

Abstract
Metastasis is considered to be responsible for 90% of cancer-related deaths. Although it is clinically evident that metastatic patterns vary by primary tumor type, the molecular mechanisms underlying the site-specific nature of metastasis are an area of active investigation. One mechanism that has emerged as an important player in this process is glycosylation, or the addition of sugar moieties onto protein and lipid substrates. Glycosylation is the most common post-translational modification, occurring on more than 50% of translated proteins. Many of those proteins are either secreted or expressed on the cell membrane, thereby making glycosylation an important mediator of cell-cell interactions, including tumor-microenvironment interactions. It has been recently discovered that alteration of glycosylation patterns influences cancer metastasis, both globally and in a site-specific manner. This review will summarize the current knowledge regarding the role of glycosylation in the tropism of cancer cells for several common metastatic sites, including the bone, lung, brain and lymph nodes.
Keywords: Organotropism, sialylation, glycosyltransferase, brain metastasis, bone metastasis, lung metastasis, lymph node metastasis
INTRODUCTION
Metastasis is estimated to be responsible for 90% of cancer mortality[1], and therefore represents a pressing clinical issue. However, in spite of significant research efforts, this process remains poorly understood, and there are few treatment options for patients who present with metastatic disease.
Common sites of metastasis include the liver, lymph nodes, bone, lung, and brain. One autopsy study of a cohort of patients with metastatic cancer of many types found that 59% had liver metastases; 53% had non-regional lymph node metastases; 44% had lung; 38% had bone, and just over 20% had brain metastases[2]. Rates for each specific secondary site differ among primary tumor types[2]. One particularly perplexing issue is that of site specificity: that is, why do secondary tumors preferentially develop in particular distant organs? A plethora of hypotheses of have been proposed, including differences in chemotactic factors, adhesive factors, and immune infiltrate, as well as host factors such as age and overall health status [3, 4], but the organotropism of metastasis remains a highly active and poorly understood area of research.
Glycosylation, the addition of sugar moieties onto various substrates, is currently a subject of much interest for its potential role in metastasis. There are thousands of glycosylated entities in cells, many of which are either secreted or present on the cell membrane [5]. Additionally, many cell-cell adhesion molecules, and molecules involved in intercellular communication, are highly glycosylated[6], making glycosylation of substantial interest when studying metastasis and the many cell-cell interactions it requires. Glycosylation can be modified at many points, as summarized in Figure 1. Alterations to all of these points have been identified in both primary and metastatic disease. Altered glycosylation is particularly important during the metastatic cascade. These more general roles have been covered extensively in previous reviews (see, for example [5, 7–9]), and will be discussed only briefly here. More recently, glycosylation has emerged as an important regulator of the organotropism of metastasis. The impact of aberrant glycosylation on cancer development is quite broad and consideration of all putative roles is beyond the scope of a single article. Therefore, this article focuses mainly on altered glycosylation in bone (section 2.1), lung (section 2.2), brain (section 2.3) and lymph node (section 2.4) metastasis, for which the evidence of a role for glycosylation in tumor progression is best developed. Other secondary sites are briefly discussed (section 2.5); however, there is a paucity of studies focusing on glycosylation in metastasis to other organs. Finally, the role of glycosylation in circulating tumor cell biology is briefly discussed in section 2.6. In particular, we emphasize secenarios for which there is evidence of a glycosylation-dependent mechanism rather than simply observations of differential glycoprotein abundance. Within these parameters, we summarize current knowledge and identify possible targets for future study regarding the role of glycosylation in directing the site specificity of metastasis.
Figure 1: An introduction to glycan structure and glycosylation reactions.
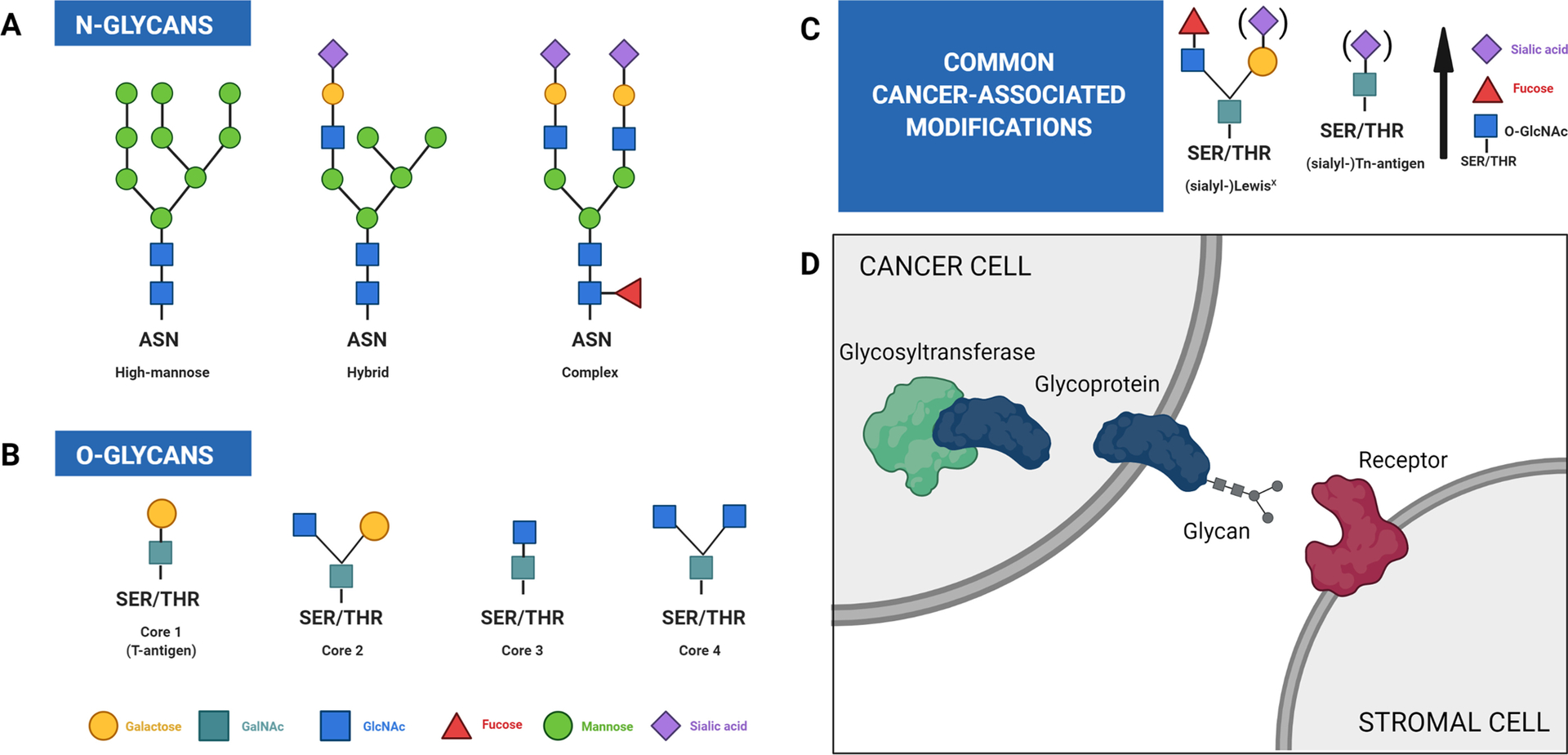
Glycosylation is a non-templated addition and there exist a vast array of possible glycan structures. Glycans can be divided into two classes, N-glycans and O-glycans. (A) N-glycans can be subdivided into three main classes, as shown here. (B) O-glycans typically consist of one of the four core structures shown here, which can be further extended and modified. Note that unmodified core 1, also known as the T-antigen, is considered a truncated glycoform and is frequently associated with cancer. (C) Several glycosylation alterations of particular importance to this review. See Table 2 for additional detail. (D) Glycosylation is a multistep process with many modifiable points. Examples include changes in the expression or activity level of glycosyltransferases, expression levels of glycoproteins, the compositions and amounts of glycans added, and the receptors with which those glycans can interact. The availability of the necessary substrates and the overall metabolic condition of the cell can also influence which glycosylation events take place. Because of the interconnected nature of this system, a perturbation at any one point has the potential to impact everything downstream, and a relatively small initial change can exert dramatic effects on the cell. Figure adapted and complied from “Glycans 1” by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates. Figure edited with BioRender.com
1. OVERVIEW OF GLYCOSYLATION AND ITS REGULATION
Glycosylation can occur on many types of biological molecules, including lipids, DNA and small organic molecules, but proteins are the most common substrates [10, 11] and the major focus of this review.
Glycosylation can take many forms. Nine of the 20 amino acids found in proteins can be potentially glycosylated, and there are 10 major monosaccharide building blocks found in mammals, all of them manufactured from glucose, which can be utilized in glycan chains [12] (Table 1). A glycosylation event can be characterized based on the type of sugar attached, the location of the attachment, the substrate receiving the sugar, and the enzyme performing the reaction. There are four main types of glycan addition to form glycoproteins: N-glycosylation, O-glycosylation, addition of glycosylphosphatidylinositol (GPI) anchors, and addition of glycosaminoglycan chains to form proteoglycans[10]. Glycans can themselves be modified by phosphorylation, sulfation, methylation, acylation, acetylation, and pyruvylation [13]. The typical processing of proteins into glycoproteins occurs in the endoplasmic reticulum (ER) and Golgi complex [11, 14]. Depending on the type of addition, the exact sequence and process of addition will vary. Selected cancer-relevant glycosylation events are summarized in Table 1 and Figure 1. The precise biochemical details of glycosylation are outside the scope of this review, but have been covered extensively elsewhere; for example, see [8, 11, 13, 15–17].
Table 1.
Selected glycosylation events
| Type | Synthesis | Key enzymes | General structure | Notes and cancer relevance |
|---|---|---|---|---|
| Oglycosylat ion [5, 10, 15, 20, 22] |
Location: Golgi complex Timing: Post-translational Sequence dependence: No consensus sequence, although a Ser/Thr residue is required and a nearby proline seems to be preferred Initial Addition: Addition of monosacchride onto the terminal oxygen of serine or threonine |
GALNT: Makes first addition of a GALNAc residue via α linkage to Ser/Thr for a mucin-type O-glycan C1GALT1 + COSMC: transferase + chaperone protein responsible for the formation of core 1 O-glycans GCNT-1/-2/-3: responsible for the synthesis of core 2 structures from core 1 Many other O-glycosyltransferases with various affinities and enzyme activities |
Common core structures designated #1–4, with 1 and 2 being the most common Core 1: the addition of β1–3Gal to O-GalNAc Core 2: addition of β1–6GlcNAc to the core 1 GalNAc Core 3 and Core 4 are larger, more complex structures and are thought to repress tumor growth |
Subdivided into mucin-type (GalNAc) and non-mucin (various moieties; includes O-GlcNAc, O-glucose, O-fuctose, O-xylose, and O-mannose). O-GlcNAc: the only reversible glycan modification; competes with phosphorylat ion at some regulatory sites. See Table 2. Elevated core 2 levels are associated with cancer, presumably because extravasatio n-promoting selectin ligands can be built from core 2 structures. Elevated levels of C1GALT1 and COSMC are associated with advanced tumors and metastasis. |
| N-glycosylat ion [5, 11, 16, 22] |
Location: Lumen of the endoplasmic reticulum Timing: Cooccurs with translation Consensus sequence: Asn-X-Ser/Thr motif, X ≠ proline; (Rarely) Asn-XCys Initial addition: Large oligosaccharide precursor, Gl3Man9GlcNAc2, which is later trimmed by a variety of glycosidases, glycosyltransfer ases, and mannosidases. In mammals, attached via GlcNAcβ1-Asn linkage. |
Oligosaccharyltransf erase (OST): The enzyme responsible for synthesis of the N-glycan oligosaccharide precursor Dolichel phosphate (Dol-P): The lipid carrier on which the N-glycan precursor is initially synthesized |
Stereotypical core includes three mannose residues and two Nacetylglucosa mine resides. (Written as: Manα1-3(Manα1-6)Manβ1-4GlcNAcβ1-4GlcNAcβ1-Asn-X-Ser/Thr) Branching patterns can be generally classified as (1) oligomannose: elongation by mannose resiudes (2) complex: elongation by GlcNAc structures and (3) hybrid: elongation by both on different parts of the core structure |
Increased size and branching of N-glycans particularly β1,6-type branching, is associated with malignant phenotypes. May help form lattices to enhance growth factor signaling and promote the formation of sialyl-LewisX antigens (see Table 2; sLeX). Altered N-glycosylation can affect RTK signaling and membrane retention. |
| Sialylation [10, 17, 22, 24] |
Location: Synthesis takes place in the cytoplasm; activation (via addition of CMP) takes place in the nucleus Linkage: Added to galactose or N-acetylgalactosa mine using α2–3 or α2–6 linkages, and can be part of a polysialic chain using α2,8 linkages. Both O- and N-glycans are typically terminated with a sialic acid at the nonreducing end. |
20 sialyltransferases, specific for the linkage type and glycan being added ST8SIA family, 6 members: attachment of the C-2 carbon of a sialic acid to the C-8 of a second sialic acid ST3GAL family, 6 members: C-2 to the C-3 ST6GAL, 2 members: C2 to ST6GALNAc, 6 members: C-2 to C-6 position of galactose to the C-6 of N-acetylgalactosamine NEU genes: encode sialidases (remove sialic acid) |
9-carbon structures carrying a negatively charged carboxylate on C1 and having a 3-carbon non-cyclic sidechain (C7–C9) Parent compounds: 2-keto-deoxynonuloso nic acid (Kdn) neuraminic acid (Neu; found only in glycosidic linkages) N-acetylneurami nic acid (Neu5Ac) |
Characteristi c feature of mammalian glycans. Critical roles in immune function and modulation and cell-cell communicati on. Capable of “masking” cell surface glycans from recognition by other cell types. Highly electronegat ive; can mediate electrostatic repulsion between cells, which has been implicated in both metastasis and neuronal development Sialylation globally increases with malignant transformati on and is correlated with increased metastatic spread. α2–6GalNAc linkages become prominent, which has a variety of signaling effects including suppression of Fas and galectin-3 apoptotic signaling, increased FAK and integrin activity, cell motility, and chemothera py resistance. O-acetylation on C-9 of sialic acid can increase or decrease depending on cancer type. Presence of Neu5Gc (not made in humans; obtained via dietary sources, e.g. red meat) provokes chronic, tumorpromoting inflammatio n and angiogenesis due to immune reaction. |
The effects of glycosylation are highly context-specific, depending on the organ or tissue in which the process occurs and the developmental stage of the organism. Additionally, there is substantial variability in glycosylation-related events between species [18]. Interestingly, many glycan deletions have little effect in vitro, but are lethal in a whole organism, indicating that important glycan functions are largely interaction-dependent rather than intrinsic [19]. Since the addition and processing of glycans is quite complex, it can be difficult to localize the exact cause of a change in glycosylation, but it is clear that a relatively small alteration in any step of the process can have serious consequences for the cell.
Alterations in glycosylation have emerged as a significant player in malignant transformation and tumor progression. These changes have been extensively reviewed elsewhere [20–24]. Briefly, abnormal glycan expression assists with invasion through the basement membrane; comprises a significant portion of cancer-associated extracellular matrix alterations [25]; contributes to tumor-induced immunosuppression [26] and drug resistance [7, 27]; increases growth factor sensitivity[22]; promotes epithelial-to-mesenchymal (EMT) programs [25, 28, 29]; encourages the formation of “tumor emboli” (cancer cell-platelet complexes) which help tumor cells survive in circulation [20]; enables cancer cells to engage in “rolling” behavior characteristic of lymphocytes[22], which is required for extravasation; and facilitates their interaction with the destination microenvironment[7, 20, 25, 30]. Selected glycoproteins and glycan structures which are strongly associated with metastasis are summarized in Table 2 for reference.
Table 2.
Cancer-relevant glycan structures
| Glycan Type | General Structure | Role in Cancer |
|---|---|---|
| Glycosaminoglycans (GAGs) and proteoglycans [10, 20, 22] |
GAGs: Long, unbranched chains of polysaccharides with molecular weights of >15 KDa. Frequently modified by sulfation. Proteoglycans: Created by the addition of GAGs onto proteins |
Key roles in creating the extracellular matrix (ECM) and maintaining the eukaryotic cell membrane. GAGs can tether ligands to RTKs, resulting in constitutive activation independent of protein/RNA levels. Increased cleavage of the proteoglycan heparan sulfate is implicated the promotion of angiogenesis and metastasis. Cleaved ectodomains from syndecan-I can promote bone metastasis. |
| Hyaluronan [22] | Large, negatively-charged polysaccharide made up of repeating [GlcAβ1–3GlcNAcβ1–4] units | Frequently highly expressed by both tumor-associated stroma and tumor cells. Can increase tissue hydration to facilitate tumor cell migration and influence tumor-matrix interactions. Known activator of CD44 signaling; essential to activate various cancer hallmark pathways, including EMT, PI3K/AKT, and MAP Kinase. |
| Lectins [2, 15, 20, 23, 24, 105] | Glycan-binding proteins. Main types include siglecs, galectins, and selectins. Sialic-acid binding immunoglobulin-like lectins (siglecs) are found on B cells, macrophages, and NK cells. Physiologic role is to dampen the immune response by masking sialoglycans on cell surfaces from other cells. Selectins (E, L, and P) have glycan ligands containing sulfate, fucose, and sialic acid. |
Siglecs can assist in tumor immune evasion and therefore are a therapeutic target of interest. Galectins bind galactose and form lattices via β1,3- or β1,4-linkages to N-acetylglucosamine; these lattices can help modulate receptor-ligand interaction. Selectin expression can influence platelet adhesion (P), homing and development of leukocytes (L) and recruitment of immune cells in response to inflammation (E and L). |
| Mucins [5, 15] | O-GalNAc glycans, synthesis details found in Table 1 | Physiological expression is restricted to the apical (lumen) side of cells; as cell polarity is lost in malignant transformation, expression becomes dysregulated. Thought to interfere with adhesive cell-cell interactions via steric and electrostatic mechanisms, which may contribute to metastasis. Several mucin-type O-glycosyltransferases have been documented as diagnostic and prognostic markers of lung and pancreatic cancer (GALNT3), contribute to breast cancer (GALNT6), and influence death receptor glycosylation in NSCLC, melanoma, and pancreatic cancer (GALNT14). CA125, commonly used as a cancer marker, is a heavily-glycosylated mucin. |
| O-GlcNAc [12, 22] | The only reversible glycan modification. Addition occurs in cytoplasm. | Has regulatory capacity similar to phosphorylation and sometimes competes for phospho-sites. Generally elevated in cancer. O-GlcNAc is found on a number of cancer-associated proteins, including cyclin D1, c-Myc, NF-κB/p65, and SNAIL. Contributes to enhancement of cancer cell metabolism. |
| (sialyl)-LewisX/A (sLeX/A) [22, 72, 105] | Produced by fucosylation of O-GalNAc core 1 (LeA) or O-GalNAc core 2 (LeX). Can be exposed by loss of the AB blood group |
Overexpressed in many cancers. One of the earliest metastasis-associated glycans to be identified. Important for selectin-lectin interaction allowing extravasation (physiologically used by leukocytes; co-opted by migratory tumor cells). Expression contributes to the formation of “tumor emboli”, platelet-tumor complexes that help protect circulating tumor cells. |
| (sialyl)-T antigen (sT) [22, 23] | O-glycan core 1 structure without any extensions. Can be modified by sialylation. |
Is considered a truncated glycoform. Rarely present in normal tissue but frequent in tumors. |
| (sialyl)-Tn antigen (sTn) [15, 22, 23] | A single GalNAc residue attached to Ser/Thr. Can be modified by sialylation; sialyl-Tn can be O-acetylated as well. Sialylated by ST6GALNAC1-4, which are α2-6 sialyltransferases, as well as a family of α2-3 sialyltransferases that act on the Galβ1-3 residues of O-GalNAc glycans. |
Is considered a truncated glycoform. Rarely present in normal tissue but frequent in tumors. Presence in cancer may be related to elevated levels of C1GALT1 and COSMC (see Table 1). |
2. THE ROLE OF GLYCOSYLATION IN SITE-SPECIFIC METASTASIS
Locations of metastasis vary greatly according to primary tumor type [31, 32]. Over the years, many hypotheses have been advanced regarding the reasons behind metastatic preferences, including Ewing’s “mechanical trapping” theory [33] and Paget’s “seed and soil,” which suggests that tumors must find a hospitable environment in order to grow [34], as well as more recent ideas about establishment of a premetastatic niche [4, 32], inflammation [4], and the contribution of host factors such as age, genetic factors, and co-morbid health conditions[4]. These theories are not mutually exclusive and we now appreciate that multiple factors can influence the patterns of metastasis. Still, identification of targetable regulators has proved elusive. Recent research has identified glycosylation as a potentially important mediator of site-specific metastasis, which raises the possibility of defined therapeutic interventions. This section provides an overview of those findings in the context of several common metastatic destination organs (summarized in Figure 2). In order to provide a more comprehensive discussion and identify possible commonalities between tumor types, we focus on metastatic destination rather than tumor of origin. The role of glycosylation specifically within various primary tumor types has been extensively reviewed; see, for example, [35, 36] (brain), [37] (breast), [38, 39] (lung), [40] (pancreatic) and [41] (prostate), among others.
Figure 2: Summary of glycosylation-associated factors identified thus far as mediators of site-specific metastasis.
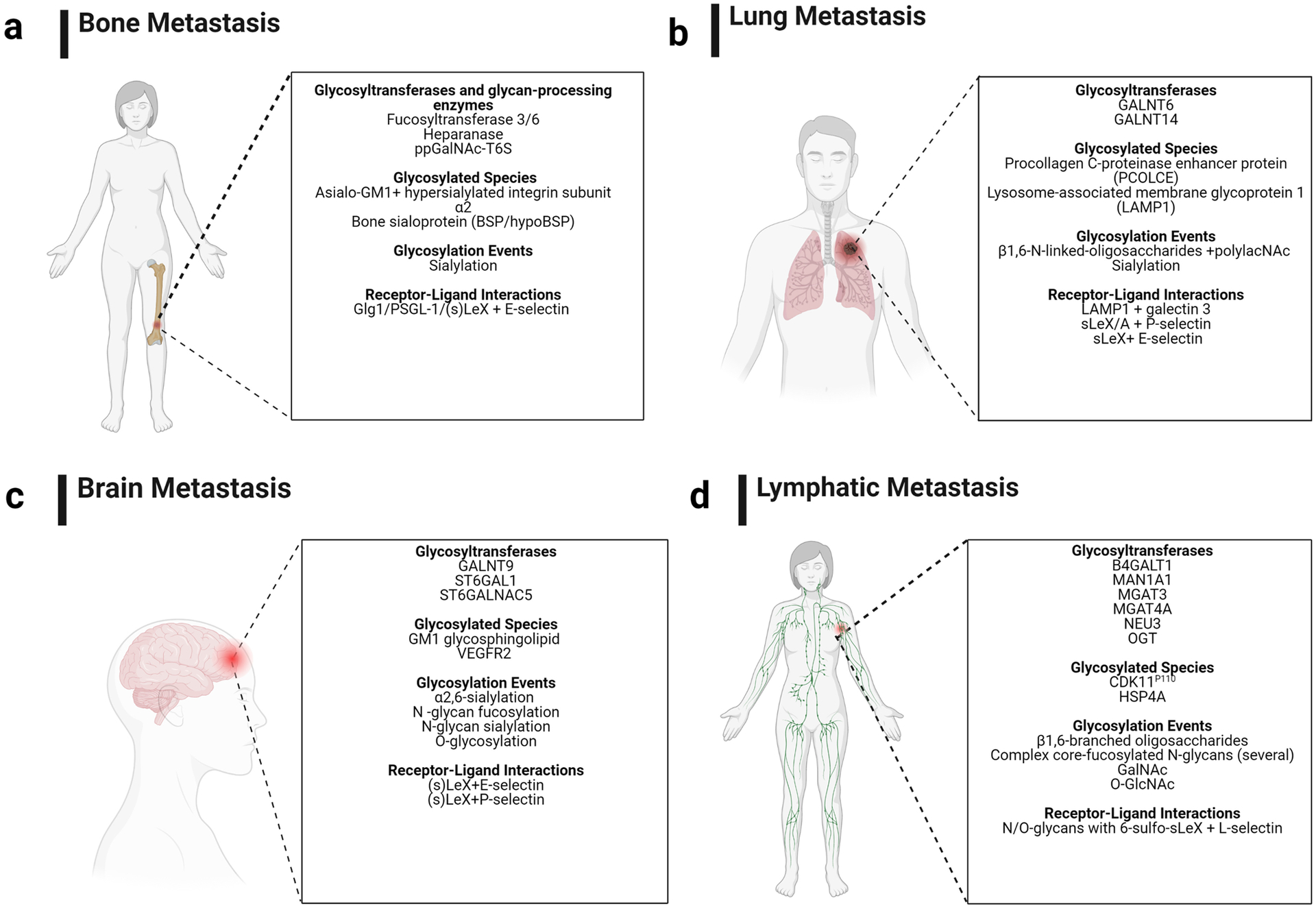
Mediators are classified according to type of alteration observed. Figure adapted and compiled from “Brain Callout (Human),” “Lung Cancer Callout,” “Metastasis to Bone Disrupts Bone Homeostasis,” and “Lymph Node Locations” by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates. Figure edited with BioRender.com
2.1. Glycosylation and bone metastasis
Bone metastasis is a frequent metastatic event for many cancer types, both early and late. It is also a source of significant patient morbidity due to pain, tumor-induced fracture and mobility loss [42]. As originally noted by Stephen Paget in the original ‘seed and soil’ hypothesis [34], and confirmed in more recent studies, breast cancer has particular affinity for bone[42]. We now know that altered glycosylation appears to play a significant role in multiple stages of metastasis to bone. Several studies have identified specific enzymes or glycoproteins that associate with development of bone metastases in clinical cohorts. Many such studies have been mostly concerned with identification of biomarkers; for example, Freire et al. (2006) identified ppGalNAc-T6, one of a family of enzymes that catalyze the first step in mucin-type O-glycan synthesis, as a negative prognostic marker for disease-free survival when found in the bone marrow of early-stage, node-negative breast cancer patients. There were no mechanistic analyses, but the researchers hypothesized that the expression of ppGalNAc-T-6 might play a role in altered O-glycan expression, especially the Tn antigen, in breast cancer [43]. Another example is bone sialoprotein (BSP), a secreted and heavily sialylated and phosphorylated protein produced by osteoblasts, osteoclasts, osteocytes, and chondrocytes. Normally, it promotes initial mineral crystal formation in bone and teeth. A form of BSP exhibiting truncated O-glycosylation (hypoBSP) is particularly associated with breast cancer metastasis [44]. In rat mammary cancer models, hypoBSP was strongly expressed in skeletal lesions but only weakly in primary tumors. A neutralizing antibody specific for hypoBSP induced remission in 80% of animals, with activity restricted to skeletal lesions. Additionally, in a cohort of 11 primary tumors and skeletal metastases from breast cancer patients, bone metastases had a higher average hypoBSP staining score than primary tumors [44], suggesting that this aberrant glycoform is biologically and therapeutically relevant for breast cancer skeletal metastases. These studies provide evidence that aberrant expression of several glycosylation-related species (a glycoprotein, a glycosyltransferase, and a glycan, respectively; see Figure 1) has a measurable impact on bone metastatic behavior. However, these studies, and others like them, often lack a mechanistic component, so it remains unclear how the abnormal glycosylation phenotype contributes to site-specific metastatic tendencies.
A small number of studies have probed the mechanism underlying the contributions of glycosylation to bone metastasis. These contributions are diverse, affecting all different stages of metastasis, and may vary according to the origin of the primary tumor. For example, heparanase, the enzyme which cleaves the proteoglycan heparan sulfate (see Table 2) increases homing of multiple myeloma cells to the bone in mice [45], which led to speculation that it may contribute to bone metastasis of solid tumors as well. It was observed that in a breast cancer model, heparanase did not directly increase bone metastasis, but instead elevated osteolytic activity [46]; the authors of that study hypothesized that release of extra growth factors from the resorbed bone may create a pro-tumor pre-metastatic niche within the bone. In prostate cancer bone metastasis, sialylation appears to be extremely important for progression. Global levels of α2,3-sialylation are elevated in bone-metastatic as compared to a non-invasive prostate cancer cell lines, and hypersialylation of the α2 subunit of integrin α2β1 increases cell adhesion capacity in vitro. Simultaneously, however, bone-metastatic prostate cancer cells express an abnormally glycosylated form of the glycosphingolipid GM1 called asialo-GM1 (AsGM1), which lacks an α2,3-sialylation mark. Mechanistic work reveals that AsGM1, but not fully-sialylated GM1, complexes with integrin α2β1 [47] via the same sialylated moiety that promotes adhesive interactions [48], and the formation of these complexes promotes invasive cell behavior [47]. Further in vitro work suggests that hypersialylated α2β1 is important for initial attachment of the prostate cancer cells, while the AsGM1-integrin complexes participate in longer-term tethering interactions to hold them there[48]. In contrast, a recent study in breast cancer [49] showed that MDA-MB-231 cells engineered (via forced expression of ST6GalNAc1) to express high levels of sialyl-Tn (sTn) (see Table 2), display reduced bone metastasis, and less osteolytic activity in vivo. In vitro, they show reduced capacity for adhesion to fibronectin or collagen I, and reduced interaction with bone marrow stem cells. S-Tn is carried primarily on integrin β1, thus in contrast to the facilitative role played by elevated sialylation in the context of integrin α2β1 in prostate cancer, the presence of excessive sTn antigen impairs the adhesion capabilities of the MDA-MB-231 cells, possibly through causing integrin dysfunction. In sum, these studies demonstrate that alterations in glycosylation can have mechanistic impacts at various stages of metastasis. However, these effects are highly nuanced; even a modification which is traditionally considered pro-metastatic (such as hypersialylation [24]) may not be in all contexts, and the pertinent mechanisms vary according to tumor type.
One glycoprotein whose relevance for bone metastasis has been exhaustively investigated is E-selectin, a glycoprotein expressed on endothelial cells that is primarily known for its involvement in leukocyte rolling and adhesion [50]. Interactions of E-selectin with its ligand sialyl-LewisX have been observed to promote metastasis for decades [50–53]. A 2008 study in prostate cancer, which primarily (90%+) metastasizes to the bone, showed that extravasation of cancer cells is mainly mediated by E-selectin ligands expressed by the tumor cells, interfacing with E-selectin on the bone endothelial cells; preferred ligands include PSGL-1 and sLeX [54]. Another study found that sialyl-LewisX (sLeX; see Table 2) is associated with bone metastasis in estrogen receptor (ER)-positive, but not ER−, breast cancer. In in vitro mechanistic studies, adherence of the ER+ cancer cells to endothelial cells under shear stress conditions is mediated by sLeX-E-selectin interaction; the authors hypothesize that this interaction may be important for bone metastasis given that E-selectin is constitutively expressed in the bone [55]. ER− cells, despite expressing sLeX and of the enzymes required for its synthesis at higher levels than ER+ tumors, appear to interact with other selectins primarily via heparan sulfate (HS) chains, which the cells also express at elevated levels, and do not bind to E-selectin under those in vitro conditions. The authors hypothesize that the context of glycan expression (i.e., ER+ vs. ER−) may be important for determining its relevance to tumor biology, and further, that ER+ tumors may express sLeX primarily on glycoproteins, while ER− tumors may incorporate it primarily into glycolipids, which would alter the functional consequences of its expression for the cell.
Entry of cancer cells into the bone marrow takes place through the sinusoidal vasculature, which expresses E-selectin and SDF-1 (the ligand for CXCR4) at high levels. Price et al. [56] identified genes associated with late relapse of breast cancer, which predominantly metastasizes to the bone, in a patient cohort. They identified seven genes, including several glycoproteins (CXCR4, CD44, SELPLG (PSGL-1), Lysosome-Associated Membrane Glycoprotein 1 [LAMP1], and the E-selectin ligand Golgi glycoprotein 1 [GLG1]) and glycosyltransferases (β-1,4-Galactosyltransferase 6 [B4GALT6], FUT4). This group was specifically interested in the trafficking of breast cancer cells through the sinusoidal vasculature during early bone metastasis. They find that E-selectin-mediated interactions are critical for initial entry of the cancer cells into the bone and interaction with the vasculature. In contrast to the findings from Julien et al. [55], E-selectin influenced bone homing in both ER+ and ER− breast cancer, though they note that ER+ and ER− cells express different subsets of E-selectin ligands. Importantly, this group confirmed E-selectin-mediated trafficking of breast cancer cells in the bone in human-in-mouse models derived from patient tumors, suggesting that these mechanisms are in fact relevant to the human disease process.
Esposito et al. (2019) further investigated the ability of E-selectin/Glg1 interactions to promote bone metastasis in mice [57]. E-selectin-null mice showed decreased bone metastatic burden from a bone-metastatic subline of human MDA-MB-231 breast cancer cells, but lung metastases were unaffected. In human ER− breast cancer, tumor Glg1 status correlated with bone metastasis-free survival, but not with metastasis-free survival to lung, liver, or brain. In the mouse model, Fut3 or Fut6-driven fucosylation of Glg1 was identified as the relevant glycosylation event on Glg1 for bone metastasis. The authors speculate that the importance of E-selectin is independent of its classic role in arresting tumor cells within the blood, and instead relates to induction of specific signaling cascades when the tumor cells interact with the bone vasculature. A complex feedback loop involving E-selectin binding, Glg1 fucosylation by Fut3/6, and Wnt pathway activation appears necessary to permit colonization and tumor expansion [57]. This provides an excellent example of the complexity of changes that can result from a relatively simply change in glycosylation activity, as well as demonstrating that glycosylation can in fact act in an organ-specific manner to promote metastasis. Of note, and in contrast to many earlier studies, these data suggest that the role of the E-selectin/Glg1 interaction is important for the later stages of metastasis, specifically colonization and outgrowth. One consideration in evaluating these studies is that there are species differences in selectin-ligand interactions; in particular, mouse E-selectin shows a wider diversity of potential ligands than human E-selectin[18]. For this reason, this mechanism will require additional investigation to verify clinical relevance.
It is clear that altered glycosylation can influence bone metastasis, both in terms of interaction with the bone vasculature for early-stage extravasation and seeding, and later colonization and outgrowth. To date, studies have identified a number of possible targets in various tumor types, including altered expression of glycoproteins or glycosyltransferases, and expression of abnormal glycan epitopes. As demonstrated by Esposito et al., relatively small changes in just a few glycosylation events and/or glycosyltransferase expression can have significant, multifaceted consequences for cancer cell behavior. Moreover, glycosylation-related aberrations may represent druggable targets. However, there exists substantial heterogeneity in the literature (which likely reflects biological heterogeneity in addition to technical differences), and detailed mechanistic investigations have only begun in the last few years. Additional substantial mechanistic work is necessary to rigorously establish targetable alterations.
2.2. Glycosylation and lung metastasis
The lung microenvironment is fertile soil for both primary lung tumors and metastatic tumors from other organs. Studies have identified a number of glycoproteins (e.g. lipocalin-2, podoplanin, tenascin C, von Willebrand factor) whose overexpression is associated with lung-specific metastases [58–61]. However, it has not yet been established whether altered glycosylation of these glycoproteins is relevant to their role in metastatic progression.
Procollagen C-proteinase enhancer protein (PCOLCE) is a potential mediator of lung metastasis identified using osteosarcoma models. It is a secreted glycoprotein with a known role in ECM remodeling, and is associated with poor prognosis in osteosarcoma. Osteosarcoma cells with shRNA-mediated PCOLCE knockdown showed reduced invasion and migration but not proliferation in vitro, and reduced lung metastasis in an orthotopic mouse model [62]. Importantly, the authors of this study identified an N-glycosylation site at Asn29 on PCOLCE which appeared essential for protein stability and secretion into the ECM. When osteosarcoma cells were transfected with either wild-type (WT) or mutant PCOLCE lacking the glycosylation site, those that received the mutant form showed impaired migratory and metastatic phenotypes. This is a relatively rare example of how glycosylation of a specific residue impacts metastatic function, but represents a good model of the type of mechanistic study needed to fully understand the role of protein modifications. However, it lacks clarity on whether this modification is important only for lung metastasis.
Other potential mediators of lung metastasis have been identified using different tumor models. The presence of β1,6-N-linked-oligosaccharides on the cell surface is a change linked with invasion and metastasis in many tumor types that preferentially metastasize to the liver and the lung[63, 64]. The site preference may be related to the possibility for several other modifications on the oligosaccharide branches (addition of Lewis antigen, α2,3-sialic acids, polylacNAc, and others) that have affinity for receptors found in the lung, such as E-selectin and galectins. Using a B16 melanoma model, which was previously shown to metastasize specifically to the lungs independent of mechanical or anatomic cues [65, 66], Krishnan et al. [67] investigated the role of β1,6-N-linked-oligosaccharides in lung-specific melanoma metastasis. In B16F10 cells, the lysosomal protein LAMP1, was identified as the substrate for a polylacNAc modification that led to interaction with galectin-3 on lung vasculature [67]. Further investigation revealed that it is polylacNAc substitution specifically on β(1,6)-N-oligosaccharides, and not on O-glycans or T/Tn antigens, that is responsible for the increased binding of galectin-3 to highly metastatic cells and lung metastatic capacity [68]. Interestingly, some previously identified cancer-associated species, such as sLeX (in contrast to [69]) and E-selectin (in contrast to [70]), did not influence lung metastasis in these studies [67]. It should also be noted that a subsequent study by this group found that whole-body loss of galectin-3 failed to attenuate lung metastasis. The authors suggest that, since galectin-3 is important for the maturation and function of many immune cell types, knock-out mice have impaired antitumor immunity which prevents any decrease in lung tumor burden [71]; more nuanced studies would be necessary to dissect the exact role of galectin-3 in lung-specific metastasis. Together, the studies highlighted here indicate that specific glycosylation events regulate lung metastasis, via several mechanisms that include interactions with the vasculature and general ECM remodeling. However, a major weakness of much of this work is that the data do not establish whether these candidates mediate lung metastasis specifically, or the metastatic process more generally. Further studies investigating this question will be needed in order to fully understand the biology of lung-specific metastasis, as well as to identify appropriate targets for therapeutic development.
As with bone metastasis, the roles of selectins and their ligands in lung metastasis are some of the more thoroughly investigated glycosylation-relevant mechanisms. Very early studies on the mediators of lung colonization also suggested that E-selectin/sLeX interactions were important for a lung colonization phenotype [72], though this should be interpreted with substantial caution given the age and technical limitations of this study. A more recent study linked E-selectin to promotion of breast to lung metastasis [70]. This, however, contrasts with the findings of Esposito et al. [57] who subsequently suggested that lung metastasis is unaffected by E-selectin deletion as described above. This is likely to be due to a difference in models; while Esposito et al. compared E-selectin knock-out and wild-type mice, Jiang et al. [70] observed overexpression of E-selectin on lung vasculature following LPS-induced systemic inflammation, and attenuation of this overexpression prevented an inflammation-associated increase in breast cancer lung metastasis. Since E-selectin expression is quite low in the lungs under standard physiological conditions [57, 70], it may be that it does not influence lung metastasis in absence of significant inflammation. Other researchers developed a therapeutic agent targeting sLeX/A, called per-O-acetylated GlcNAcβ1,3Galβ-O-naphthalenemethanol (AcGnG-NM)[69, 73], which they tested in a mouse model[69]. AcGnG-NM acts as a synthetic decoy of an intermediate in sLeX biosynthesis, thereby preventing cells from decorating glycoproteins with sLeX and thus attenuating the selectin-ligand interaction that contributes to metastatic spread. Murine Lewis lung carcinoma (LLC) cells positive for sLeX/A bound to constructs containing P-selectin but not L- or E-selectin, and this was blocked by AcGnG-NM. Mice subcutaneously injected with LLC cells and subsequently treated with AcGnG-NM demonstrated a 3-fold decrease in the micrometastatic burden in the lung. Since the primary tumor size was not affected, this appears to be a metastasis-specific impact[69].
Sialylation represents an additional glycosylation-related process which is modified in both lung and bone metastasis. Because of the pleiotropic roles of sialylation in metastasis [24, 27], Büll et al. [74] tested the efficacy of P-3Fax-Neu5Ac, a peracetylated analogue of sialic acid that acts as an inhibitory glycomimetic, for reducing metastasis in a mouse model of melanoma. They found a significant reduction of lung metastases after tail vein injection in the treatment group, either when using cancer cells pre-treated with the inhibitor, or when mice were treated with melanoma-targeted (anti-TRP1) nanoparticles carrying the inhibitor. The nanoparticle-conjugated formulation demonstrated prolonged inhibition of sialylation in vitro. In vivo, mice were pre-treated with nanoparticles one hour before receiving tumor cells via tail vein injection, and treated a second time with the nanoparticles approximately 16h later; this brief treatment course was sufficient to reduce lung colonization at 14 days post-injection. In this model, they did not observe metastases in other organs, so it is difficult to know whether this effect is truly lung-specific, but it does demonstrate the potential of targeting abnormal glycosylation to treat metastasis. In a follow-up experiment, the researchers investigated whether there was a difference between growth inhibition in melanotic and amelanotic (which downregulate TRP-1) lesions; they found that treatment with the targeted nanoparticles carrying the glycomimetic specifically inhibited development of melanotic lesions, and that the glycomimetic increased both TRP-1 expression and nanoparticle uptake in the melanotic lesions, suggesting a potential positive feedback loop that could be therapeutically useful.
In addition to identification of glycosylated targets, progress has been made in identifying critical enzymes. Polypeptide N-acetyl galactosaminyl transferases (GALNTs) are a family of enzymes responsible for O-linked N-acetyl galactosamine (GalNAc) addition. Song et al. (2016) identified GALNT14 as a mediator of breast to lung metastasis[75] and dissected multiple ways that GALNT14 activity could enhance metastatic behavior. According to patient microarray data, GALNT14 is the only GALNT family member whose expression in primary breast tumors was significantly associated with distant metastasis-free survival. Specifically, patients with GALNT14 expression in their primary tumors had higher risk of metastasis to lung but not brain or bone. Knockdown of GALNT14 in lung-seeking breast cancer sublines reduced both O-GalNAc glycosylation and metastatic spread to the lungs in a mouse model, which could be rescued by restoring GALNT14 expression. Overall, the authors suggested that KRas/PI3K stimulated, c-Jun driven upregulation of GALNT14 results in a survival advantage during the micrometastatic phase by making cells resistant to anti-metastatic BMP signaling, potentially via inactivating O-GalNAcylation of the BMP receptor. As the tumor reaches the macrometastatic phase, GALNT14 enhances CXCL1 production through an as yet unknown post-translational mechanism which recruits macrophages to help form a permissive environment, and also renders the cancer cells more receptive to macrophage-derived FGF growth signaling [75]. Importantly, this paper demonstrates that change in one glycosylation regulator can contribute to multiple stages of metastasis tailored to the microenvironment of a specific organ.
Overall, the current data indicate that aberrant glycosylation is relevant for lung metastasis. Some mediators – notably E-selectin and sialylation – are undoubtedly shared with other metastatic sites. However, it does appear that the precise interaction partners and mechanisms underlying these effects are frequently organ-specific, which again supports the hypothesis of site-specific metastatic mechanisms, and gives credence to the idea that the highly contextual nature of glycosylation and its alterations make it a promising target of study for this purpose.
2.3. Glycosylation and brain metastasis
Brain metastasis represents a serious clinical challenge due to significant associated morbidity and mortality and lack of effective treatment options. Its incidence is on the rise, likely as a consequence of better primary tumor control and improved imaging techniques[76]. Unlike other metastatic sites, where the study of the impact of glycosylation remains in its early stages, glycosylation and glycogene changes are well-established in the brain setting; it was established in the 1980s and 1990s that brain tumors show aberrant ganglioside expression and shedding (summarized in [77]), and altered glycosphingolipid expression. Initial studies focused on primary brain tumors. These are reviewed by Moskal et al. [35] and will not be repeated here. A number of glycoproteins also seem to mediate brain metastasis (see, for example, [78–82]). As noted for glycoproteins that mediate metastasis at other sites, however, altered glycosylation has not yet been specifically linked to their pro-metastatic functions.
More recent studies have begun to elucidate roles for glycosylation enzymes in brain metastasis. Pangeni et al. [83] identified GALNT9 (encodes GALNAC-T9, a transferase responsible for the initial step in mucin O-glycan synthesis [Table 1, Figure 1]) as heavily methylated in brain metastases but infrequently methylated in primary breast tumors. Loss of protein expression was associated with worse prognosis for breast cancer patients. This suggests that impairment of O-glycosylation, the result of GALNT9 loss, may play a role in breast-to-brain metastasis. Another enzyme, the α2–6 sialyltransferase ST6GALNAC5, was identified in metastatic breast cancer cells in a landmark paper by the Massagué group [84]. Affinity testing using a lectin that binds α2,6-sialylated groups revealed substantial binding in both mammary tumors and brain lesions formed by brain-metastatic BrM2 cells in mice, as well as in 6 out of 12 of human brain metastatic patient samples tested, but low or no binding in patient lung metastasis samples. Modulation of ST6GALNAC5 confirmed that its role is exclusive to brain metastasis, and that it specifically impacts infiltration of cancer cells through the blood-brain barrier. Importantly, transduction of a lung-metastatic derivative of MDA-MB-231 breast cancer cells with ST6GALNAC5 resulted in increased brain micrometastases, suggesting that ST6GALNAC5 is important for the initial invasion of the brain, while other unknown mediators are necessary for growth into macrometastases [84]. Thus, those authors suggest that glycogenes with restricted expression patterns might be important organ-specific mediators of metastatic colonization and emphasizes that mediators may be stage- as well as organ-specific. It should be noted that another group attempted to verify this finding using an in vitro BBB model derived from human cell lines and the BrM2 cell line [85]; in that model, BrM2 cells and cells engineered to overexpression ST6GALNAC5 specifically demonstrated reduced adhesion to the BBB and no change in transmigration capacity. The authors suggest that this is due to the use of a human-on-human model rather than a human-in-mouse model as in the Massagué study, and hypothesize that the relevance of ST6GALNAC5 may stem from adaptation of the human cancer cells to the mouse brain environment. Regrettably, this hypothesis was not investigated further, nor were any other potential mediators identified.
Interestingly, given the importance of E-selectin/ligand interactions at other metastatic sites, there is evidence that NSCLC cells produce a variety of cytokines and other factors which destroy the endothelial cell glycocalyx to expose adhesion molecules, including E-selectin, during brain metastasis[86]. The endothelial glycocalyx is a dense layer of transmembrane and membrane-bound species bound to the endothelium by various glycoproteins and proteoglycans; it forms a “mesh” in which various adhesion molecules such as E- and P-selectin are normally embedded. Brain-metastatic lung cancer cells rapidly destroy this layer upon invasion, exposing E-selectin. The cancer cells, which tend to express high levels of E-selectin ligands such as sLeX antigens, can then more effectively interface with the endothelial cells for transmigration of the BBB. The authors additionally speculate that long-term exposure to cancer cell-derived cytokines drives increased expression of E-selectin by the vasculature, which would further facilitate brain metastasis [86]. Of note, different cell subtypes expose and interact with different selectins. While both adenocarcinoma and squamous cell models expose E-selectin, adenocarcinoma cell models additional expose P-selectin. A second group investigating NSCLC [87] found that the interaction between LeX and E-selectin is critical for NSCLC brain metastasis. Importantly, considering the known species-specific differences in selectin/ligand interactions [18], they verify the expression of E-selectin and LeX (which is quite rare in the normal human cortex) in brain metastasis tissue samples from patients, supporting a role for this interaction in human disease as well as animal models.
The blood-brain barrier (BBB) represents a unique challenge for brain-metastatic cancer cells, and a number of studies have shown that altered glycosylation is particularly important for cancer cell transmigration of the BBB. This barrier, composed of a basal lamina, endothelial cells, astroglia, pericytes and perivascular macrophages, separates the brain and its internal extracellular fluid from the normal systemic blood flow[88]. It is characterized by high electric resistance, a lack of permeability, and the presence of many tight junctions and complex adhesion junctions [89]. Adding to the complexity, the so-called “brain-tumor barrier” (BTB) is in many ways distinct from the non-pathologic BBB. Tumor-associated changes to the microenvironment and the vasculature result in loss of selectivity between the blood and extracellular fluid, and abnormal, non-uniform permeability that can facilitate further cancer cell invasion and/or inhibit effective drug delivery [90]. In order to form a brain metastasis, cancer cells must breach this barrier; there have been several studies suggesting that glycosylation changes may be important for endowing cancer cells with this ability. One paper found that inhibition of the synthesis of GM1 glycosphingolipid decreased adhesive abilities of MDA-MB-231 and MCF-7 cells to a human cord-blood derived model of the BBB by approximately 40%, suggesting that glycosphingolipids may play a role in cancer cell adhesion to the BBB [91]. Although not in a cancer setting, another group investigating retention differences of IgG antibodies against amyloid beta (for treatment of Alzheimers) found that the presence of an α2,6-sialylation on the FAB chain of an IgG antibody drug known as mAb 4G8 played a key role in retention of the drug by inhibiting efflux back across the BBB. Removal of that glycan restored efflux to normal levels [92]. This suggests that α2,6-sialylation may play an important role in the breaching of the BBB, in addition to the role in brain-specific colonization identified by Massagué[84]. Additionally, the glycan species LeX and sLeX have been identified as important facilitators of NSCLC invasion through the BBB [93], similar to their well-documented roles in extravasation in other contexts[5]. Several integrins, such as α5β3, α5β8 [94], and VLA-4 [95], are also strongly associated with brain metastases, likely by contributing to adhesive tumor-endothelial cell interactions that destabilize the BBB. It should be noted, however, that the role of glycosylation in these processes has not yet been confirmed, and further that several of these mechanisms are not brain-exclusive (see [95–97].
Because glycosylation is such a well-established factor in brain tumor growth, there has been significant interest in specifically identifying the relevant glycan species. This is technically demanding, due to the difficulty of separating and studying the various glycan isomers[89]. The Mechref group performed a series of studies examining the role of glycosylation in breast-to-brain metastasis [88, 89, 98]. First, they performed NGS transcriptome and gene ontology analysis on a panel of cell lines including a primary brain cancer and five metastatic breast cell lines [98]. Twelve glycosylation-related genes were found to be differentially expressed in a brain-seeking line as compared to the rest of the panel, including genes relevant to sialylation, fucosylation, synthesis of complex glycan structures, and glycan anchors. The upregulated genes included the previously-identified[84] sialyltransferase ST6GALNAC5, as well as the related ST6GAL1, and several genes within the integrin pathway (IGTA2, IGTA3, IGTA6)[98]. Subsequent studies by this group focus on the development of precise mass-spectrometry methods for identification of specific glycan species that are present in altered abundance in brain-seeking metastatic breast cancer. These studies identify 46 isomers of 23 glycan species with unique expression patterns in brain-seeking MDA-MB-231BR cells; of those, 24 were sialylated, supporting the critical role of this modification for brain metastasis. Further, α2,6-sialylated N-glycans were particularly enriched in both the MDA-MB-231BR and CRL-1620, a primary brain cancer [89]. The authors speculate that this indicates that these glycans are particularly important for both the breaching of the BBB and subsequent colonization. A second study restricted to membrane N-glycans demonstrated that the most important global change in metastatic cell lines seemed to be N-glycan sialylation, while altered fucosylation was second-most common[88]. Such detailed biochemical analyses confirm the role of sialylation in brain metastasis and identify certain glycans for further investigation with regard to their specific biological role.
One final characteristic of brain metastatic tumors which deserves mention is their penchant for metabolic abnormalities, which have potential to produce glycosylation abnormalities due to alterations in available metabolites for glycosylation reactions. Brain-metastatic tumors tend to display altered sphingolipid metabolism [99], altered amino acid metabolism [100], and some are highly glycolytic [100]. A very recent study also suggests that fatty acid synthesis is upregulated in brain-metastatic breast cancer cells, and is in fact required for successful brain metastasis, though this study relied primarily on intracranial injection models and therefore reveals more about metabolic requirements for growth within the brain than for brain-specific metastasis [101]. However, that finding is supported by the identification of altered lipid metabolism downstream of PI3K as an important mediator of specifically of outgrowth, rather than dissemination, of brain-metastatic tumors[102]. Brain-metastatic tumors are also more sensitive to perturbations in environmental metabolite availability than metastatic cells at other sites. For example, one study found that brain metastatic breast cancer cells were more sensitive to hyper- or hypoglycemia than bone-metastatic cells. In response to glucose deprivation, they experience substantial changes in cell morphology, decreases in colony formation capacity, and downregulation of proteins which are normally overexpressed, including secreted VEGF and integrin β3 [103]. Demonstrating the link between metabolic and glycosylation abnormalities, this study also showed that both bone- and brain-metastatic derivatives of MDA-MB-231 cells expressed exclusively an immature, unglycosylated form of VEGFR2 under normal physiologic glucose conditions, while the parental cells expressed a mix of a fully-glycosylated and unglycosylated form of the protein. The metastatic derivatives required hyperglycemic conditions to produce the fully glycosylated form[103]. This avenue of investigation is fairly new, and much work remains to be done regarding both metabolic abnormalities, and any potential link to glycosylation, in the brain metastatic context. However, the early data are encouraging, especially given the sensitivity of brain metastatic cells to metabolic changes, which might represent a vulnerability for these otherwise difficult tumors.
In summary, the role of abnormal glycosylation in the context of brain metastasis is, in many ways, much more developed than for other metastatic sites, though many controversies and open questions remain. It is interesting to note, again, that several mechanisms are common between the brain and other metastatic sites, such as mediators of BBB transmigration which also participate in extracranial extravasation, E-selectin-mediated interactions, and sialylation. Indeed, Valiente et al. [80] note that it is likely that some of the mechanisms identified in brain-metastatic tumors are relevant for other sites, but that the unique selective pressures of the brain microenvironment select for cells which display more exaggerated versions of those phenotypes. Given the sheer number of glycosylation-relevant mediators of primary and metastatic brain tumor growth, it seems likely that this is the case for glycosylation-related phenotypes. The current picture is both promising and complex, and as with all research on brain metastasis, the limitations of our current model systems (which rely largely on in vitro BBB surrogates and mouse models, which do not easily or fully recapitulate human brain metastasis [104]) should be considered. Altogether, however, abnormal glycosylation is clearly a frequent, biologically consequential event in brain metastasis with substantial potential for therapeutic targeting.
2.4. Glycosylation and lymph node metastasis
Lymph node metastasis is a common early event in cancer and is considered a predictor of poor prognosis. Many studies have identified altered glycosylation events in lymphatic metastases. It is known that L-selectin-mediated interactions between leukocytes and lymphatic vessels are critically important for migration of leukocytes into the lymph nodes[105], and that even moderate reduction of L-selectin can substantially impair leukocyte trafficking[26]. Interaction between lymphocytes and the lymph node high endothelial venule (HEV) appears to depend specifically on interaction between L-selectin and O- and N-glycans carrying sulfated glycans such as 6-sulfo-sLeX [16, 105]. A few studies have suggested that L-selectin expression on cancer cells may facilitate lymphatic metastasis, although this appears to be tumor-type specific (reviewed in [106]).
Because of the clinical relevance of lymphatic metastasis, many studies have been conducted to identify potential markers, which have revealed that altered glycosylation is commonly associated with lymphatic metastasis across a variety of tumor types. A 1991 study [107] found that breast cancer patients whose primary tumors stained positive for HPA lectin, which binds N-acetyl-galactosamine (GalNAc), are more likely to develop axillary lymph node metastases. Similarly, increased β1,6-branched oligosaccharides correlate with lymph node metastasis and overall poor prognosis in a breast cancer cohort [108]. A more recent study using MALDI-IMS to detect N-glycan species identified F(6)A4G4Lac1, a core-fucosylated tetra-antennary glycan containing a single polylactosamine arm, as associated with lymphatic metastasis and poor prognosis in breast cancer patients and animal models [109]. A few studies in breast cancer have provided more mechanistic data. One in vitro study using co-culture of lymphatic endothelial cells with either a weakly metastatic (MCF7) or highly metastatic (MDA-MB-231) breast cancer cell line identified “metastasis-specific” genes which were uniquely upregulated in the lymphatic endothelial cells in response to co-culture with the highly metastatic cell line. Among them were MAN1A1, a mannosidase, and MGAT4A, a glycosyltransferase (see Table 2). In both that model and a similar model using weakly- or highly-metastatic prostate cancer cell lines, E-selectin was also upregulated [110]. Another study probing primary tumor samples from patients with invasive ductal carcinoma of the breast found that O-GlcNAc (see Table 1 and Figure 1) levels were globally reduced in tumors from patients with lymph node metastases. This was accompanied by a reduction in OGT protein expression (the enzyme responsible for O-GlcNAcylation), but no change in OGA, which removes the mark. Within the metastatic group, tumors from patients who had >3 positive lymph nodes demonstrated reduced global O-GlcNAc as compared to those with only 1–3 positive lymph nodes. Which proteins species carried O-GlcNAc modifications also varied between the metastatic and non-metastatic groups; interestingly, the authors note that proteins involved in glycolysis and the pentose-phosphate pathway were particularly likely to display discordant O-GlcNAc status [111].
Similar correlative studies have been conducted in other tumor types as well. In endometrial cancer, primary tumor samples from node-positive patients had lower levels of a complex core-fucosylated N-glycan species (named (Hex)2(HexNAc)2(Deoxyhexose)1+(Man)3(GlcNAc)2) than primary tumor samples from node-negative patients [112]. In head and neck squamous cell carcinoma, increased expression of the ganglioside-specific sialidase (a glycosyltransferase enzyme that removes sialic acid) NEU3 correlated with lymph node metastasis [113]. Elevated expression of the glycosyltransferase B4GALT1 correlates with lymph node metastasis in pancreatic cancer, specifically through increased N-glycosylation of CDK11P110, which stabilizes the CDK and promotes tumor progression and chemoresistance [114]. In oral squamous cell carcinoma, expression of the pentose phosphate pathway enzyme glucose-6-phosphate dehydrogenase (G6PD) in the primary tumor is associated with lymphatic metastasis. Mechanistic investigation using G6PD-deficient cells demonstrated reduced lymphatic metastases in an orthotopic mouse model, and revealed that G6PD loss results in elevated expression of the glycosyltransferase MGAT3, which specifically increased the bisecting GlcNAc-branched N-glycosylation of E-cadherin. This suggests that the opposite – reduction of MGAT3 and reduced bisecting GlcNAc-brached N-glycosylation of E-cadherin – might encourage lymphatic metastasis in the tumor setting [115]. Concordantly, in a mouse model of hepatocellular carcinoma, miRNA-mediated downregulation of MGAT3, and the resultant decrease in bisecting β1,4-GlcNAc-branched N-glycosylation, is associated with elevated lymphatic metastasis [116].
These studies strongly suggest that altered glycosylation is relevant in lymphatic metastasis of a variety of tumor types. However, most of them are simply correlative, with little or no mechanistic exploration, and whether any of these events are explicitly required for lymph node metastasis is not assessed. Additionally, these studies do not explore the impact of these alterations of the frequency of metastasis at other sites. Given that lymphatic metastasis is often an intermediate event between primary tumor growth and the establishment of distant metastases, it must be considered that the altered glycosylation events identified by these studies may simply be associated with general metastatic or invasive behavior of tumor cells, rather than lymph node-specific organotropism.
One recent study has identified a glycosylation-dependent mechanism specific to lymphatic metastases. Gu et al. [117] investigated the role of B cells in the promotion of breast cancer lymphatic metastasis. They find that orthotopic mammary tumors in mouse models induce B-cell accumulation in the tumor-draining lymph nodes, and that pathogenic IgG from those B cells promotes lymphatic, but not bone, lung, or primary tumor growth. Pathogenic IgG is found to bind HSP4A. Interestingly, tumor-associated HSP4 is aberrantly N-glycosylated; removing this N-glycosylation abrogates the interaction of pathogenic IgG and HSP4A. The binding event between N-glycosylated HSP4 and pathogenic IgG activates the CXCR4/SDF1α pathway via NF-κB. Of note, CXCR4/SDF1α has been previously implicated in the attraction of tumor cells to distant organs during metastasis[118]. Indeed, breast cancer patients with lymph node metastases showed higher levels of serum anti-HSP4 IgG than either non-metastatic cases or healthy controls. Overall, this paper intriguingly demonstrates that interaction between an abnormal glycoform of HSP4A and pathogenic IgG activates pro-metastatic signaling and specifically promotes a premetastatic niche in the lymph nodes.
2.5. Glycosylation at other secondary sites
There is some evidence that glycosylation may impact metastasis to other secondary organs in addition to the four outlined above, but there exists a paucity of studies on the topic. Peritoneal metastasis in ovarian cancer is dependent on P-selectin/sLeX interactions, which slow the cancer cell “rolling” sufficiently as compared to non-metastatic cells to allow metastasis to take place [119]. A study in mice injected subcutaneously with human neuroblastoma samples found that ovarian metastases, but not liver metastases, displayed elevated E-selectin binding capacity which was specifically ablated by removal of α2,3-sialylation, which is more common on glycolipids and suggests that glycolipid-mediated selectin interactions might be key for this metastatic event [120]. Several older studies have suggested that interaction of E-selectin with β1,6-branched N-oligosaccharides carrying sLe antigens is important for liver metastasis [71], and particularly that modulation of sLe antigen addition through manipulation of the fucosyltransferases responsible for the final addition can modulate liver metastasis in animal models of liver metastasis after intrasplenic injection[52]. A more recent study investigated the impact of galectin-3 on the creation of the pre-metastatic niche and consequent liver metastasis. Reticker-Flynn et al. [121] established a spontaneous mouse metastasis model using cells derived from KRas-mutant, p53-knockout lung adenocarcinoma cells. The livers of tumor-bearing mice exhibited increased galectin-3 levels that was derived from recruited myeloid cells, even before detectable metastasis. Galectin-3 has particular affinity for the T-antigen (see Table 2) but is unable to bind more extensively glycosylated forms of the protein. Comparison of glycosyltransferase expression between metastatic and nonmetastatic cell lines showed that GCNT3, an enzyme responsible for the addition of a β(1,6)-GlcNAc to the T antigen, is under-expressed in metastatic cell lines, while ST6GalNAc4, which is responsible for the addition of an α(2,6)-NeuAc to sialyl-T antigens, is overexpressed. These changes prevent branching (GCNT3) and cap elongation (ST6GalNAc4), leading to increased presentation of the T antigen without expression changes. Genetic manipulation confirmed that reintroduction of GCNT3 or knockdown of ST6GalNAc4 reduced galectin-3 binding. Importantly, ST6GalNAc4 knock-down cells showed a 95% decrease in liver metastases after splenic injection in mice. Overall, the recruitment of galectin-3+ myeloid cells to the pre-metastatic niche might represent a mechanism which aids with both extravasation and colonization [121]. It is important to note, however, that although this study utilized a lung-to-liver metastasis model, organ specificity was not directly tested, so although it appears that this mechanism is relevant to liver metastasis, it remains unknown whether it is recapitulated in other sites.
There has also been interest in characterizing altered glycosylation within hematologic malignancies. Although most of these cancers do not “metastasize” in the typical sense, there are examples of aberrant glycosylation leading to increased spread. Common glycosylation alterations in hematologic malignancies match those reported in solid tumors, including fucosylation, sialylation, and abnormal bisecting GlcNAc, exposure of Tn antigen, and alterations in MUC1 expression (reviewed in [122]). Notably, MGAT3, the enzyme responsible for the addition of bisecting GlcNAc residues, has been reported to be elevated in blast-crisis stage chronic myeloid leukemia and multiple myeloma, and experimental upregulation led to increased spleen colonization by K562 leukemia cells [123]. Multiple myeloma (MM), a cancer of plasma cells that is known to traffic into the bone marrow and participate in metastasis-like behavior throughout the axial skeleton, exhibits altered glycosylation that in many cases mirrors that of bone-metastatic solid tumors. As noted earlier (see ‘Bone Metastasis’), elevated expression of heparanase, the enzyme that cleaves heparan sulfate (see Table 2), increases homing of MM cells to the bone in mice [45]. Additionally, altered sialylation plays a substantial role in the bone-homing behavior of multiple myeloma cells. Two sialyltransferases, ST3GAL1 and ST3GAL6, are associated with poor prognosis in MM patients[124]. ST3GAL6, which participates in the synthesis of sLeX, (see Table 2), is highly expressed in MM. Knockdown of ST3GAL6 interferes with adhesion and migration in vitro and bone marrow homing and survival by MM cells in vivo, putatively because the presence of sLeX glycans on selectin ligands such as P-selectin glycoprotein ligand 1 (PSGL-1) is necessary for interaction with E- and P-selectins on the bone vasculature [125]. Because E-selectin is strongly expressed on bone vasculature, the authors hypothesize that this interaction is primarily E-selectin mediated, which parallels the E-selectin-mediated interaction observed in breast-to-bone metastasis [57]. That same selectin ligand, PSGL-1, is essential for hematogenous metastasis of lymphomas [122]. Interestingly, application of the same glycomimetic E-selectin inhibitor which showed potential for reducing bone metastasis in the Esposito paper [57] also reduces bone homing in mouse models of multiple myeloma [124]; this raises the possibility that some glycosylation-dependent bone-homing mechanisms may be relevant across otherwise disparate tumor types. A follow-up paper [126] suggests that pharmacological, global inhibition of sialylation (both by administration of the inhibitor in vivo and pre-treatment of multiple myeloma cells to avoid toxicity) reduces homing of multiple myeloma cells to the bone and improves animal survival in a normally-aggressive mouse model and acts synergistically with bortezomib, the standard of care for multiple myeloma. Desialylation reduces E-selectin-mediated affinity, as well as the interactions between integrin α4β7 and MADCAM1 and integrin α4β1 and VCAM1, primarily through desialylation of the α4 subunit, which are additionally important for the homing process. Interestingly, administration of the sialylation inhibitor in vitro reversed stromal cell-mediated, but not endothelial cell-mediated, resistance to bortezomib. Together, these studies demonstrate that there exist intriguing commonalities in glycosylation-dependent mechanisms between solid tumor metastasis and the spread of hematologic cancers.
2.6. Glycosylation in circulating tumor cells
Circulating tumor cells (CTCs) – tumor cells which have escaped from solid-organ tumor sites and entered the bloodstream – are a subject of intense interest, for clinical applications (both prognostic/diagnostic, and intervention-focused) as well as for their role in the biology of metastasis [127]. CTCs that are able to survive in circulation for long enough to reach a distant site and extravasate are the cells from which distant metastases develop. Recently, there has been recognition of the role that glycosylation plays in the biology and behavior of circulating tumor cells. CTCs are thought to use selectin/ligand interactions similar to physiological leukocyte rolling in order to interact with blood vessels[22]. Interestingly, several studies have shown that cancer cells express distinct fucosyltransferaes (FUTs), which are critical enzymes for the production of selectin ligands, as compared to leukocytes[128]. A huge number of studies have probed the expression of selectin ligands and the landscape of cancer cell-endothelial cell interactions. Which selectin and ligand predominate is largely tumor-type specific, and due to space constraints, the nuances of this will not be discussed here; many excellent reviews have been published, including [128, 129]. Importantly, considering the prevalence of animal research, selectin-ligand interactions are also highly species specific; one study which performed a panel of analyses investigating the interaction of various human cancer cell lines with human or mouse E-selectin concluded that (1) different ligands are functional in static vs. dynamic flow conditions; (2) mouse and human E-selectin interact with different ligands, and (3) mouse E-selectin interacts with a wider variety of ligands than does human E-selectin[18]. Taken together, these results suggest that meticulous experimental design which considers both flow conditions and species compatibility will be necessary to elucidate the most clinically-relevant selectin/ligand binding events.
Several studies have shown that cancer cells are capable of using alternative ligands for selectin binding. For example, hypo-glycosylated MUC1 has been found to interact with E- and P- (but not L-) selectin [130], and HS and chondroitin sulfate (CS) – both GAGs (see Table 2) – have been shown to act as P- and L-selectin ligands; in particular, over-sulfated forms of both have been found in breast cancer cells and display higher-affinity selectin interactions than their normally-sulfated counterparts due to ionic interactions. Patient data shows that overexpression of CS-sulfating enzymes correlates with increased risk of distant metastasis [131]. Finally, CD44, which is highly associated with cancer, can act as an E-selectin ligand under dynamic flow conditions. The CD44s isoform is expressed by leukocytes and can act as an E-selectin ligand in that context, but the glycosylation marks present on that isoform differ significantly from those present on cancer-associated CD44. One group has specifically identified CD44v3 and CD44v4/5, as E-selectin ligands on human breast cancer cells[132]. In sum, cancer cells are capable of leveraging a number of non-canonical selectin ligands to aid their survival in the bloodstream and their interaction with endothelial cells, platelets, and others as they engage in hematogenous metastasis.
More recently, the role of abnormal blood flow in tumor biology has also been a subject of interest. Abnormal blood flow is a common characteristic in and around tumors due to changes in vascular permeability and vessel geometry. It is more common in organs with substantial vessel branching and turning, such as the lungs and liver, which also happen to be common metastatic sites. The luminal side of endothelial cells is coated in a “glycocalyx” composed of many glycosylated species, including sialic acids and proteoglycans. “Disturbed flow” is known to wear down the glycocalyx over time. One group has demonstrated that under abnormal blood flow conditions, both pro-survival clustering behavior and endothelial cell interactions of CTCs are increased[133]. This was found to be due to global erosion of the glycocalyx; however, it was not directly dependent on increased E-selectin exposure, as previously published by Rai et al. [86]. Overall, it is clear that CTC survival and interaction with endothelial cells are highly dependent on glycosylation; hopefully, further studies will elucidate more tumor-specific mechanisms that can be leveraged for cancer therapy.
GLYCAN TARGETING AS A THERAPEUTIC APPROACH
Given its wide-ranging impact on cancer development and progression, targeting glycosylation for therapeutic purposes is an active area of research. Several recent reviews provide extensive detail [8, 134–136]; some relevant therapies are highlighted here. Preclinically, fluorinated analogs of sialic acid and fucose can be used to inhibit sialyl- and fucosyltransferases, and have been shown to reduce sLeX expression and leukocyte rolling behavior[137]. Soyasaponin-I [138] and AL10 [139], both sialyltransferase inhibitors, inhibited cancer cell metastasis in mouse models of breast cancer and lung cancer, respectively. Additionally, a small-molecule inhibitor of the O-glycosyltransferase ppGalNAc-T-3 has been identified[140]. ppGalNAc-T-3 has been broadly implicated in metastasis; in in vitro studies, the inhibitor was capable of blocking invasive behavior by breast cancer cells[140]. Finally, there is substantial interest in using antibodies against cancer-associated glycoforms to improve drug targeting, and a growing recognition that which protein glycoform a cell expresses can have consequences for therapeutic response[135].
On the clinical side, there have been a handful of efforts to harness cancer-associated abnormal glycosylation. One such effort, an sTn-KLH vaccine for metastatic breast cancer (KLH is a carrier added for immunogenicity), was abandoned due to lack of clinical benefit[141]. However, recent results have been more promising. Despite the failure of the sTn-KLH vaccine, interest in developing glycan vaccines for cancer has not abated; a number of vaccines against cancer-associated glycans (for example: MUC1, GD2/GD3, CEA,sLeA) in early clinical trials for a variety of cancer types as of this writing. So far, none have been outstandingly effective, but some have demonstrated modest positive results[142]. GMI-1271, the E-selectin antagonist employed by both Esposito [57] and Price[56], has completed phase I safety trials in healthy patients, and is currently in testing for use in acute myeloid leukemia and multiple myeloma [135]. In the neuroblastoma setting, there are a number of trials investigating antibodies to GD2, a disialoganglioside found on nearly all neuroblastoma tumors, as monotherapy and in combination with various immunotherapeutic strategies [23, 143]. The same is true across many cancer types for MUC1, a heavily glycosylated mucin protein that is frequently overexpressed in cancer and associated with metastasis [135], as well as a number of other cancer-associated glycans[142]. For a comprehensive discussion of the state of the art of glycan-directed therapies, see Smith et al. [142]. It should also be noted that there is significant interest in identifying CTC-specific glycan epitopes for more sensitive, specific CTC identification as a noninvasive surveillance strategy. The success of these attempts has been mixed, and work is ongoing [144–146]. Altogether, development of glycosylation-related therapies is rapidly advancing; however additional basic and translational research will be needed in order to identify and exploit effective targets, particularly in the case of site-specific metastasis.
CONCLUSIONS
Glycosylation is a ubiquitous and highly impactful form of post-translational modification that is getting increasing attention as a contributor to both primary tumor and metastatic biology. Although modifications in glycans were at first thought to be random, it has become clear that they are actually highly controlled and, due to their prevalence on the cell surface and in the ECM, can impact a wide variety of cell-cell communication processes, as well as intrinsic processes. Modifications in various aspects of glycosylation, including alterations in glycosyltransferase activity, glycoprotein expression, and the specific glycan species present (see Figure 1), have demonstrated relevance throughout the metastatic cascade, and particularly with respect to the later stages such as colonization and outgrowth. Abnormal glycosylation is diagnostically, prognostically, and increasingly therapeutically relevant to treatment of cancer. Metastasis already represents a significant morbidity and mortality burden for cancer patients; as treatments for primary tumors continue to improve and patients live longer, it will become increasingly important to also control metastatic outgrowths. One persistent mystery of metastasis is how, exactly, cancer cells develop a predilection for particular secondary organ sites. This problem is receiving a substantial influx of attention, particularly as advances in technology enable improved tracking and analysis of organotropism [102]. As summarized in Figure 2, alterations in glycosylation - due to changes in glycosyltransferase activity, levels of specific glycan marks, and/or the glycosylation status of critical proteins - is a consequential mediator of organ-specific metastasis. Recent investigations have recognized mediators that are shared across organ sites, as well as glycosylation events that appear highly specific for a given organ microenvironment. However, particularly outside of the brain, our understanding of the specific mechanistic role of differential glycosylation in organotropism remains in its early stages. Future work should focus on delineating additional organ-specific, mechanistically-relevant glycosylation changes and determining which of these could be therapeutically exploited.
Acknowledgements:
This work was supported by 1F31CA265070-01 (Bindeman) and T-32 CA009592 (Chen). The authors would like to thank Julie Rhoades, PhD, for providing feedback, and Demond Williams for the helpful discussions.
Footnotes
Publisher's Disclaimer: This AM is a PDF file of the manuscript accepted for publication after peer review, when applicable, but does not reflect post-acceptance improvements, or any corrections. Use of this AM is subject to the publisher’s embargo period and AM terms of use. Under no circumstances may this AM be shared or distributed under a Creative Commons or other form of open access license, nor may it be reformatted or enhanced, whether by the Author or third parties. See here for Springer Nature’s terms of use for AM versions of subscription articles: https://www.springernature.com/gp/open-research/policies/accepted-manuscript-terms
Conflicts of interest/Competing interests: The authors have no relevant conflicts of interest to declare.
REFERENCES
- 1.Chaffer CL, & Weinberg RA (2011). A perspective on cancer cell metastasis. Science, 331(6024), 1559–1564. 10.1126/science.1203543 [DOI] [PubMed] [Google Scholar]
- 2.Budczies J, Von Winterfeld M, Klauschen F, Bockmayr M, Lennerz JK, Denkert C, Wolf T, Warth A, Dietel M, Anagnostopoulos I, Weichert W, Wittschieber D, & Stenzinger A (2015). The landscape of metastatic progression patterns across major human cancers. Oncotarget, 6(1), 570–583. 10.18632/oncotarget.2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liotta LA (2001). An attractive force in metastasis. Nature, 410(6824), 24–25. 10.1038/35065180 [DOI] [PubMed] [Google Scholar]
- 4.Welch DR, & Hurst DR (2019). Defining the hallmarks of metastasis. Cancer Research, 79(12), 3011–3027. 10.1158/0008-5472.can-19-0458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oliveira-Ferrer L, Legler K, & Milde-Langosch K (2017). Role of protein glycosylation in cancer metastasis. Seminars in Cancer Biology, 44, 141–152. 10.1016/j.semcancer.2017.03.002 [DOI] [PubMed] [Google Scholar]
- 6.Varki A, & Gagneux P (2017). Biological functions of glycans. In Varki A, Cummings RD, Esko JD, et al. (eds.), Essentials of Glycobiology 3rd ed. Cold Spring Harbor Laboratory Press. 10.1101/glycobiology.3e.007 [DOI] [Google Scholar]
- 7.Josic D, Martinovic T, & Pavelic K (2019). Glycosylation and metastases. Electrophoresis, 40(1), 140–150. 10.1002/elps.201800238 [DOI] [PubMed] [Google Scholar]
- 8.Reily C, Stewart TJ, Renfrow MB, & Novak J (2019). Glycosylation in health and disease. Nature Reviews Nephrology, 15(6), 346–366. 10.1038/s41581-019-0129-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vajaria BN, & Patel PS (2017). Glycosylation: A hallmark of cancer? Glycoconjugate Journal, 34(2), 147–156. 10.1007/s10719-016-9755-2 [DOI] [PubMed] [Google Scholar]
- 10.Prestegard JH, Liu J, & Widmalm G (2017). Oligosaccharides and polysaccharides. In Varki A, Cummings RD, Esko JD, et al. (eds.), Essentials of Glycobiology 3rd ed. Cold Spring Harbor Laboratory Press. 10.1101/glycobiology.3e.003 [DOI] [PubMed] [Google Scholar]
- 11.Rini JM, & Esko JD (2017). Glycosyltransferases and glycan-processing enzymes. In Varki A, Cummings RD, Esko JD, et al. (eds.), Essentials of Glycobiology 3rd ed. Cold Spring Harbor Laboratory Press. 10.1101/glycobiology.3e.006 [DOI] [Google Scholar]
- 12.Stowell SR, Ju T, & Cummings RD (2015). Protein glycosylation in cancer. Annual Review of Pathology: Mechanisms of Disease, 10(1), 473–510. 10.1146/annurev-pathol-012414-040438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freeze HH, Hart GW, & Schnaar RL (2017). Glycosylation precursors. In Varki A, Cummings RD, Esko JD, et al. (eds.), Essentials of Glycobiology 3rd ed. Cold Spring Harbor Laboratory Press. 10.1101/glycobiology.3e.005 [DOI] [PubMed] [Google Scholar]
- 14.Varki A (2016). Biological roles of glycans. Glycobiology, 27(1), 3–49. 10.1093/glycob/cww086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brockhausen I, & Stanley P (2017). O-GalNAc glycans. In Varki A, Cummings RD, Esko JD, et al. (eds.), Essentials of Glycobiology 3rd ed. Cold Spring Harbor Laboratory Press. 10.1101/glycobiology.3e.010 [DOI] [PubMed] [Google Scholar]
- 16.Stanley P, Taniguchi N, & Aebi M (2017). N-glycans. In Varki A, Cummings RD, Esko JD, et al. (eds.), Essentials of Glycobiology 3rd ed. Cold Spring Harbor Laboratory Press. 10.1101/glycobiology.3e.009 [DOI] [Google Scholar]
- 17.Varki A, Schnaar RL, & Schauer R (2017). Sialic acids and other nonulosonic acids. In Varki A, Cummings RD, Esko JD, et al. (eds.), Essentials of Glycobiology 3rd ed. Cold Spring Harbor Laboratory Press. 10.1101/glycobiology.3e.015 [DOI] [PubMed] [Google Scholar]
- 18.Starzonek S, Maar H, Labitzky V, Wicklein D, Rossdam C, Buettner FFR, Wolters-Eisfeld G, Guengoer C, Wagener C, Schumacher U, & Lange T (2020). Systematic analysis of the human tumor cell binding to human vs. murine E- and P-selectin under static vs. dynamic conditions. Glycobiology, 30(9), 695–709. 10.1093/glycob/cwaa019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Varki A (2017). Biological roles of glycans. Glycobiology, 27(1), 3–49. 10.1093/glycob/cww086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodrigues JG, Balmana M, Macedo JA, Pocas J, Fernandes A, de-Freitas-Junior JCM, Pinho SS, Gomes J, Magalhaes A, Gomes C, Mereiter S, & Reis CA (2018). Glycosylation in cancer: selected roles in tumour progression, immune modulation and metastasis. Cellular Immunology, 333, 46–57. 10.1016/j.cellimm.2018.03.007 [DOI] [PubMed] [Google Scholar]
- 21.Dongre A, & Weinberg RA (2019). New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nature Reviews Molecular Cell Biology, 20(2), 69–84. 10.1038/s41580-018-0080-4 [DOI] [PubMed] [Google Scholar]
- 22.Varki A, Kannagi R, Toole B, & Stanley P (2017). Glycosylation changes in cancer. In Varki A, Cummings RD, Esko JD, et al. (eds.), Essentials of Glycobiology 3rd ed. Cold Spring Harbor Laboratory Press. 10.1101/glycobiology.3e.047 [DOI] [PubMed] [Google Scholar]
- 23.Ho WL, Hsu WM, Huang MC, Kadomatsu K, & Nakagawara A (2016). Protein glycosylation in cancers and its potential therapeutic applications in neuroblastoma. Journal of Hematology & Oncology, 9(1), 100. 10.1186/s13045-016-0334-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodrigues E, & Macauley MS (2018). Hypersialylation in cancer: modulation of inflammation and therapeutic opportunities. Cancers (Basel), 10(6). 10.3390/cancers10060207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cox TR (2021). The matrix in cancer. Nature Reviews Cancer, 21(4), 217–238. 10.1038/s41568-020-00329-7 [DOI] [PubMed] [Google Scholar]
- 26.Ku AW, Muhitch JB, Powers CA, Diehl M, Kim M, Fisher DT, Sharda AP, Clements VK, OĽoughlin K, Minderman H, Messmer MN, Ma J, Skitzki JJ, Steeber DA, Walcheck B, Ostrand-Rosenberg S, Abrams SI, & Evans SS (2016). Tumor-induced MDSC act via remote control to inhibit L-selectin-dependent adaptive immunity in lymph nodes. eLife, 5. 10.7554/elife.17375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dobie C, & Skropeta D (2021). Insights into the role of sialylation in cancer progression and metastasis. British Journal of Cancer, 124(1), 76–90. 10.1038/s41416-020-01126-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu J, Isaji T, Im S, Fukuda T, Hashii N, Takakura D, Kawasaki N, & Gu J (2014). β-galactoside α2,6-sialyltranferase 1 promotes transforming growth factor-β-mediated epithelial-mesenchymal transition. Journal of Biological Chemistry, 289(50), 34627–34641. 10.1074/jbc.m114.593392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Do Nascimento JCF, Beltrão EIC, & Rocha CRC (2020). High FUT3 expression is a marker of lower overall survival of breast cancer patients. Glycoconjugate Journal, 37(2), 263–275. 10.1007/s10719-020-09914-2 [DOI] [PubMed] [Google Scholar]
- 30.Laubli H, & Borsig L (2019). Altered cell adhesion and glycosylation promote cancer immune suppression and metastasis. Front Immunol, 10, 2120. 10.3389/fimmu.2019.02120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen DX, Bos PD, & Massagué J (2009). Metastasis: from dissemination to organ-specific colonization. Nature Reviews Cancer, 9(4), 274–284. 10.1038/nrc2622 [DOI] [PubMed] [Google Scholar]
- 32.Peinado H, Zhang H, Matei IR, Costa-Silva B, Hoshino A, Rodrigues G, Psaila B, Kaplan RN, Bromberg JF, Kang Y, Bissell MJ, Cox TR, Giaccia AJ, Erler JT, Hiratsuka S, Ghajar CM, & Lyden D (2017). Premetastatic niches: Organ-specific homes for metastases. Nature Reviews Cancer, 17(5), 302–317. 10.1038/nrc.2017.6 [DOI] [PubMed] [Google Scholar]
- 33.Ewing J (1928). Neoplastic diseases: A treatise on tumours. British Journal of Surgery, 16(61), 174–175. 10.1002/bjs.1800166126 [DOI] [Google Scholar]
- 34.Paget S (1889). The distribution of secondary growths in cancer of the breast. The Lancet, 133(3421), 571–573. 10.1016/s0140-6736(00)49915-0 [DOI] [PubMed] [Google Scholar]
- 35.Moskal JR, Kroes RA, & Dawson G (2009). The glycobiology of brain tumors: Disease relevance and therapeutic potential. Expert Review of Neurotherapeutics, 9(10), 1529–1545. 10.1586/ern.09.105 [DOI] [PubMed] [Google Scholar]
- 36.Veillon L, Fakih C, Abou-El-Hassan H, Kobeissy F, & Mechref Y (2018). Glycosylation Changes in Brain Cancer. ACS Chem Neurosci, 9(1), 51–72. 10.1021/acschemneuro.7b00271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kolbl AC, Andergassen U, & Jeschke U (2015). The Role of Glycosylation in Breast Cancer Metastasis and Cancer Control. Front Oncol, 5, 219. 10.3389/fonc.2015.00219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jia L, Zhang J, Ma T, Guo Y, Yu Y, & Cui J (2018). The Function of Fucosylation in Progression of Lung Cancer. Frontiers in Oncology, 8. 10.3389/fonc.2018.00565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang S, Xia J, Yang Z, Xu M, & Li S (2021). Lung cancer molecular mutations and abnormal glycosylation as biomarkers for early diagnosis. Cancer Treat Res Commun, 27, 100311. 10.1016/j.ctarc.2021.100311 [DOI] [PubMed] [Google Scholar]
- 40.Munkley J (2019). The glycosylation landscape of pancreatic cancer. Oncol Lett, 17(3), 2569–2575. 10.3892/ol.2019.9885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Samaržija I (2021). Post-Translational Modifications That Drive Prostate Cancer Progression. Biomolecules, 11(2). 10.3390/biom11020247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mundy GR (2002). Metastasis to bone: Causes, consequences and therapeutic opportunities. Nature Reviews Cancer, 2(8), 584–593. 10.1038/nrc867 [DOI] [PubMed] [Google Scholar]
- 43.Freire T, Berois N, Sóñora C, Varangot M, Barrios E, & Osinaga E (2006). UDP-N-acetyl-D-galactosamine:polypeptideN-acetylgalactosaminyltransferase 6 (ppGalNAc-T6) mRNA as a potential new marker for detection of bone marrow-disseminated breast cancer cells. International Journal of Cancer, 119(6), 1383–1388. 10.1002/ijc.21959 [DOI] [PubMed] [Google Scholar]
- 44.Zepp M, Kovacheva M, Altankhuyag M, Westphal G, Berger I, Gather KS, Hilbig H, Neuhaus J, Hänsch GM, Armbruster FP, & Berger MR (2018). IDK1 is a rat monoclonal antibody against hypoglycosylated bone sialoprotein with application as biomarker and therapeutic agent in breast cancer skeletal metastasis. The Journal of Pathology: Clinical Research, 4(1), 55–68. 10.1002/cjp2.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang Y, Macleod V, Bendre M, Huang Y, Theus AM, Miao H-Q, Kussie P, Yaccoby S, Epstein J, Suva LJ, Kelly T, & Sanderson RD (2005). Heparanase promotes the spontaneous metastasis of myeloma cells to bone. Blood, 105(3), 1303–1309. 10.1182/blood-2004-06-2141 [DOI] [PubMed] [Google Scholar]
- 46.Kelly T, Suva LJ, Huang Y, Macleod V, Miao H-Q, Walker RC, & Sanderson RD (2005). Expression of heparanase by primary breast tumors promotes bone resorption in the absence of detectable bone metastases. Cancer Research, 65(13), 5778–5784. 10.1158/0008-5472.can-05-0749 [DOI] [PubMed] [Google Scholar]
- 47.Van S (2009). AsialoGM1 and integrin α2β1 mediate prostate cancer progression. International Journal of Oncology, 35(4). 10.3892/ijo_00000381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Slambrouck S, Groux-Degroote S, Krzewinski-Recchi M-A, Cazet A, Delannoy P, & Wim (2014). Carbohydrate-to-carbohydrate interactions between α2,3-linked sialic acids on α2 integrin subunits and asialo-GM1 underlie the bone metastatic behaviour of LNCAP-derivative C4–2B prostate cancer cells. Bioscience Reports, 34(5), 546–557. 10.1042/bsr20140096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujita R, Hamano H, Kameda Y, Arai R, Shimizu T, Ota M, Sato D, Kobayashi H, Iwasaki N, & Takahata M (2019). Breast cancer cells expressing cancer-associated sialyl-Tn antigen have less capacity to develop osteolytic lesions in a mouse model of skeletal colonization. Clinical & Experimental Metastasis, 36(6), 539–549. 10.1007/s10585-019-09999-6 [DOI] [PubMed] [Google Scholar]
- 50.Nakahara S, & Raz A (2008). Biological modulation by lectins and their ligands in tumor progression and metastasis. Anticancer Agents Medicinal Chemistry, 8(1), 22–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Biancone L (1996). Redirection of tumor metastasis by expression of E-selectin in vivo. Journal of Experimental Medicine, 183(2), 581–587. 10.1084/jem.183.2.581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weston BW, Hiller KM, Mayben JP, Manousos GA, Bendt KM, Liu R, & Cusack JC (1999). Expression of human α(1,3)fucosyltransferase antisense sequences inhibits selectin-mediated adhesion and liver metastasis of colon carcinoma cells. Cancer Research, 59(9), 2127–2135. [PubMed] [Google Scholar]
- 53.Yamada N, Chung YS, Takatsuka S, Arimoto Y, Sawada T, Dohi T, & Sowa M (1997). Increased sialyl Lewis A expression and fucosyltransferase activity with acquisition of a high metastatic capacity in a colon cancer cell line. British Journal of Cancer, 76(5), 582–587. 10.1038/bjc.1997.429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barthel SR, Gavino JD, Wiese GK, Jaynes JM, Siddiqui J, & Dimitroff CJ (2008). Analysis of glycosyltransferase expression in metastatic prostate cancer cells capable of rolling activity on microvascular endothelial (E)-selectin. Glycobiology, 18(10), 806–817. 10.1093/glycob/cwn070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Julien S, Ivetic A, Grigoriadis A, Qize D, Burford B, Sproviero D, Picco G, Gillett C, Papp SL, Schaffer L, Tutt A, Taylor-Papadimitriou J, Pinder SE, & Burchell JM (2011). Selectin ligand Sialyl-Lewis x antigen drives metastasis of hormone-dependent breast cancers. Cancer Research, 71(24), 7683–7693. 10.1158/0008-5472.can-11-1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Price TT, Burness ML, Sivan A, Warner MJ, Cheng R, Lee CH, Olivere L, Comatas K, Magnani J, Kim Lyerly H, Cheng Q, McCall CM, & Sipkins DA (2016). Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Science Translational Medicine, 8(340), 340ra73–340ra73. 10.1126/scitranslmed.aad4059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Esposito M, Mondal N, Greco TM, Wei Y, Spadazzi C, Lin S-C, Zheng H, Cheung C, Magnani JL, Lin S-H, Cristea IM, Sackstein R, & Kang Y (2019). Bone vascular niche E-selectin induces mesenchymal–epithelial transition and Wnt activation in cancer cells to promote bone metastasis. Nature Cell Biology, 21(5), 627–639. 10.1038/s41556-019-0309-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tyagi A, Sharma S, Wu K, Wu S-Y, Xing F, Liu Y, Zhao D, Deshpande RP, D’Agostino RB, & Watabe K (2021). Nicotine promotes breast cancer metastasis by stimulating N2 neutrophils and generating pre-metastatic niche in lung. Nature Communications, 12(1). 10.1038/s41467-020-20733-9 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Ichikawa J, Ando T, Kawasaki T, Sasaki T, Shirai T, Tsukiji N, Kimura Y, Aoki K, Hayakawa K, Suzuki-Inoue K, Saitoh M, & Haro H (2020). Role of platelet C-type lectin-like receptor 2 in promoting lung metastasis in osteosarcoma. Journal of Bone and Mineral Research, 35(9), 1738–1750. 10.1002/jbmr.4045 [DOI] [PubMed] [Google Scholar]
- 60.Oskarsson T, Acharyya S, Zhang XHF, Vanharanta S, Tavazoie SF, Morris PG, Downey RJ, Manova-Todorova K, Brogi E, & Massagué J (2011). Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nature Medicine, 17(7), 867–874. 10.1038/nm.2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang Q, Liu W, Fan J, Guo J, Shen F, Ma Z, Ruan C, Guo L, Jiang M, & Zhao Y (2020). von Willebrand factor promotes platelet-induced metastasis of osteosarcoma through activation of the VWF-GPIb axis. Journal of Bone Oncology, 25, 100325–100325. 10.1016/j.jbo.2020.100325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang S, Zhong L, Li Y, Xiao D, Zhang R, Liao D, Lv D, Wang X, Wang J, Xie X, Chen J, Wu Y, & Kang T (2019). Up-regulation of PCOLCE by TWIST1 promotes metastasis in osteosarcoma. Theranostics, 9(15), 4342–4353. 10.7150/thno.34090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dennis JW, Laferte S, Waghorne C, Breitman ML, & Kerbel RS (1987). Beta 1–6 branching of Asn-linked oligosaccharides is directly associated with metastasis. Science, 236, 582.+. 10.1126/science.2953071 [DOI] [PubMed] [Google Scholar]
- 64.Hiraizumi S, Takasaki S, Ohuchi N, Harada Y, Nose M, Mori S, & Kobata A (1992). Altered glycosylation of membrane glycoproteins associated with human mammary carcinoma. Japanese journal of cancer research : Gann, 83(10), 1063–1072. 10.1111/j.1349-7006.1992.tb02723.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fidler IJ, & Nicolson GL (1976). Organ selectivity for implantation survival and growth of B16 melanoma variant tumor lines. Journal of the National Cancer Institute, 57(5), 1199–1202. 10.1093/jnci/57.5.1199 [DOI] [PubMed] [Google Scholar]
- 66.Fidler IJ, & Nicolson GL (1977). Fate of recirculating B16 melanoma metastatic variant cells in parabiotic syngeneic recipients: Brief communication. Journal of the National Cancer Institute, 58(6), 1867–1872. 10.1093/jnci/58.6.1867 [DOI] [PubMed] [Google Scholar]
- 67.Krishnan V, Bane SM, Kawle PD, Naresh KN, & Kalraiya RD (2005). Altered melanoma cell surface glycosylation mediates organ specific adhesion and metastasis via lectin receptors on the lung vascular endothelium. Clinical & Experimental Metastasis, 22(1), 11–24. 10.1007/s10585-005-2036-2 [DOI] [PubMed] [Google Scholar]
- 68.Srinivasan N, Bane SM, Ahire SD, Ingle AD, & Kalraiya RD (2009). Poly N-acetyllactosamine substitutions on N- and not O-oligosaccharides or Thomsen–Friedenreich antigen facilitate lung specific metastasis of melanoma cells via galectin-3. Glycoconjugate Journal, 26(4), 445–456. 10.1007/s10719-008-9194-9 [DOI] [PubMed] [Google Scholar]
- 69.Brown JR (2006). A disaccharide-based inhibitor of glycosylation attenuates metastatic tumor cell dissemination. Clinical Cancer Research, 12(9), 2894–2901. 10.1158/1078-0432.ccr-05-2745 [DOI] [PubMed] [Google Scholar]
- 70.Jiang M, Xu X, Bi Y, Xu J, Qin C, & Han M (2014). Systemic inflammation promotes lung metastasis via E-selectin upregulation in mouse breast cancer model. Cancer Biology & Therapy, 15(6), 789–796. 10.4161/cbt.28552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.More SK, Srinivasan N, Budnar S, Bane SM, Upadhya A, Thorat RA, Ingle AD, Chiplunkar SV, & Kalraiya RD (2015). N-glycans and metastasis in galectin-3 transgenic mice. Biochemical and Biophysical Research Communications, 460(2), 302–307. 10.1016/j.bbrc.2015.03.030 [DOI] [PubMed] [Google Scholar]
- 72.Martín-Satué M, de Castellarnau C, & Blanco J (1999). Overexpression of a(1,3)-fucosyltransferase VII is sufficient for the acquisition of lung colonization phenotype by human lung adenocarcinoma Hal-24Luc cells. British Journal of Cancer, 80(8), 1169–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fuster MM, Brown JR, Wang L, & Esko JD (2003). A disaccharide precursor of sialyl lewis X inhibits metastatic potential of tumor cells. Cancer Research, 63, 2775–2781. [PubMed] [Google Scholar]
- 74.Büll C, Boltje TJ, Van Dinther EAW, Peters T, De Graaf AMA, Leusen JHW, Kreutz M, Figdor CG, Den Brok MH, & Adema GJ (2015). Targeted delivery of a sialic acid-blocking glycomimetic to cancer cells inhibits metastatic spread. ACS Nano, 9(1), 733–745. 10.1021/nn5061964 [DOI] [PubMed] [Google Scholar]
- 75.Song K-H, Park MS, Nandu TS, Gadad S, Kim S-C, & Kim M-Y (2016). GALNT14 promotes lung-specific breast cancer metastasis by modulating self-renewal and interaction with the lung microenvironment. Nature Communications, 7(1), 13796. 10.1038/ncomms13796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Leone JP, & Leone BA (2015). Breast cancer brain metastases: The last frontier. Experimental Hematology & Oncology, 4(1). 10.1186/s40164-015-0028-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kroes RA, Dawson G, & Moskal JR (2007). Focused microarray analysis of glyco-gene expression in human glioblastomas. Journal of Neurochemistry, 103(s1), 14–24. 10.1111/j.1471-4159.2007.04780.x [DOI] [PubMed] [Google Scholar]
- 78.Münsterberg J, Loreth D, Brylka L, Werner S, Karbanova J, Gandrass M, Schneegans S, Besler K, Hamester F, Robador JR, Bauer AT, Schneider SW, Wrage M, Lamszus K, Matschke J, Vashist Y, Uzunoglu G, Steurer S, Horst AK, et al. (2020). ALCAM contributes to brain metastasis formation in non-small-cell lung cancer through interaction with the vascular endothelium. Neuro-Oncology, 22(7), 955–966. 10.1093/neuonc/noaa028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gong X, Hou Z, Endsley MP, Gronseth EI, Rarick KR, Jorns JM, Yang Q, Du Z, Yan K, Bordas ML, Gershan J, Deepak P, Geethadevi A, Chaluvally-Raghavan P, Fan Y, Harder DR, Ramchandran R, & Wang L (2019). Interaction of tumor cells and astrocytes promotes breast cancer brain metastases through TGF-β2/ANGPTL4 axes. npj Precision Oncology, 3(1). 10.1038/s41698-019-0094-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Valiente M, Anna, Jin X, Chen Q, Xiang, Derek, Jamie, Mark, Jason, Brogi E, & Massagué J (2014). Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell, 156(5), 1002–1016. 10.1016/j.cell.2014.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Klotz R, Thomas A, Teng T, Han SM, Iriondo O, Li L, Restrepo-Vassalli S, Wang A, Izadian N, Mackay M, Moon B-S, Liu KJ, Ganesan SK, Lee G, Kang DS, Walmsley CS, Pinto C, Press MF, Lu W, et al. (2020). Circulating tumor cells exhibit metastatic tropism and reveal brain metastasis drivers. Cancer Discovery, 10(1), 86–103. 10.1158/2159-8290.cd-19-0384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Klotz R, & Yu M (2020). Insights into brain metastasis: Recent advances in circulating tumor cell research. Cancer Reports. 10.1002/cnr2.1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pangeni RP, Channathodiyil P, Huen DS, Eagles LW, Johal BK, Pasha D, Hadjistephanou N, Nevell O, Davies CL, Adewumi AI, Khanom H, Samra IS, Buzatto VC, Chandrasekaran P, Shinawi T, Dawson TP, Ashton KM, Davis C, Brodbelt AR, et al. (2015). The GALNT9, BNC1 and CCDC8 genes are frequently epigenetically dysregulated in breast tumours that metastasise to the brain. Clinical Epigenetics, 7(1). 10.1186/s13148-015-0089-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bos PD, Zhang XHF, Nadal C, Shu W, Gomis RR, Nguyen DX, Minn AJ, Van De Vijver MJ, Gerald WL, Foekens JA, & Massagué J (2009). Genes that mediate breast cancer metastasis to the brain. Nature, 459(7249), 1005–1009. 10.1038/nature08021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Drolez A, Vandenhaute E, Delannoy C, Dewald J, Gosselet F, Cecchelli R, Julien S, Dehouck M-P, Delannoy P, & Mysiorek C (2016). ST6GALNAC5 expression decreases the interactions between breast cancer cells and the human blood-brain barrier. International Journal of Molecular Sciences, 17(8), 1309. 10.3390/ijms17081309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rai S, Nejadhamzeeigilani Z, Gutowski NJ, & Whatmore JL (2015). Loss of the endothelial glycocalyx is associated with increased E-selectin mediated adhesion of lung tumour cells to the brain microvascular endothelium. Journal of Experimental & Clinical Cancer Research, 34(1). 10.1186/s13046-015-0223-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jassam SA, Maherally Z, Smith JR, Ashkan K, Roncaroli F, Fillmore HL, & Pilkington GJ (2016). TNF-α enhancement of CD62E mediates adhesion of non–small cell lung cancer cells to brain endothelium via CD15 in lung-brain metastasis. Neuro-Oncology, 18(5), 679–690. 10.1093/neuonc/nov248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Peng W, Mirzaei P, Zhu R, Zhou S, & Mechref Y (2020). Comparative membrane N-glycomics of different breast cancer cell lines to understand breast cancer brain metastasis. Journal of Proteome Research, 19(2), 854–863. 10.1021/acs.jproteome.9b00664 [DOI] [PubMed] [Google Scholar]
- 89.Peng W, Goli M, Mirzaei P, & Mechref Y (2019). Revealing the biological attributes of N-glycan isomers in breast cancer brain metastasis using porous graphitic carbon (PGC) liquid chromatography-tandem mass spectrometry (LC-MS/MS). Journal of Proteome Research, 18(10), 3731–3740. 10.1021/acs.jproteome.9b00429 [DOI] [PubMed] [Google Scholar]
- 90.Bailleux C, Eberst L, & Bachelot T (2021). Treatment strategies for breast cancer brain metastases. British Journal of Cancer, 124(1), 142–155. 10.1038/s41416-020-01175-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Drolez A, Vandenhaute E, Julien S, Gosselet F, Burchell J, Cecchelli R, Delannoy P, Dehouck M-P, & Mysiorek C (2016). Selection of a relevant in vitro blood-brain barrier model to investigate pro-metastatic features of human breast cancer cell lines. PLoS ONE, 11(3), e0151155. 10.1371/journal.pone.0151155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Finke JM, Ayres KR, Brisbin RP, Hill HA, Wing EE, & Banks WA (2017). Antibody blood-brain barrier efflux is modulated by glycan modification. Biochim Biophys Acta Gen Subj, 1861(9), 2228–2239. 10.1016/j.bbagen.2017.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jassam SA, Maherally Z, Ashkan K, Pilkington GJ, & Fillmore HL (2019). Fucosyltransferase 4 and 7 mediates adhesion of non-small cell lung cancer cells to brain-derived endothelial cells and results in modification of the blood–brain-barrier: In vitro investigation of CD15 and CD15s in lung-to-brain metastasis. Journal of Neuro-Oncology, 143(3), 405–415. 10.1007/s11060-019-03188-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fares J, Kanojia D, Rashidi A, Ulasov I, & Lesniak MS (2020). Genes that mediate metastasis across the blood–brain barrier. Trends in Cancer, 6(8), 660–676. 10.1016/j.trecan.2020.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.García-Martín AB, Zwicky P, Gruber T, Matti C, Moalli F, Stein JV, Francisco D, Enzmann G, Levesque MP, Hewer E, & Lyck R (2019). VLA-4 mediated adhesion of melanoma cells on the blood–brain barrier is the critical cue for melanoma cell intercalation and barrier disruption. Journal of Cerebral Blood Flow & Metabolism, 39(10), 1995–2010. 10.1177/0271678x18775887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Li XQ, Lu JT, Tan CC, Wang QS, & Feng YM (2016). RUNX2 promotes breast cancer bone metastasis by increasing integrin alpha5-mediated colonization. Cancer Letters, 380(1), 78–86. 10.1016/j.canlet.2016.06.007 [DOI] [PubMed] [Google Scholar]
- 97.Schneider JG, Amend SR, & Weilbaecher KN (2011). Integrins and bone metastasis: Integrating tumor cell and stromal cell interactions. Bone, 48(1), 54–65. 10.1016/j.bone.2010.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Peng W, Zhu R, Zhou S, Mirzaei P, & Mechref Y (2019). Integrated transcriptomics, proteomics, and glycomics reveals the association between up-regulation of sialylated N-glycans/integrin and breast cancer brain metastasis. Scientific Reports, 9(1). 10.1038/s41598-019-53984-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hawkins CC, Ali T, Ramanadham S, & Hjelmeland AB (2020). Sphingolipid metabolism in glioblastoma and metastatic brain tumors: a review of sphingomyelinases and sphingosine-1-phosphate. Biomolecules, 10(10), 1357. 10.3390/biom10101357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lv Y, Ma X, Du Y, & Feng J (2021). Understanding patterns of brain metastasis in triple-negative breast cancer and exploring potential therapeutic targets. OncoTargets and Therapy, Volume 14, 589–607. 10.2147/ott.s293685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ferraro GB, Ali A, Luengo A, Kodack DP, Deik A, Abbott KL, Bezwada D, Blanc L, Prideaux B, Jin X, Possada JM, Chen J, Chin CR, Amoozgar Z, Ferreira R, Chen IX, Naxerova K, Ng C, Westermark AM, et al. (2021). Fatty acid synthesis is required for breast cancer brain metastasis. Nature Cancer. 10.1038/s43018-021-00183-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jin X, Demere Z, Nair K, Ali A, Ferraro GB, Natoli T, Deik A, Petronio L, Tang AA, Zhu C, Wang L, Rosenberg D, Mangena V, Roth J, Chung K, Jain RK, Clish CB, Vander Heiden MG, & Golub TR (2020). A metastasis map of human cancer cell lines. Nature, 588(7837), 331–336. 10.1038/s41586-020-2969-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Adham SAI, Al Rawahi H, Habib S, Al Moundhri MS, Viloria-Petit A, & Coomber BL (2014). Modeling of hypo/hyperglycemia and their impact on breast cancer progression related molecules. PLoS ONE, 9(11), e113103. 10.1371/journal.pone.0113103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Masmudi-Martín M, Zhu L, Sanchez-Navarro M, Priego N, Casanova-Acebes M, Ruiz-Rodado V, Giralt E, & Valiente M (2021). Brain metastasis models: What should we aim to achieve better treatments? Advanced Drug Delivery Reviews, 169, 79–99. 10.1016/j.addr.2020.12.002 [DOI] [PubMed] [Google Scholar]
- 105.Cummings RD, & McEver RP (2017). C-type lectins. In Varki A, Cummings RD, Esko JD, et al. (eds.), Essentials of Glycobiology 3rd ed. Cold Spring Harbor Laboratory Press. 10.1101/glycobiology.3e.034 [DOI] [PubMed] [Google Scholar]
- 106.Ding D, Yao Y, Zhang S, Su C, & Zhang Y (2017). C-type lectins facilitate tumor metastasis. Oncology Letters, 13(1), 13–21. 10.3892/ol.2016.5431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Brooks SA, & Leathem AJ (1991). Prediction of lymph node involvement in breast cancer by detection of altered glycosylation in the primary tumour. Lancet, 338(8759), 71–4. 10.1016/0140-6736(91)90071-v [DOI] [PubMed] [Google Scholar]
- 108.Handerson T, Camp R, Harigopal M, Rimm D, & Pawelek J (2005). Beta1,6-branched oligosaccharides are increased in lymph node metastases and predict poor outcome in breast carcinoma. Clin Cancer Res, 11(8), 2969–73. 10.1158/1078-0432.Ccr-04-2211 [DOI] [PubMed] [Google Scholar]
- 109.Herrera H, Dilday T, Uber A, Scott D, Zambrano JN, Wang M, Angel PM, Mehta AS, Drake RR, Hill EG, & Yeh ES (2019). Core-Fucosylated Tetra-Antennary N-Glycan Containing A Single N-Acetyllactosamine Branch Is Associated with Poor Survival Outcome in Breast Cancer. Int J Mol Sci, 20(10). 10.3390/ijms20102528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Oliveira-Ferrer L, Milde-Langosch K, Eylmann K, Rossberg M, Müller V, Schmalfeldt B, Witzel I, Wellbrock J, & Fiedler W (2020). Mechanisms of Tumor-Lymphatic Interactions in Invasive Breast and Prostate Carcinoma. Int J Mol Sci, 21(2). 10.3390/ijms21020602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jiang K, Gao Y, Hou W, Tian F, Ying W, Li L, Bai B, Hou G, Wang PG, & Zhang L (2016). Proteomic analysis of O-GlcNAcylated proteins in invasive ductal breast carcinomas with and without lymph node metastasis. Amino Acids, 48(2), 365–74. 10.1007/s00726-015-2089-8 [DOI] [PubMed] [Google Scholar]
- 112.Mittal P, Briggs M, Klingler-Hoffmann M, Kaur G, Packer NH, Oehler MK, & Hoffmann P (2021). Altered N-linked glycosylation in endometrial cancer. Anal Bioanal Chem, 413(10), 2721–2733. 10.1007/s00216-020-03039-z [DOI] [PubMed] [Google Scholar]
- 113.Shiga K, Takahashi K, Sato I, Kato K, Saijo S, Moriya S, Hosono M, & Miyagi T (2015). Upregulation of sialidase NEU3 in head and neck squamous cell carcinoma associated with lymph node metastasis. Cancer Sci, 106(11), 1544–53. 10.1111/cas.12810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen Y, Su L, Huang C, Wu S, Qiu X, Zhao X, Meng Q, Meng YM, Kong X, Wang M, Liu C, & Wong PP (2021). Galactosyltransferase B4GALT1 confers chemoresistance in pancreatic ductal adenocarcinomas by upregulating N-linked glycosylation of CDK11(p110). Cancer Lett, 500, 228–243. 10.1016/j.canlet.2020.12.006 [DOI] [PubMed] [Google Scholar]
- 115.Wang Y, Li Q, Niu L, Xu L, Guo Y, Wang L, & Guo C (2020). Suppression of G6PD induces the expression and bisecting GlcNAc-branched N-glycosylation of E-Cadherin to block epithelial-mesenchymal transition and lymphatic metastasis. Br J Cancer, 123(8), 1315–1325. 10.1038/s41416-020-1007-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Huang H, Liu Y, Yu P, Qu J, Guo Y, Li W, Wang S, & Zhang J (2018). MiR-23a transcriptional activated by Runx2 increases metastatic potential of mouse hepatoma cell via directly targeting Mgat3. Sci Rep, 8(1), 7366. 10.1038/s41598-018-25768-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gu Y, Liu Y, Fu L, Zhai L, Zhu J, Han Y, Jiang Y, Zhang Y, Zhang P, Jiang Z, Zhang X, & Cao X (2019). Tumor-educated B cells selectively promote breast cancer lymph node metastasis by HSPA4-targeting IgG. Nat Med, 25(2), 312–322. 10.1038/s41591-018-0309-y [DOI] [PubMed] [Google Scholar]
- 118.Zlotnik A, Burkhardt AM, & Homey B (2011). Homeostatic chemokine receptors and organ-specific metastasis. Nat Rev Immunol, 11(9), 597–606. 10.1038/nri3049 [DOI] [PubMed] [Google Scholar]
- 119.Li S-S, Ip CKM, Tang MYH, Tang MKS, Tong Y, Zhang J, Hassan AA, Mak ASC, Yung S, Chan T-M, Ip PP, Lee CL, Chiu PCN, Lee LTO, Lai H-C, Zeng J-Z, Shum HC, & Wong AST (2019). Sialyl lewis x-P-selectin cascade mediates tumor–mesothelial adhesion in ascitic fluid shear flow. Nature Communications, 10(1). 10.1038/s41467-019-10334-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hänel L, Gosau T, Maar H, Valentiner U, Schumacher U, Riecken K, Windhorst S, Hansen NO, Heikaus L, Wurlitzer M, Nolte I, Schlüter H, & Lange T (2018). Differential Proteome Analysis of Human Neuroblastoma Xenograft Primary Tumors and Matched Spontaneous Distant Metastases. Sci Rep, 8(1), 13986. 10.1038/s41598-018-32236-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Reticker-Flynn NE, & Bhatia SN (2015). Aberrant glycosylation promotes lung cancer metastasis through adhesion to galectins in the metastatic niche. Cancer Discovery, 5(2), 168–181. 10.1158/2159-8290.cd-13-0760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pang X, Li H, Guan F, & Li X (2018). Multiple roles of glycans in hematological malignancies. Frontiers in Oncology, 8. 10.3389/fonc.2018.00364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yoshimura M, Ihara Y, Ohnishi A, Ijuhin N, Nishiura T, Kanakura Y, Matsuzawa Y, & Taniguchi N (1996). Bisecting N-acetylglucosamine on K562 cells suppresses natural killer cytotoxicity and promotes spleen colonization. Cancer Research, 56(2), 412–418. [PubMed] [Google Scholar]
- 124.Natoni A, Bohara R, Pandit A, & O’Dwyer M (2019). Targeted approaches to inhibit sialylation of multiple myeloma in the bone marrow microenvironment. Frontiers in Bioengineering and Biotechnology, 7. 10.3389/fbioe.2019.00252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Glavey SV, Manier S, Natoni A, Sacco A, Moschetta M, Reagan MR, Murillo LS, Sahin I, Wu P, Mishima Y, Zhang Y, Zhang W, Zhang Y, Morgan G, Joshi L, Roccaro AM, Ghobrial IM, & O’Dwyer ME (2014). The sialyltransferase ST3GAL6 influences homing and survival in multiple myeloma. Blood, 124(11), 1765–1776. 10.1182/blood-2014-03-560862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Natoni A, Farrell ML, Harris S, Falank C, Kirkham-Mccarthy L, Macauley MS, Reagan MR, & O’Dwyer M (2020). Sialyltransferase inhibition leads to inhibition of tumor cell interactions with E-selectin, VCAM1, and MADCAM1, and improves survival in a human multiple myeloma mouse model. Haematologica, 105(2), 457–467. 10.3324/haematol.2018.212266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Peixoto A, Cotton S, Santos LL, & Ferreira JA (2021). The Tumour Microenvironment and Circulating Tumour Cells: A Partnership Driving Metastasis and Glycan-Based Opportunities for Cancer Control. Adv Exp Med Biol, 1329, 1–33. 10.1007/978-3-030-73119-9_1 [DOI] [PubMed] [Google Scholar]
- 128.Fabricius H, Starzonek S, & Lange T (2021). The Role of Platelet Cell Surface P-Selectin for the Direct Platelet-Tumor Cell Contact During Metastasis Formation in Human Tumors. Front Oncol, 11, 642761. 10.3389/fonc.2021.642761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Geng Y, Marshall JR, & King MR (2012). Glycomechanics of the metastatic cascade: tumor cell-endothelial cell interactions in the circulation. Ann Biomed Eng, 40(4), 790–805. 10.1007/s10439-011-0463-6 [DOI] [PubMed] [Google Scholar]
- 130.Geng Y, Takatani T, Yeh K, Hsu JW, & King MR (2013). Targeting Underglycosylated MUC1 for the Selective Capture of Highly Metastatic Breast Cancer Cells Under Flow. Cell Mol Bioeng, 6(2), 148–159. 10.1007/s12195-013-0282-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Martinez P, Vergoten G, Colomb F, Bobowski M, Steenackers A, Carpentier M, Allain F, Delannoy P, & Julien S (2013). Over-sulfated glycosaminoglycans are alternative selectin ligands: insights into molecular interactions and possible role in breast cancer metastasis. Clin Exp Metastasis, 30(7), 919–31. 10.1007/s10585-013-9592-7 [DOI] [PubMed] [Google Scholar]
- 132.Shirure VS, Liu T, Delgadillo LF, Cuckler CM, Tees DF, Benencia F, Goetz DJ, & Burdick MM (2015). CD44 variant isoforms expressed by breast cancer cells are functional E-selectin ligands under flow conditions. Am J Physiol Cell Physiol, 308(1), C68–78. 10.1152/ajpcell.00094.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mensah SA, Nersesyan AA, Harding IC, Lee CI, Tan X, Banerjee S, Niedre M, Torchilin VP, & Ebong EE (2020). Flow-regulated endothelial glycocalyx determines metastatic cancer cell activity. Faseb j, 34(5), 6166–6184. 10.1096/fj.201901920R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Costa AF, Campos D, Reis CA, & Gomes C (2020). Targeting glycosylation: A new road for cancer drug discovery. Trends in Cancer. 10.1016/j.trecan.2020.04.002 [DOI] [PubMed] [Google Scholar]
- 135.Mereiter S, Balmana M, Campos D, Gomes J, & Reis CA (2019). Glycosylation in the era of cancer-targeted therapy: Where are we heading? Cancer Cell, 36(1), 6–16. 10.1016/j.ccell.2019.06.006 [DOI] [PubMed] [Google Scholar]
- 136.Liu H, Ma L, Lin J, Cao B, Qu D, Luo C, Huang W, Han L, Xu H, Wu Z, Xu R, & Zhang D (2020). Advances in molecular mechanisms of drugs affecting abnormal glycosylation and metastasis of breast cancer. Pharmacological Research, 155, 104738. 10.1016/j.phrs.2020.104738 [DOI] [PubMed] [Google Scholar]
- 137.Rillahan CD, Antonopoulos A, Lefort CT, Sonon R, Azadi P, Ley K, Dell A, Haslam SM, & Paulson JC (2012). Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nature Chemical Biology, 8(7), 661–668. 10.1038/nchembio.999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hsu C-C, Lin T-W, Chang W-W, Wu C-Y, Lo W-H, Wang P-H, & Tsai Y-C (2005). Soyasaponin-I-modified invasive behavior of cancer by changing cell surface sialic acids. Gynecologic Oncology, 96(2), 415–422. 10.1016/j.ygyno.2004.10.010 [DOI] [PubMed] [Google Scholar]
- 139.Chiang C-H, Wang C-H, Chang H-C, More SV, Li W-S, & Hung W-C (2010). A novel sialyltransferase inhibitor AL10 suppresses invasion and metastasis of lung cancer cells by inhibiting integrin-mediated signaling. Journal of Cellular Physiology, n/a-n/a. 10.1002/jcp.22068 [DOI] [PubMed] [Google Scholar]
- 140.Song L, & Linstedt AD (2017). Inhibitor of ppGalNAc-T3-mediated O-glycosylation blocks cancer cell invasiveness and lowers FGF23 levels. eLife, 6. 10.7554/elife.24051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zeichner SB (2012). The failed Theratope vaccine: 10 years later. The Journal of the American Osteopathic Association, 112(8), 482–483. 10.7556/jaoa.2012.112.8.482 [DOI] [PubMed] [Google Scholar]
- 142.Smith BAH, & Bertozzi CR (2021). The clinical impact of glycobiology: targeting selectins, Siglecs and mammalian glycans. Nature Reviews Drug Discovery. 10.1038/s41573-020-00093-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sait S, & Modak S (2017). Anti-GD2 immunotherapy for neuroblastoma. Expert Review of Anticancer Therapy, 17(10), 889–904. 10.1080/14737140.2017.1364995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kölbl AC, Hiller RA, Ilmer M, Liesche F, Heublein S, Schröder L, Hutter S, Friese K, Jeschke U, & Andergassen U (2015). Glycosyltransferases as marker genes for the quantitative polymerase chain reaction-based detection of circulating tumour cells from blood samples of patients with breast cancer undergoing adjuvant therapy. Mol Med Rep, 12(2), 2933–8. 10.3892/mmr.2015.3732 [DOI] [PubMed] [Google Scholar]
- 145.Neves M, Azevedo R, Lima L, Oliveira MI, Peixoto A, Ferreira D, Soares J, Fernandes E, Gaiteiro C, Palmeira C, Cotton S, Mereiter S, Campos D, Afonso LP, Ribeiro R, Fraga A, Tavares A, Mansinho H, Monteiro E, et al. (2019). Exploring sialyl-Tn expression in microfluidic-isolated circulating tumour cells: A novel biomarker and an analytical tool for precision oncology applications. N Biotechnol, 49, 77–87. 10.1016/j.nbt.2018.09.004 [DOI] [PubMed] [Google Scholar]
- 146.Saldova R, Reuben JM, Abd Hamid UM, Rudd PM, & Cristofanilli M (2011). Levels of specific serum N-glycans identify breast cancer patients with higher circulating tumor cell counts. Ann Oncol, 22(5), 1113–1119. 10.1093/annonc/mdq570 [DOI] [PubMed] [Google Scholar]