

Abstract
Muscular dystrophies are approximately 50 devastating, untreatable monogenic diseases leading to progressive muscle degeneration and atrophy. Gene correction of transplantable cells using CRISPR/Cas9-based tools is a realistic scenario for autologous cell replacement therapies to restore organ function in many genetic disorders. However, muscle stem cells have so far lagged behind due to the absence of methods to isolate and propagate them and their susceptibility to extensive ex vivo manipulations. Here, we show that mRNA-based delivery of SpCas9 and an adenine base editor results in up to >90% efficient genome editing in human muscle stem cells from many donors regardless of age and gender and without any enrichment step. Using NCAM1 as an endogenous reporter locus expressed by all muscle stem cells and whose knockout does not affect cell fitness, we show that cells edited with mRNA fully retain their myogenic marker signature, proliferation capacity, and functional attributes. Moreover, mRNA-based delivery of a base editor led to the highly efficient repair of a muscular dystrophy-causing SGCA mutation in a single selection-free step. In summary, our work establishes mRNA-mediated delivery of CRISPR/Cas9-based tools as a promising and universal approach for taking gene edited muscle stem cells into clinical application to treat muscle disease.
Keywords: MT: DNA editing, human muscle stem cells, CRISPR/Cas9, gene editing, base editing, muscular dystrophy, stem cell therapy, mRNA delivery
Graphical abstract
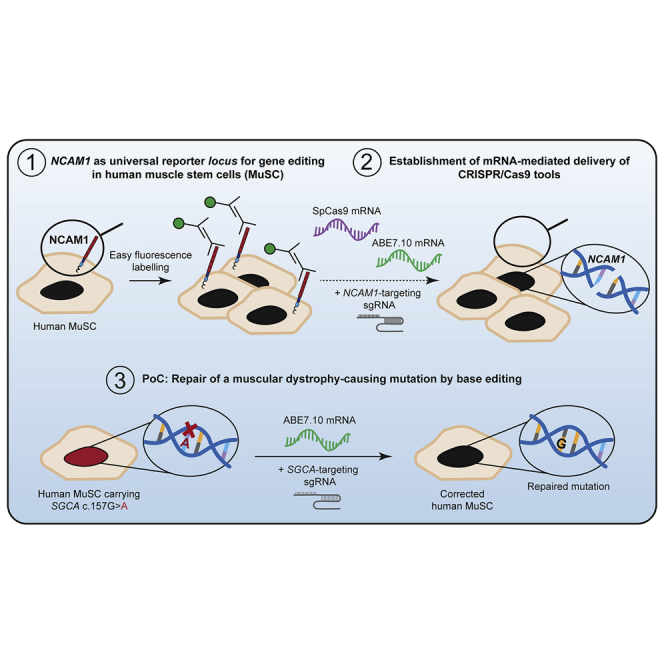
Gene-repaired muscle stem cells (MuSCs) are an ideal source for autologous cell therapies to treat muscular dystrophy. We established highly efficient, reproducible, and safe gene editing of human primary MuSCs via the mRNA-mediated delivery of CRISPR/Cas enzymes and validated our approach by repairing a muscular dystrophy-causing SGCA mutation with >80% efficiency.
Introduction
Cell and gene therapies are on their way to becoming a realistic option for patients with muscular dystrophies (MDs).1, 2, 3 MDs are a group of approximately 50 severe, debilitating monogenic disorders leading to a progressive inability to use muscles for daily activities up to the dependence on respiratory support in some patients.4 So far, treatment consists of supportive measures, but correcting the underlying genetic defect in a sufficient number of cells in muscles would be a cure.
Adult skeletal muscle can regenerate thanks to a dedicated pool of muscle stem cells (MuSCs), also called satellite cells due to their localization in a very specialized anatomical niche beneath the basal lamina that surrounds multinucleated muscle fibers.5 They have the capacity to self-renew and to give rise to committed progenitors and subsequently myofibers through the activation of a differentiation cascade involving key myogenic transcription factors (Figure S1). Due to the scarcity of MuSCs, various methods have been developed to produce myogenic cells from various sources to use in cell replacement therapies for MDs. The allogeneic transplantation of myoblasts or mesangioblasts has so far failed to achieve any clinical benefit.1 Induced pluripotent stem cells (iPSCs) are an attractive and unlimited source of healthy cells, and several protocols are in place to differentiate them into transplantable myogenic progenitor-like cells.6,7 However, iPSC-derived muscle cells do not yet meet quality and safety criteria for transplantation into patients. Overall, allogeneic transplantation requires immunosuppression or cloaking of transplantable cells,8,9 adding another level of uncertainty, and it has not yet been shown for iPSC-derived muscle cells. In addition, MuSCs are irreplaceable in muscle regeneration;10,11 thus, a long-term therapeutic effect of a stem cell therapy would require that the transplanted cells can reconstitute the MuSC compartment.
Primary MuSCs would therefore be more predictable as a source for autologous transplantation as there is little chance that immunomodulation would be needed and they would likely give rise to both healthy, stable muscle fibers and new quiescent MuSCs.12,13 However, using MuSCs in autologous cell replacement therapies for MDs requires ex vivo correction of the disease-causing mutation before transplantation to ensure a sustained healthy muscle turnover by the grafted cells.
CRISPR/Cas systems allow for unprecedented precision and relative ease in targeting defined regions of the genome. Since the original description of Cas9 as an RNA-guided endonuclease,14 numerous directed evolution and mutagenesis approaches have resulted in Cas enzymes with enhanced specificity, and thus increased safety with respect to on- versus off-target editing profiles.15,16 In addition, engineered Cas9-fusion proteins such as base and prime editors have enabled highly precise rewriting of the genome independent of cellular DNA repair pathway, cell-cycle phase or an exogenous DNA template.17,18 One example is adenine base editors (ABEs), which convert A to G nucleotides at target genomic loci by combining the RNA-guided DNA-binding capacity of Cas9 with the enzymatic activity of an evolved tRNA adenosine deaminase.19
Using plasmid-based ex vivo delivery of an ABE, we previously achieved a nearly 100% correction of a G>A MD-causing mutation in the gene encoding α-sarcoglycan (SGCA c.157G>A) in primary human MuSCs. The corrected cells exhibited their full myogenic and regenerative potential in xenografts.20 However, plasmid-based delivery carries a risk of transgene integration and leads to a relatively long exposure to gene-modifying enzymes, which in turn increases the probability of off-target mutagenesis. mRNA-mediated transgene delivery could provide transient expression while eliminating the risk of transgene integration.
We aimed to explore the suitability of mRNA-based ex vivo delivery of CRISPR/Cas9 tools into human MuSCs to perform targeted genome modifications. In a systematic way, we established NCAM1 as an easily accessible and universal endogenous reporter locus to test gene editing approaches in primary MuSCs in a donor- and mutation-independent manner. NCAM1 is a membrane protein with extracellular epitopes easily detected by immunostaining and flow cytometry and is expressed in all cultured human MuSCs and myoblasts.21
Results
mRNA nucleofection results in close to 100% transfection efficiency of primary human MuSCs with minimal toxicity
To establish a protocol for mRNA-mediated transgene delivery into human primary MuSCs, we first used mRNA encoding green fluorescent protein (GFP). The nucleofection of GFP mRNA in a range of 0.2–2 μg/150,000 cells resulted in >98% transfection efficiency of human MuSCs, outperforming plasmid-based delivery (Figures 1A and 1B). The nucleofection of GFP mRNA also led to highly homogeneous and mRNA dose-dependent transgene expression levels not seen after plasmid transfection (Figures 1A, 1C, and 1D). Cell fitness and viability are paramount if edited cells are intended for use in transplantation therapies. To determine the parameters resulting in high transfection efficiency and minimal cellular toxicity, we compared eight different nucleofection programs (Figure 1E). We found that a transfection efficiency of close to 100% could be achieved robustly using program 1. We therefore used this nucleofection program for subsequent experiments. We further learned that the transfection efficiency of close to 100% could be maintained while increasing the cell viability to >80% with program 5 (as compared to untransfected cells) (Figure 1E).
Figure 1.

mRNA nucleofection enables close to 100% transfection of human MuSCs with minimal toxicity
(A) Representative histograms showing GFP fluorescence intensity of MuSCs transfected with GFP-encoding plasmid or mRNA in a range of doses per 150,000 cells at day 1 after nucleofection. (B) Transfection efficiency of MuSCs transfected with GFP encoding plasmid or mRNA in a range of concentrations (n = 5, means ± SDs). (C) Median GFP fluorescence intensity of MuSCs transfected with GFP encoding mRNA (n = 5, medians ± SDs). (D) Representative fluorescence images of MuSCs transfected with GFP encoding plasmid or mRNA in a range of concentrations; scale bar: 400 μm. (E) Effect of different nucleofection programs (decreasing strength top to bottom) on MuSC transfection efficiency and viability. Cell viability was determined for programs resulting in close-to-100% transfection efficiency (1–5) and was normalized to untransfected cells (U) (n ≥ 3; means ± SDs); p value calculated with the Kruskal-Wallis test and a post hoc Dunn’s test. ∗p < 0.05; ∗∗∗p < 0.001. Untr., untransfected.
mRNA-mediated delivery of SpCas9 results in highly efficient gene editing in MuSCs from many donors
To develop and systematically assess a pipeline for mRNA-mediated delivery of gene editing tools to primary human MuSCs, we aimed to establish a universal readout system relevant to MuSCs from all donors. For that purpose, we designed a strategy to target the gene encoding neural cell adhesion molecule 1 (NCAM1), a membrane protein expressed by all human MuSCs and myoblasts with an extracellular domain that is easy to detect in living cells (Figures 2A, 2B, and S2A). We delivered mRNA encoding Streptococcus pyogenes Cas9 (SpCas9) to MuSCs from six donors of different ages and genders (Tables 1 and S1). The transfection of a range of SpCas9 mRNA concentrations and an single-guide RNA (sgRNA) targeting NCAM1 exon 3 at a constant ratio resulted in the efficient formation of small insertions and deletions (indels) at the expected DNA double-strand break (DSB) site, with the highest editing rate observed for 2 μg of SpCas9 mRNA (Figures S2B–S2D). Gene editing led to comparable rates of NCAM1 protein knockout as assessed by immunofluorescence staining and flow cytometry (Figures 2C–2E and S2E). We selected this concentration to further validate NCAM1 as a suitable locus to assess present and future editing strategies and their impact on human MuSCs. We investigated the kinetics of NCAM1 protein knockout and its effects on viability and proliferative potency. We assessed whether there was a shift in the percentage of edited cells with extended cultivation times after nucleofection (Figure S2A). We observed an increase in the percentage of edited MuSCs from day 2 to day 8 after nucleofection for all of the donors, reaching indel rates of up to >90% at day 8 (Figures 2F and S2F). Consistently, NCAM1-positive cells decreased between days 4 and 6 after nucleofection and remained constant thereafter (Figure 2G).
Figure 2.

mRNA-mediated SpCas9 delivery results in efficient knockout of NCAM1 in human MuSCs
(A) NCAM1 knockout strategy targeting exon 3. Expected cleavage site, dotted line. (B) Experiment setup. MuSCs were transfected with SpCas9 mRNA with or without sgRNA and processed for DNA, flow cytometry, and microscopy analysis. NCAM1 extracellular epitopes are easily detectable via immunolabeling. (C) Representative images of NCAM1 protein knockout in MuSCs, day 9 post-transfection; scale bar: 100 μm. (D) Flow cytometry analysis of NCAM1 expression in edited MuSCs and controls, day 8 post-transfection. (E) Quantification of D; n = 6 donors; means ± SDs. (F) Predicted editing efficiency (ICE) at days 2 and 8 post-transfection (n = 6; means ± SDs). (G) Percentage of NCAM1+ cells at days 4, 6, and 8 after transfection assessed by flow cytometry for a subset of the samples from (E) (n = 3; means ± SDs). p value calculated with Wilcoxon matched-pairs signed rank test. ∗p < 0.05. Untr., untransfected.
Table 1.
MuSC donors used in this study
| Donor ID | Age at biopsy (y) | Gender (m/f) | Muscle histology | MD mutation |
|---|---|---|---|---|
| A | 21 | m | normal histology | no |
| B | 15 | m | normal histology | no |
| C | 24 | f | normal histology | no |
| D | 31 | m | normal histology | no |
| E | 23 | m | normal histology | heterozygous RYR1 c.14545G>A |
| F | 43 | m | normal histology | heterozygous SGCA c.157G>A |
| G | 72 | f | normal histology | no |
| H | 61 | m | normal histology | no |
| I | 43 | f | normal histology | no |
| J | 50 | f | normal histology | no |
| K | 13 | m | normal histology | no |
| L | 32 | m | normal histology | no |
| M | 24 | m | normal histology | FHL1 c.687delA |
mRNA-mediated delivery of ABE7.10 results in highly efficient selection-free base editing of human MuSCs
Base editors are considered relatively safe for therapeutic applications. We therefore investigated mRNA-based delivery of ABE7.10.19 We designed an sgRNA to mutate the splice donor site of NCAM1 exon 7 (Figure 3A). Triggering exon 7 skipping via disruption of the canonical 5′ splice site should induce a frameshift and the appearance of a premature stop codon in exon 8, thus leading to a knockout of NCAM1. mRNA-delivery of ABE7.10 and the corresponding sgRNA resulted in >90% A to G conversion of the target adenine located in the center of the consensus ABE activity window (protospacer positions 4–8), at protospacer position 5 (A5), as confirmed via amplicon sequencing (Figures 3B–3D). A neighboring adenine at protospacer position 8 (A8) was co-edited in up to 22% of the sequencing reads. In contrast, plasmid-based experiments using the identical splice donor targeting strategy resulted in a lower mean editing efficiency and lower rates of A5 versus A8 to G conversion (Figure S3). However, the described nucleotide changes at A5 and A8 did not result in a clear NCAM1 protein knockout (Figure S4A). We analyzed the splicing patterns around NCAM1 exon 7 and found that disruption of the canonical splice donor site by base editing resulted in several intron retention and truncated splice isoforms, some of which encode in-frame transcripts (Figures S4B and S4C).
Figure 3.

mRNA-mediated ABE7.10 delivery leads to almost 100% disruption of an NCAM1 splice donor in human MuSCs
(A) NCAM1 exon 7 splice donor site (GT, orange) targeting strategy via ABE7.10 (editing window: dotted rectangle). Target adenine: A5. (B) Schematic overview of experimental workflow. MuSCs were transfected with ABE7.10 mRNA and then processed for gDNA analysis. (C) Representative Sanger-sequencing chromatogram of edited (bottom) compared to non-edited (top) MuSCs. The dotted rectangle indicates the target adenine, A5. (D) Percentage of next-generation sequencing (NGS) reads with A > G conversion of adenines located at protospacer position 5 (A5) and 8 (A8) (n = 3, means ± SDs). Untr., untransfected.
Human MuSCs retain their myogenic and proliferative properties following mRNA-mediated gene and base editing
To determine whether mRNA-mediated delivery of gene editing tools or knockout of NCAM1 altered the myogenic or proliferative properties of human MuSCs, we analyzed the expression profiles of myogenic and proliferation markers of passage-matched unedited and edited cells from the same donor. Purity of the MuSC populations remained constant and higher than 95% as determined by the myogenic marker Desmin (DES) and counterstained with the fibroblast marker TE7. The myogenic transcription factors PAX7, MYF5, and the proliferation marker Ki-67 were similarly expressed in edited and unedited MuSCs. Ki-67-positive proliferating MuSCs varied between cell populations from 30% to 60% before and after editing (Figures 4A, 4B, S5, S6A, and S6B). We next assessed the differentiation capacity of edited MuSCs in vitro with fusion assays. All of the edited and unedited MuSC populations gave rise to multinucleated myotubes with the typical striated pattern (Figures 4C, 4D, and S6C). Fusion indices remained constant between donor- and passage-matched untransfected cells, and cells edited by SpCas9 or ABE7.10 mRNA and the respective sgRNA (Figures 4E and 4F).
Figure 4.

Human MuSCs preserve their myogenic properties after mRNA-mediated editing of NCAM1
(A) Representative confocal microscopy images of MuSCs stained for DES, PAX7, and MYF5 after NCAM1 knockout using SpCas9 mRNA (2 μg, day 5 post-transfection). Scale bar: 50 μm. (B) Fold change of the percentage of cells expressing myogenic and proliferation markers at day 5 after transfection of SpCas9 mRNA with or without sgRNA (compared to passage and donor-matched untransfected cells) (n = 6, means ± SDs; unpaired t test; no significant changes). (C) Representative confocal microscopy images of MuSCs differentiated into myotubes after NCAM1 knockout using SpCas9 mRNA, and immunostained for myosin heavy chain (MYHC). Scale bar: 50 μm. (D) Representative confocal microscopy images of MuSCs differentiated into myotubes after NCAM1 editing using ABE7.10 mRNA and immunostained for MYHC. Scale bar: 50 μm. (E and F) Fusion indices calculated from (C) and (D); means ± SDs (n = 2–3 donors and ≥200 nuclei per sample); p value calculated with Friedman test with Dunn’s multiple comparison test; no significant changes. Nuclei were counterstained with Hoechst (blue). Untr., untransfected.
mRNA-based ABE delivery efficiently corrects the SGCA c.157G>A MD-causing mutation in human MuSCs
The MD-causing SGCA c.157G>A mutation can be repaired highly efficiently in human MuSCs by plasmid-based delivery of ABE7.10 and a suitable sgRNA, following the enrichment of transfected cells using a fluorescence reporter.20 We asked whether a comparable repair efficacy could be achieved through the mRNA-mediated delivery of ABE7.10 in the absence of any selection marker. We transfected human MuSCs carrying a heterozygous SGCA c.157G>A mutation with ABE7.10 mRNA and the corresponding sgRNA (Figure 5A). mRNA-mediated base editing efficiently repaired the mutation, resulting in c.157G nucleotide rates of >80%. We found bystander editing of the adenine located at protospacer position 10 (outside the consensus editing window) in a very low percentage of reads, <0.4%. We did not detect reads containing indels specific for mRNA-transfected or edited samples (Figures 5B, 5C, and S7).
Figure 5.

mRNA-mediated delivery of ABE7.10 repairs the SGCA c.157G>A mutation in human MuSCs without selection
(A) ABE strategy to repair the SGCA c.157G>A mutation. The target adenine (A6, red) is located in protospacer position 6, in the center of the ABE editing window (dotted line). (B) EditR analysis of nucleotide rates at each protospacer position in MuSCs from a heterozygous SGCA c.157G>A mutation carrier transfected with 3 μg ABE7.10 mRNA with or without sgRNA. (C) Percentage of amplicon sequencing reads containing c.157G, bystander A > G editing of a neighboring adenine (A10), and indels. Carrier MuSCs untransfected or transfected with 3 μg ABE7.10 mRNA, with or without sgRNA (n = 3 repeats, means ± SDs).
Discussion
Bringing autologous gene-corrected primary MuSCs into clinical application for the restoration of defined muscles in MD is a foreseeable development that requires a thorough understanding of the necessary gene editing steps, their safety profile, and a highly standardized and validated procedure. mRNA-mediated delivery provides a platform for time-restricted and integration risk-free exogenous expression of gene-modifying enzymes for therapeutic genome editing applications. We demonstrate highly efficient gene editing of primary human MuSCs from a variety of donors from different age groups (15–75 years) and genders using mRNA-mediated delivery of CRISPR/Cas9-based tools. Nucleofection allows the delivery of mRNA to almost 100% of the cells, resulting in highly uniform transgene expression levels and eliminating the need for reporter genes or enrichment steps to obtain a homogeneous population of gene edited primary MuSCs. It remains to be explored whether similar mRNA transfection efficiency and cell viability can be achieved using different electroporation systems.
Establishing an optimized pipeline for each delivery method and gene editing tool across MuSCs derived from many donors requires a target locus that is universally accessible, provides an easy readout, and whose functional knockout at the protein level does not interfere with cell fitness, thereby enabling a thorough analysis of edited cells. NCAM1 is the receptor for the Zika virus22 and mutations in NCAM1 have been associated with defective brain connectivity and schizophrenia.23 In muscles, NCAM1 is found at the membrane of all human MuSCs and myoblasts,24 and its extracellular epitopes are easily detectable by immunolabeling. Mutations in NCAM1 have not been associated with muscle disease. We found that MuSCs bearing an edited NCAM1 gene did not show a selective advantage or disadvantage regardless of editing method and NCAM1 protein expression status. Moreover, using NCAM1 as a model locus, we show that mRNA nucleofection, exogenous expression of SpCas9, or ABE7.10, as well as editing did not affect the myogenic gene expression signature or proliferation capacity of MuSCs from all of the donors.
We observed a >90% rate of T > C conversion (A > G on the opposite strand) of the second nucleotide at the consensus 5′ splice site of NCAM1 exon 7 by base editing. ABE-mediated disruption of splice donors has been shown to correlate well with protein loss.25 Skipping of NCAM1 exon 7 from the mRNA should induce a frameshift with the subsequent introduction of a premature stop codon and nonsense-mediated mRNA decay. However, the mutation of this splice donor site resulted in only a minor reduction in the percentage of NCAM1-expressing cells. The presence of several truncated and intron retention isoforms in base edited MuSCs suggests the use of cryptic splice sites upon the disruption of the canonical splice donor. Some of those splice isoforms are in-frame and predicted to result in the translation of a full-length protein with some changes in the amino acid sequence in this region.
Ultimately, our aim is to establish a pipeline to correct MD-causing mutations whereby methodological aspects other than the specific gene editing enzyme and sgRNA sequences are applicable to MuSCs from all or most donors. Using the mRNA-mediated delivery of ABE7.10 to precisely repair the SGCA c.157G>A mutation in cells from a heterozygous carrier, we were able to achieve high c.157G nucleotide rates of >80% without taking advantage of a selection marker. Increasing mRNA stability by optimizing the UTR and 5′ cap configuration or by using modified RNA bases, as well as using an ABE variant with a faster adenosine deamination kinetics such as ABE8e or ABE8,26,27 may increase the editing rates, but they may also influence off-target events. Very high on-target editing efficiencies are particularly desirable in the context of cell replacement therapies for genetic muscle disease, whereby the transplanted cells need to reconstitute the diseased tissue. However, the trade-off between on-target editing rates and specificity requires particular attention in any clinical context. Off-target nomination with relevant cell-based assays and in silico predictions, followed by next-generation sequencing-based validation approaches could be the way to go to bring autologous gene edited MuSCs to patients.
We focused on editing MuSCs ex vivo. In vivo gene repair of myonuclei and MuSCs using mRNA encoding CRISPR/Cas-based tools will be a future perspective. Lipid nanoparticle (LNP)-mediated delivery of mRNA in vivo has been introduced in the mRNA-based vaccines against SARS-CoV-2 and was also shown in the context of ABE of the PCSK9 gene to treat hypercholesterolemia in non-human primates.28, 29, 30 The first clinical trial of systemic in vivo mRNA-based delivery of CRISPR/Cas9 components is presently performed to target the liver of patients with hereditary transthyretin amyloidosis.31,32 However, skeletal muscle accounts for approximately 40% of adult body mass, and systemic LNP-mediated mRNA delivery to target tissues other than the liver is still a challenge.
To treat degenerative genetic muscle disease, both in vivo gene repair and cell-based gene therapy approaches are complementary. Autologous transplantation of MuSCs has been shown to result in the functional improvement of the swallowing muscles in oculopharyngeal MD patients. These cells were not genetically corrected and therefore only of limited long-term therapeutic value.33 Transplanting autologous healthy MuSCs could have a lifelong therapeutic effect. Indeed, despite their limited availability and local migratory potential, it is becoming increasingly clear that well-defined and highly myogenic MuSCs could be a feasible, safe, and efficacious cell source for autologous cell replacement therapies to treat muscle-wasting disorders.13,20,34, 35, 36, 37 Obtaining large numbers of highly regenerative ex vivo expanded primary MuSCs for autologous therapies is still a challenge, but significant progress has been made in recent years.13,34,38,39 The number of cells needed for the functional reconstitution of human muscles has yet to be determined and may depend on many factors related to both the fitness of the transplanted cells and the environment of the recipient muscle. Eye-closure muscles, finger flexors, and distal leg muscles all may profit from sustainable autologous transplantations.
In summary, we show that mRNA is an ideal substrate to deliver gene editing tools to primary human MuSCs, enabling dose-dependent and integration risk-free transgene expression and genome editing in cells from a wide range of donors without affecting their proliferative, myogenic, and differentiation properties. Our results bring gene edited human MuSCs one step closer to clinical application in transplantation therapies to treat skeletal muscle disease.
Materials and methods
Study approval
The research use of human material was approved by regulatory agencies (EA2/051/10 and EA2/175/17, Charité – Universitätsmedizin Berlin), and written informed consent was obtained from donors or legal guardians.
mRNA and sgRNA
Codon-optimized ABE7.10 mRNA based on the amino acid sequence of ABE7.10_4.120 was purchased from AmpTec (Hamburg, Germany). SpCas9 and GFP mRNA were purchased from Aldevron (North Dakota, USA). sgRNA were purchased from Integrated DNA Technologies (IDT) (Iowa, USA) or Synthego (California, USA). The sgRNA sequences are listed in Table S2.
Vectors
To obtain a fully codon-optimized vector for ABE7.10 expression in mammalian cells, the previously described ABE7.10_4.1 plasmid containing a T2A-Venus reporter and a human U6 promoter-driven sgRNA expression cassette20 was digested with PacI and BglII to excise the TadA heterodimer and the 5′ region of SpCas9(D10A). The corresponding codon-optimized sequence (GeneArt Gene Optimizer, Thermo Fisher Scientific, Massachusetts, USA) synthesized as a gBlock Gene Fragment (IDT; Table S2) with 20 nucleotide homology arms was inserted using the Gibson Assembly Mix (New England Biolabs, Massachusetts, USA). The new vector was called ABE7.10co_4.1. All of the plasmid-based ABE experiments described here were performed using ABE7.10co_4.1, although for simplicity, we refer to it as ABE7.10. sgRNA cloning was performed as described.20 Briefly, ABE7.10_4.1co was digested with BplI and oligos containing the spacer sequence, and overhangs to the BplI-digested vector backbone were annealed and ligated. All of the constructs were confirmed via Sanger sequencing. Oligos and sgRNAs used for cloning are listed in Table S3.
Primary MuSC isolation and culture
MuSC isolation was performed as described.13,20,34 Primary MuSC populations used in this study are listed in Tables 1 and S1. Primary MuSCs were cultured in Skeletal Muscle Cell Growth Medium (SMCGM, Provitro, Germany) enriched with supplement mix (Provitro) at 37°C in at humidified incubator with 5% CO2. For cell passaging, MuSCs were washed with Dulbecco’s phosphate-buffered saline (DPBS) (Thermo Fisher Scientific) and detached with 0.25% trypsin-EDTA (Provitro) or TrypLE Express (Gibco, Thermo Fisher Scientific) at 37°C for 5 min.
Plasmid transfection and fluorescence-activated cell sorting (FACS)
MuSCs were seeded 1 day before transfection at a density of 75,000 cells/9.5 cm2 in SMCGM. Lipofectamine 3000 (Thermo Fisher Scientific) was applied according to the manufacturer’s instructions to transfect 1 μg of plasmid DNA into 75,000 cells. The medium was changed 1 day after transfection. On the second day, the cells were harvested in SMCGM supplemented with 100 μg/mL Primocin (Invivogen, Massachusetts, USA) and Venus+ cells were selected using the FACSAria Fusion Cell Sorter (BD Biosciences, New Jersey, USA). Venus+ cells were plated and cultured in SMCGM with Primocin (100 μg/μL) for 2 days before further expansion without antibiotics.
Nucleofection
Primary MuSCs were harvested using TrypLE Express, spun down for 5 min at 200 × g, and washed once with DPBS. After a second spin down, the supernatant was removed and the cells were resuspended in P5 Primary Cell Nucleofector Solution (Lonza, Switzerland) already premixed with mRNA at a concentration of 7.5 × 106 cells per milliliter. For 3 μg of gene editing molecule-encoding mRNA, 2 μg of 5′/3′ end-modified sgRNA (1:0.67 ratio) were added to a 20 μL reaction. The cells were electroporated with the Amaxa 4D Nucleofector (Lonza) using the X Unit with 16-well nucleofection cuvettes using the program EY-100 (Except for Figure 1E: EY-100 [1], EO-100 [2], DU-100 [3]; DI-100 [4]; CX-100 [5]; CM-100 [6]; CF-100 [7]; CB-100 [8]). Afterward, 80 μL of prewarmed SMCGM was added to each cuvette and the cells were transferred to a single well of a 6-well plate containing 2 mL of prewarmed SMCGM. The cell culture medium was changed the day after.
Flow cytometry analysis and viability assessment
Data from GFP mRNA transfected cells were acquired with the FACSCanto flow cytometry analyzer (BD Biosciences) 18–24 h after nucleofection. Cultured primary MuSCs were detached with TripLE Express for 5 min at 37°C. The viability of 0.2 μg GFP mRNA transfected and untransfected MuSCs was evaluated by recording the total number of events detected per 60 s at constant speed (cells from all of the samples were collected in the same manner and resuspended in identical volumes). These counts were normalized to the untransfected control for each individual cell line used in this experiment. For NCAM1 (CD56) immunostaining, cultured primary MuSCs were detached with TripLE Express for 5 min at 37°C and centrifuged at 200 × g for 5 min (all of the centrifugation steps from here on were carried out in this way) at room temperature. After removal of the supernatant, cells were resuspended in 2 mL ice-cold staining buffer (1% bovine serum albumin [BSA] in DPBS) and centrifuged once more at 4°C. The buffer was aspirated, and the cell pellet was resuspended in ice-cold staining buffer containing the anti-NCAM1/CD56 primary antibody (Table S4) at a concentration of 0.1 μg/μL, except for the unstained control. All of the samples were incubated for 15 min at 4°C with gentle shaking every 5 min. After incubation, cells were washed twice with ice-cold staining buffer and incubated with AlexaFluor 647-conjugated secondary antibody (Invitrogen, Germany) diluted 1:500 in ice-cold staining buffer for 15 min at 4°C in the dark, with gentle shaking every 5 min. The samples were washed 3 times with ice-cold staining buffer, resuspended in ice-cold DPBS, and filtered through a 40 μm cell strainer cap into FACS tubes for flow cytometric analysis. Data were acquired with the LSCFortessa Flow Cytometer (BD Biosciences). All of the flow cytometry data were analyzed using the FlowJo Single Cell analysis tool version 10.7.1 (BD Biosciences).
Genomic DNA extraction and genome editing analysis via ICE and EditR
Genomic DNA (gDNA) was isolated with Agencourt AMPure XP beads (Beckman Coulter, California, USA) as described.20 For target site amplification, Phusion or Q5 High Fidelity DNA Polymerase (New England Biolabs) were used in combination with the primers SDF29 + SDF30, CS94 + CS95 or CS149 + CS152, and HE24 + HE25 for NCAM1 exon 3, NCAM1 exon 7, and SGCA exon 2, respectively. PCR products were purified using the NucleoSpin Gel and PCR Clean-up Kit (MACHERY-NAGEL, Germany). Sanger sequencing was performed by LGC Genomics (Germany), and the resulting chromatograms were analyzed with ICE (Synthego, version 2.0; https://www.synthego.com) and EditR40 (version 1.0.10).
Amplicon sequencing
PCR amplification of the target sites was carried out using Q5 High Fidelity DNA Polymerase (New England Biolabs) with primers CS149 + CS152 (Table S2) for NCAM1 exon 7 and HE255 + HE256 for SGCA exon 2. PCR products were run on an agarose gel to confirm their size and purity. After gel extraction using the NucleoSpin Gel and PCR Clean-up Kit (MACHERY-NAGEL), DNA concentration was measured with the Qubit fluorometer (Invitrogen). Samples were sent to GENEWIZ (Germany) for next-generation sequencing (Amplicon EZ service) using an Illumina MiSeq platform and 250-bp paired-end reads. Raw sequencing data are available at the NCBI Sequence Read Archive (SRA) under the Bioproject accession number PRJNA809319 (https://www.ncbi.nlm.nih.gov/sra/PRJNA809319). Results were analyzed using CRISPResso2.41 The following parameters were applied: Editing tool—base editors; sequencing design—paired-end reads; minimum homology for alignment to an amplicon—60%; base editor output—A > G; center of the quantification window (relative to the 3′ end of the provided sgRNA)—−10; quantification window size (base pair)—10; minimum average read quality (phred33 scale)—>30; minimum single base pair quality (phred33 scale)—no filter; replace bases with N that have a quality lower than (phred33 scale)—no filter; exclude base pairs from the left side of the amplicon sequence for the quantification of the mutations—15 bp; exclude base pairs from the right side of the amplicon sequence for the quantification of the mutations—15 bp.
RT-PCR and splice isoform identification
Total RNA was isolated from cultured MuSCs using TRIzol (Thermo Fisher Scientific) following the manufacturer's instructions. cDNA synthesis was performed using the QuantiTect Reverse Transcription kit (Qiagen, Hilden, Germany). RT-PCR was performed with primers CS184 + CS186 (Table S2) and Q5 High Fidelity DNA Polymerase (New England Biolabs). The identity of the resulting bands was confirmed by Sanger sequencing following gel extraction. Alternatively, the RT-PCR reaction was used for subcloning using the CloneJET PCR Cloning Kit (Thermo Fisher), and the ligation mix was transformed into electrocompetent DH10B E. coli bacteria. Colony PCR was performed according to the kit's instructions, and the resulting amplicons were cleaned up and sent for Sanger sequencing at LGC Genomics (Germany).
Immunostaining
Cells were plated on 8-well μ-Slides (ibidi, Germany) (8,000–10,000 cells/cm2) and fixed 1 day after with a 3.7% formaldehyde solution in DPBS for 10 min at room temperature. Cells seeded for the differentiation assay were fixed 3–4 days after inducing myoblast fusion. Cells were permeabilized using 0.2% Triton X-100 in DPBS (except for cells stained with the anti-NCAM1 antibody) and blocked with 5% BSA in DPBS for 1 h at room temperature. Primary antibodies were incubated overnight at 4°C as indicated in Table S4. Alexa Fluor 488- or 568-conjugated secondary antibodies (Invitrogen) were incubated for 1 h at room temperature at a 1:500 dilution in DPBS. Nuclei were counterstained with Hoechst 33342 dye (Invitrogen). Images were acquired with the laser scanning confocal microscope LSM 700 (Carl Zeiss Microscopy GmbH, Germany) and the DMI6000 fluorescence microscope (Leica, Germany). Images were processed with the ZEN 2.3 software (Carl Zeiss Microscopy GmbH), ImageJ (Wayne Rasband, NIH, Bethesda, MD, USA) and Adobe Illustrator 2021. A total of ≥150 nuclei were counted per sample to calculate percentage values for myogenic and proliferation markers. After differentiation of MuSCs, the fusion index was calculated as the percentage of nuclei within myotubes versus the total number of nuclei. A myotube was defined as a muscle cell containing ≥2 nuclei. For fusion index analysis, nuclei were counted from 2 separate wells (≥200 nuclei counted per well) and from at least 3 distinct images captured at 20× magnification.
Statistics
All of the experiments were performed in at least three biologically different replicates unless otherwise indicated in the figure legend. Details about the group size and statistical tests are described in the corresponding figure legend. All of the statistical analyses and graphs were performed using GraphPad Prism software (version 8.0). Graphs show the means ± SDs where applicable.
Acknowledgments
This study was supported by the Gisela Krebs Foundation (grant to H.E.), the German Research Foundation (DFG; grant no. SP 1152/12-1), the Helmholtz Society through the Pre-GoBio program and the Helmholtz Validation Fund (HVF) through grants to S. Spuler, and through the Mighty Maje Initiative of the Deutsche Muskelschwund-Hilfe e.V. (German patient organization for muscle diseases) with a grant to C.S. We thank all of the muscle biopsy donors. We thank Janine Kieshauer and Dominique Braumann for their help in biopsy processing and cell characterization. We thank Andreas Spuler for performing the muscle biopsy procedures. We thank Stephanie Meyer-Liesener and Stefanie Haafke for excellent technical assistance.
Author contributions
C.S., S.D.F., and H.E. designed the experiments. C.S. and S.D.F. conducted and analyzed the experiments. A.M. developed the methods for the human MuSC isolation, propagation, and characterization. S.M. helped with the experimental design. C.S., S.D.F., H.E., and S. Spuler discussed the results. S. Spuler acquired the funding. H.E. and S. Spuler wrote the manuscript, with contributions from C.S. and S.D.F. H.E. and S. Spuler designed the study and coordinated the project.
Declaration of interests
S. Spuler and A.M. are inventors on a pending patent application (2016/030371)—technology for primary human MuSC isolation and manufacturing. S. Spuler, C.S. and H.E. are co-inventors on a pending patent application (European Patent Office 21 160 696.7) relevant to this publication.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.02.016.
Contributor Information
Simone Spuler, Email: simone.spuler@charite.de.
Helena Escobar, Email: helena.escobar@charite.de.
Supplemental information
References
- 1.Biressi S., Filareto A., Rando T.A. Stem cell therapy for muscular dystrophies. J. Clin. Invest. 2020;130:5652–5664. doi: 10.1172/JCI142031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boyer O., Butler-Browne G., Chinoy H., Cossu G., Galli F., Lilleker J.B., Magli A., Mouly V., Perlingeiro R.C.R., Previtali S.C., et al. Myogenic cell transplantation in genetic and acquired diseases of skeletal muscle. Front. Genet. 2021;12:702547. doi: 10.3389/fgene.2021.702547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ausems C.R.M., van Engelen B.G.M., van Bokhoven H., Wansink D.G. Systemic cell therapy for muscular dystrophies: the ultimate transplantable muscle progenitor cell and current challenges for clinical efficacy. Stem Cell Rev. Rep. 2021;17:878–899. doi: 10.1007/s12015-020-10100-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dowling J.J., Weihl C.C., Spencer M.J. Molecular and cellular basis of genetically inherited skeletal muscle disorders. Nat. Rev. Mol. Cell Biol. 2021;22:713–732. doi: 10.1038/s41580-021-00389-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mauro A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chal J., Oginuma M., Al Tanoury Z., Gobert B., Sumara O., Hick A., Bousson F., Zidouni Y., Mursch C., Moncuquet P., et al. Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat. Biotechnol. 2015;33:962–969. doi: 10.1038/nbt.3297. [DOI] [PubMed] [Google Scholar]
- 7.Darabi R., Arpke R.W., Irion S., Dimos J.T., Grskovic M., Kyba M., Perlingeiro R.C. Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell. 2012;10:610–619. doi: 10.1016/j.stem.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deuse T., Hu X., Gravina A., Wang D., Tediashvili G., De C., Thayer W.O., Wahl A., Garcia J.V., Reichenspurner H., et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat. Biotechnol. 2019;37:252–258. doi: 10.1038/s41587-019-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Luzy I.R., Law K.C.L., Moriarty N., Hunt C.P.J., Durnall J.C., Thompson L.H., Nagy A., Parish C.L. Human stem cells harboring a suicide gene improve the safety and standardisation of neural transplants in Parkinsonian rats. Nat. Commun. 2021;12:3275. doi: 10.1038/s41467-021-23125-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sambasivan R., Yao R., Kissenpfennig A., Van Wittenberghe L., Paldi A., Gayraud-Morel B., Guenou H., Malissen B., Tajbakhsh S., Galy A. Pax7-expressing satellite cells are indispensable for adult skeletal muscle regeneration. Development. 2011;138:3647–3656. doi: 10.1242/dev.067587. [DOI] [PubMed] [Google Scholar]
- 11.Lepper C., Partridge T.A., Fan C.M. An absolute requirement for Pax7-positive satellite cells in acute injury-induced skeletal muscle regeneration. Development. 2011;138:3639–3646. doi: 10.1242/dev.067595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins C.A., Olsen I., Zammit P.S., Heslop L., Petrie A., Partridge T.A. Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell. 2005:289–301. doi: 10.1016/j.cell.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 13.Marg A., Escobar H., Karaiskos N., Grunwald S.A., Metzler E., Kieshauer J., Sauer S., Pasemann D., Malfatti E., Mompoint D., et al. Human muscle-derived CLEC14A-positive cells regenerate muscle independent of PAX7. Nat. Commun. 2019;10:5776. doi: 10.1038/s41467-019-13650-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Slaymaker I.M., Gao L., Zetsche B., Scott D.A., Yan W.X., Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Komor A.C., Kim Y.B., Packer M.S., Zuris J.A., Liu D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533:420–424. doi: 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A·T to G·C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Escobar H., Krause A., Keiper S., Kieshauer J., Müthel S., de Paredes M.G., Metzler E., Kühn R., Heyd F., Spuler S. Base editing repairs an SGCA mutation in human primary muscle stem cells. JCI Insight. 2021;6:e145994. doi: 10.1172/jci.insight.145994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Illa I., Leon-Monzon M., Dalakas M.C. Regenerating and denervated human muscle fibers and satellite cells express neural cell adhesion molecule recognized by monoclonal antibodies to natural killer cells. Ann. Neurol. 1992;31:46–52. doi: 10.1002/ana.410310109. [DOI] [PubMed] [Google Scholar]
- 22.Srivastava M., Zhang Y., Chen J., Sirohi D., Miller A., Zhang Y., Chen Z., Lu H., Xu J., Kuhn R.J., et al. Chemical proteomics tracks virus entry and uncovers NCAM1 as Zika virus receptor. Nat. Commun. 2020;11:3896. doi: 10.1038/s41467-020-17638-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hildebrandt H., Mühlenhoff M., Oltmann-Norden I., Röckle I., Burkhardt H., Weinhold B., Gerardy-Schahn R. Imbalance of neural cell adhesion molecule and polysialyltransferase alleles causes defective brain connectivity. Brain. 2009;132:2831–2838. doi: 10.1093/brain/awp117. [DOI] [PubMed] [Google Scholar]
- 24.Capkovic K.L., Stevenson S., Johnson M.C., Thelen J.J., Cornelison D.D. Neural cell adhesion molecule (NCAM) marks adult myogenic cells committed to differentiation. Exp. Cell Res. 2008;314:1553–1565. doi: 10.1016/j.yexcr.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kluesner M.G., Lahr W.S., Lonetree C.L., Smeester B.A., Qiu X., Slipek N.J., Claudio Vázquez P.N., Pitzen S.P., Pomeroy E.J., Vignes M.J., et al. CRISPR-Cas9 cytidine and adenosine base editing of splice-sites mediates highly-efficient disruption of proteins in primary and immortalized cells. Nat. Commun. 2021;12:2437. doi: 10.1038/s41467-021-22009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richter M.F., Zhao K.T., Eton E., Lapinaite A., Newby G.A., Thuronyi B.W., Wilson C., Koblan L.W., Zeng J., Bauer D.E., et al. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020;38:883–891. doi: 10.1038/s41587-020-0453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaudelli N.M., Lam D.K., Rees H.A., Solá-Esteves N.M., Barrera L.A., Born D.A., Edwards A., Gehrke J.M., Lee S.J., Liquori A.J., et al. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020;38:892–900. doi: 10.1038/s41587-020-0491-6. [DOI] [PubMed] [Google Scholar]
- 28.Musunuru K., Chadwick A.C., Mizoguchi T., Garcia S.P., DeNizio J.E., Reiss C.W., Wang K., Iyer S., Dutta C., Clendaniel V., et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature. 2021;593:429–434. doi: 10.1038/s41586-021-03534-y. [DOI] [PubMed] [Google Scholar]
- 29.Rothgangl T., Dennis M.K., Lin P.J.C., Oka R., Witzigmann D., Villiger L., Qi W., Hruzova M., Kissling L., Lenggenhager D., et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat. Biotechnol. 2021;39:949–957. doi: 10.1038/s41587-021-00933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pilkington E.H., Suys E.J.A., Trevaskis N.L., Wheatley A.K., Zukancic D., Algarni A., Al-Wassiti H., Davis T.P., Pouton C.W., Kent S.J., et al. From influenza to COVID-19: lipid nanoparticle mRNA vaccines at the frontiers of infectious diseases. Acta Biomater. 2021;131:16–40. doi: 10.1016/j.actbio.2021.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.First systemic CRISPR agent in humans. Nat. Biotechnol. 2020;38:1364. doi: 10.1038/s41587-020-00773-8. [DOI] [PubMed] [Google Scholar]
- 32.Gillmore J.D., Gane E., Taubel J., Kao J., Fontana M., Maitland M.L., Seitzer J., O'Connell D., Walsh K.R., Wood K., et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 2021;385:493–502. doi: 10.1056/NEJMoa2107454. [DOI] [PubMed] [Google Scholar]
- 33.Périé S., Trollet C., Mouly V., Vanneaux V., Mamchaoui K., Bouazza B., Marolleau J.P., Laforêt P., Chapon F., Eymard B., et al. Autologous myoblast transplantation for oculopharyngeal muscular dystrophy: a phase I/IIa clinical study. Mol. Ther. 2014;22:219–225. doi: 10.1038/mt.2013.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marg A., Escobar H., Gloy S., Kufeld M., Zacher J., Spuler A., Birchmeier C., Izsvak Z., Spuler S. Human satellite cells have regenerative capacity and are genetically manipulable. J. Clin. Invest. 2014;124:4257–4265. doi: 10.1172/JCI63992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia S.M., Tamaki S., Lee S., Wong A., Jose A., Dreux J., Kouklis G., Sbitany H., Seth R., Knott P.D., et al. High-yield purification, preservation, and serial transplantation of human satellite cells. Stem Cell Rep. 2018;10:1160–1174. doi: 10.1016/j.stemcr.2018.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barruet E., Garcia S.M., Striedinger K., Wu J., Lee S., Byrnes L., Wong A., Xuefeng S., Tamaki S., Brack A.S., et al. Functionally heterogeneous human satellite cells identified by single cell RNA sequencing. Elife. 2020;9:e51576. doi: 10.7554/eLife.51576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Novak J.S., Mázala D.A.G., Nearing M., Hindupur R., Uapinyoying P., Habib N.F., Dickson T., Ioffe O.B., Harris B.T., Fidelia-Lambert M.N., et al. Human muscle stem cells are refractory to aging. Aging Cell. 2021;20:e13411. doi: 10.1111/acel.13411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gilbert P.M., Havenstrite K.L., Magnusson K.E.G., Sacco A., Leonardi N.A., Kraft P., Nguyen N.K., Thrun S., Lutolf M.P., Blau H.M. Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science. 2010;329:1078–1081. doi: 10.1126/science.1191035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quarta M., Brett J.O., DiMarco R., De Morree A., Boutet S.C., Chacon R., Gibbons M.C., Garcia V.A., Su J., Shrager J.B., et al. An artificial niche preserves the quiescence of muscle stem cells and enhances their therapeutic efficacy. Nat. Biotechnol. 2016;34:752–759. doi: 10.1038/nbt.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kluesner M.G., Nedveck D.A., Lahr W.S., Garbe J.R., Abrahante J.E., Webber B.R., Moriarity B.S. EditR: a method to quantify base editing from sanger sequencing. CRISPR J. 2018;1:239–250. doi: 10.1089/crispr.2018.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E., et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019;37:224–226. doi: 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.