

Summary
Classic approaches to characterizing cell cycle leverage chemicals or altered nucleotide pools, which could impact chromatin states at specific phases of the cell cycle. Such approaches could induce metabolic alterations and/or DNA damage, which could reshape protein recruitment and histone modifications. In this protocol, we describe ways to fix and sort cells across the cell cycle based on their DNA content. We further detail immunoprecipitation and library preparation, allowing analysis of the epigenome by chromatin immunoprecipitation sequencing (ChIP-seq) for small numbers of cells.
For complete details on the use and execution of this protocol, please refer to Van Rechem et al. (2021).
Subject areas: Cell isolation, Flow Cytometry/Mass Cytometry, ChIPseq, Molecular Biology, Chromatin immunoprecipitation (ChIP)
Graphical abstract
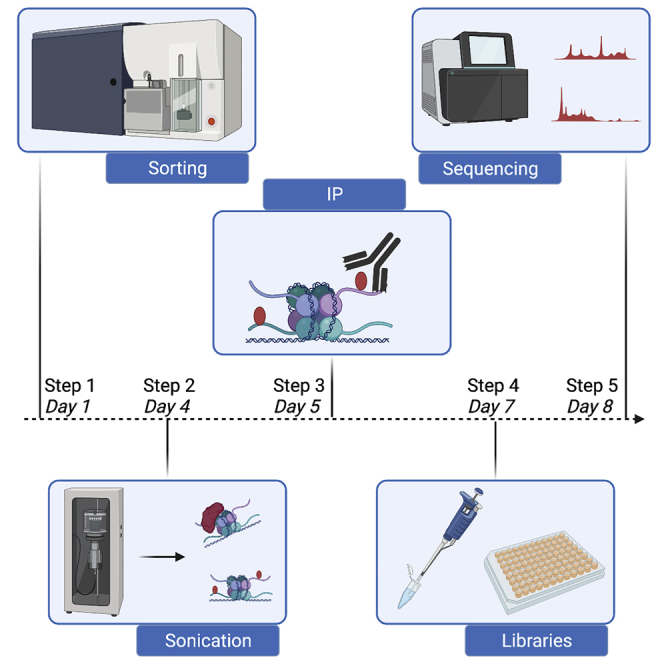
Highlights
-
•
Drugs are not used to arrest cells at different cell cycle phases
-
•
This protocol is adapted to small amounts of chromatin for ChIP-seq
-
•
This protocol can be adapted to separate additional cell cycle phases
-
•
This protocol can be adapted to any cycling cells and other histone modifications
Classic approaches to characterizing cell cycle leverage chemicals or altered nucleotide pools, which could impact chromatin states at specific phases of the cell cycle. Such approaches could induce metabolic alterations and/or DNA damage, which could reshape protein recruitment and histone modifications. In this protocol, we describe ways to fix and sort cells across the cell cycle based on their DNA content. We further detail immunoprecipitation and library preparation, allowing analysis of the epigenome by chromatin immunoprecipitation sequencing (ChIP-seq) for small numbers of cells.
Before you begin
Experimental conditions described in this protocol apply to hTERT RPE-1 cells. However, we have also used this protocol successfully in HEK293T cells.
All buffers in the tables (buffers section) need to be prepared in advance and stored at the reported temperature.
Optimize sonication
Timing: 2 days to few weeks
This optimization step can take longer for two main reasons: (1) cells growth rate and (2) difficulty to sonicate certain cell types. One might realize that even the longest tested sonication time is not enough and restart over with adjusted sonication time to test.
Note: while we optimize the sonication with unsorted cells, sorted cells can be easier to sonicate and may need less sonication time (∼5 min less). Because of the time and price of sorting (especially if using a facility) we do not optimize with sorted cells, but one could decide to do so.
-
1.
Plate cells in one 15 cm2 plate in order to obtain 70% confluency at 48 h (∼1 × 107 cells per plate).
-
2.
Add formaldehyde to the medium at a final concentration of 1%, swirl to mix and incubate at 37°C for 13 min.
CRITICAL: formaldehyde should be freshly opened and is toxic, use gloves and dispose properly.
-
3.
Add 1.25 M glycine pH 2.5 at a final concentration of 0.125 M to stop the crosslink, swirl to mix and incubate for 2 min minimum.
Note: during the quenching the medium should turn bright yellow.
Note: the efficacy of glycine is improved by low pH.
-
4.
Remove the medium and wash the cells with cold 1× PBS.
-
5.
Collect the cells with a lifter with 1× PBS and transfer in a conical tube. Centrifuge for 2 min at 100 g at 4°C.
Note: use a cell lifter instead of the scraper which damage less the cells.
-
6.
Resuspend the cells in 1 mL cellular lysis buffer freshly complemented with protease inhibitors (cocktail tablets EDTA free). Incubate 5 min on ice and centrifuge for 2 min at 100 g at 4°C.
-
7.
Discard the supernatant by aspirating or pipetting and resuspend the cells in 300 μL of nuclear lysis buffer freshly complemented with protease inhibitors (cocktail tablets EDTA free). This corresponds to ∼1 × 107 cells in total.
Note: the amount of nuclear lysis buffer can be adjusted according to the number of cells.
-
8.
Sonication: we use the QSonica Q800R system with the chiller turned on. Transfer the 300 μL of chromatin in 0.5 mL thin wall PCR tubes. Sonicate at amplitude 70% 15 s ON 45 s OFF for 20 min of total time ON.
Note: we used 0.5 ml thin wall PCR tubes (BRAND 781312) but any equivalent (thin wall) will work.
Note: we found this sonicator optimal for DNA sonication, however equivalent bath sonicators could be used and will need adapting for the sonication time and amplitude.
Note: if you do not know how much sonication your cells will need, sonicate for 10 min, remove few microliters from the tube, add 5 extra min, remove few microliters, and so on up to 40 min of sonication time, and test all these times following the next steps.
Note: do not over sonicate, the size will not decrease further but the chromatin will start to be altered and the ChIP will be less efficient.
Note: the bath should never get hot.
-
9.
Add 3 μL of chromatin and 1 μL of proteinase K to 65 μL of elution buffer, incubate at 65°C for 4–16 h in a benchtop thermomixer shaking at 1,000 rpm.
-
10.
Add 65 μL of phenol/chloroform/isoamyl alcohol 25:24:1, vortex, centrifuge for 1 min at 20,000 g at 18°C–25°C.
-
11.
Take 20 μL of the upper phase, mix with appropriate dye and run on a 1% agarose gel. The chromatin should be under 300 bp. If there is a big tail, the chromatin is not sonicated enough (see Figure 1). Troubleshooting 1.
Note: use a DNA loading dye with only one tracking color that runs at a high molecular to avoid using a dye running at the same size as your expected chromatin as it would hide it.
Figure 1.

Example of agarose gel profiles after sonication
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-H3K4me1, 0.2 μg | Abcam | Cat# ab8895, RRID:AB_306847 |
| Anti-H3K4me2, 0.1 μg | Abcam | Cat# ab32356, RRID:AB_732924 |
| Anti-H3K4me3, 0.2 μg | Millipore | Cat# 07-473, RRID:AB_1977252 |
| Anti-H3K27Ac, 0.2 μg | Active Motif | Cat# 39133, RRID:AB_2561016 |
| Anti-H3K9Ac, 0.15 μg | Abcam | Cat# ab4441, RRID:AB_2118292 |
| Anti-H3K27me3, 0.15 μg | Millipore | Cat# 07-449, RRID:AB_310624 |
| Anti-H3K9me3, 0.2 μg | Abcam | Cat# ab8898, RRID:AB_306848 |
| Anti-H3K36me3, 0.2 μg | Abcam | Cat# ab9050, RRID:AB_306966 |
| Anti-H3K9me1, 0.2 μg | Abcam | Cat# ab8896, RRID:AB_732929 |
| Anti-H3K9me2, 0.2 μg | Abcam | Cat# ab1220, RRID:AB_449854 |
| Anti-H3K36me1, 0.2 μg | Cell Signaling Technology | Cat# 14111, RRID:AB_2798395 |
| Anti-H3K36me2, 0.2 μg | Abcam | Cat# ab9049, RRID:AB_1280939 |
| Chemicals, peptides, and recombinant proteins | ||
| Dulbecco’s Modified Eagle’s Medium - High Glucose | Sigma-Aldrich | Cat# D5648 |
| FBS | GIBCO | Cat# 26140-079 |
| Penicillin-Streptomycin | Life Technologies | Cat# 15140122 |
| L-Glutamine | Life Technologies | Cat# 25030-081 |
| Hoechst 33342 | Thermo Fisher Scientific | Cat# H3570 |
| Trypsin-0.25% EDTA | Life Technologies | Cat# 2520056 |
| Formaldehyde | Electron Microscopy Sciences | Cat# 15686 |
| Ribonuclease A | Sigma-Aldrich | Cat# R4875 |
| Proteinase K | Sigma-Aldrich | Cat# P6556 |
| AMPure XP | Beckman Coulter | Cat# A63881 |
| Dynabeads Protein A | Thermo Fisher Scientific | Cat# 10001D |
| Dynabeads Protein G | Thermo Fisher Scientific | Cat # 10003D |
| Protein A–Agarose Fast Flow | Sigma-Aldrich | Cat # P3476-5ML |
| Critical commercial assays | ||
| TruSeq ChIP Library Preparation Kit Set A | Illumina | Cat# IP-202-1012 |
| TruSeq ChIP Library Preparation Kit Set B | Illumina | Cat# IP-202-1024 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat # Q32851 |
| High Sensitivity D1000 ScreenTape | Agilent | Cat # 5067-5584 |
| High Sensitivity D1000 Reagents | Agilent | Cat # 5067-5585 |
| Deposited data | ||
| Raw data | (Van Rechem et al., 2021) | GEO: GSE175752 |
| Experimental models: Cell lines | ||
| hTERT RPE-1 | Nicholas Dyson’s lab | N/A |
| Other | ||
| FACS sorter | N/A | N/A |
| Magnetic stand for 1.5 mL tubes | N/A | N/A |
| Magnetic stand for 96 wells plate | N/A | N/A |
| Sonicator | QSonica | Q800R |
| Qubit | Thermo Fisher Scientific | N/A |
| Nanodrop | Thermo Fisher Scientific | 2000c |
| TapeStation or Bioanalyser | N/A | N/A |
| Thermocycler | N/A | N/A |
Materials and equipment
Antibodies
Here is a list of antibodies we have optimized for this protocol. We specify which dilution IP buffer worked best for ChIP Sequencing and the amount of antibody to use. To get good sequencing results, some antibodies needed several IPs to be pooled together and some more chromatin in the IP. This could be due to the amount of histone modifications present in the genome and/or to the efficacy of these antibodies. When nothing is specified, we used our standard conditions (0.5 μg of chromatin and 0.2 μg of antibody per IP).
H3K4me1 ab8895 lot GR193882-1, Dilution IP buffer High Triton.
H3K4me2 ab32356 lot GR209821-1, Dilution IP buffer High Triton, 0.1 μg of antibody per IP.
H3K4me3 Millipore 07-473 lot 2648189 Dilution IP buffer Low Triton.
H3K27Ac Active Motif 39133 lot 31814008, Dilution IP buffer High Triton.
H3K9Ac ab4441 lot GR224698-1, Dilution IP buffer High Triton, 0.15 μg of antibody per IP.
H3K27me3 Millipore 07-449 lot 26532203, Dilution IP buffer High Triton, 0.15 μg of antibody per IP.
H3K9me3 ab8898 lot GR30928-1, Dilution IP buffer High Triton.
H3K36me3 ab9050 lot GR10860-1, Dilution IP buffer High Triton.
H3K9me1 ab8896-100 lot 815309, Dilution IP buffer High Triton. Pool two IPs.
H3K9me2 ab1220 lot GR32351-2, Dilution IP buffer High Triton. Pool three IPs using 1 μg of chromatin per IP.
H3K36me1 Cell Signaling 14111S ref 03/2017 lot 1, Dilution IP buffer High Triton.
H3K36me2 ab9049 lot GR316128-1, Dilution IP buffer High Triton. Pool two IPs.
Buffers
All buffers need to be filtered.
Cellular Lysis buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| PIPES pH 8 (500 mM) | 5 mM | 2 mL |
| KCl | 85 mM | 1.27 g |
| NP-40 | 0.5% | 1 mL |
| ddH2O | n/a | up to total |
| Total | n/a | 200 mL |
Store at 4°C for up to a year
Nuclear Lysis buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris pH 8 (1 M) | 50 mM | 5 mL |
| EDTA pH 8 (100 mM) | 10 mM | 10 mL |
| SDS (10%) | 1% | 10 mL |
| ddH2O | n/a | 75 mL |
| Total | n/a | 100 mL |
Store at 18°C–25°C for up to a year
Dilution IP buffer Low Triton
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris pH 8 (1 M) | 16.7 mM | 8.35 mL |
| EDTA pH 8 (100 mM) | 1.2 mM | 6 mL |
| NaCl (5 M) | 167 mM | 16.7 mL |
| SDS (10%) | 0.1% | 5 mL |
| Triton X100 | 0.24% | 1.2 mL |
| ddH2O | n/a | 462.75 mL |
| Total | n/a | 500 mL |
Store at 4°C for up to a year
Dilution IP buffer High Triton
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris pH 8 (1 M) | 16.7 mM | 8.35 mL |
| EDTA pH 8 (100 mM) | 1.2 mM | 6 mL |
| NaCl (5 M) | 167 mM | 16.7 mL |
| SDS (10%) | 0.1% | 5 mL |
| Triton X100 | 1.84% | 9.2 mL |
| ddH2O | n/a | 454.75 mL |
| Total | n/a | 500 mL |
Store at 4°C for up to a year
TSE buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris pH 8 (1 M) | 20 mM | 10 mL |
| EDTA pH 8 (100 mM) | 2 mM | 10 mL |
| NaCl (5 M) | 500 mM | 50 mL |
| Triton X100 | 1% | 5 mL |
| SDS (10%) | 0.1% | 5 mL |
| ddH2O | n/a | 430 mL |
| Total | n/a | 500 mL |
Store at 18°C–25°C for up to a year
LiCl buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris pH 8 (1 M) | 100 mM | 50 mL |
| LiCl | 500 mM | 10.6 g |
| Sodium deoxycholate | 1% | 5 g |
| NP-40 | 1% | 5 mL |
| ddH2O | n/a | up to total |
| Total | n/a | 50 mL |
Store at 18°C–25°C for up to a year
TE buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Tris pH 8 (1 M) | 10 mM | 5 mL |
| EDTA pH 8 (100 mM) | 1 mM | 5 mL |
| ddH2O | n/a | 490 mL |
| Total | n/a | 500 mL |
Store at 18°C–25°C for up to a year
Elution buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| NaHCO3 | 50 mM | 0.1 g |
| SDS (10%) | 1% | 2.5 mL |
| NaCl (5 M) | 140 mM | 700 μL |
| ddH2O | n/a | up to total |
| Total | n/a | 25 mL |
Store at 18°C–25°C for up to 1 month
Glycine
| Reagent | Final concentration | Amount |
|---|---|---|
| Glycine | 1.25 M | 23.46 g |
| ddH2O | n/a | up to 250 mL |
| Total | n/a | 250 mL |
Store at 18°C–25°C for up to a year
Note: add ddH2O to ∼200 mL, adjust the pH to 2.5 with HCl, add the remaining ddH2O to 250 mL.
Step-by-step method details
Cell sorting
During this step cells are fixed and sorted based on DNA content. Depending on the number of cells, their cell cycle distribution, and how much chromatin you will need, this step can be repeated as needed and samples pooled together (this explains the broad range in timing).
-
1.
Seed 2.5 × 106 RPE in four 15 cm2 plates for 48 h.
Note: cells need to be at ∼70% confluence when harvested.
-
2.
Treat the cells with Hoechst 33342 adding 1/1,000 directly on the medium, swirl to mix and incubate the cells at 37°C for 1 h. Troubleshooting 2.
Note: incubation time will need to be adjusted depending on the growth rate of the cells (30 min for HEK293T for example).
Note: Hoechst can be toxic for certain cells, after incubation if cells look different (i.e., rounded) less Hoechst should be used.
-
3.
Shake off the loosely attached mitotic cells by tapping the side of the plates. Collect the medium in 50 mL conical and centrifuge in order to pellet the cells in mitosis that could be less adherent. Transfer the spun medium in another tube as it will be used in steps 5 and 9, leave enough medium behind to make sure not to aspirate the pelleted mitotic cells.
-
4.
Rinse the cells with 1× PBS and add 2.5 mL of trypsin per 15 cm2 plate.
Note: a single cell suspension is critical for the sorting.
-
5.
Resuspend the cells in the spun medium from step 3, for four 15 cm2 plates use 36 mL of medium, transfer in the 50 mL conical tube containing the pellet of mitotic cells.
-
6.
Crosslink the cells by adding 1 mL of 37% formaldehyde to the medium (1% final), incubate at 37°C for 13 min.
Note: formaldehyde should be freshly opened and is toxic, use gloves and dispose properly.
-
7.
Stop the fixation by adding 3.7 mL of glycine 1.25 M pH 2.5 (0.125 M final), incubate for at least 2 min at 18°C–25°C.
Note: the medium should turn bright yellow.
-
8.
Centrifuge the cells, wash the cell pellet with 1× PBS and centrifuge again.
-
9.
Resuspend the cell pellet in 2 mL of spun medium from step 3.
Note: the sorting step is more efficient when the cells have been resuspended in their original medium containing Hoechst, especially if the sorting step takes several hours.
-
10.
Place the cells on ice until sorting.
Note: fixed cells can be kept on ice for few hours before sorting.
-
11.
Sort the cells with a BD FACS Fusion using the BV421-A laser. A typical sort is represented in Figure 2.
Figure 2.

Gating example for cell sorting
Gating: place G1 (from the base of the large peak (1N) to 50% of the slope on the right), then G2/M (from the base of the second peak (2N) and encompassing the entire peak), and finally early and late S phase (the S fractions are equally divided in the middle). Troubleshooting 3 and 4.
Note: any sorter with a laser that can read Hoechst can be used. The containers to hold and collect the cells will depend on the sorter.
Note: all cells are sorted in four fractions and the lysis volume adjusted on the number of cells in step 14.
Note: The amount of cells needed will depend on the yield of chromatin obtained and on the number of IPs the experimentalist wants to do with the chromatin. Several sorts can be pooled together is necessary.
-
12.
Transfer the cells to 15 mL conical tubes and centrifuge for 15 min at 1,000 g at 4°C .
-
13.
Lyse the cells in 200 μL cellular lysis buffer complemented with fresh proteinase inhibitors (cocktail tablets EDTA free), incubate for 5 min on ice, centrifuge for 5 min at 2,000 g at 4°C .
-
14.
Remove the supernatant and resuspend the pellet in nuclear lysis buffer complemented with fresh proteinase inhibitors (cocktail tablets EDTA free), adjusting for the size of the pellet. For an estimated volume we use 10 μL for 300,000 cells, 25 μL for 1 million, 60 μL for 4 million.
Pause point: keep at −80°C (the unsonicated chromatin will be stable for many years).
Sonication
During this step the collected chromatin will be sheared at an expected size of < 300 pb, roughly one to two nucleosomes and some linker DNA.
-
15.
Turn on the QSonica Q800R chiller. Transfer the chromatin in 0.5 mL thin wall PCR tubes. Sonicate according to the time determined in the optimization step.
Note: sorted cells can be easier to sonicate, therefore, they may need less sonication time. Do not over sonicate as the ChIP will be less efficient. The size will not decrease further but the chromatin will start to be altered.
Note: the sonication bath should never get hot.
Note: Steps 16–18 are optional but recommended. While the sonication efficiency should always be the same, we advise to check the sonication every time to make sure that this step worked properly before proceeding to the immunoprecipitation step. During these steps store the remaining chromatin at −80°C.
-
16.
Add 3 μL of chromatin and 1 μL of proteinase K to 65 μL of elution buffer, incubate at 65°C for 4–16 h in a benchtop thermomixer shaking at 1,000 rpm.
-
17.
Add 65 μL of phenol/chloroform/isoamyl alcohol 25:24:1, vortex, centrifuge for 1 min at full speed at 18°C–25°C.
-
18.
Take 20 μL of the upper phase, mix with appropriate dye and run on a 1% agarose gel. The chromatin should be under 300 bp. If there is a big tail, the chromatin is not sonicated enough (see Figure 1).
Note: use DNA loading dye with only one tracking color that runs at a high molecular to avoid using a dye running at the same size as your expected chromatin as it hides the chromatin.
Note: the efficiency of sonication for cells within different cell cycle phases is identical.
-
19.
Transfer the sonicated chromatin to 1.5 mL tubes and centrifuge for 10 min at 20,000 g at 4°C to clear the debris. Transfer the supernatant in a clean tube leaving any potential pellet behind.
Note: cold chromatin (just thaw or kept on ice) can present some SDS precipitates which will be pelleted during the centrifugation. Before proceeding with step 19, leave the chromatin at 18°C–25°C for few minutes to allow SDS to go back into solution.
Note: we recommend keeping the chromatin in low bind DNA tubes, however regular tubes will work as well.
Note: the expected chromatin concentration (DNA concentration from Nanodrop) is 300–1,000 ng/μL. Troubleshooting 5.
Immunoprecipitation
During this step, the chromatin will be immunoprecipitated with your antibodies of choice. Some antibodies require specific conditions to work their best. When in doubt, we recommend testing two different dilution IP buffers (low and high triton). We have incorporated a section with the antibodies we tested, and their optimal conditions in the material section.
-
20.
Prebind the beads with the antibody. In a 200 μL tube: add 100 μL dilution IP buffer complemented with fresh protease inhibitors (cocktail tablets EDTA free), 2.5 μL protein A or G magnetic beads (prewashed and resuspended in dilution IP buffer, protein A for rabbit polyclonal and G for mouse monoclonal antibodies), and 0.2 μg of antibody. Incubate at 4°C for 6 h under rotation.
Note: PCR strip tubes are useful at this step as they are easier to handle and allow to vortex several tubes at once in this and subsequent steps.
Note: the incubation time can be increased.
Note: the amount of antibody can vary depending on efficacy.
-
21.
Pool the chromatin in a 1.5 mL tube: per IP include 0.5 μg of chromatin in 10 μL of nuclear lysis buffer with fresh protease inhibitors added (cocktail tablets EDTA free), also include 0.5 μg of chromatin for input (if using this to generate baseline input sequencing profiles) and 0.5 μg for pipetting error.
Note: example for 5 IPs + input + pipetting error: 5 × 0.5 + 0.5 + 0.5 = 2.5 μg of chromatin in 5 × 10 +10 +10 = 70 μL of nuclear lysis buffer. Note that the volume of chromatin will be deducted to the volume of nuclear lysis buffer: if 2.5 μg of chromatin represent 5 μL then 65 μL of nuclear lysis buffer will be needed.
Note: the amount of chromatin per IP can vary, we have used 1 μg for some antibodies (see material section).
Note: the chromatin concentration is based on DNA concentration taken on a Nanodrop.
-
22.
Take 10 μL of the pooled chromatin (diluted in nuclear lysis buffer in the previous step). This will serve as the input, store at 4°C until further use.
-
23.
Preclear step. Bring the remaining of the pooled chromatin to 100 μL per IP with dilution IP buffer complemented with fresh protease inhibitors (cocktail tablets EDTA free) (low or high triton depending on the IP condition).
Add 2.5 μL of protein A agarose beads per IP (prewashed and resuspended in dilution IP buffer), incubate at 4°C for 2 h under rotation.
Centrifuge and transfer the chromatin in another tube leaving the beads behind.
Add 2.5 μL of protein A (or G) magnetic beads per IP (prewashed and resuspended in dilution IP buffer), incubate at 4°C for 2 h under rotation.
Note: while protein A or G magnetic beads are used accordingly to the antibody that will be used for the IP, protein A agarose are used regardless of the IP. If using protein G for the IP, one could choose to use protein G agarose but this will not improve the preclear.
-
24.
Quickly spin down the tubes with beads/antibodies (from step 20) and place them on a magnetic rack before removing the supernatant. Add 100 μL of precleared chromatin per tube, incubate at 4°C for 12–16 h with rotation.
-
25.
Proceed to the washes. After a quick spin place the tubes on a magnet and remove the supernatant. For each wash, add 100 μL of buffer, vortex thoroughly, spin quickly, place on the magnetic rack and remove the buffer. Perform 6 washes: twice dilution IP buffer, once TSE buffer, once LiCl buffer, twice TE buffer. All washes are performed at 18°C–25°C. Troubleshooting 6.
-
26.
Elution. After the last wash, add 50 μL of elution buffer to the beads, transfer in a 1.5 mL tube, add 1 μL of RNAseA 100 μg/μL and incubate at 37°C for 30 min in a benchtop thermomixer shaking at 1,000 rpm. Add 1 μL of proteinase K (10 mg/mL) and incubate at 37°C for 1 h in a benchtop thermomixer shaking at 1,000 rpm. If pooled chromatin has been kept at 4°C for an input preparation, add 40 μL of elution buffer and proceed as for the IPs from this step.
Note: if pooling IPs together (see material section) it should be done at that step using 50 μL of elution buffer for all IPs.
Note: the elution buffer should be less than a month old.
-
27.
Decrosslinking. Remove the samples from the beads (quick spin and magnetic rack) and incubate at 65°C for 4 h in a benchtop thermomixer shaking at 1,000 rpm.
-
28.
DNA purification. Add 125 μL of Agencourt AMPure XP to both the de-crosslinked IPs and input, incubate them for 10 min at 18°C–25°C, then place them on the magnet and remove the supernatant.
The tubes are left on the magnet and 450 μL of freshly prepped 70% EtOH is added, then remove and repeat for a second time.
The last step is to wait for the beads to dry on the magnet before resuspending them in 40 μL of H2O. They are then incubated for 1 min at 18°C–25°C before being placed on the magnet. IP or input material is then removed from the beads.
Note: In order to know when the beads are dry, we recommend looking under the light. The beads are dry when they are no longer shiny. However, do not wait longer than needed. If the beads dry too much (the smear against the magnet looks like it is “cracking”) the recovery will be less efficient (see example in Figure 3).
Figure 3.

Examples of AMPure beads ready or not to be resuspended in water for DNA purification
Libraries preparation for sequencing
During this step the DNA will be prepped to be sequenced with the Illumina technology.
To avoid any exogenous DNA contamination work on a clean bench with clean pipetmans and sterile filter tips.
We used the TruSeq ChIP Sample Preparation kits and implemented few protocol modifications annotated below. The Illumina protocol can be found here: https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/samplepreps_truseq/truseqchip/truseq-chip-sample-prep-guide-15023092-b.pdf.
We are annotating the main steps below as an overview, please refer to the guide except when noted otherwise.
-
29.
Quantify each ChIP DNA using a Qubit dsDNA HS Assay Kit. We used 1 ng of DNA per library prep (or the entire IP if the concentration is below the threshold of detection). Place each DNA in a well of a 96 well plate and bring the volume up to 50 μL with H2O.
-
30.
Perform end repair (page 11 of the Illumina PDF listed above).
-
31.
Adenylate 3′ ends (page 14 of the Illumina PDF listed above).
-
32.
Ligate adapters (page 16 of the Illumina PDF listed above).
-
33.
Go directly to Enrich DNA fragments step (page 25 of the Illumina PDF listed above) omitting Purify ligation products (we purify using a different method after the PCR step) and stopping at step 5 (page 27 of the Illumina PDF listed above).
Note: we typically perform 13 cycles of PCR.
Note: we do not perform the washes step after the PCR, this step is replaced by a double-sided SPRI purification with AMPure beads as noted thereafter.
-
34.
Add 30 μL of AMPure beads to the 50 μL PCR reaction, incubate at 18°C–25°C for 10 min.
-
35.
Place the tubes on the magnetic stand. Keep the sample: transfer 77 μL of the sample in new wells. Discard the beads.
-
36.
Add 12 μL of AMPure beads, incubate at 18°C–25°C for 10 min.
-
37.
Place the plate on the magnetic stand. Discard the sample: take off 80 μL of the sample and discard. Keep the beads.
-
38.
Perform two washes leaving the plate on the magnetic stand: add 200 μL of fresh prepped 80% EtOH, remove, repeat and dry the beads (as before, not too much).
-
39.
Resuspend the beads in 17.5 μL of resuspension buffer, incubate at 18°C–25°C for 1 min.
-
40.
Place the plate on the magnetic stand. Transfer 15 μL of each library in the final plate.
-
41.
Validate the libraries using a TapeStation or a Bioanalyzer and proceed to sequencing.
Note: a typical TapeStation result is presented in Figure 4.
Figure 4.

TapeStation analyses for a typical ChIP-seq library performed with this protocol
Note: libraries should yield at a minimum 1,000 ng/mL. Troubleshooting 7.
Expected outcomes
Expected outcomes were annotated in the protocol. The final libraries should yield at a minimum 1,000 ng/mL (Qubit concentration).
Limitations
The main limitation to this protocol is the availability of antibodies that work for ChIP-seq. Some antibodies will be more effective than others with the lower amounts of chromatin being used. We recommend purchasing consistent lots that work when possible. Modifying the Dilution IP buffer as annotated in this protocol can lead to successful sequencing results. Another option for a better outcome is to modify the amount of SDS in the Nuclear Lysis buffer, bringing it down to as low as 0.2%. Such chromatin will require further sonication and the Dilution IP buffers will need to be adapted in order for the IP to be done in 0.2% SDS (bring the SDS at 0.2% in the Dilution IP buffers). Of note, this protocol has only been successfully tested for histone and histone marks, additional adaptations might be necessary for other proteins. Troubleshooting 8.
Troubleshooting
Problem 1
Before you begin step 11. Clearly visible DNA fragments above 300 bp after sonication.
Potential solution
Sonicate more (see Figure 1).
Problem 2
Step 2. Cells change shape after Hoechst treatment.
Potential solution
Hoechst is toxic to the cells, reduce the concentration.
Problem 3
Step 11. Cells not really visible with the laser exciting Hoechst.
Potential solution
Increase the time/concentration of Hoechst treatment.
Problem 4
Step 11. Cells are clumpy during sorting.
Potential solution
Make sure to resuspend the cells in a single cell suspension and dilute more the cells before sort (sort in a higher volume). A cell strainer can be used at this step to remove any non-single cell.
Problem 5
Step 19. Chromatin concentration too low.
Potential solution
Decrease the volume of Nuclear Lysis buffer when resuspending the cells.
Problem 6
Step 25. The magnetic beads lose their magnetism (slide down the tube when on the magnetic stand).
Potential solution
Increase the speed of the washes / reduce to one wash with TE instead of two.
Problem 7
Step 41. Libraries with very low amount of DNA.
Potential solution
Multiply the number of IPs and pool them together at the elution step (#26).
Try another antibody or another Dilution IP buffer (see limitations section).
The numbers of PCR cycles in step 33 can be increased slightly (∼2 cycles), however too many cycles will create PCR duplicates that will need to be removed from the sequencing results. We therefore recommend to multiple the number of IPs instead.
Problem 8
Limitations. Too much background / no peaks when assessing the ChIP-seq on IGV or equivalent.
Potential solution
Increase the number of washes and vortex more.
Try another antibody or another Dilution IP buffer (see limitations section).
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Capucine Van Rechem (cvrechem@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
This study is supported by R01GM097360 and R35GM144131 (J.R.W.), NIH/NCI Cancer Center Support Grant P30 CA006927 (J.R.W.), and the American Lung Association Lung Cancer Discovery Award.
Author contributions
C.V.R. and J.R.W. worked together to optimize the conditions that are described in this methods article. C.V.R. conducted the methods.
Declaration of interests
In the past year, J.R.W. has consulted for Qsonica (manufacturer of Q800R system). J.R.W. has served as a consultant or advisor for Salarius Pharmaceuticals, Daiichi Sankyo, Inc., and VYNE Therapeutics. J.R.W. has also received sponsored research from Salarius Pharmaceuticals. C.V.R. declares no competing interests.
Contributor Information
Johnathan R. Whetstine, Email: johnathan.whetstine@fccc.edu.
Capucine Van Rechem, Email: cvrechem@stanford.edu.
Data and code availability
The published article includes all datasets and codes generated during this study.
References
- Van Rechem C., Ji F., Chakraborty D., Black J.C., Sadreyev R.I., Whetstine J.R. Collective regulation of chromatin modifications predicts replication timing during cell cycle. Cell Rep. 2021;37:109799. doi: 10.1016/j.celrep.2021.109799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The published article includes all datasets and codes generated during this study.