromosomes and provide additional insights into potential mechanisms underlying sex differences in cancer.
Anti-Cancer Immunity
Normal tissue stroma and systemic immunity are a higher scale of safeguard against cancer as compared to cellular responses to genotoxic stress. Tissue maintenance and immunity are both substantially affected by age and sex, as are cancer rates. It is likely that sex differences in chronic inflammation, cellular senescence and waning immunity all contribute to the distinct male and female increases in cancer as a function of age. Sex differences in aging and immunity are well-described[65]. In eutherian species, sex differences in immunity are likely the result of selective pressures exerted by invasive placentation and long-lived fetal microchimeric cells on the evolution of female immune function[66]. Male and female differences in immunity are evident in vaccine responses, susceptibility and severity of infectious diseases, wound healing, and auto-immune disease[65]. Therefore, similar difference should be expected in anti-cancer immunity (Figure 2).
The innate immune system comprising natural killer (NK) cells, granulocytes, dendritic cells, type I Interferon signaling, and complement are the first line of defense against pathogens and cells lacking appropriate “self” signals. Males and females differ in the abundance of cellular components of innate immunity. Males have higher numbers of NK cells. Females have higher numbers of macrophages and neutrophils, which also demonstrate greater phagocytic activity[67] but lower numbers of type II innate lymphoid cells, which are suggested to be mechanistically related to their increased risk of autoimmune disease[68]. Type I IFN responses to TLR7 ligands are more robust in females[69, 70], which has important effects on tumor surveillance by the innate immune system. These effects include, direct tumor suppressive effects of type I IFNs on cell proliferation and apoptosis[71, 72], on the maturation and function of dendritic cells[73] and NK cells[74], as well as the inhibition of immune suppressive functions of myeloid-derived suppressor cells[75] and T regulatory cells[76]. A critical role fortype I IFN signaling in NK cell mediated anti-tumor immunity was initially demonstrated in murine cancer models involving NK cell depletion and genetic disruption of IFN signaling[77, 78]. IFN effects on NK cell cytotoxic activities are both direct[79, 80] and indirect through IFN-stimulated IL15 secretion by DCs[81]. IL15 in turn, is essential for full maturation of cytolytic NK cell functions[82]
Normal sex differences in adaptive immunity would be predicted to impact on responses to cancer-neo-antigens. Females exhibit higher numbers of circulating CD4 T cells and B cells, while males possess higher levels of CD8 T cells [83–85]. Upon in vitro stimulation, female human peripheral blood mononuclear cells exhibit higher numbers of activated CD4 T cells, which also express higher levels of IFNγ as compared to male cells, while male cells express higher levels of IL-17[86].
Sex differences in immunity can be traced to sex chromosome-encoded genes and gonadal hormone actions, as outlined below.
Role of sex chromosomes in immunity
Multiple genes in the innate and type I IFN response pathway are encoded by X alleles, such as the pattern recognition receptors TLR7 and 8. TLR7 is expressed at higher levels in female cells and activation of TLR7 elicits a stronger type I IFN response as compared to males[69, 70]. In addition, interleukin 2 (IL-2) receptor-γ chain, IL-3 receptor-α chain, IL-13 receptor-α chains, IL-1 receptor-associated kinase 1 (IRAK1), FOXP3 and MiRNAs 18 and 19 are all X chromosome alleles with immune regulatory functions[87, 88].
The importance of sex chromosomes to adaptive immune function is highlighted by the effects of sex chromosome aneuploidies on autoimmune disease in Klinefelter’s Disease (XXY) and Turner’s syndrome (XO). Males with Klinefelter’s syndrome have immune profiles that more closely resemble that of females with increased CD4 T cells and CD4/CD8 ratios as well as increased numbers of B cells and immunoglobin concentrations compared to XY males[89]. Females with Turner’s syndrome have decreased T and B cell numbers and lower immunoglobin concentrations as compared to XX females and are more similar to males[90].
Role of sex hormones in immunity
Sex hormones are potent and complex regulators of immune response. In general, estrogens are positive regulators of immunity, progesterone is anti-inflammatory, and androgens are immune-suppressive. Estrogens, progesterone, and androgens all exert their effects through their cognate receptors, which are widely expressed by immune cells. Specific cells and transcriptional targets of sex hormones in immune regulation have been most completely identified using in vitro human cellular and mouse models of the known sex differences in immunity. Important to innate immunity, estrogens increase IFNγ secretion and cytotoxicity of NK cells[91], enhance DC maturation and antigen presentation[92], and increase the expression of TLRs 4, 7 and 9, and IFNα[93, 94]. In contrast, androgen treatment reduces NK cell numbers in mice[95] and decreases the expression of TNF, while increasing IL10 and transforming growth factor-β (TGFβ) in macrophages[96].
Across multiple species, low and high doses of estrogen have differing effects on adaptive immunity. Low doses increase cell-mediated immunity, while high doses increase humoral immunity[97]. Specifically, estrogen treatment in patients increases the numbers of regulatory T cells[98] and B cells[99]. Testosterone effects on immunity have been studied in men with and without androgen deficiencies. Compared to males with normal testosterone levels, men with low androgens have increased antibody titers and CD4/CD8 T cell ratio, as well as higher NK cell numbers, like profiles seen in women[100, 101].
Impact of sex bias on tumor immunity and immunotherapy
Clinical responses to immune checkpoint inhibition emphasize the importance of considering sex differences in immunity for optimal therapy development and rigorous evaluation of efficacy. Meta-analyses of response to immune checkpoint inhibition alone in NSCLC and melanoma indicate that greater benefit is experienced by male patients compared to their female counterparts[102, 103]. When combined with chemotherapy, ICI for advanced stage NSCLC was more beneficial for females[104].
The immunological basis for these varying results is not completely understood, but they are associated with substantial differences in the immune cell components of male and female tumors and the mechanisms of immune evasion. Female tumors possessed higher abundance of activated dendritic cells, B cells, and multiple T cell subsets with greater clonality of the TCR-repertoire compared to male tumors[105]. Female tumors also contained higher numbers of myeloid-derived suppressor cells (MDSCs) and evidence of greater T cell dysregulation. These findings suggest that initially, female patients mount a great anti-tumor immune response than male patients and that immune evasion therefore requires inhibition of T cell effector functions. This view is supported by the differences in recurrent driver mutations in melanoma, which suggest that common drivers are more effectively cleared by female immune mechanisms as compared to males[106]. MDSCs are now recognized as essential targets in ICI. Their immune suppressive functions are enhanced by estrogen treatment and thus, their greater abundance in female tumors, in the setting of higher estrogen, may be critically important to the lower efficacy of ICI in females[107] and suggests that estrogen antagonism may be an important adjunct to ICI for females.
Concluding Remarks
That cell-intrinsic differences in cell cycle activity, metabolism, and epigenetics are evident from the time of fertilization should lead us to expect that they will be present in cancer cells too. During normal sexual differentiation these differences arise through genetic and epigenetic mechanisms, just like cancer phenotypes do. Moreover, sex differences are not limited to the cellular mechanisms we briefly reviewed. Most notably, they include substantial differences in systemic metabolism, angiogenesis, and those pathways that underlie sex differences in longevity. Consequently, we should expect that treatments targeting these mechanisms will differ in efficacy and toxicity along a spectrum of sex differences (Figure 3). To move forward we need to (i) more fully evaluate sex and gender as independent and interacting variables in determining cancer risks, cancer biology, clinical presentation, efficacy and toxicity of treatments, and survivorship issues; (ii) recognize that the epidemiology of sex differences alone warrants adaptation of clinical trial design to detect sex differences in response to treatment. Oncology has a long and proud history of rigorous empiricism to move patient care forward and we should not wait for more complete understanding of the mechanisms underlying sex differences before acting through prospective clinical trial design and careful analysis of data to detect sex effects. We also need to (iii) work to address and resolve controversies that surround sex and gender science. The data regarding the prevalence of sex differences in cancer are consistent and significant, yet there are those who chose to “not believe” that sex differences in cancer exist or matter. This is an obstacle to advancement and is a disappointing and inappropriate response from members of a community that are pursuing “evidence-based” science and medicine.
Figure 3:
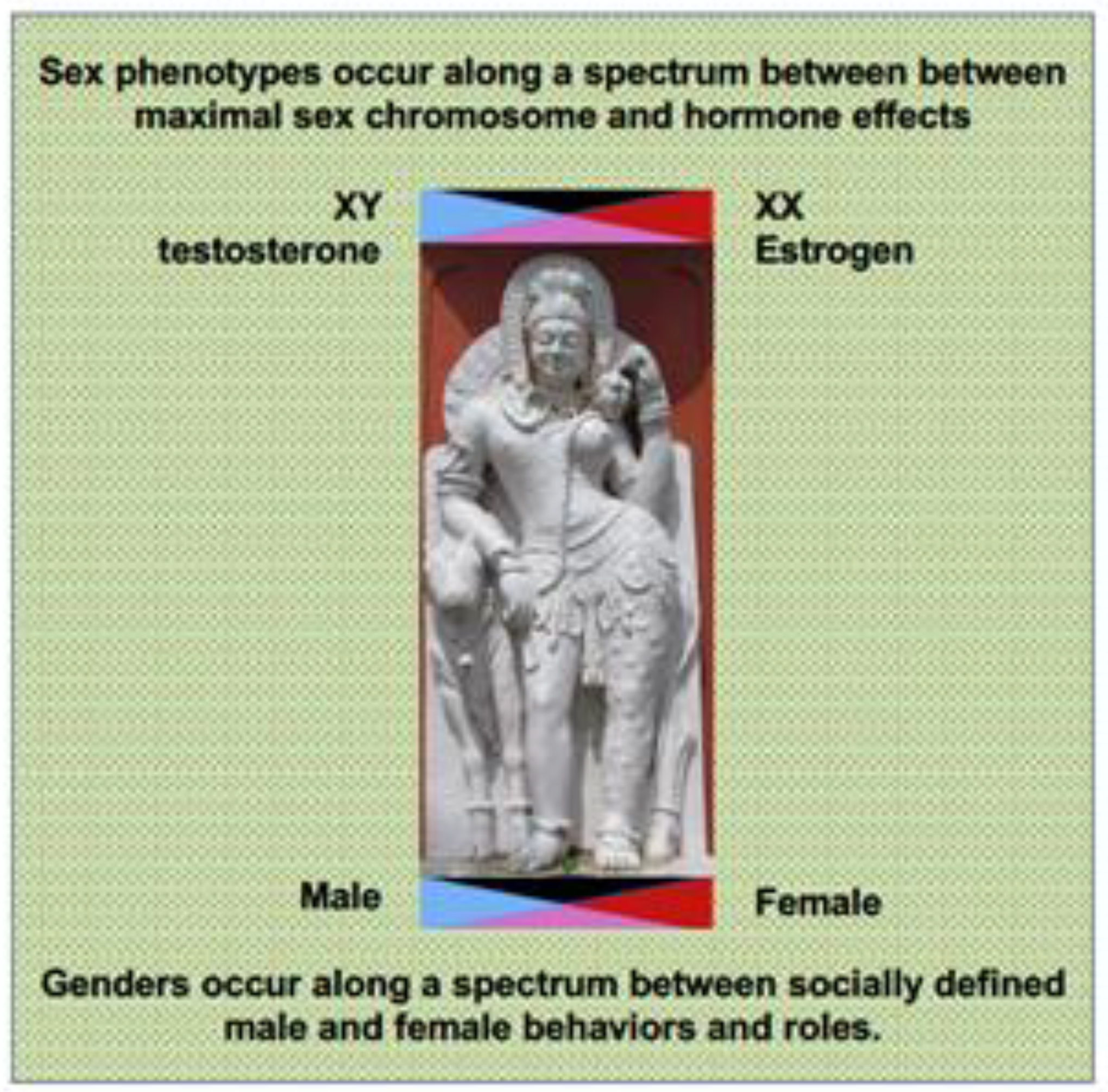
The spectrum of sex and gender differences affect multiple aspects of normal developmental biology, aging and disease. These differences will need to be completely cataloged and characterized to determine which have significant impact on cancer incidence, treatment response, and survival.
In conclusion, for the sake of patients and the relevance of our science to the human condition, it will be essential to ensure that all basic, translational and clinical research is appropriately designed and powered to detect sex differences and that the data are specifically and appropriately analyzed for those differences, when they exist (Outstanding Questions).
Outstanding Questions.
How to best translate sex differences in cancer biology into clinical oncology remains to be determined. As sex effects in cancer are most likely to exist along a spectrum in which there may distinct but overlapping distributions of male and female phenotypes, sex-adapted treatments are likely to require more than simple assignment of patient sex alone.
Inadequate consideration of sex as a biological variable in drug development and administration has resulted in significant harm to patients and is likely to have missed agents that may have sex-biased efficacy and/or toxicity. How to disseminate best practices for incorporating sex as a biological variable into all phases of oncology research for greater understanding, efficiency and harm reduction is currently unclear. Recognition that cell sex, animal sex, and patient sex must be accounted at all times, and that experimental design must include power calculations to detect interactions between sex and other experimental variables will be key.
How to best imbue science and medicine with the profound biology of sex differences without eliciting negative responses and resistance that arise from: a) gender-sex misconceptions; b) misinterpretation and mis-placed value judgements regarding what the data mean for male-female equality in education, opportunity, and success; and 3) dogmatic adherence to univariate, linear, and “canonical” characterization of cellular mechanisms; is unclear and is likely to remain a challenge.
Highlights.
Sex differences in cancer incidence and survival are the norm and evident across cancer types, around the globe, in all genetic ancestries, and in all age groups.
Increasingly, sex differences in cancer relevant cellular and systems biology are being described.
Sex differences in mutational burden, DNA repair, epigenetics, metabolism, tumor suppressor activity, cell cycle regulation, and immunity are reported.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dong M, et al. , Sex Differences in Cancer Incidence and Survival: A Pan-Cancer Analysis. Cancer Epidemiol Biomarkers Prev, 2020. 29(7): p. 1389–1397. [DOI] [PubMed] [Google Scholar]
- 2.Monterroso P, et al. , Racial/ethnic and sex differences in young adult malignant brain tumor incidence by histologic type. Cancer Epidemiol, 2021. 76: p. 102078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scelo G, et al. , Variability of Sex Disparities in Cancer Incidence over 30 Years: The Striking Case of Kidney Cancer. Eur Urol Focus, 2018. 4(4): p. 586–590. [DOI] [PubMed] [Google Scholar]
- 4.Sun T, Warrington NM, and Rubin JB, Why does Jack, and not Jill, break his crown? Sex disparity in brain tumors. Biol Sex Differ, 2012. 3: p. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carey JL, et al. , Drugs and Medical Devices: Adverse Events and the Impact on Women’s Health. Clin Ther, 2017. 39(1): p. 10–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(CDC), U.S.C.f.D.C., National Health and Nutrition Examination Survey (NHANES) III Data Exploration System. 2007.
- 7.Horak P, et al. , Assigning evidence to actionability: An introduction to variant interpretation in precision cancer medicine. Genes Chromosomes Cancer, 2021. [DOI] [PubMed] [Google Scholar]
- 8.Rubin JB, et al. , Sex differences in cancer mechanisms. Biol Sex Differ, 2020. 11(1): p. 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang W, et al. , Sex differences in GBM revealed by analysis of patient imaging, transcriptome, and survival data. Sci Transl Med, 2019. 11(473). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopes-Ramos CM, et al. , Gene Regulatory Network Analysis Identifies Sex-Linked Differences in Colon Cancer Drug Metabolism. Cancer Res, 2018. 78(19): p. 5538–5547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naqvi S, et al. , Conservation, acquisition, and functional impact of sex-biased gene expression in mammals. Science, 2019. 365(6450). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grant EJ, et al. , Solid Cancer Incidence among the Life Span Study of Atomic Bomb Survivors: 1958–2009. Radiat Res, 2017. 187(5): p. 513–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu WL, et al. , The incidence of leukemia, lymphoma and multiple myeloma among atomic bomb survivors: 1950–2001. Radiat Res, 2013. 179(3): p. 361–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drozd V, et al. , A Search for Causes of Rising Incidence of Differentiated Thyroid Cancer in Children and Adolescents after Chernobyl and Fukushima: Comparison of the Clinical Features and Their Relevance for Treatment and Prognosis. Int J Environ Res Public Health, 2021. 18(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levin RJ, et al. , Incidence of thyroid cancer surrounding Three Mile Island nuclear facility: the 30-year follow-up. Laryngoscope, 2013. 123(8): p. 2064–71. [DOI] [PubMed] [Google Scholar]
- 16.RE C, et al. , New Malignancies Among Cancer Survivors: SEER Cancer Registries, 1973–2000. National Cancer Institute. 2006. [Google Scholar]
- 17.Bertram JS and Craig AW, Specific induction of bladder cancer in mice by butyl-(4-hydroxybutyl)-nitrosamine and the effects of hormonal modifications on the sex difference in response. Eur J Cancer, 1972. 8(6): p. 587–94. [DOI] [PubMed] [Google Scholar]
- 18.Thomas-Ahner JM, et al. , Gender differences in UVB-induced skin carcinogenesis, inflammation, and DNA damage. Cancer Res, 2007. 67(7): p. 3468–74. [DOI] [PubMed] [Google Scholar]
- 19.Lee SM, et al. , The Effect of Sex on the Azoxymethane/Dextran Sulfate Sodium-treated Mice Model of Colon Cancer. J Cancer Prev, 2016. 21(4): p. 271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kadekar S, et al. , Gender differences in chemical carcinogenesis in National Toxicology Program 2-year bioassays. Toxicol Pathol, 2012. 40(8): p. 1160–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wischhusen JW, et al. , Clinical Factors Associated with Urinary Tract Cancer in Individuals with Lynch Syndrome. Cancer Epidemiol Biomarkers Prev, 2020. 29(1): p. 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bucksch K, et al. , Cancer risks in Lynch syndrome, Lynch-like syndrome, and familial colorectal cancer type X: a prospective cohort study. BMC Cancer, 2020. 20(1): p. 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amadou A, Achatz MIW, and Hainaut P, Revisiting tumor patterns and penetrance in germline TP53 mutation carriers: temporal phases of Li-Fraumeni syndrome. Curr Opin Oncol, 2018. 30(1): p. 23–29. [DOI] [PubMed] [Google Scholar]
- 24.Lopez-Otin C, et al. , The hallmarks of aging. Cell, 2013. 153(6): p. 1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fischer KE and Riddle NC, Sex Differences in Aging: Genomic Instability. J Gerontol A Biol Sci Med Sci, 2018. 73(2): p. 166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Machiela MJ, et al. , Characterization of large structural genetic mosaicism in human autosomes. Am J Hum Genet, 2015. 96(3): p. 487–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Podolskiy DI, et al. , Analysis of cancer genomes reveals basic features of human aging and its role in cancer development. Nat Commun, 2016. 7: p. 12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen J, et al. , Peripheral blood mitochondrial DNA copy number, length heteroplasmy and breast cancer risk: a replication study. Carcinogenesis, 2015. 36(11): p. 1307–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalsbeek AMF, et al. , Mitochondrial genome variation and prostate cancer: a review of the mutational landscape and application to clinical management. Oncotarget, 2017. 8(41): p. 71342–71357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nandakumar P, et al. , Nuclear genome-wide associations with mitochondrial heteroplasmy. Sci Adv, 2021. 7(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borras C, et al. , Mitochondria from females exhibit higher antioxidant gene expression and lower oxidative damage than males. Free Radic Biol Med, 2003. 34(5): p. 546–52. [DOI] [PubMed] [Google Scholar]
- 32.Lin C, et al. , Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell, 2009. 139(6): p. 1069–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williamson LM and Lees-Miller SP, Estrogen receptor alpha-mediated transcription induces cell cycle-dependent DNA double-strand breaks. Carcinogenesis, 2011. 32(3): p. 279–85. [DOI] [PubMed] [Google Scholar]
- 34.Polkinghorn WR, et al. , Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov, 2013. 3(11): p. 1245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Di Sante G, et al. , Hormone-induced DNA damage response and repair mediated by cyclin D1 in breast and prostate cancer. Oncotarget, 2017. 8(47): p. 81803–81812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh V, et al. , Targeting the TLK1/NEK1 DDR axis with Thioridazine suppresses outgrowth of androgen independent prostate tumors. Int J Cancer, 2019. 145(4): p. 1055–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Karanika S, et al. , Targeting DNA Damage Response in Prostate Cancer by Inhibiting Androgen Receptor-CDC6-ATR-Chk1 Signaling. Cell Rep, 2017. 18(8): p. 1970–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Al-Ubaidi FL, et al. , Castration therapy results in decreased Ku70 levels in prostate cancer. Clin Cancer Res, 2013. 19(6): p. 1547–56. [DOI] [PubMed] [Google Scholar]
- 39.Li L, et al. , Androgen receptor inhibitor-induced “BRCAness” and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci Signal, 2017. 10(480). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crowe DL and Lee MK, New role for nuclear hormone receptors and coactivators in regulation of BRCA1-mediated DNA repair in breast cancer cell lines. Breast Cancer Res, 2006. 8(1): p. R1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Medunjanin S, et al. , Transcriptional activation of DNA-dependent protein kinase catalytic subunit gene expression by oestrogen receptor-alpha. EMBO Rep, 2010. 11(3): p. 208–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goodwin JF, et al. , DNA-PKcs-Mediated Transcriptional Regulation Drives Prostate Cancer Progression and Metastasis. Cancer Cell, 2015. 28(1): p. 97–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schiewer MJ, et al. , Dual roles of PARP-1 promote cancer growth and progression. Cancer Discov, 2012. 2(12): p. 1134–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang F, et al. , Poly(ADP-ribose) polymerase 1 is a key regulator of estrogen receptor alpha-dependent gene transcription. J Biol Chem, 2013. 288(16): p. 11348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang C, et al. , MDC1 functionally identified as an androgen receptor co-activator participates in suppression of prostate cancer. Nucleic Acids Res, 2015. 43(10): p. 4893–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zou R, et al. , MDC1 Enhances Estrogen Receptor-mediated Transactivation and Contributes to Breast Cancer Suppression. Int J Biol Sci, 2015. 11(9): p. 992–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Broestl L and Rubin JB, Sexual Differentiation Specifies Cellular Responses to DNA Damage. Endocrinology, 2021. 162(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bruder C, et al. , Estimating lifetime and 10-year risk of lung cancer. Prev Med Rep, 2018. 11: p. 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.White A, et al. , A review of sex-related differences in colorectal cancer incidence, screening uptake, routes to diagnosis, cancer stage and survival in the UK. BMC Cancer, 2018. 18(1): p. 906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haupt S and Haupt Y, Cancer and Tumour Suppressor p53 Encounters at the Juncture of Sex Disparity. Front Genet, 2021. 12: p. 632719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Haupt S, et al. , Sex disparities matter in cancer development and therapy. Nat Rev Cancer, 2021. 21(6): p. 393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dunford A, et al. , Tumor-suppressor genes that escape from X-inactivation contribute to cancer sex bias. Nat Genet, 2017. 49(1): p. 10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Edgren G, et al. , Enigmatic sex disparities in cancer incidence. Eur J Epidemiol, 2012. 27(3): p. 187–96. [DOI] [PubMed] [Google Scholar]
- 54.Kaneko S and Li X, X chromosome protects against bladder cancer in females via a KDM6A-dependent epigenetic mechanism. Sci Adv, 2018. 4(6): p. eaar5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andricovich J, et al. , Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell, 2018. 33(3): p. 512–526 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haupt S, et al. , Identification of cancer sex-disparity in the functional integrity of p53 and its X chromosome network. Nat Commun, 2019. 10(1): p. 5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parodi S, et al. , Loss of whole chromosome X predicts prognosis of neuroblastoma patients with numerical genomic profile. Pediatr Blood Cancer, 2019. 66(5): p. e27635. [DOI] [PubMed] [Google Scholar]
- 58.Abruzzese E, et al. , Monosomy X as a recurring sole cytogenetic abnormality associated with myelodysplastic diseases. Cancer Genet Cytogenet, 1997. 93(2): p. 140–6. [DOI] [PubMed] [Google Scholar]
- 59.Shahrabi S, et al. , Sex chromosome changes in leukemia: cytogenetics and molecular aspects. Hematology, 2018. 23(3): p. 139–147. [DOI] [PubMed] [Google Scholar]
- 60.Kool M, et al. , Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One, 2008. 3(8): p. e3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Duijf PH, Schultz N, and Benezra R, Cancer cells preferentially lose small chromosomes. Int J Cancer, 2013. 132(10): p. 2316–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Willis-Owen SAG, et al. , Y disruption, autosomal hypomethylation and poor male lung cancer survival. Sci Rep, 2021. 11(1): p. 12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lysiak M, et al. , Deletions on Chromosome Y and Downregulation of the SRY Gene in Tumor Tissue Are Associated with Worse Survival of Glioblastoma Patients. Cancers (Basel), 2021. 13(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buscheck F, et al. , Y-chromosome loss is frequent in male renal tumors. Ann Transl Med, 2021. 9(3): p. 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Klein SL and Flanagan KL, Sex differences in immune responses. Nat Rev Immunol, 2016. 16(10): p. 626–38. [DOI] [PubMed] [Google Scholar]
- 66.Natri H, et al. , The Pregnancy Pickle: Evolved Immune Compensation Due to Pregnancy Underlies Sex Differences in Human Diseases. Trends Genet, 2019. 35(7): p. 478–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Scotland RS, et al. , Sex differences in resident immune cell phenotype underlie more efficient acute inflammatory responses in female mice. Blood, 2011. 118(22): p. 5918–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Russi AE, et al. , Cutting edge: c-Kit signaling differentially regulates type 2 innate lymphoid cell accumulation and susceptibility to central nervous system demyelination in male and female SJL mice. J Immunol, 2015. 194(12): p. 5609–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Berghofer B, et al. , TLR7 ligands induce higher IFN-alpha production in females. J Immunol, 2006. 177(4): p. 2088–96. [DOI] [PubMed] [Google Scholar]
- 70.Griesbeck M, et al. , Sex Differences in Plasmacytoid Dendritic Cell Levels of IRF5 Drive Higher IFN-alpha Production in Women. J Immunol, 2015. 195(11): p. 5327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Takaoka A, et al. , Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature, 2003. 424(6948): p. 516–23. [DOI] [PubMed] [Google Scholar]
- 72.Zitvogel L, et al. , Type I interferons in anticancer immunity. Nat Rev Immunol, 2015. 15(7): p. 405–14. [DOI] [PubMed] [Google Scholar]
- 73.Diamond MS, et al. , Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med, 2011. 208(10): p. 1989–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee CK, et al. , Distinct requirements for IFNs and STAT1 in NK cell function. J Immunol, 2000. 165(7): p. 3571–7. [DOI] [PubMed] [Google Scholar]
- 75.Pace L, et al. , APC activation by IFN-alpha decreases regulatory T cell and enhances Th cell functions. J Immunol, 2010. 184(11): p. 5969–79. [DOI] [PubMed] [Google Scholar]
- 76.Sisirak V, et al. , Impaired IFN-alpha production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res, 2012. 72(20): p. 5188–97. [DOI] [PubMed] [Google Scholar]
- 77.Rautela J, et al. , Loss of Host Type-I IFN Signaling Accelerates Metastasis and Impairs NK-cell Antitumor Function in Multiple Models of Breast Cancer. Cancer Immunol Res, 2015. 3(11): p. 1207–17. [DOI] [PubMed] [Google Scholar]
- 78.Dunn GP, et al. , A critical function for type I interferons in cancer immunoediting. Nat Immunol, 2005. 6(7): p. 722–9. [DOI] [PubMed] [Google Scholar]
- 79.Swann JB, et al. , Type I IFN contributes to NK cell homeostasis, activation, and antitumor function. J Immunol, 2007. 178(12): p. 7540–9. [DOI] [PubMed] [Google Scholar]
- 80.Mizutani T, et al. , Conditional IFNAR1 ablation reveals distinct requirements of Type I IFN signaling for NK cell maturation and tumor surveillance. Oncoimmunology, 2012. 1(7): p. 1027–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mattei F, et al. , IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J Immunol, 2001. 167(3): p. 1179–87. [DOI] [PubMed] [Google Scholar]
- 82.Lucas M, et al. , Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity, 2007. 26(4): p. 503–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lee BW, et al. , Age- and sex-related changes in lymphocyte subpopulations of healthy Asian subjects: from birth to adulthood. Cytometry, 1996. 26(1): p. 8–15. [DOI] [PubMed] [Google Scholar]
- 84.Lisse IM, et al. , T-lymphocyte subsets in West African children: impact of age, sex, and season. J Pediatr, 1997. 130(1): p. 77–85. [DOI] [PubMed] [Google Scholar]
- 85.Uppal SS, Verma S, and Dhot PS, Normal values of CD4 and CD8 lymphocyte subsets in healthy indian adults and the effects of sex, age, ethnicity, and smoking. Cytometry B Clin Cytom, 2003. 52(1): p. 32–6. [DOI] [PubMed] [Google Scholar]
- 86.Zhang MA, et al. , Peroxisome proliferator-activated receptor (PPAR)alpha and -gamma regulate IFNgamma and IL-17A production by human T cells in a sex-specific way. Proc Natl Acad Sci U S A, 2012. 109(24): p. 9505–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fish EN, The X-files in immunity: sex-based differences predispose immune responses. Nat Rev Immunol, 2008. 8(9): p. 737–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pinheiro I, Dejager L, and Libert C, X-chromosome-located microRNAs in immunity: might they explain male/female differences? The X chromosome-genomic context may affect X-located miRNAs and downstream signaling, thereby contributing to the enhanced immune response of females. Bioessays, 2011. 33(11): p. 791–802. [DOI] [PubMed] [Google Scholar]
- 89.Kocar IH, et al. , The effect of testosterone replacement treatment on immunological features of patients with Klinefelter’s syndrome. Clin Exp Immunol, 2000. 121(3): p. 448–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bianchi I, et al. , The X chromosome and immune associated genes. J Autoimmun, 2012. 38(2–3): p. J187–92. [DOI] [PubMed] [Google Scholar]
- 91.Nakaya M, Tachibana H, and Yamada K, Effect of estrogens on the interferon-gamma producing cell population of mouse splenocytes. Biosci Biotechnol Biochem, 2006. 70(1): p. 47–53. [DOI] [PubMed] [Google Scholar]
- 92.Paharkova-Vatchkova V, Maldonado R, and Kovats S, Estrogen preferentially promotes the differentiation of CD11c+ CD11b(intermediate) dendritic cells from bone marrow precursors. J Immunol, 2004. 172(3): p. 1426–36. [DOI] [PubMed] [Google Scholar]
- 93.Rettew JA, Huet YM, and Marriott I, Estrogens augment cell surface TLR4 expression on murine macrophages and regulate sepsis susceptibility in vivo. Endocrinology, 2009. 150(8): p. 3877–84. [DOI] [PubMed] [Google Scholar]
- 94.Seillet C, et al. , The TLR-mediated response of plasmacytoid dendritic cells is positively regulated by estradiol in vivo through cell-intrinsic estrogen receptor alpha signaling. Blood, 2012. 119(2): p. 454–64. [DOI] [PubMed] [Google Scholar]
- 95.Hou J and Zheng WF, Effect of sex hormones on NK and ADCC activity of mice. Int J Immunopharmacol, 1988. 10(1): p. 15–22. [DOI] [PubMed] [Google Scholar]
- 96.D’Agostino P, et al. , Sex hormones modulate inflammatory mediators produced by macrophages. Ann N Y Acad Sci, 1999. 876: p. 426–9. [DOI] [PubMed] [Google Scholar]
- 97.Straub RH, The complex role of estrogens in inflammation. Endocr Rev, 2007. 28(5): p. 521–74. [DOI] [PubMed] [Google Scholar]
- 98.Singh RP and Bischoff DS, Sex Hormones and Gender Influence the Expression of Markers of Regulatory T Cells in SLE Patients. Front Immunol, 2021. 12: p. 619268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lu FX, et al. , The strength of B cell immunity in female rhesus macaques is controlled by CD8+ T cells under the influence of ovarian steroid hormones. Clin Exp Immunol, 2002. 128(1): p. 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Malkin CJ, et al. , The effect of testosterone replacement on endogenous inflammatory cytokines and lipid profiles in hypogonadal men. J Clin Endocrinol Metab, 2004. 89(7): p. 3313–8. [DOI] [PubMed] [Google Scholar]
- 101.Page ST, et al. , Effect of medical castration on CD4+ CD25+ T cells, CD8+ T cell IFNgamma expression, and NK cells: a physiological role for testosterone and/or its metabolites. Am J Physiol Endocrinol Metab, 2006. 290(5): p. E856–63. [DOI] [PubMed] [Google Scholar]
- 102.Conforti F, et al. , Cancer immunotherapy efficacy and patients’ sex: a systematic review and meta-analysis. Lancet Oncol, 2018. 19(6): p. 737–746. [DOI] [PubMed] [Google Scholar]
- 103.Conforti F, et al. , Sex-based differences in response to anti-PD-1 or PD-L1 treatment in patients with non-small-cell lung cancer expressing high PD-L1 levels. A systematic review and meta-analysis of randomized clinical trials. ESMO Open, 2021. 6(5): p. 100251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Conforti F, et al. , Sex-based heterogeneity in response to lung cancer immunotherapy: a systematic review and meta-analysis. J Natl Cancer Inst, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Conforti F, et al. , Sex-Based Dimorphism of Anticancer Immune Response and Molecular Mechanisms of Immune Evasion. Clin Cancer Res, 2021. 27(15): p. 4311–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Castro A, et al. , Strength of immune selection in tumors varies with sex and age. Nat Commun, 2020. 11(1): p. 4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chakraborty B, et al. , Inhibition of estrogen signaling in myeloid cells increases tumor immunity in melanoma. J Clin Invest, 2021. 131(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen X, et al. , Sex difference in neural tube defects in p53-null mice is caused by differences in the complement of X not Y genes. Dev Neurobiol, 2008. 68(2): p. 265–73. [DOI] [PubMed] [Google Scholar]
- 109.Delbridge ARD, et al. , Loss of p53 Causes Stochastic Aberrant X-Chromosome Inactivation and Female-Specific Neural Tube Defects. Cell Rep, 2019. 27(2): p. 442–454 e5. [DOI] [PubMed] [Google Scholar]
- 110.Waskar M, et al. , Drosophila melanogaster p53 has developmental stage-specific and sex-specific effects on adult life span indicative of sexual antagonistic pleiotropy. Aging (Albany NY), 2009. 1(11): p. 903–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kfoury N, et al. , Cooperative p16 and p21 action protects female astrocytes from transformation. Acta Neuropathol Commun, 2018. 6(1): p. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sun T, et al. , Sexually dimorphic RB inactivation underlies mesenchymal glioblastoma prevalence in males. J Clin Invest, 2014. 124(9): p. 4123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rockwell NC, et al. , Sex- and mutation-specific p53 gain-of-function activity in gliomagenesis. Cancer Res Commun, 2021. 1(3): p. 148–163. [DOI] [PMC free article] [PubMed] [Google Scholar]