

Abstract
Background:
Altered kinase localization is gaining appreciation as a mechanism of cardiovascular disease. Previous work suggests GSK-3β localizes to and regulates contractile function of the myofilament. We aimed to discover GSK-3β’s in vivo role in regulating myofilament function, the mechanisms involved, and the translational relevance.
Methods:
Inducible cardiomyocyte-specific GSK-3β knockout (KO) mice and LV myocardium from non-failing and failing human hearts were studied.
Results:
Skinned cardiomyocytes from KO mice failed to exhibit calcium sensitization with stretch indicating a loss of Length-Dependent Activation (LDA), the mechanism underlying the Frank Starling Law. Titin acts as a length sensor for LDA, and KO mice had decreased titin stiffness compared to Control mice, explaining the lack of LDA. KO mice exhibited no changes in titin isoforms, titin phosphorylation, or other thin filament phosphorylation sites known to affect passive tension or LDA. Mass spectrometry identified several z-disc proteins as myofilament phospho-substrates of GSK-3β. Agreeing with the localization of its targets, GSK-3β that is phosphorylated at Y216 binds to the z-disc. We showed pY216 was necessary and sufficient for z-disc binding using adenoviruses for wildtype (WT), Y216F, and Y216E GSK-3β in neonatal rat ventricular cardiomyocytes. One of GSK-3β’s z-disc targets, abLIM-1, binds to the z-disc domains of titin that are important for maintaining passive tension. Genetic knockdown of abLIM-1 via siRNA in human engineered heart tissues resulted in enhancement of LDA, indicating abLIM-1 may act as a negative regulator that is modulated by GSK-3β. Lastly, GSK-3β myofilament localization was reduced in LV myocardium from failing human hearts, which correlated with depressed LDA.
Conclusions:
We identified a novel mechanism by which GSK-3β localizes to the myofilament to modulate LDA. Importantly, z-disc GSK-3β levels were reduced in heart failure patients, indicating z-disc localized GSK-3β is a possible therapeutic target to restore the Frank-Starling mechanism in heart failure patients.
Subject Codes: Heart Failure, Contractile Function, Proteomics, Myocardial biology
Introduction
A major mechanism driving the pathobiology of cardiovascular disease involves dysregulation of kinases and is frequently observed as either aberrant activation or deactivation1–3. There has been increasing appreciation of another layer of complexity in kinase regulation involving subcellular pools of the enzyme whose activities are independently controlled4, 5. Further, these separate pools can have different phospho-targets and thus behave like functionally distinct kinases4. While these kinase pools can complicate attempts to alter enzyme activity therapeutically, they also provide an opportunity to alter function more selectively and avoid off-target effects. Traditional experimental approaches, however, may not detect dysregulation of small, localized pools of a kinase.
Glycogen synthase kinase 3β (GSK-3β) is a prolific cytosolic serine/threonine kinase. In the heart, GSK-3β acts as a negative regulator of hypertrophic signaling, preventing translocation of proteins to the nucleus that activate hypertrophic gene expression6–9. There is a lack of consensus on whether GSK-3β is protective or harmful in heart failure6, 10–12, possibly because there exists a subcellular pool of GSK-3β with unexplored actions. Indeed, we previously found that GSK-3β activity was depressed in a dog model of heart failure concurrent with cardiac dyssynchrony13, which typically arises from conduction abnormalities and results in premature activation of one region of the ventricular wall13. The observed decrease in GSK-3β activity correlated with depressed myofilament calcium sensitivity, which could be rescued in vitro with exposure to exogenous GSK-3β. Furthermore, GSK-3β was uncoupled from Akt, its canonical upstream de-activator, suggesting an independently regulated pool of GSK-3β13. However, there is no direct evidence that GSK-3β localizes to the sarcomere or regulates sarcomere function in vivo.
Changes in myofilament calcium sensitivity are frequently observed in heart failure due to altered phosphorylation of thin filament proteins14. However, dependent on the specific heart failure etiology15–17, comorbidities18 , and treatments19, myofilament calcium de-sensitization and over-sensitization are both observed. Unfortunately, both situations are detrimental, with de-sensitization resulting in hypo-contractility and worsening of the weakened heart while over-sensitization can cause arrhythmias and slowed relaxation15. In addition, calcium sensitivity is dynamically regulated in response to acute changes, such as stretch. Length-Dependent Activation (LDA) is the mechanism by which stretch increases calcium sensitivity in cardiomyocytes and underlies the organ level Frank-Starling Law20, 21 that allows the heart to respond to changes in blood volume on a beat-to-beat basis. Animal models of heart failure exhibit a depressed Frank-Starling response5, 22, however in humans this is less clear, as Frank-Starling/LDA has been found to be both diminished23–25 and unaffected26, 27 by heart failure.
It is unknown whether GSK-3β regulates myofilament function in vivo, whether it does so through a localized pool at the myofilament, the mechanisms involved, and whether this occurs in humans. In this study we directly address these questions using inducible cardiomyocyte-specific GSK-3β knockout mice, engineered heart tissue, and human left ventricular (LV) tissue from non-failing (NF) rejected donor hearts and heart failure patients. Confirming our hypothesis, ablation of GSK-3β in vivo results in reduction in LDA and interestingly, passive tension, a property of titin which has been established as a facilitator of LDA28. Exogenous treatment of skinned myocytes with recombinant GSK-3β can increase calcium sensitivity at long sarcomere lengths and increase passive tension. We also found that GSK-3β’s z-disc phosphorylation target, abLIM-1, binds to the z-disc region of titin (Z1Z2 domains) and modulates LDA. Importantly, in human heart failure GSK-3β mis-localizes away from the z-disc, which correlates with an absent LDA. GSK-3β localizes to the sarcomeric z-disc via its own phosphorylation at Y216, which reveals a novel mechanism for targeting this kinase to specific myofilament targets. Overall, this study uncovers a novel subcellular pool of GSK-3β that is critical for maintaining normal contractile function.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Study Approval
Human study protocols were approved by the IRB at the Cleveland Clinic and Loyola University Chicago. All patients gave informed consent. Animal studies were approved by the Loyola University Chicago Health Sciences Division and University of Cincinnati Institutional Animal Care and Use Committees.
Mice
Cardiomyocyte-specific, conditional GSK-3βflox/flox, α-MHC Mer Cre Mer (MCM) + mice6 (a kind gift from Dr. Thomas Force and rederived at Jackson Laboratories) and littermate controls, GSK-3βflox/flox α-MHC MCM -, were generated from several crosses of GSK-3βflox/flox and α-MHC Mer Cre Mer (MCM) strains, both on the C57BL/6J background. Mice were backcrossed with wild type C57BL/6J mice every 10–12 generations. Cardiac Myosin Binding Protein-C (cMyBP-C) null mice, cMyBP-Ct/t 29, 30 and Cardiomyocyte-specific cMyBP-CΔC0C1f, α-MHC mice31 were generated as described and characterized previously. Approximately equal numbers of male and female mice, age 12–14 weeks, were used in all experiments.
Mass Spectrometry
Left ventricle (LV) tissue from CON (n=4) and GSK-3β KO (n=5) mice were enriched for the myofilament, digested in trypsin, desalted, and phospho-enriched using titanium oxide beads. Samples were analyzed by LC-MS/MS using Data-Independent Acquisition (DIA) (Orbitrap Fusion Lumos Tribrid Mass Spectrometer). Data were processed using DIA-Umpire and then underwent Median-based normalization.
X-ray Diffraction
X-ray diffraction patterns were collected from freshly skinned mouse muscle strips using the small-angle instrument at BioCAT beamline 18ID at the Advanced Photon Source, Argonne National Laboratory32. Sarcomere length was adjusted by laser diffraction using a 4-mW HeNe laser. The data were analyzed using data reduction programs belonging to the MuscleX software package developed at BioCAT33, as described previously34.
Engineered Heart Tissues (EHTs)
EHTs were made by seeding iPSC-CMs and human adult cardiac fibroblasts onto decellularized porcine ventricular scaffolds as previously reported35. Two weeks after seeding, EHTs were treated with scrambled or abLIM-1 siRNA for 4 hours. 72 hours after treatment, passive and active mechanics of EHTs were measured as previously described36.
Statistics
Variables are expressed as mean ± standard error. Statistical significance was calculated in GraphPad Prism (version 9) by t-test, one-way analysis of variance, or two-way analysis of variance followed by the Tukey or Mann-Whitney post-hoc tests, as indicated. Normality (by Shapiro Wilk test) and equal variance (by F-test for t-tests or Brown-Forsythe test for ANOVAs) were tested prior to all parametric tests (Table S1). Correlations between categorical variables was performed with linear regression. P-values less than 0.05 were considered statistically significant. A priori power analyses were performed based on data from published studies13, 37, 38 and pilot experiments.
Further details are provided in the Supplemental Methods.
Results
GSK-3β modulates length-dependent activation in vivo
We utilized a cardiomyocyte-specific inducible GSK-3β knockout mouse to assess whether GSK-3β affects myofilament function in vivo. To evaluate knockdown efficiency, we prepared two sets of protein samples from the LV, those processed from whole tissue and those enriched for the myofilament (see Figure S1 for specificity of this enrichment)39. We detected significant GSK-3β in the myofilament-enriched samples from GSK-3βfl/fl/Cre- tamoxifen-treated control mice (CON, Figure 1A–C) suggesting there is a pool of GSK-3β which localizes to the myofilament. In the GSK-3βfl/fl/Cre+ tamoxifen-treated knockout mice (KO), there was an ~70% reduction in GSK-3β in both the whole tissue and myofilament-enriched samples (Figure 1A–C). At 12 weeks of age (used for all studies), there were no in vivo functional or structural differences between CON and KO mice as assessed by echocardiography (Table S2), except a mild hypertrophy in the anterior wall, consistent with GSK-3β’s role as an inhibitor of hypertrophy.
Figure 1. Genetic ablation of GSK-3β results in loss of length-dependent activation.
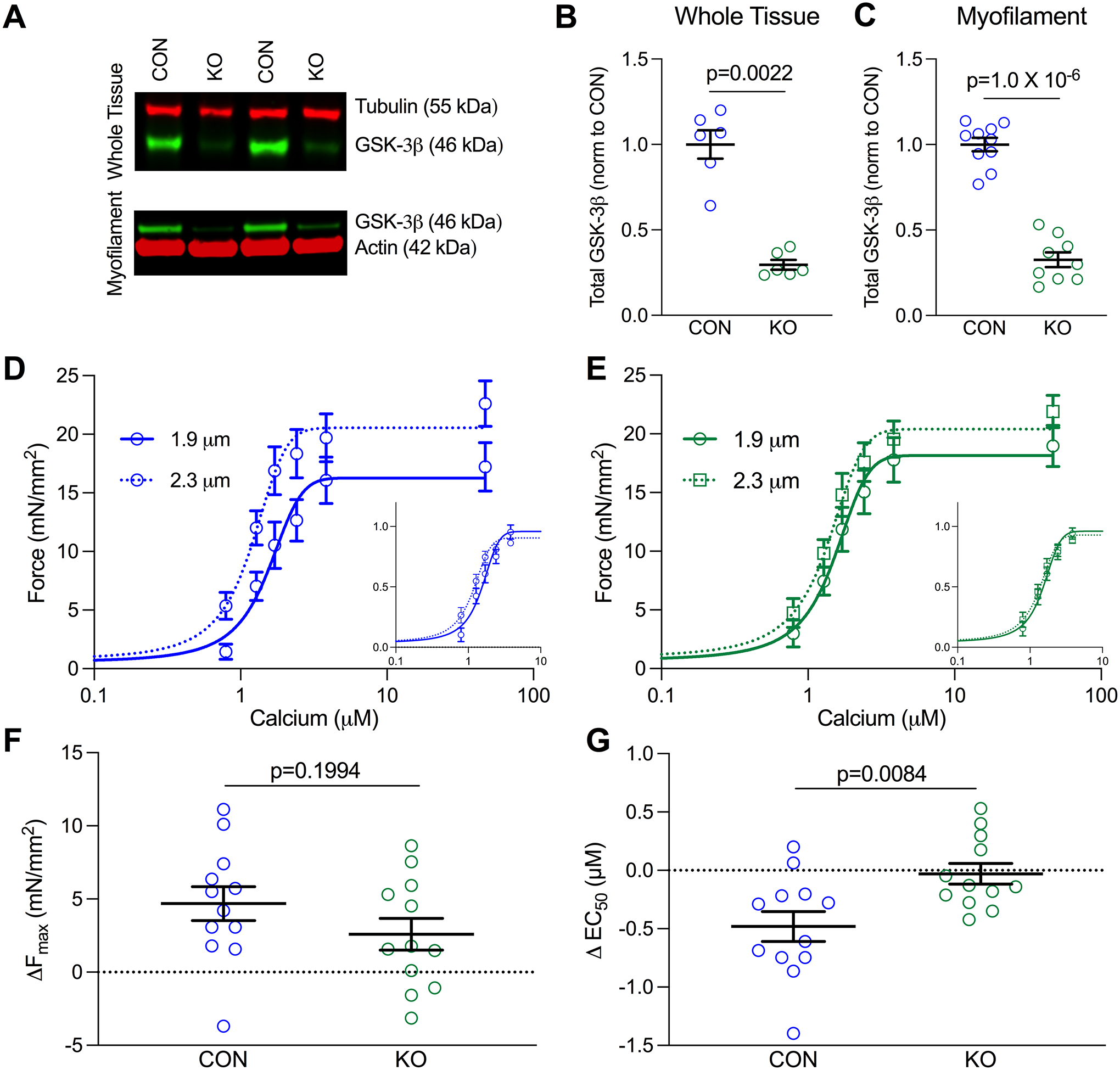
(A) Example western blots of GSK-3β in LV whole tissue lysis (top) and myofilament-enriched (bottom) samples from GSK-3β knockout (KO) and littermate control (CON) mice. (B) Quantification of GSK-3β in whole tissue (p=0.0022 n=6/group) and (C) myofilament-enriched (p=1.0 × 10−6, n-values: CON=10, KO = 9) samples. GSK-3β was normalized to either tubulin (whole tissue) or actin (myofilament) and further normalized to CON, shown as mean ± SEM. P-values were calculated by Mann-Whitney t-test for the whole tissue comparison. (D) Mean force as a function of calcium concentration and fitted curves for skinned myocytes from CON and (E) KO mice in which curves were measured at a sarcomere length (SL) of 1.9 μm and then 2.3 μm. Normalized curves are shown in the bottom right of each graph to emphasize shifts in calcium sensitivity. (F) Delta Fmax and (G) EC50 for CON and GSK-3β KO mice between SL 1.9 and 2.3 μm (p=0.0084 by unpaired T-test, n=12 myocytes from 4 CON and 3 KO mice).
We previously found decreased GSK-3β activity in a dog model of ventricular mechanical dyssynchrony13 that has out-of-phase stress-strain relationships. Thus, we hypothesized GSK-3β may be involved in a mechano-transduction pathway at the myofilament, one of the most critical being Length Dependent Activation (LDA)40. Thus, we performed force-calcium measurements at a sarcomere length (SL) of 1.9 μm (short) then stretched the myocyte to SL=2.3 μm (long) in skinned myocytes isolated from LV myocardium from CON and KO mice (n=4–5 mice/group, 2–4 cells/mouse) (Figure 1D–E). Stretching CON myocytes increased both Fmax and calcium sensitivity (p=6.4×10−4, p=4.97×10−4 respectively, via two-way repeated measures ANOVA, Figure 1F–G, Table S3), the expected effect of LDA. Conversely, myocytes from GSK-3β KO mice exhibited no significant change in either Fmax or EC50 with stretch. Additionally, there was no statistical difference in calcium sensitivity between the two groups at the short SL (p=0.33). Thus, GSK-3β is critical for the ability of the myofilament to adequately respond to stretch, and its genetic removal ablates LDA.
To ensure this was a direct effect of GSK-3β, we next tested whether exogenous GSK-3β would similarly enhance calcium sensitivity at long sarcomere lengths while having no impact at short sarcomere lengths. To avoid potential rundown from four subsequent activations, we performed paired pre- and post-GSK-3β treatments separately at short and long sarcomere lengths in CON and KO mice. Exogenous GSK-3β treatment (0.1 μg for 15 minutes) had no effect in myocytes at a short sarcomere length (n= 8 cells from 3 mice/group), but increased calcium sensitivity when the myocytes were stretched to a long sarcomere length (n= 11 myocytes from 4 mice; p=0.037 by paired t-test) (Figure S2). Interestingly, when analyzed by two-way ANOVA, there was no interaction between genotype and treatment, hence the data were analyzed by a paired t-test. This result indicates exogenous GSK-3β can enhance LDA in both CON and KO myocytes and suggests the functional impact of GSK-3β is not saturated in normal hearts.
GSK-3β is required for maintaining passive tension
We next sought to explore the mechanism of GSK-3β’s impact on LDA. We first showed by western blot that phosphorylation sites on cardiac troponin I (S23/24) and cardiac myosin binding protein-C (cMyBP-C) (S273, S282, S302) that can impact LDA41 are unchanged in the GSK-3β KO mice (Figure S3). There is evidence LDA is impacted by changes in interfilament spacing, which describes the distance between thick and thin filaments. When the sarcomere is stretched, interfilament lattice spacing decreases, promoting force generation42. Using synchrotron small angle x-ray diffraction, we measured interfilament lattice spacing (d10) in isolated, skinned papillary muscles in relaxing solution at both short and long SLs. Lattice spacing significantly decreased with stretch, but there were no statistical differences between GSK-3β CON and KO fibers (Figure 2A), eliminating it as a plausible mechanism.
Figure 2. GSK-3β KO mice have increased titin compliance.
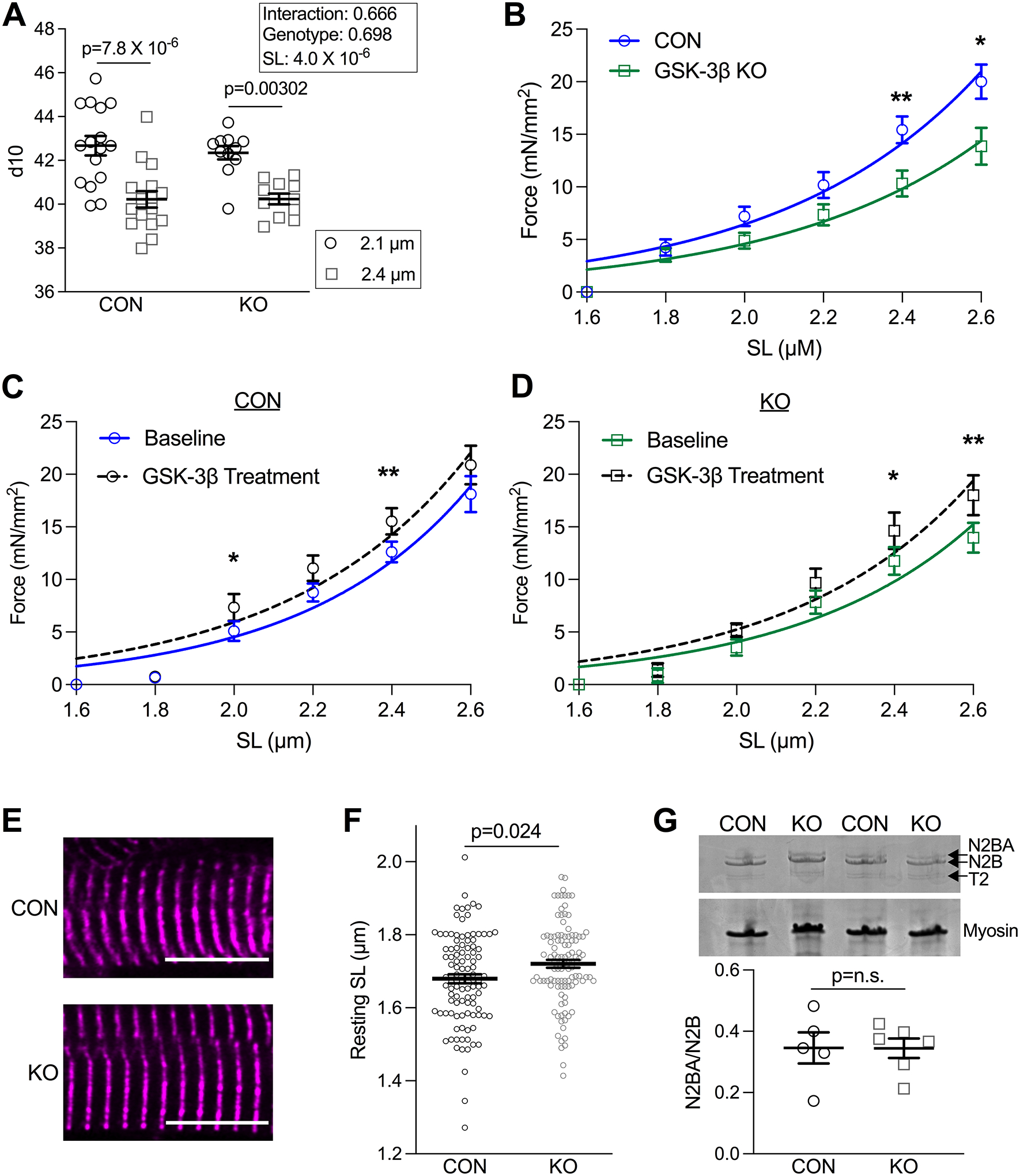
(A) Summary d10 (lattice spacing) collected by x-ray diffraction data from skinned papillary muscle fibers from CON (16 fibers from 6 animals) and GSK-3β KO mice (11 fibers from 4 animals). Fibers were measured at SL 2.1 and 2.4 μm. (effect of SL p=4.0 × 10−6 by repeated measures 2-way ANOVA with Sidak’s multiple pair-wise comparison test). (B) Passive tension measured in CON and GSK-3β KO mice at SLs from 1.6 to 2.6 μm. (p-values determined by unpaired parametric t-test, with the exception of data collected at 2.0, which was determined by Mann-Whitney T-test). Passive tension measured in (C) CON (n=3 mice/4–5 cells per mouse; p-values: 1.8=0.88W, 2.0=0.022W, 2.2=0.060P, 2.4=0.011W, 2.6=0.15P) and (D) KO mice (n=3 mice/4–5 cells per mouse; p-values: 1.8=0.69W, 2.0=0.080W, 2.2=0.20P, 2.4=0.043P, 2.6=0.0073P) at baseline and with GSK-3β treatment. P-values were determined by paired t-test, pParametric test, WNon-parametric Wilcoxon Test. (E) Representative images of fixed tissue from CON and KO mice in which the z-disc is designated with α-actinin (n=30 cells/animal from 3 animals/group). Scale bar = 10 um. (F) Summary data of resting SL (μm) (p=0.024 by Mann-Whitney t-test) shown as means ± SEM. (G) Representative Coomassie stained gel with quantification below of CON and GSK-3β KO mice in which titin isoforms N2BA, N2B, degraded titin (T2), and myosin are indicated by arrows (n= 5–6 mice/group). P-value was determined by Mann-Whitney-test.
As the molecular spring responsible for passive tension43 and resting sarcomere length, titin acts as a “stretch sensor” for LDA. For example, transgenic expression of a more compliant titin isoform depressed LDA28. To assess whether titin may be contributing to GSK-3β’s modulation of LDA, we assessed passive tension in CON and KO myocytes. Myocytes from KO mice had significantly decreased passive tension at long SLs (2.4 and 2.6 μm, p=0.0083 and p=0.016 by unpaired t-test, n=4 mice/group, 3–4 cells/mouse, Figure 2B, Table S4). If GSK-3β indeed modulates LDA via passive tension, recombinant GSK-3β should be able to increase passive tension similar to its effect on calcium sensitivity at long sarcomere lengths. We performed passive tension experiments in CON and GSK-3β KO myocytes before and after treatment with exogenous GSK-3β (0.1 μg GSK-3β for 5 minutes). GSK-3β treatment significantly increased passive tension in both CON and KO myocytes (calculated by paired t-test, n= 3 mice/group, 4–5 cells/mouse, Figure 2C–D).
In addition to passive tension and consistent with a more compliant titin, resting sarcomere length was also increased in GSK-3β KO myocytes compared to CON myocytes (n=3 mice/group, 30 cells/mouse, p=0.024 by Mann-Whitney test, Figure 2E–F). Together, these results indicate that loss of GSK-3β from the myofilament results in a more compliant titin, which is known to depress LDA.
There are two isoforms of titin, the stiff N2B and more compliant N2BA44, and it is possible the observed change in passive tension was due to a switch in the relative expression of these two isoforms. We prepared45 LV samples from CON and KO mice and quantified the ratio of N2BA/N2B (n=5–6/group, Figure 2G) and found the isoform composition to be unchanged. Thus, the decrease in passive tension in KO cells was likely due to reduced phosphorylation of GSK-3β’s myofilament targets.
Exogenous GSK-3β cannot rescue function in the absence of cMyBP-C
We next determined whether genetic removal of a protein in the LDA pathway, specifically, cardiac myosin binding protein-C (cMyBP-C), would block the ability of exogenous GSK-3β to rescue calcium sensitivity at a long SL. LDA requires myosin binding protein-C46, whose C-terminus binds titin47 and N-terminus binds the thin filament48, 49. Importantly, we observed no evidence of a direct interaction of GSK-3β and cMyBP-C (i.e., no change in cMyBP-C phosphorylation in the KO mice), so here we are testing whether GSK-3β’s functional impact lies upstream of cMyBP-C.
We used two genetically engineered mouse lines, one with complete ablation of cMyBP-C (cMyBP-C KO)30 and one transgenic expressing a truncated cMyBP-C lacking the N-terminal C0 and C1 domains that interact with the thin filament (ΔC0-C1f)31. Both strains have been previously studied, although we confirmed their cMyBP-C expression profiles here (Figure S4A). We measured force-calcium relationships before and after exogenous GSK-3β treatment (n=3 mice/group, 3 cells/mouse). While GSK-3β increased calcium sensitivity at long SLs in Con myocytes, it failed to affect calcium sensitivity in the cMyBP-C KO or cMyBP-CΔC0C1f mice (Figure S4B–G). Together, these findings indicate cMyBP-C, and particularly the N-terminus, is downstream of GSK-3β’s titin-based modulation of calcium sensitivity. However, the precise role of cMyBP-C in LDA has not been fully elucidated, so it is possible cMyBP-C acts in a separate parallel pathway to the titin-based mechanism and that GSK-3β is involved in both.
GSK-3β phosphorylates z-disc proteins
To further understand the mechanism by which GSK-3β modulates LDA, we next sought to identify the myofilament phospho-targets of GSK-3β. Mass spectrometry analysis of phospho-enriched myofilament samples from GSK-3β CON (n=4) and KO mice (n=5) identified 981 phospho-sites on 305 proteins (Table S5). Using cutoffs of p < 0.05 and log2FC < −0.5, we found decreased phosphorylation at 9 S/T residues on 8 proteins in the KO mice, including the structural z-disc protein abLIM-150, and z-disc affiliated proteins Supervillin51, Synaptopodin52, and SPEG (Figure 3A–B), suggesting they are GSK-3β targets. Two phosphorylation sites increased (log2FC>0.5, HSPB6 and B3AT). We did not detect changes in phosphorylation of any thin filament proteins known to impact calcium sensitivity as well as any changes in titin phosphorylation that could be linked to the decreased passive tension.
Figure 3. GSK-3β phosphorylates z-disc proteins.
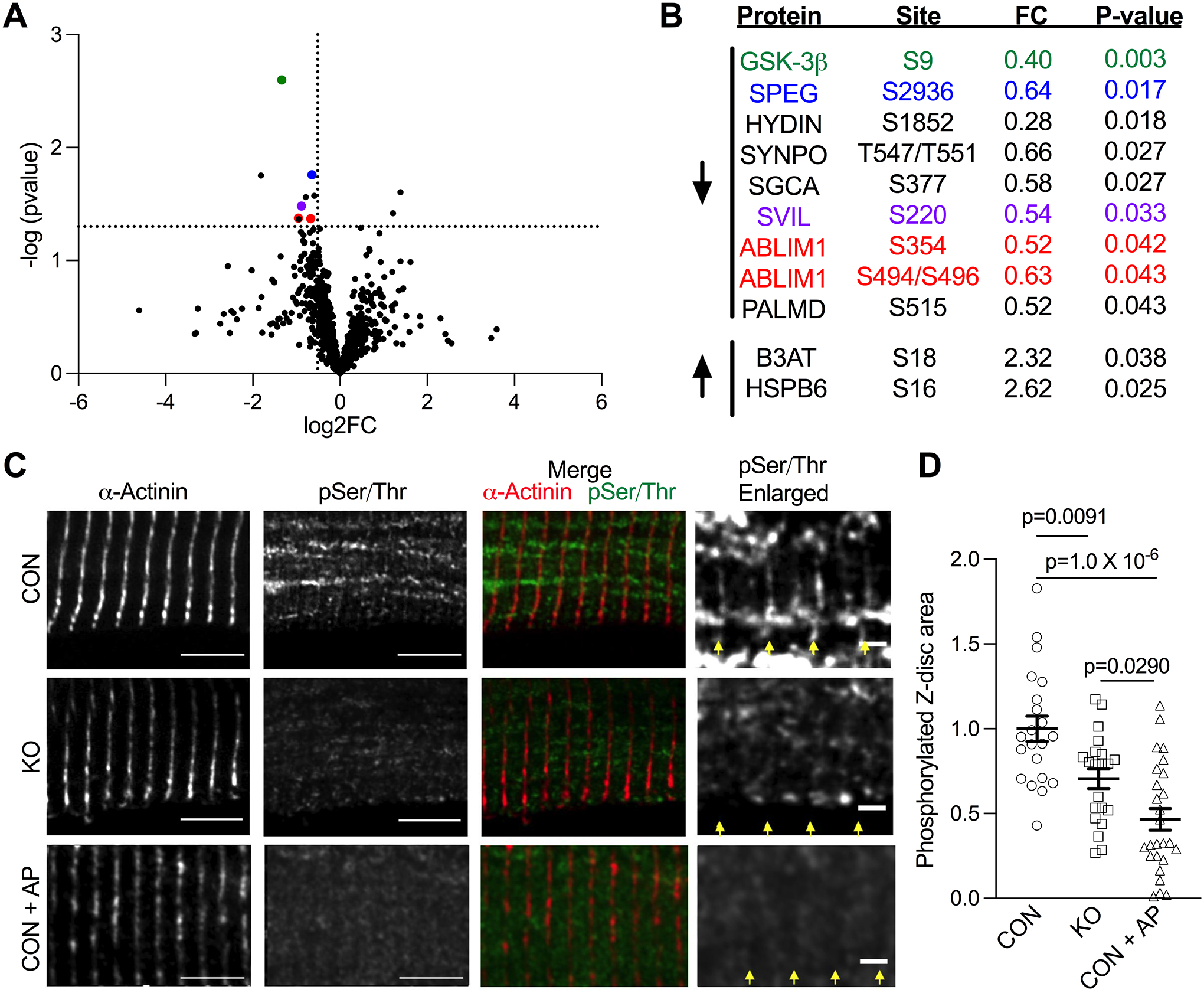
(A) Volcano plot depicting phosphorylation sites in CON (n=4) and GSK-3β KO (n=5) mice identified by mass spectrometry. Cut-offs for p-value are displayed at p=0.05 and for a fold change of a 30% decrease. (B) Significantly downregulated and upregulated sites. Proteins shown to localize to the z-disc are color coded to their respective points on the volcano plot. (C) Representative immunofluorescence from CON and GSK-3β KO LV tissue in which samples were probed for the z-disc marker ⍺-actinin (red in merged image) and phosphorylated Serine/Threonine residues (green in merged image). A subset of CON tissue was pre-incubated with alkaline phosphatase to act as a positive control (Phosphatase). Scale bar = 5 μm (2 μm in enlarged images). Yellow arrows delineate z-discs. (D) Quantification of Z-disc area containing pSer/Thr staining (normalized to control). n=3 mice, 21–26 cells/group, 10 z-discs/cell. P-values were calculated via one-way ANOVA with Tukey’s multiple comparison test.
The mass spectrometry results indicated GSK-3β’s myofilament targets are primarily at the z-disc in vivo, which we verified with immunofluorescence on LV skinned myocytes from CON and KO mice (n=3), using antibodies against phosphorylated serine/threonine residues, and α-actinin. As a positive control, a subset of CON myocytes was incubated with alkaline phosphatase. We used the ⍺-actinin channel to delineate the z-disc and create a region of interest to quantify the pSer/Thr signal. The area of z-disc pSer/Thr staining significantly decreased in GSK-3β KO mice compared to CON animals (p=0.0091, Figure 3C–D). Decreased phosphorylated z-disc area was also observed in cells incubated with alkaline phosphatase (p=1.0 × 10−6).
Phosphorylation at Y216 targets GSK-3β to the myofilament
Consistent with its targets, we also found GSK-3β localizes to the myofilament z-disc. Myocytes isolated from human NF LV myocardium were skinned prior to plating/fixing to remove the confounding presence of the cytosolic pool of GSK-3β. While total GSK-3β displayed a mild co-localization with α-actinin, pY216 GSK-3β displayed a much stronger localization to the z-disc. Comparatively, pS9 GSK-was not at the z-disc and instead appeared to localize to the intercalated disc (Figure 4A). This intercalated disc localization was not unique to the pS9 antibody, however, as all GSK-3β antibodies localized to the intercalated disc (Figure S5). Immunofluorescence images for IgG and secondary antibody only controls to test for non-specific binding in human myocytes are shown in Figure S6.
Figure 4. GSK-3β associates with the myofilament which is mediated by Y216 phosphorylation.
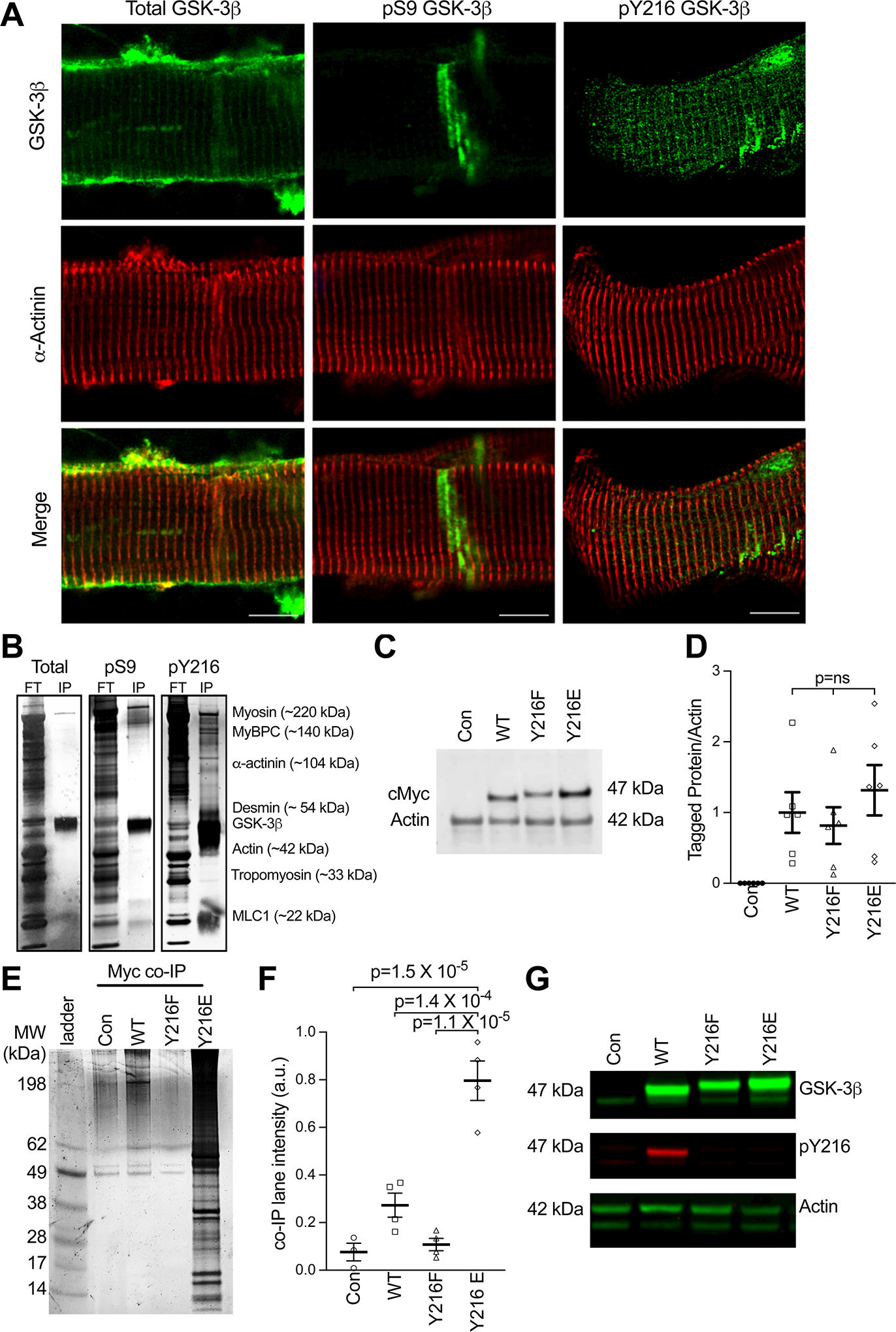
(A) Representative immunofluorescence of human non failing (NF) ventricular myocytes. Total, pS9 and pY219 GSK-3β are depicted in green and counterstained with α-actinin (red). Scale bars = 10 μm. (B) Silver-stained gel depicting flow through (FT) and eluted immunoprecipitation (IP) for myofilament enriched NF sample co-immunoprecipitated with either total, pS9 or pY216 GSK-3β antibodies. Labels on the right correspond to hypothesized myofilament proteins and molecular weights. (C) Example western blot for cMyc for Con (un-transduced) neonatal rat ventricular cardiomyocytes (NRVMs) and those transduced with Myc-tagged WT GSK-3β (WT), Y216F GSK-3β, and Y216E GSK-3β. (D) Summary data from data in panel B, means ± SEM (n = 6/group). P-values were calculated via ordinary one-way ANOVA with Tukey’s multiple comparisons. (E) Representative silver-stained gel depicting co-immunoprecipitation of proteins pulled down (Myc co-IP) with a myc tag antibody in control, WT, Y216F, and Y216E groups. (F) Summary data of co-IP experiment, showing total lane intensity (mean ± SEM) (n = 4/group, statistics by non-parametric one-way ANOVA (Kruskal Wallis) with Dunn’s multiple comparisons). (G) Representative western blot from panel E, depicting total GSK-3β, pY216, and actin in myofilament enriched NRVMs.
That phosphorylation at Y216 on GSK-3β increased its affinity for the myofilament was further shown by immunoprecipitation experiments in human NF LV myocardium enriched for the myofilament. Using total GSK-3β, pS9 GSK-3β, or pY216 GSK-3β as bait, total and pS9 GSK-3β did not pull-down myofilament proteins, while pY216 GSK-3β showed high levels of the primary myofilament proteins (Figure 4B). Because of the strong association of myofilament proteins for each other, this result does not indicate a specific binding partner of pY216 GSK-3β. As total GSK-3β should include pY216 GSK-3β, it was surprising there was little interaction between total GSK-3β and the myofilament. One explanation is that Y216 phosphorylation makes up a small percentage of total GSK-3β, however, it is also possible this is due to differences between the primary antibodies.
To confirm whether phosphorylation at Y216 is necessary and sufficient for GSK-3β’s association with the myofilament, we generated myc-tagged adenoviruses (so the same primary antibody could be used in each) for wildtype (WT) GSK-3β, phospho-null (Y216F) GSK-3β, and phospho-mimetic (Y216E) GSK-3β and transduced them into neonatal rat ventricular myocytes (NRVMs). Probing for the myc-tag revealed equal expression in the transduced NRVMs (Figure 4C–D). Co-immunoprecipitation in myofilament-enriched samples showed Y216E GSK-3β strongly bound to the myofilament, while almost no binding was observed with the phospho-null Y216F GSK-3β (Figure 4E–F). A modest amount of myc-WT GSK-3β also associated with the myofilament, which we attributed to endogenous Y216 phosphorylation, as shown by western blot (Figure 4G).
abLIM-1 interacts with titin at the z-disc and is required for normal LDA
Of the GSK-3β phospho-targets we identified by mass spectrometry, abLIM-1 was of specific interest because: one, multiple phosphorylation sites on the protein were decreased in the KO mice indicating strong regulation by GSK-3β (Figure 5A), two, we previously identified abLIM-1 as a target of GSK-3β in a dog model of heart failure13, and three, LIM domains are involved in stress sensing53. No statistical differences in protein (n=5) (Figure 5B–C) or transcript levels (n=4) (Figure S7A) of abLIM-1 were detected in KO mice. Immunofluorescence in CON and KO myocytes showed abLIM-1 localizes to the z-disc (Figure 5D) as previously50 and is solely a sarcomeric protein, as abLIM-1 was only detected in the myofilament fraction and absent from the soluble, primarily cytosolic, fraction (n= 3 CON, 4 KO) (Figure S7B–C). Immunofluorescence images for IgG and secondary antibody only controls to test for non-specific binding in mouse myocytes are shown in Figure S8.
Figure 5. abLIM-1 localizes to the z-disc and modulates passive stiffness via titin.
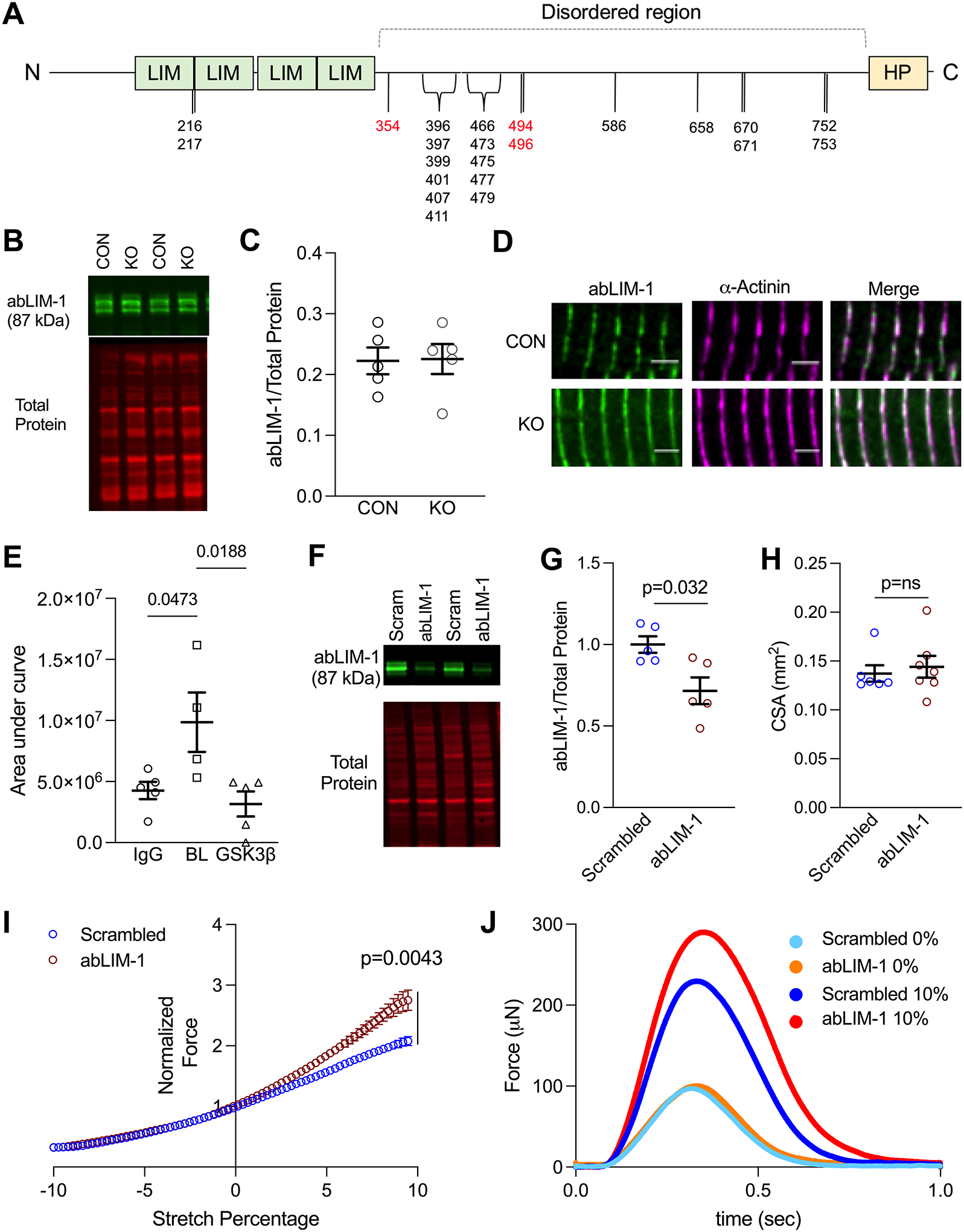
(A) Graphic of abLIM-1 domains and phosphorylation sites. Sites depicted in black are unchanged in GSK-3β KO mice, and sites depicted in red were reduced. (B) Representative western blot of abLIM-1 in whole tissue lysis of CON and GSK-3β KO mice. (C) abLIM-1 normalized to total protein in CON and KO mice (n=5). Statistics were calculated via Mann-Whitney test (D) abLIM-1 (green) and α-actinin (pink) staining in CON and GSK-3β KO mouse LV. (E) Quantification (area under the curve) of Z1Z2 titin peptides non-specifically bound (IgG control) and pulled down by GST-tagged abLIM-1, at baseline (BL) and with GSK-3β pre-treatment (GSK) obtained by mass spectrometry. n values are as follows: IgG=5, BL=4, GSK-3β =5. P-values were calculated via one-way ANOVA and Tukey’s multiple comparison test (F) Representative western blot of abLIM-1 and total protein stain in scrambled and abLIM-1 EHTS (G) Quantification of abLIM-1/total protein, normalized to scrambled (n=5, p=0.032). P-value was calculated by Mann-Whitney test. (H) Cross-sectional area of scrambled and abLIM-1 EHTs. P-value was calculated via unpaired Mann-Whitney test (I) Twitch forces in EHTs treated with either scrambled (blue) or abLIM-1 (red) siRNA. Twitch forces were measured at 72 steps between a -10% (of total EHT length) slack to +10% stretch. Twitch forces are normalized to 0% stretch. (n values: Scrambled =6, abLIM-1= 7). p =0.0043 refers to the difference between groups at 10% stretch, calculated by unpaired t-test (J) Representative traces of force (μN) at 0% (light blue = scrambled, orange = abLIM-1) and 10% (dark blue = scrambled, red = abLIM-1) stretch in a scrambled and abLIM-1 siRNA treated EHT.
The N-terminus of titin, specifically the Z1Z2 domains and z-repeats, localizes to the z-disc54. The Z1Z2 domains bind to telethonin (TCAP), a complex that is important for stress-sensing at the z-disc55 and involved in maintaining passive tension56. As passive tension was decreased in the GSK-3β KO mice, we hypothesized abLIM-1 interacts with the Z1Z2 domains of titin at the z-disc. We performed co-immunoprecipitation experiments in which we incubated recombinant GST-tagged abLIM-1 with Z1Z2 peptides (a kind gift from Dr. Siegfried Labeit). Using the GST-tag on abLIM-1 as bait, we were able to identify Z1Z2 in the elutants via mass spectrometry (Figure 5E), indicating that abLIM-1 can bind the Z1Z2 domains on titin. To determine whether this interaction can be regulated by GSK-3β, we treated abLIM-1 with GSK-3β and found this ablated its interaction with Z1Z2 (n=4–5 samples/group; p=0.0188 by ANOVA as previous studies have indicated IP-MS/MS experiments in the myofilament can be analyzed by parametric statistical tests57).
We next sought to determine mechanistically whether abLIM-1 is involved in LDA signaling in the myocyte. We generated engineered human heart tissues (EHT)35 by seeding de-cellularized myocardium with human induced pluripotent stem cell cardiomyocytes (hiPSC-CMs) and allowed them to mature for two weeks. The EHTs were then treated with either scrambled siRNA or abLIM-1 siRNA, which resulted in a 30% knockdown of abLIM-1 (p=0.032, Figure 5F–G). The abLIM-1 treatment did not result in alteration of cross-sectional area of the EHTs (Figure 5H). EHTs were paced at 1 Hz in Tyrodes solution while simultaneously measuring isometric twitch force. To measure LDA, the EHT was slacked to −10% of the tissue culture length and then stretched to +10%, all while continuing 1 Hz pacing (n = 6 scrambled, 7 abLIM-1). Both scrambled and abLIM-1 siRNA treated EHTs exhibited increased twitch force with stretch, however the EHTs with reduced abLIM-1 showed enhanced length sensitivity compared with the scrambled group, with a divergence occurring around 2% stretch (Figure 5I). The greatest difference in force occurred at 10% stretch (p=0.0043) (Figure 5J). These experiments indicate that abLIM-1 binds titin at the z-disc to depress passive tension and thus LDA, and that its phosphorylation by GSK-3β relieves this inhibition and results in normal function.
Myofilament GSK-3β is decreased in heart failure samples with dampened LDA
We next determined whether myofilament-localized GSK-3β plays a role in human heart failure. We used LV myocardium from human NF rejected donor hearts (n=19) and explanted hearts from HF patients (n=22, demographics in Table 1). These 41 samples were collected from two biobanks (Loyola University Chicago and Cleveland Clinic). Myofilament GSK-3β was significantly reduced in myocardium from HF patients compared to NF hearts, while whole tissue GSK-3β remained unchanged when normalized to either the total protein stain (Figure 6A–B) or the actin band of the total protein stain (Figure S9). No statistical differences were detected in phosphorylated GSK-3β (pS9 and pY216 normalized to total GSK-3β) in either whole tissue or the myofilament (Figure S10A–E).
Table 1.
Patient characteristics for Non-failing and Heart Failure samples.
| Non-failing | Heart failure | P-value | |
|---|---|---|---|
| n | 19 | 22 | - |
| Age (years) | 58.3 ± 8.6 | 53.9 ± 2.71 | 0.21 |
| % White | 94.7% | 72.2%2 | 0.063 |
| % Female | 42.1% | 40.9% | 0.94 |
| % Ischemic | 0% | 63.6% | <0.0001 |
| LVEF (%) | 62.1 ± 2.2%3 | 21.4 ± 2.0%4 | <0.0001 |
Age, gender, ethnicity, ischemia, and left ventricular ejection fraction (LVEF) for NF and HF LV samples. P-values were calculated as follows: Age (unpaired t-test), Ethnicity (Chi-Square), Sex (Chi-Square), Ischemia (Chi-Square), LVEF (Mann-Whitney Test).
Superscripts represent categories with missing values (1 4 missing values, 2 4 missing values, 3 7 missing values, 4 11 missing values).
Missing values have been excluded from statistical analysis.
Figure 6. Myofilament-localized GSK-3β and LDA is reduced in human heart failure.
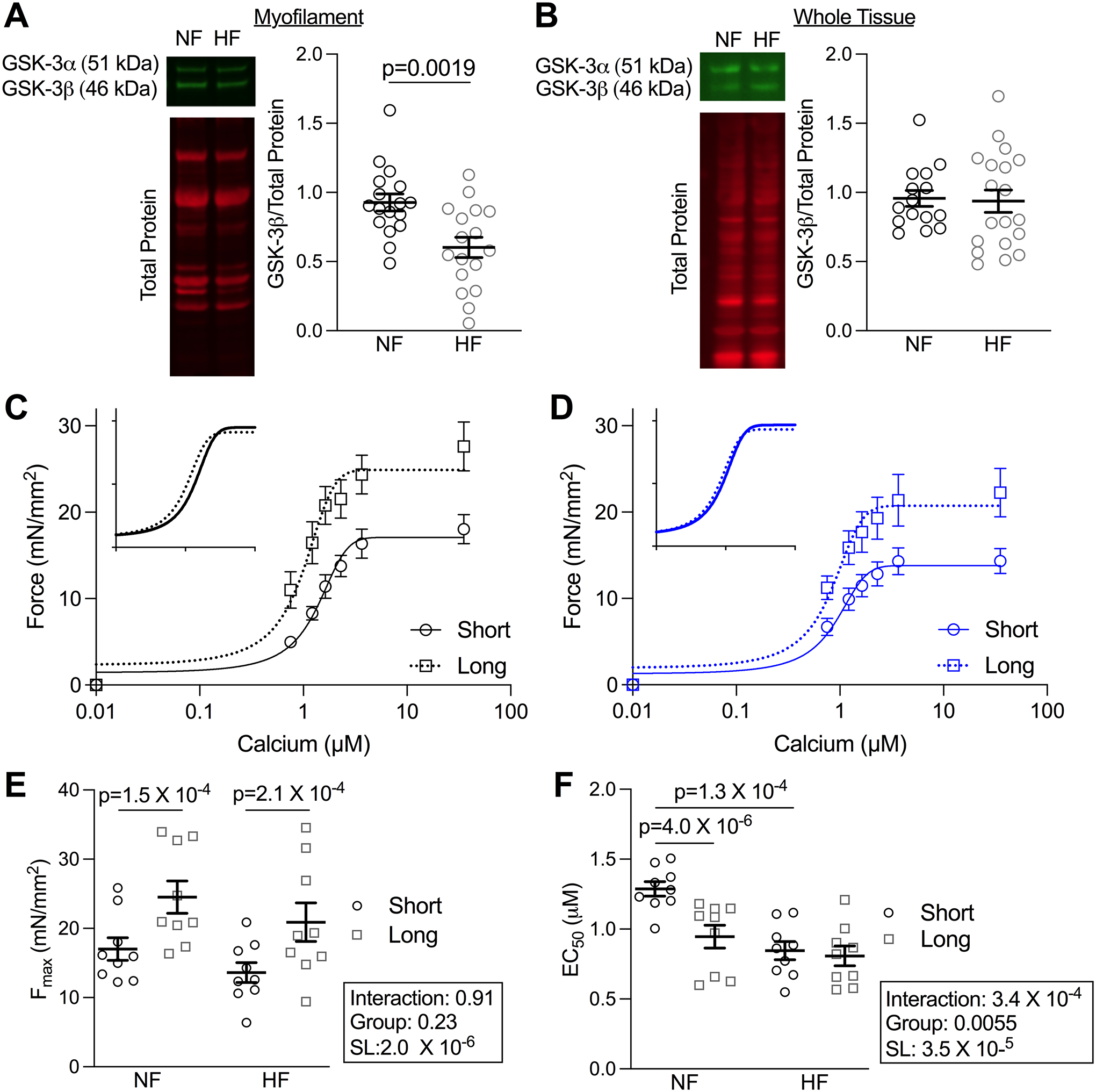
(A) Example blots and quantification of myofilament-enriched (n=17, p=0.0019 by unpaired t-test) and (B) Whole tissue preparation (n values: NF=15, HF=19) of LV from human NF and HF samples. (C) Mean force as a function of calcium concentration and fitted curves for skinned myocytes from the LV of NF and (D) HF patients from which measurements were taken at short (1.9 μm) and long (2.3 μm) SL. (E) Fmax and (F) EC50 depicted as mean± SEM (n = 9 myocytes from 3 patients). P-values were calculated from two-way repeated measures ANOVA with pair-wise comparisons.
Despite observing no statistical difference in whole tissue GSK-3β between the groups, we selected the samples with the highest and lowest levels of whole tissue GSK-3β (n = 5–7 cells/ 3 hearts per group) and measured force-calcium relationships at long SL. There were no statistical differences between these groups (Figure S11), supporting that whole tissue (primarily cytosolic) GSK-3β is not responsible for modulating sarcomeric function.
Lastly, we tested whether HF patients with diminished sarcomere-localized GSK-3β had depressed LDA as the mouse model would predict. We measured force-calcium relationships at both short and long SLs in NF and HF LV (n= 3 hearts/group, 3 cells/heart). Both NF and HF groups experienced an increase in Fmax with stretch; however, while we observed increased calcium sensitivity in NF myocytes when increasing SL, this effect was entirely absent in the HF group (Figure 6C–F, Table S6). These data suggest z-disc localized GSK-3β is lost in human heart failure, which correlates with a loss of LDA.
Discussion
GSK-3β is a central kinase in multiple critical signaling pathways in the cardiomyocyte, and recent evidence13, 58 suggests it also regulates sarcomere contractile function, although direct evidence is lacking. To address this gap, we utilized an inducible cardiomyocyte-specific GSK-3β KO mouse model. We discovered GSK-3β is essential for normal length-dependent activation (LDA), the ability of the myocyte to respond to stretch and a critical component of the Frank-Starling mechanism, resulting in calcium desensitization at longer sarcomere lengths. Mechanistically, this is via decreased titin-based passive tension observed in the KO mice, which is a critical “length sensor” for LDA. GSK-3β localizes to the sarcomere z-disc via its own phosphorylation at Y216 and phosphorylates several z-disc proteins, including actin binding LIM protein 1 (abLIM-1). Almost nothing is known about abLIM-1 in the heart, but we have found that it localizes to the z-disc where it can bind to the Z1Z2 domains of titin, and this binding is blocked by GSK-3β phosphorylation. Furthermore, abLIM-1 acts as an inhibitor of passive tension and LDA, which is relieved by GSK-3β. Importantly, in LV myocardium from human HF patients there was reduced sarcomeric GSK-3β (but not whole tissue GSK-3β) and diminished LDA compared to NF samples. As reduced LDA in HF is detrimental, altering Y216 phosphorylation to restore sarcomeric GSK-3β localization is a potential therapeutic strategy for restoring the Frank-Starling mechanism in heart failure patients.
GSK-3β modulates length-dependent activation via titin
The Frank-Starling law of the heart states that the stroke volume of the LV increases as LV volume increases59, allowing the heart to respond to changes in volume on a beat-to-beat basis. This behavior stems from the response of cardiomyocytes to stretch, which results in an increase in force production, termed length-dependent activation (LDA). LDA, and thus the Frank-Starling mechanism, can become depressed in heart failure23–27. While most studies are conducted in patients with end stage heart failure, animal models have shown that this mechanism may be lost in the earlier stages, prior to hypertrophy and fibrosis5. Despite the fact it was initially discovered over a century ago – the molecular mechanisms of LDA are still unclear, which may explain why some studies have found the Frank-Starling mechanism to be unaltered in heart failure26, 27. There are several hypotheses supported by the existing work, and these may not be exclusive, since such a critical behavior could warrant redundant pathways to operate. There are currently three primary mechanisms for explaining LDA20: 1) decreased inter-filament spacing from sarcomeric stretch brings myosin heads closer to actin to increase the likelihood of crossbridge formation, 2) phosphorylation/regulation of thin filament proteins with stretch increases calcium sensitivity, and 3) stretch is sensed by titin strain, resulting in structural rearrangement that favors crossbridge formation.
We were able to rule out the first two mechanisms by which GSK-3β regulates LDA, as lattice spacing was not altered in GSK-3β KO mice and phospho-proteomics did not identify any phosphorylation sites connected to LDA or calcium sensitivity20. Instead, our data implicate the titin-based mechanism. Titin’s ability to create passive tension directly correlates with an enhanced LDA response28, 60, 61; and myocytes with a loss of titin compliance have a blunted LDA just as we observed in the GSK-3β KO mice. Additionally, incubation with recombinant GSK-3β increased passive tension. How, then, does GSK-3β modify titin compliance? GSK-3β did not affect titin itself, as MS revealed GSK-3β did not phosphorylate any titin residues, and there was no change in titin isoforms. Thus, GSK-3β must regulate the interaction between titin and z-disc proteins.
GSK-3β facilitates LDA via abLIM-1
While a great deal remains unknown about titin’s structure, function, and interacting partners at the z-disc, there are studies that support the hypothesis that altering titin’s interaction with z-disc proteins can alter passive tension. For example, the Z1Z2 domains of titin, which are located in its z-disc region, are highly compliant in isolation, but are stabilized via z-disc binding partners54.
Of the GSK-3β phosphorylated proteins identified in our phospho-proteomics screen, we were most interested in abLIM-1. abLIM-1 has been the focus of very few studies, only one of which identified it in the heart50. However, several proteins of the same family, which contain LIM domains, such as Muscle LIM protein (MLP) and Four and a half LIM domain protein (FHL1 and FHL2) have been shown to be stress/strain sensors62. Indeed, this work establishes abLIM-1 as a critical z-disc protein in maintaining normal function. First, abLIM-1 interacts with the Z1Z2 domains of titin in vitro which also interact with thelethonin (TCAP), an interaction important for stress-sensing, maintaining passive tension55, 56, and likely LDA although this has not yet been shown.
Similar to TCAP, abLIM-1 is also required for normal stress sensing at the z-disc, as siRNA-induced reduction in abLIM-1 significantly increased LDA. The direction of this impact was somewhat surprising, as we anticipated facilitation or enhancement of LDA by abLIM-1, and that its reduction would lead to depressed LDA. However, these results show that abLIM-1 is not simply required for the LDA mechanism to proceed but modulates the response of the cell to stretch by acting as a “brake” on stretch sensing and LDA. This regulatory paradigm is not unprecedented, as there are other proteins in cardiac EC-coupling that inhibit activity in a phosphorylation-dependent manner, such as cMyBP-C and phospholamban. In fact, our experiments with recombinant Z1Z2 and abLIM-1 show that GSK-3β ablates their interaction and relieves abLIM-1’s inhibition of passive tension and LDA. These results introduce a new player in sarcomere mechano-sensing, and future studies are warranted to understand the structural basis for abLIM-1’s novel functional role and how its phosphorylation alters this function.
GSK-3β localizes through pY216
It is likely that declining myofilament GSK-3β contributes to decreased LDA and Frank-Starling mechanism observed in the failing human heart5, 24, 26. As GSK-3β can directly enhance calcium sensitivity at long SLs, it is an intriguing candidate for a HF therapeutic. Indeed, a therapeutic that could increase calcium sensitivity at long SLs while not affecting it at short or resting SLs would be highly beneficial, especially since LDA takes place within 5 ms21 so it can regulate calcium sensitivity during a single beat. A straight-forward calcium sensitizer would increase contractility but would also slow relaxation. However, a therapeutic that increased calcium sensitivity at long SLs when the heart is filled with blood just prior to systole would enhance contractility, and as the LV chamber volume decreases during systole and myocyte SL shortens calcium sensitivity would decrease, aiding relaxation. Indeed, compounds that aim to enhance sarcomere-based contractility have frequently resulted in depressed relaxation63, creating a benefit-cost tradeoff.
However, GSK-3β is such a promiscuous signaling kinase64 that broadly elevating GSK-3β would likely result in catastrophic off target effects. Thus, the ability to manipulate strictly myofilament-localized GSK-3β is imperative, which we found is regulated by its phosphorylation at Y216. Several kinases have been reported to phosphorylate Y216, including Fyn65, PYK266, MEK167, and as an auto-phosphorylation event68. Generally, tyrosine kinases phosphorylate fewer targets than serine/threonine kinases69, thus the large number of kinases targeting Y216 suggests dynamic regulation. Whether targeting the Y216 phosphorylation site directly or by modulating the activity of an upstream kinase would be a useful therapeutic approach to restore myofilament GSK-3β and thus LDA in heart failure will need to be established in future studies.
Conclusion
We have identified a novel mechanism by which GSK-3β regulates LDA, arising from its z-disc localization and phosphorylation targets, most likely abLIM-1, a new player in sarcomere function. While LDA and the Frank-Starling mechanism were described well over a century ago, many aspects of the mechanism remain unresolved. This study reveals new kinase regulation of LDA and suggests myofilament GSK-3β is a strong therapeutic candidate to rescue the depressed LDA that is observed in heart failure. Furthermore, these results add to the small number of studies demonstrating that the z-disc is not merely a structural scaffold for the thin filaments but is also capable of regulating active contraction.
Supplementary Material
Novelty and Significance.
What is Known?
GSK-3β can target myofilament proteins and alter calcium sensitivity in vitro.
Stretch increases myofilament calcium sensitivity through a process known as length-dependent activation that is not well understood but involves titin-based strain.
Length-dependent activation is reduced in heart failure.
What New Information Does this Article Contribute?
GSK-3β phosphorylates myofilament proteins at the z-disc and contributes to length-dependent activation in vivo.
GSK-3β’s effect on length-dependent activation occurs through interaction of its myofilament target, abLIM-1, with the z-disc region of titin.
Loss of myofilament GSK-3β in human heart failure correlates with decreased length-dependent activation.
Length-dependent activation (LDA) refers to the process by which increased sarcomere length results in increased myofilament calcium sensitivity. While much work has been done to elucidate the mechanism by which LDA occurs, a clear picture remains elusive. Our in vivo work shows that the primarily cytosolic kinase GSK-3β localizes to the myofilament and alters LDA via titin-based strain. We provide evidence that GSK-3β’s z-disc substrate, actin binding LIM domain protein 1 (abLIM-1) is a negative regulator of LDA. Additionally, abLIM-1 is a novel binding partner to titin’s z-disc bound Z1Z2 domains. In human heart failure myofilament GSK-3β is diminished in samples that also have reduced LDA. These findings suggest that LDA can be regulated by protein interactions at the z-disc, which could be beneficial for targeting the diminished LDA response that can occur in heart failure.
Acknowledgements
We would like to thank the patients and organ donors who donated the samples used in this project. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357.
Sources of Funding
This work was supported by the National Institutes of Health (R01HL136737 to J.A.K., 9P41GM103622 to T.I., R01HL130357, R01HL105826, R01AR078001, and R01HL143490 to S.S), the American Heart Association (831515 to M.J.S., 111POST7210031 to M.P., 19TPA34830084 to S.S., and 14SDG20380148 to J.A.K.), and the National Science Foundation (1653160 to S.G.C). This study utilized equipment obtained with the support of a shared instrumentation grant S10OD028449 from the National Institutes of Health to Loyola University Chicago.
Nonstandard Abbreviations and Acronyms
- GSK-3β
Glycogen Synthase Kinase 3β
- CRT
Cardiac Resynchronization Therapy
- cMyBP-C
Cardiac Myosin binding protein-C
- MCM
Mer cre mer
- LV
Left ventricle
- abLIM-1
Actin-binding LIM protein 1
- NRVM
Neonatal rat ventricular cardiomyocytes
- EHT
Engineered Heart Tissue
- hi-PSC
Human induced pluripotent stem cell
Footnotes
Disclosures
J.A.K. and S.S. provided consulting and conducted collaborative studies with various pharmaceutical companies, but all such work is unrelated to the content of this manuscript. S.G.S. holds equity ownership in Propria LLC, which has licensed technology used in the research reported in this publication. No other disclosures reported.
References
- 1.Sakaguchi T, Takefuji M, Wettschureck N, Hamaguchi T, Amano M, Kato K, Tsuda T, Eguchi S, Ishihama S, Mori Y, Yura Y, Yoshida T, Unno K, Okumura T, Ishii H, Shimizu Y, Bando YK, Ohashi K, Ouchi N, Enomoto A, Offermanns S, Kaibuchi K and Murohara T. Protein Kinase N Promotes Stress-Induced Cardiac Dysfunction Through Phosphorylation of Myocardin-Related Transcription Factor A and Disruption of Its Interaction With Actin. Circulation. 2019;140:1737–1752. [DOI] [PubMed] [Google Scholar]
- 2.Ai X, Curran JW, Shannon TR, Bers DM and Pogwizd SM. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ Res. 2005;97:1314–22. [DOI] [PubMed] [Google Scholar]
- 3.Fuller SJ, Osborne SA, Leonard SJ, Hardyman MA, Vaniotis G, Allen BG, Sugden PH and Clerk A. Cardiac protein kinases: the cardiomyocyte kinome and differential kinase expression in human failing hearts. Cardiovasc Res. 2015;108:87–98. [DOI] [PubMed] [Google Scholar]
- 4.Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, Jo SH, Danner T, Zhang M, Rainer PP, Bedja D, Kirk JA, Ranek MJ, Dostmann WR, Kwon C, Margulies KB, Van Eyk JE, Paulus WJ, Takimoto E and Kass DA. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature. 2015;519:472–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Komamura K, Shannon RP, Ihara T, Shen YT, Mirsky I, Bishop SP and Vatner SF. Exhaustion of Frank-Starling mechanism in conscious dogs with heart failure. American Journal of Physiology-Heart and Circulatory Physiology. 1993;265:H1119–H1131. [DOI] [PubMed] [Google Scholar]
- 6.Woulfe KC, Gao E, Lal H, Harris D, Fan Q, Vagnozzi R, DeCaul M, Shang X, Patel S, Woodgett JR, Force T and Zhou J. Glycogen synthase kinase-3beta regulates post-myocardial infarction remodeling and stress-induced cardiomyocyte proliferation in vivo. Circ Res. 2010;106:1635–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhai P and Sadoshima J. Glycogen synthase kinase-3beta controls autophagy during myocardial ischemia and reperfusion. Autophagy. 2012;8:138–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morisco C, Zebrowski D, Condorelli G, Tsischlis P, Vatner SF and Sadoshima J. The Akt-Glycogen Synthase Kinase 3b Pathway Regulates Transcription of Atrial Natriuretic Factor Induced by b-Adrenergic Receptor Stimulation in Cardiac Myocytes. The Journal of Biological Chemistry. 2000;275:14466–14475. [DOI] [PubMed] [Google Scholar]
- 9.Haq S, Choukroun G, Kang ZB, Ranu H, Matsui T, Rosenzweig A, Molkentin JD, Alessandrini A, Woodgett J, Hajjar R, Michael A, Force T Glycogen synthase kinase-3beta is a negative regulator of cardiomyocyte hypertrophy. The Journal of Cell Biology. 2000;151:117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Antos CL, McKinsey TA, Frey N, Kutschke W, McAnally J, Shelton JM, Richardson JA, Hill JA and Olson EN. Activated glycogen synthase-3b suppresses cardiac hypertrophy in vivo. PNAS. 2001;99:907–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsuda T, Zhai P, Maejima Y, Hong C, Gao S, Tian B, Goto K, Takagi H, Tamamori-Adachi M, Kitajima S and Sadoshima J. Distinct roles of GSK-3a and GSK-3b phosphoryltion in the heart under pressure overload. PNAS. 2008;105:20900–20905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirotani S, Zhai P, Tomita H, Galeotti J, Marquez JP, Gao S, Hong C, Yatani A, Avila J and Sadoshima J. Inhibition of glycogen synthase kinase 3beta during heart failure is protective. Circ Res. 2007;101:1164–74. [DOI] [PubMed] [Google Scholar]
- 13.Kirk JA, Holewinski RJ, Kooij V, Agnetti G, Tunin RS, Witayavanitkul N, de Tombe PP, Gao WD, Van Eyk J and Kass DA. Cardiac resynchronization sensitizes the sarcomere to calcium by reactivating GSK-3beta. J Clin Invest. 2014;124:129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gordon AM, Homsher E and Regnier M. Regulation of Contraction in Striated Muscle. Physiological Reviews. 2000;80:853–924. [DOI] [PubMed] [Google Scholar]
- 15.Robinson P, Griffiths PJ, Watkins H and Redwood CS. Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res. 2007;101:1266–73. [DOI] [PubMed] [Google Scholar]
- 16.Ren X, Hensley N, Brady M and Gao WD. The genetic and molecular bases for hypertrophic cardiomyopathy: the role for calcium sensitization. Journal of Cardiothoracic and Vascular Anesthesia 2018. [DOI] [PubMed] [Google Scholar]
- 17.Wolff MR, Buck SH, Stoker SW, Greaser ML and Mentzer RM. Myofibrillar calcium sensitivity of isometric tension is increased in human dilated cardiomyopathies: role of altered beta-adrenergically mediated protein phosphorylation. J Clin Invest. 1996;98:167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papadaki M, Holewinski RJ, Previs SB, Martin TG, Stachowski MJ, Li A, Blair CA, Moravec CS, Van Eyk JE, Campbell KS, Warshaw DM and Kirk JA. Diabetes with heart failure increases methylglyoxal modifications in the sarcomere, which inhibit function. JCI Insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kass DA and Solaro RJ. Mechanisms and use of calcium-sensitizing agents in the failing heart. Circulation. 2006;113:305–15. [DOI] [PubMed] [Google Scholar]
- 20.de Tombe PP, Mateja RD, Tachampa K, Ait Mou Y, Farman GP and Irving TC. Myofilament length dependent activation. J Mol Cell Cardiol. 2010;48:851–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mateja RD and de Tombe PP. Myofilament length-dependent activation develops within 5 ms in guinea-pig myocardium. Biophys J. 2012;103:L13–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gill RM, Jones BD, Corbly AK, Ohad DG, Smith GD, Sandusky GE, Christe ME, Wang J and Shen W. Exhaustion of the Frank-Starling mechanism in conscious dogs with heart failure induced by chronic coronary microembolization. Life sciences. 2006;79:536–44. [DOI] [PubMed] [Google Scholar]
- 23.Sequeira V, Wijnker PJ, Nijenkamp LL, Kuster DW, Najafi A, Witjas-Paalberends ER, Regan JA, Boontje N, Ten Cate FJ, Germans T, Carrier L, Sadayappan S, van Slegtenhorst MA, Zaremba R, Foster DB, Murphy AM, Poggesi C, Dos Remedios C, Stienen GJ, Ho CY, Michels M and van der Velden J. Perturbed length-dependent activation in human hypertrophic cardiomyopathy with missense sarcomeric gene mutations. Circ Res. 2013;112:1491–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwinger RH, Boehm M, Koch A, Schmidt U, Morano I, Eissner HJ, Uberfuhr P, Reichart B and Erdmann E. The failing human heart is unable to use the Frank-Starling mechanism. Circulation Research. 1994;74:959–969. [DOI] [PubMed] [Google Scholar]
- 25.Kitzman D,W, Higginbotham M,B, Cobb Frederick R, Sheikh Khalid H and Sullivan Martin J. Exercise intolerance in patients with heart failure and preserved left ventricular systolic function: Failure of the Frank-Starling mechanism. Journal of the American College of Cardiology. 1991;17:1065–1072. [DOI] [PubMed] [Google Scholar]
- 26.Holubarsch C, Ruf T, Goldstein DJ, Ashton RC, Nickl W, Pieske B, Pioch K, Luedemann J, Wiesner S, Hasenfuss G, Posival H, Just H and Burkhoff D. Existence of the Frank-Starling mechanism in the failing human heart. Investigations on the organ, tissue, and sarcomere levels. Circulation. 1996;94:683–689. [DOI] [PubMed] [Google Scholar]
- 27.Weil J, Eschenhagen T, Hirt S, Magnussen O, Mittmann C, REmmers U and Scholz H. Preserved Frank-Starling mechanism in human end stage heart failure. Cardiovascular Research. 1998;37:541–548. [DOI] [PubMed] [Google Scholar]
- 28.Ait-Mou Y, Hsu K, Farman GP, Kumar M, Greaser ML, Irving TC and de Tombe PP. Titin strain contributes to the Frank-Starling law of the heart by structural rearrangements of both thin- and thick-filament proteins. Proc Natl Acad Sci U S A. 2016;113:2306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lynch TL t, Ismahil MA, Jegga AG, Zilliox MJ, Troidl C, Prabhu SD and Sadayappan S. Cardiac inflammation in genetic dilated cardiomyopathy caused by MYBPC3 mutation. J Mol Cell Cardiol. 2017;102:83–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McConnell BK, Jones KA, Fatkin D, Arroyo LH, Lee RT, Aristizabal O, Turnbull DH, Georgakopoulos D, Kass D, Bond M, Niimura H, Schoen FJ, Conner D, Fischman DA, Seidman CE and Seidman JG. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J Clin Invest. 1999;104:1235–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lynch TL t, Kumar M, McNamara JW, Kuster DWD, Sivaguru M, Singh RR, Previs MJ, Lee KH, Kuffel G, Zilliox MJ, Lin BL, Ma W, Gibson AM, Blaxall BC, Nieman ML, Lorenz JN, Leichter DM, Leary OP, Janssen PML, de Tombe PP, Gilbert RJ, Craig R, Irving T, Warshaw DM and Sadayappan S. Amino terminus of cardiac myosin binding protein-C regulates cardiac contractility. J Mol Cell Cardiol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fischetti R, Stepanov S, Rosenbaum G, Barrea R, Black E, Gore D, Heurich R, Kondrashkina E, Kropf AJ, Wang S, Zhang K, Irving TC and Bunker GB. The BioCAT undulator beamline 18ID: a facility for biological non-crystalline diffraction and X-ray absorption spectroscopy at the Advanced Photon Source. Journal of Synchrotron Radiation. 2004;11:399–405. [DOI] [PubMed] [Google Scholar]
- 33.Jiratrakanvong J, Shao J, Menendez M, Li X, Li J, Ma W, Agam G and Irving T. MuscleX: software suite for diffraction X-ray imaging V1.13.1. 2018.
- 34.Ma W, Gong H and Irving T. Myosin Head Configurations in Resting and Contracting Murine Skeletal Muscle. Int J Mol Sci. 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwan J, Kwaczala AT, Ryan TJ, Bartulos O, Ren Y, Sewanan LR, Morris AH, Jacoby DL, Qyang Y and Campbell SG. Anisotropic engineered heart tissue made from laser-cut decellularized myocardium. Sci Rep. 2016;6:32068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sewanan LR, Schwan J, Kluger J, Park J, Jacoby DL, Qyang Y and Campbell SG. Extracellular Matrix From Hypertrophic Myocardium Provokes Impaired Twitch Dynamics in Healthy Cardiomyocytes. JACC Basic Transl Sci. 2019;4:495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirk JA, Chakir K, Lee KH, Karst E, Holewinski RJ, Pironti G, Tunin RS, Pozios I, Abraham TP, de Tombe P, Rockman HA, Van Eyk JE, Craig R, Farazi TG and Kass DA. Pacemaker-induced transient asynchrony suppresses heart failure progression. Sci Transl Med. 2015;7:319ra207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin TG, Myers VD, Dubey P, Dubey S, Perez E, Moravec CS, Willis MS, Feldman AM and Kirk JA. Cardiomyocyte Contractile Impairment in Heart Failure Results from Reduced BAG3-mediated Sarcomeric Protein Turnover. bioRxiv. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Arrell DK, Neverova I, MFraser H, Marban E and Van Eyk JE. Proteomic analysis of pharmacologically preconditioned cardiomyocytes reveals novel phosphorylation of myosin light chain 1. Circulation Research. 2001;89:480–487. [DOI] [PubMed] [Google Scholar]
- 40.Konhilas JP, Irving TC and de Tombe PP. Frank-Starling law of the heart and the cellular mechanisms of length-dependent activation. Pflugers Arch. 2002;445:305–10. [DOI] [PubMed] [Google Scholar]
- 41.Kumar M, Govindan S, Zhang M, Khairallah RJ, Martin JL, Sadayappan S and de Tombe PP. Cardiac Myosin-binding Protein C and Troponin-I Phosphorylation Independently Modulate Myofilament Length-dependent Activation*. Journal of Biological Chemistry. 2015;290:29241–29249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martyn DA, Adhikari BB, Regnier M, Gu J, Xu S and Yu LC. Response of Equatorial X-Ray Reflections and Stiffness to Altered Sarcomere Length and Myofilament Lattice Spacing in Relaxed Skinned Cardiac Muscle. Biophysical Journal. 2004;86:1002–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Linke WA. Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res. 2008;77:637–48. [DOI] [PubMed] [Google Scholar]
- 44.Hutchinson KR, Saripalli C, Chung CS and Granzier H. Increased myocardial stiffness due to cardiac titin isoform switching in a mouse model of volume overload limits eccentric remodeling. Journal of Molecular and Cellular Cardiology. 2015;79:104–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lahmers S, Wu Y, Call DR, Labeit S and Granzier H. Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res. 2004;94:505–13. [DOI] [PubMed] [Google Scholar]
- 46.Hanft LM, Fitzsimons DP, Hacker TA, Moss RL and McDonald KS. Cardiac MyBP-C phosphorylation regulates the Frank-Starling relationship in murine hearts. J Gen Physiol. 2021;153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freiburg A and Gautel M. A molecular map of the interactions between titin and myosin-binding protein C. Implications for sarcomeric assembly in familial hypertrophic cardiomyopathy. European journal of biochemistry. 1996;235:317–23. [DOI] [PubMed] [Google Scholar]
- 48.Harris SP, Belknap B, Van Sciver RE, White HD and Galkin VE. C0 and C1 N-terminal Ig domains of myosin binding protein C exert different effects on thin filament activation. Proceedings of the National Academy of Sciences. 2016;113:1558–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mun JY, Previs MJ, Yu HY, Gulick J, Tobacman LS, Beck Previs S, Robbins J, Warshaw DM and Craig R. Myosin-binding protein C displaces tropomyosin to activate cardiac thin filaments and governs their speed by an independent mechanism. Proceedings of the National Academy of Sciences. 2014;111:2170–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roof DJ, Hayes A, Adamian A, Chishti AH and LI T. Molecular Characterization of abLIM, a Novel Actin-binding and Double Zinc Finger Protein. J Cell Biol. 1997;138:575–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hedberg-Oldfors C, Meyer R, Nolte K, Abdul Rahim Y, Lindberg C, Karason K, Thuestad IJ, Visuttijai K, Geijer M, Begemann M, Kraft F, Lausberg E, Hitpass L, Gotzl R, Luna EJ, Lochmuller H, Koschmieder S, Gramlich M, Gess B, Elbracht M, Weis J, Kurth I, Oldfors A and Knopp C. Loss of supervillin causes myopathy with myofibrillar disorganization and autophagic vacuoles. Brain. 2020;143:2406–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Asanuma K, Kim K, Oh J, Giardino L, Chabanis S, Faul C, Reiser J and Mundel P. Synaptopodin regulates the actin-bundling activity of α-actinin in an isoform-specific manner. Journal of Clinical Investigation. 2005;115:1188–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoshijima M Mechanical stress-strain sensors embedded in cardiac cytoskeleton: Z disk, titin, and associated structures. Am J Physiol Heart Circ Physiol. 2006;290:H1313–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kollar V, Szatmari D, Grama L and Kellermayer MS. Dynamic strength of titin’s Z-disk end. J Biomed Biotechnol. 2010;2010:838530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gregorio CC, Trombitas K, Centner T, Kolmerer B, Stier G, Kunke K, Suzuki K, Obermayr F, Herrmann B, Granzier H, Sorimachi H and Labeit S. The NH2 terminus of titin spans the Z-disc: its interaction with a novel 19-kD ligand (T-cap) is required for sarcomeric integrity. The Journal of Cell Biology. 1998;143:1013–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee EH, Gao M, Pinotsis N, Wilmanns M and Schulten K. Mechanical strength of the titin Z1Z2-telethonin complex. Structure. 2006;14:497–509. [DOI] [PubMed] [Google Scholar]
- 57.Rudolph F, Fink C, Huttemeister J, Kirchner M, Radke MH, Lopez Carballo J, Wagner E, Kohl T, Lehnart SE, Mertins P and Gotthardt M. Deconstructing sarcomeric structure-function relations in titin-BioID knock-in mice. Nat Commun. 2020;11:3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kuster DW, Sequeira V, Najafi A, Boontje NM, Wijnker PJ, Witjas-Paalberends ER, Marston SB, Dos Remedios CG, Carrier L, Demmers JA, Redwood C, Sadayappan S and van der Velden J. GSK3beta phosphorylates newly identified site in the proline-alanine-rich region of cardiac myosin-binding protein C and alters cross-bridge cycling kinetics in human: short communication. Circ Res. 2013;112:633–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Solaro RJ. Mechanisms of the Frank-Starling law of the heart: the beat goes on. Biophysical journal. 2007;93:4095–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cazorla O, Wu Y, Irving TC and Granzier H. Titin-Based Modulation of Calcium Sensitivity of Active Tension in Mouse Skinned Cardiac Myocytes. Circulation Research. 2001;88:1028–1035. [DOI] [PubMed] [Google Scholar]
- 61.Methawasin M, Hutchinson KR, Lee E-J, Smithill JE, Saripalli C, Hidalgo CG, Ottenheijm CAC and Granzier H. Novel Mouse Model Attenuates the Frank-Starling Mechanism But Has a Beneficial Effect on Diastole. Circulation. 2014;129:1924–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li A, Ponten F and dos Remedios CG. The interactome of LIM domain proteins: the contributions of LIM domain proteins to heart failure and heart development. Proteomics. 2012;12:203–25. [DOI] [PubMed] [Google Scholar]
- 63.Hajjar RJ, Schmidt U, Helm P and Gwathmey JK. Ca++ Sensitizers Impair Cardiac Relaxation in Failing Human Myocardium. Journal of Pharmacology and Experimental Therapeutics. 1997;280:247–254. [PubMed] [Google Scholar]
- 64.Beurel E, Grieco SF and Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. 2015;148:114–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lesort M, Jope RS and Johnson GVW. Insulin transiently increases tau phosphorylation involvement of glycogen synthase kinase-3beta and Fyn tyrosine kinase. J Neurochem. 1999;72:576–584. [DOI] [PubMed] [Google Scholar]
- 66.Hartigan JA, Xiong WC and Johnson GVW. Glycogen synthase kinase 3beta is tyrosine phosphorylated by PYK2. Biochem Biophys Res Commun. 2001;284:485–489. [DOI] [PubMed] [Google Scholar]
- 67.Takahashi-Yanaga F, Shiraishi F, Hirata M, Miwa Y, Morimoto S and Sasaguri T. Glycogen synthase kinase-3β is tyrosine-phosphorylated by MEK1 in human skin fibroblasts. Biochemical and Biophysical Research Communications. 2004;316:411–415. [DOI] [PubMed] [Google Scholar]
- 68.Wang Q, Fiol C, DePaoli-Roach A and Roach P. Glycogen Synthase Kinase-3b is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. The Journal of Biological Chemistry. 1994;269:14566–14574. [PubMed] [Google Scholar]
- 69.Stachowski MJ, Holewinski RJ, Grote E, Venkatraman V, Van Eyk JE and Kirk JA. Phospho-Proteomic Analysis of Cardiac Dyssynchrony and Resynchronization Therapy. Proteomics. 2018;18:e1800079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.