

Abstract
Intestinal microbiota play an important role in the life of amphibians and its composition may vary by developmental stage. In this study, 16S rRNA high‐throughput sequencing was used to profile the intestinal microbiota of Hynobius maoershanensis, which exclusively inhabit the Maoer Mountain swamp at an altitude of approximately 2,000 m. We characterized the bacterial composition, structure, and function of the microbiota of H. maoershanensis at different developmental stages. The alpha diversity was not markedly different for the Simpson, Shannon, Ace, and Sobs indices of microbes. The beta diversity revealed that there were age‐related differences in the structure of the intestinal microbes of H. maoershanensis, specifically, at the phylum level. Bacteroidetes and Proteobacteria were the dominant bacteria present in the adult stage, and the relative abundance of Bacteroidetes was significantly higher compared with that of tadpoles. Firmicutes and Proteobacteria were the dominant phylum during the tadpole stage and their relative abundance was significantly higher compared with the adult period. Functional analysis revealed that the pathways associated with organismal systems and metabolism were significantly enriched in the adults, whereas human diseases, genetic information processing, and cellular processes were more enriched in the hindlimb bud stage. Human diseases and environmental information processing were more enriched in the forelimb bud stage at KEGG pathway level 1. Possibilities for the observed discrepancies include the adaptation to eating habits and the remodeling of the intestines during development. We speculated that H. maoershanensis adults may be more suitable to a high‐fiber diet, whereas the tadpoles are associated with a carnivorous diet. Our study provides evidence of variations in the intestinal microbiota during development in amphibians, highlighting the influence of historical developments on the intestinal microbiota and an increased understanding of the importance of physiological characteristics in shaping the intestinal microbiota of amphibians. These data will help us formulate more effective protection measures for H. maoershanensis.
Keywords: developmental stage, Hynobius maoershanensis, intestinal microbiota
Hynobius maoershanensis (Urodela: Hynobiidae). The larvae of H. Maoershanensis live an aquatic life, whereas the adults live amphibiously. Due to the sharp decline of its population, it is the key protection wildlife in national nature reserve in China.

1. INTRODUCTION
Vertebrates have a complex symbiotic relationship with their intestinal microbiota, which regulate metabolism, immune function, and energy balance to play a vital role in maintaining health (Hwang et al., 2015; Lapébie et al., 2019). For example, Bacteroidetes, which exist widely in the ecosystem, utilize thousands of enzyme combinations to decompose polysaccharides, convert them into pyruvate and release energy that can be directly used by the host organism (Lapébie et al., 2019; Thomas et al., 2011). Firmicutes, which are widespread in the gastrointestinal tract, can produce byproducts through fermentation, such as short chain fatty acids, that are directly absorbed through the host bowel wall (den Besten et al., 2013). However, higher levels of Bacteroidetes and Firmicutes can increase insulin resistance by modulating the levels of glucagon‐like peptide I, which will lead to a decrease in the efficiency of glucose uptake and utilization (Hwang et al., 2015). Intestinal flora is not always beneficial to the body and can often cause disease. For example, one of the principal risk factors for gastric cancer is Helicobacter pylori infection, which can lead to the accumulation of DNA damage in genes at the ends of chromosome arms, resulting in various genotoxic effects (Koeppel et al., 2015).
Intestinal microbes are affected by many factors. Studies have revealed the relationship between diet and intestinal microbiota; for example, herbivorous and carnivorous animals have different gut microbial characteristics. The intestines of herbivores are dominated by cellulose‐ and lignin‐decomposing bacteria (Rumenococcus, Clostridium, etc.) (Li et al., 2021; Zhang et al., 2019), whereas the intestines of carnivores are mainly concentrated with protein‐ and lipid‐degrading bacteria (Peptostreptococcus) (David et al., 2014; Gillman et al., 2020; Tu et al., 2005). The intestinal microbiota of omnivorous animals, such as wild birds, are primarily associated with Firmicutes (50%) and Proteobacteria (25%) (Kirsten et al., 2018). Feeding habits not only affect the superiority of intestinal bacteria but also the abundance. In a study of intestinal microbes based on nine diets of 62 species of insects, those that fed on rotted wood had the highest abundance of gut microbes, whereas insects that fed on pollen wood exhibited the lowest (Colman et al., 2012). The intestinal microbiota are also related to changes in dietary resources. When aliments were plentiful, Ruminococcaceae (90%) were the main species of the African buffalo; however, under food restriction conditions, Solibacillus (72%) was the predominant species (Couch et al., 2021). Marked differences in the structure of intestinal microbiota caused by dietary transformation are also reflected in amphibians; with gastropods as the primary food source, the intestinal microbiota of fire salamanders were Bacteroidea (47.8%) and Firmicutes (32.1%) (Wang et al., 2021), whereas the intestinal microbiota were primarily Proteobacteria and Firmicutes in tadpoles which are mainly phytophagous and that of insectivorous adults were mainly Firmicutes and Bacteroides (Kohl et al., 2013). These studies suggest that the abundance of intestinal microbiota is closely related to diet.
Age is closely related to the intestinal microbiota of animals. The reorganization of intestinal microbiotas caused by aging is primarily related to changes in the structure and physical/chemical properties of the digestive system (Bonder et al., 2016; Chen, Cui, et al., 2021; Chen, Li, et al., 2021; Vangay et al., 2018; Zhang et al., 2021). For instance, the stripping and shedding of old intestines during the metamorphic development of some mosquitoes can lead to complete or nearly complete elimination of intestinal bacteria, resulting in the absence of bacteria in the intestines of newly emerged adults (Moll et al., 2001). Amphibians complete the transformation of their digestive tracts from a long and simple non‐acidic stomach to a short and complex acidic stomach through a metamorphosis process (Zhang et al., 2020). The close relationship between the structure of the digestive tract and dietary changes can lead to changes in the intestinal microbiomes (Schreiber et al., 2005). Studies have shown that the intestinal microbiota of frogs change from fish‐like (Proteobacteria, Firmicutes) during the tadpole stage to an amniotic membrane (Firmicutes, Bacteroides) phenotype during the frog stage (Kohl et al., 2013; Zhang et al., 2018). In addition, changes in age result in altered metabolic levels, which affect the structure of the intestinal microbiomes. For example, a Japanese study showed that people over 100 years old contain higher levels of secondary bile acid (produced by intestinal bacteria metabolism) compared with 40‐year‐olds and those under the age of 100. This significantly reduces the risk of aging diseases compared with those of the elderly under 100 years old (Rimal & Patterson, 2021).
A large number of studies involving microbiota have focused on mammals and fish (Frese et al., 2015; Li et al., 2021; Sullam et al., 2012), whereas studies on amphibians have been primarily focused on tailless amphibians (Chai et al., 2018; Kohl et al., 2013; Vences et al., 2016; Weng et al., 2016). Few studies have been conducted on the intestinal microbiotas of tailed amphibians in populations living under artificial feeding conditions (Zhang et al., 2018). There have been a few studies on the intestinal microbiota of wild populations, especially tailed amphibians that are rare species. Currently, amphibians are suffering from a rapid decline and are most susceptible to diseases in their ecosystem (Greener et al., 2020). Because amphibians play important roles in the ecosystem, studying their symbiotic microbiota may lead to strategies for their protection (Alford, 2011; Vences et al., 2016). Therefore, it is necessary to expand intestinal microbiota studies with more species.
Hynobius maoershanensis primarily lives in the surrounding area of the alpine swamp and the vegetation nearby consists primarily of hemlock forest and mountaintop dwarf forest (Chen, Cui, et al., 2021; Chen, Li, et al., 2021; Jiang & Jiang, 2006). The larvae of H. maoershanensis live an aquatic life, whereas the adults live amphibiously. Their diets and digestive systems undergo significant changes during development (Chen, Cui, et al., 2021; Chen, Li, et al., 2021; Ning et al., 2021). Based on the significant changes during development, we hypothesized that there are distinct differences in the structure and function of the intestinal microbiota during the three developmental stages (forelimb bud stage tadpoles, hindlimb bud stage tadpoles and adults) of the tailed amphibian, H. maoershanensis. We identified the influencing factors of the intestinal microbes in the ontogeny of H. maoershanensis, which result in changes in the intestinal microbiota during the development of amphibians, especially tailed amphibians. Our findings provide a reference for monitoring the health and physiological conditions of wild amphibians.
2. MATERIALS AND METHODS
2.1. Sample collection and preservation
Hynobius maoershanensis belongs to the Hynobius hynobius genus and only lives in an area around the high mountain marshes of Xingan County, Guangxi Zhuang Autonomous Region (25°52′N, 110°24′E) at an altitude of 1,950–2,000 m.
Samples were obtained from the cloaca of 31 adults, 12 hindlimb bud tadpoles, and 30 forelimb bud tadpoles of H. maoershanensis in December 2019 (Table 1). Anal swabs were collected by nondestructive sampling. The cloaca was wiped with an alcohol swab before inserting a sterile cotton swab and rotating for three to five circles and then placed into a sterile preservation tube (Colston et al., 2015). All samples were frozen immediately after collection, transported to the laboratory, and stored at −80℃ (Song et al., 2018). All H. maoershanensis were returned to their original location after sampling.
TABLE 1.
Host information of 73 anal swab samples of H. maoershanensis
| Adult | Hindlimb bud stage | Forelimb bud stage | |
|---|---|---|---|
| Total length (mm) | 165.04 ± 12.42 | 63.96 ± 7.88 | 27.42 ± 36.61 |
| Weight (g) | 21.09 ± 5.00 | 1.82 ± 0.67 | 0.23 ± 0.17 |
2.2. DNA extraction, amplification, and sequencing
Total bacterial genome DNA from all samples was extracted using the E.Z.N.A.® Soil DNA kit (Omega Bio‐tek). The V3–V4 hypervariable region of the 16S rRNA gene was amplified by the polymerase chain reaction (GeneAmp 9700; ABI) using the general bacterial primers (338F, 5′‐ACTCCTACGGGAGGCAGCAG‐3′; 806R, 5′‐GGACTACHVGGGTWTCTAAT‐3′) (Mori et al., 2014). The initial PCR was performed using Transgen ap221–02 TransStart® FastPfu Fly DNA Polymerase. A 20 μl reaction mixture contained 10 ng template DNA, 4 μl 5× FastPfu buffer, 2 μl 2.5 mM dNTPs, 0.8 μl of each primer (5 μM), and 0.2 μl bovine serum albumin. The PCR products were extracted from a 2% agarose gel and purified using an AxyPrep DNA Gel Extraction Kit (Axygen Biosciences). The purified PCR fragments were collected and adjusted to an equal molar concentration (Majorbio BioPharm Technology Co., Ltd, Shanghai provide sequencing services) and the paired ends were sequenced (2 × 300) using an Illumina Miseq platform (Illumina).
2.3. Data analysis
On the basis of the overlap between PE reads, the paired reads were merged into a single sequence. The quality of the reads and the effect of the merge were filtered by in a quality control step. According to the barcodes and primer sequences at the beginning and end of the sequence, the effective sequences were obtained and the sequence direction was corrected. The data were flattened according to the minimum number of sample sequences. The base was filtered if the tail mass value was less than 20 and set to a window of 50 bp. The back‐end base was cut from the window if the average quality value in the window was lower than 20. The reads below 50 bp were filtered after quality control and the reads containing an “N” base were removed. The minimum overlap length was 10 bp, the maximum mismatch ratio of the overlap region was 0.2, the allowable mismatch number of the barcode was 0 and the maximum mismatch number of the primers was 2 (Trimmomatic software, Illumina) (Raman et al., 2019).
The original fastq file uses flash for pair‐end double‐ended sequence splicing. Uparse (version number: 7.0.1090; http://www.drive5.com/uparse/) operational taxonomic unit (OTU) clustering and Qiime (version number: 1.9.1; http://qiime.org/install/index.html) were used. The water abundance table for each taxonomic species was generated and the beta diversity distance was calculated. An RDP classifier (version 2.11; https://sourceforge.net/projects/rdp‐classifier/) was used for sequence classification. Using the Greengenes database (version 135; http://greengenes.secondgenome.com/), each 16S rRNA gene sequence was classified using rRNA database alignment.
The alpha diversity indices (Shannon index, Simpson index, ACE index, and Chao index) were calculated using the mothur program (version v.1.30.1; http://www.mothur.org/wiki/Schloss_SOP#Alpha_diversity). The diversity distance matrix of beta was calculated by Qiime (http://qiime.org/install/index.html). Principal coordinate analysis (PCoA) was used to calculate and visualize weighted and unweighted UniFrac distance matrices. Nonparametric Anosim analysis was used to test whether the difference between groups was significantly greater compared with that within groups. The Wilcoxon rank‐sum test and FDR‐adjusted p values were used to test the difference of genes, and KEGG pathways (http://picrust.github.io/picrust/) were compared between groups to predict their function. All data were analyzed using the Majorbio I‐Sanger Cloud Platform (http://www.i‐sanger.com).
3. RESULTS
3.1. Sequence quality evaluation
A total of 3,329,196 sequences of the hypervariable V3–V4 region of the 16S rRNA gene were obtained from 73 fecal samples, of which 1,073,747 were valid, and the average length of the sequences were 417 bp. The sample sequences were leveled in accordance with the minimum number of sample sequences (CD14:28144). A total of 1,035 OTUs were clustered using a sequence similarity of 97%. The rank abundance, rarefaction, and alpha diversity index was constructed based on these OTUs and showed that the depth of the sequencing results was sufficient (Figure 1; Table 2).
FIGURE 1.

Rarefaction curves and rank‐abundance distribution curves
TABLE 2.
Alpha diversity index of H. maoershanensis gut microbiota (a: forelimb bud stage and hindlimb bud stage b: forelimb bud stage and adults c: hindlimb bud stage and adults)
| Hindlimb bud stage | Forelimb bud stage | p‐value | Q‐value (FDR) | |
|---|---|---|---|---|
| (a) | ||||
| Sobs | 19.167 ± 9.98 | 14.467 ± 5.847 | .108 | 0.396 |
| Shannon | 0.932 ± 0.529 | 0.799 ± 0.262 | .237 | 0.396 |
| Simpson | 0.551 ± 0.255 | 0.569 ± 0.151 | .351 | 0.396 |
| Ace | 21.437 ± 10.5 | 19.768 ± 15.004 | .396 | 0.396 |
| Adult | Forelimb bud stage | p‐value | Q‐value (FDR) | |
|---|---|---|---|---|
| (b) | ||||
| Sobs | 15.419 ± 7.898 | 14.467 ± 5.847 | .558 | 0.558 |
| Shannon | 0.959 ± 0.343 | 0.799 ± 0.262 | .337 | 0.531 |
| Simpson | 0.475 ± 0.129 | 0.569 ± 0.151 | .030 | 0.120 |
| Ace | 20.744 ± 10.651 | 19.768 ± 15.004 | .399 | 0.531 |
| Adult | Hindlimb bud stage | p value | Q‐value (FDR) | |
|---|---|---|---|---|
| (c) | ||||
| Sobs | 15.419 ± 7.898 | 19.167 ± 9.98 | .188 | 0.751 |
| Shannon | 0.959 ± 0.343 | 0.932 ± 0.529 | .989 | 1.000 |
| Simpson | 0.475 ± 0.129 | 0.551 ± 0.255 | 1.000 | 1.000 |
| Ace | 20.744 ± 10.651 | 21.437 ± 10.5 | .807 | 1.000 |
3.2. Analysis of alpha and beta diversity
The alpha diversity analysis, based on 1035 OTUs (Table 2), revealed that there were no significant differences in the Shannon, Simpson, ACE, and Sobs indices between the three stages (FDR > 0.05) (Table 2), which indicates a similar diversity. PCoA based on unweighted (R 2 = 0.109, FDR < 0.001) and weighted (R 2 = 0.458, FDR < 0.001) UniFrac distances by the Adonis test showed that the intestinal microbiota were highly aggregated by different developmental stages. This suggests that there were differences in the structure of the intestinal microbiota in H. maoershanensis (Figure 2).
FIGURE 2.

Beta diversity difference in gut microbiota at different developmental stages. (a) unweighted‐unfair; (b) weighted‐infair
3.3. Difference in the composition and structure of intestinal microbiota
The OTUs (n = 1,035) obtained from the samples consisted of 29 phyla and 214 microbial families. The most dominant phyla were Proteobacteria (35.04%), Firmicutes (33.19%), and Bacteroidetes (19.57%). The most dominant families were Oxalobacteraceae (14.46%), Flavobacteriaceae (12.43%), and Peptostreptococcaceae (10.15%) (Figure 3). Considering the different stages of development, the proportion of intestinal microbiotas differed remarkably in the three stages. At the phylum level, the top three phylum with the highest enrichment of forelimb bud stage were Firmocutes, Proteobacteria, and Fusobacteria, whereas those in the hindlimb bud stage and adults were Firmicutes, Proteobacteria, and Bacteroidetes (Figure 4). At the family level, the top three families with the highest enrichment in the forelimb and hindlimb bud stage were Peptostreptococcaceae, Clostridiaceae, and Enterobacteriaceae, whereas the adults consisted of Oxalobacteraceae, Flavobacteriaceae, and Pseudomonadaceae (Figure 5). Based on a Wilcoxon rank‐sum test, the relative abundance of Firmicutes and Fusobacteria in the tadpole stages were markedly different compared with that in the adult stage (FDR < 0.001), whereas Bacteroidetes were more enriched in adults (FDR < 0.001) at the phylum level (Figure 6). At the family level, Peptostreptococcaceae was more enriched in the tadpole stage compared with the adults, especially in the hindlimb bud stage (FDR < 0.001); Clostridiaceae were enriched in the tadpole stage compared with the adults, especially in the forelimb bud stage. In contrast, the relative abundance of Oxalobacteraceae and Flavobacteriaceae in the adults was significantly higher than that in tadpoles (FDR < 0.001) (Figure 7). The relative proportions of the other bacterial phyla and families are listed in Appendices S1 and S2.
FIGURE 3.
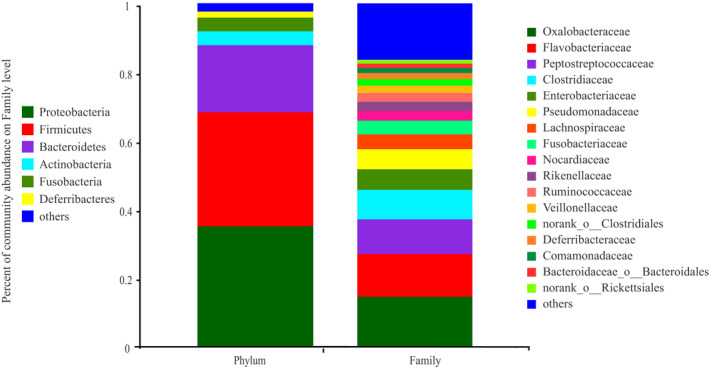
Gut microbiota composition at the phylum and family level
FIGURE 4.

Gut microbiota composition at the phylum level
FIGURE 5.

Gut microbiota composition at the family level
FIGURE 6.
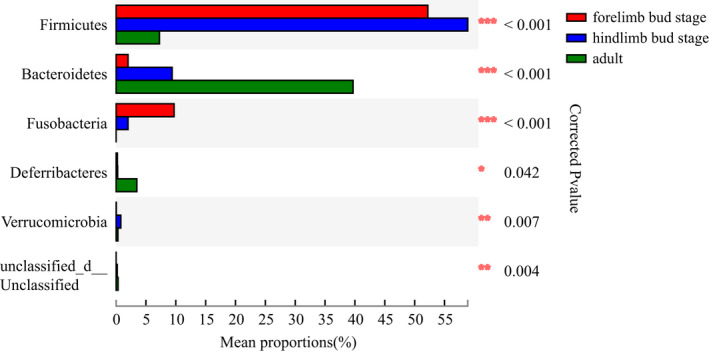
The gut microbiota composition and difference at the phylum level at different developmental stages
FIGURE 7.
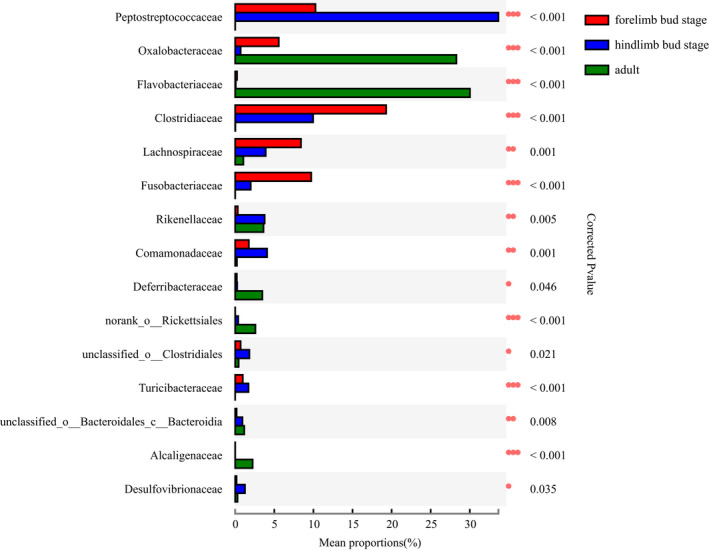
The gut microbiota composition and difference at the family level at different developmental stages
Lefse analysis was used to detect differences in the relative abundance of microbiota at different levels to further identify shifts in composition at different stages. The results showed that there were 25 intestinal bacterial taxa with higher abundance in this group of which one taxa was from the forelimb bud stage, 16 were from the hindlimb bud stage, and eight were from the adults. The Peptostreptococcaceae family, the Betaproteobacteria class, the Oxalobacteraceae family, and the Burkholderiates order were the major taxa contributing to these differences (Figure 8).
FIGURE 8.
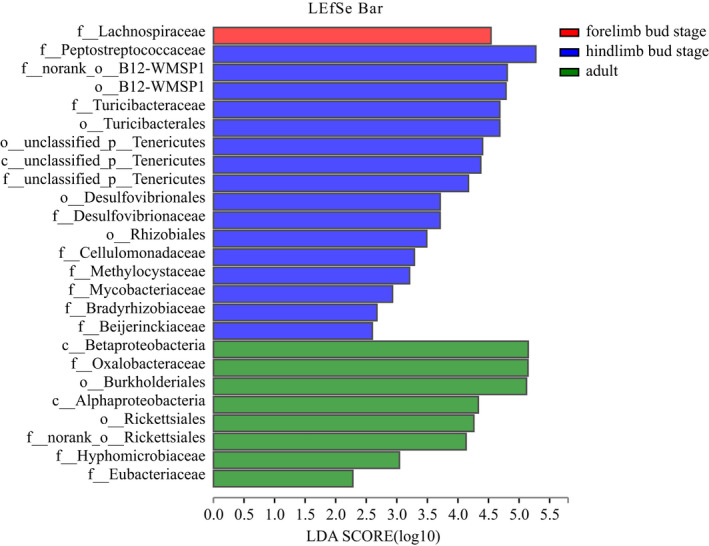
The LEfSe of gut microbiota abundance
3.4. Differences in the functional distribution of intestinal flora
The Mann–Whitney U test was used to identify differences in KEGG pathways to predict the function of the intestinal microbiota at different developmental stages. Pathways associated with organismal systems and metabolism were significantly enriched in adults, whereas genetic information processing and cellular processes were more enriched in the hindlimb bud stage. Human diseases and environmental information processing were more enriched in the forelimb bud stage at KEGG pathway level 1 (Table 3). For KEGG pathway level 2, pathways associated with amino acid metabolism, cancer, carbohydrate metabolism, and cardiovascular disease were significantly enriched in adults, whereas pathways associated with biosynthesis of other secondary metabolites were enriched in the hindlimb bud stage (Appendix S3).
TABLE 3.
Differences in KEGG Pathways Level 1
| Pathway in level 1 | Relative abundance (%) | Mann–Whitney U test | |||
|---|---|---|---|---|---|
| Forelimb bud stage | Hindlimb bud stage | Adult | z | p (FDR) | |
| Metabolism | 38.43 ± 0.36 | 37.06 ± 0.38 | 40.12 ± 0.37 | −4.631 | .000 |
| Human Diseases | 25.58 ± 0.03 | 24.99 ± 0.03 | 24.84 ± 0.03 | −4.501 | .000 |
| Genetic Information Processing | 15.57 ± 0.40 | 18.04 ± 0.49 | 15.64 ± 0.43 | −4.386 | .000 |
| Environmental Information Processing | 16.01 ± 1.24 | 15.27 ± 1.14 | 15.1 ± 1.21 | −3.073 | .002 |
| Cellular Processes | 3.64 ± 0.23 | 3.92 ± 0.25 | 3.48 ± 0.22 | −4.111 | .000 |
| Organismal Systems | 0.77 ± 0.05 | 0.72 ± 0.04 | 0.82 ± 0.05 | −3.982 | .000 |
| Unclassified | 0.00 ± 1.01 | 0.00 ± 1.03 | 0.00 ± 0.94 | −4.645 | .000 |
4. DISCUSSION
Based on our study, the beta diversity analysis of the intestinal microbiota of H. maoershanensis showed differences in age, whereas the alpha diversity revealed a higher degree of similarity. The results were similar to that of the northern leopard frog (Lithobates pipiens) (Kohl et al., 2013). Adults of H. maoershanensis exhibited a greater abundance of Oxalobacteraceae and Flavorbacteriaceae. In terms of function, the organic systems and metabolism pathways were significantly enriched. The hindlimb bud stage exhibited a higher abundance of Peptostreptococcaceae, and the functional pathways of genetic information processing and cellular processes were enriched, whereas the forelimb bud stage contained a higher abundance of Clostridiaceae and Enterobacteriaceae. In terms of predicting function, the human diseases, environmental information processing approach was more enriched. The differences of structure and function of intestinal microbiotas of H. maoershanensis based on the different developmental stages were consistent with our predictions and similar results have been reported for other amphibians, such as the northern leopard frog (Lithobates pipiens) (Kohl et al., 2013), Bufo gargarizans (Chai et al., 2018), which may be related to age and age‐based dietary changes. Generally, in mammals, amphibians, or even some insects, gut microbes will undergo morphological, physical, and chemical changes concomitantly with the development of the digestive system (Colman et al., 2012; Kohl et al., 2013; Ley et al., 2008; Moll et al., 2001). During the development of amphibians, there will be changes in the living environment (from completely aquatic to terrestrial life) and adults exhibit a higher diversity and richness of intestinal microbiota compared with tadpoles (Chai et al., 2018; Kohl et al., 2013). However, the alpha diversity of adults and tadpoles did not show significant differences, which may be related to the sampling time of adults. We sampled H. maoershanensis during underwater breeding activities. At this time, the habitat of the adult is consistent with that of the tadpole. As with other species, there was no significant difference in the alpha diversity of intestinal microbiotas at different stages of ontogeny and the reasons for this may be diverse (Chen, Cui, et al., 2021; Chen, Li, et al., 2021; Kohl et al., 2013).
At present, there have been no definitive studies on the feeding habits of H. maoershanensis. Studies have shown a decrease in the Firmicutes:Bacteroidetes ratio during the adaptation to a high‐fiber diet, whereas an increase in the ratio tends result from high‐protein food consumption (De Filippo et al., 2010). In this study, the abundance of Firmicutes (adult: 7.27% ± 11.67%, hindlimb bud stage: 58.9% ± 28.41% and forelimb bud stage: 52.19% ± 31.03%) and Bacteroides (adult; 39.69% ± 21.4%, hindlimb bud stage: 9.38% ± 16.37%, and forelimb bud stage: 1.99% ± 2.7%) were significantly different at different developmental stages (p < .001, FDR < 0.001). The Firmicutes:Bacteroidetes ratio was the highest during the forelimb bud stage (26.226), followed by the hindlimb bud stage (6.279), and the adult stage (0.183). In addition, there was a higher abundance of Oxalobacteriaceae in the adult intestines (28.28) (p < .001, FDR < 0.001), a type of bacteria that mainly digests oxalic acid which is enriched in plant foods (Green et al., 2007; Yu et al., 2021). In addition, the adult intestine had a significantly higher Bacteroide content (39.69% ± 21.4%) (p < .001, FDR < 0.001) compared with the tadpole stages, because the bacteria in this phylum have the ability to degrade and utilize host mucin glycosides, especially when there is a lack of food‐derived substrates in the intestine. These are considered bacteria that are adapted to food‐deficient environments (Flint et al., 2012; Sonnenburg et al., 2005). Therefore, it is speculated that the increase of Bacteroides in the adult intestine of H. maoershanensis may result from a nutrient‐poor environment. Based on these factors, we speculate that the H. maoershanensis adults may be more adapted to a high‐fiber diet with a higher proportion of plant foods. The adult H. maoershanensis may be facing a severe shortage of food. Compared with adults, for the sake of the nutritional requirements for maximum growth of the organism during the tadpole stage, the intestinal microbiotas of the tadpole exhibited higher levels of Peptostreptococcaceae, Clostridiaceae and Fusobacteriaceae (Soverini et al., 2016), which have higher proteolysis and amino acid metabolism activities, indicating that H. maoershanensis in the tadpole stage may prefer a carnivorous diet. To some extent, this is consistent with the view that tadpoles have a diverse diet (Bennett, 1992; Karasov, 1992). The functional differences of intestinal microbiota at different developmental stages might be attributed to the changes of diet and development (Chen, Cui, et al., 2021; Chen, Li, et al., 2021; Moya & Ferrer, 2016). For example, the relative abundance of Clostridiaceae was more enriched in the forelimb bud stage, the infections with bacteria of the genus Sarcina (family Clostridiaceae) in human and animal were associated with gastric dilation and emphysematous gastritis (Owens et al., 2021), consistent with the functional prediction that human disease were more enriched in the forelimb bud stage. The prediction of intestinal microbial function of H. maoershanensis needs to be further studied.
There are two points worth noting. First, H. maoershanensis adults contain a significantly higher abundance of Flavobacteriaceae (up to 30%) compared with the tadpole stages. Second, there was no significant difference in the relative abundance of Pseudomonadaceae during the three developmental stages, which accounted for a high proportion in each group (Adult: 3.78% ± 10.7%, hindlimb bud stage: 0.28% ± 0.5%, and forelimb bud stage: 8.13% ± 20.07%). Flavobacterium are pathogens associated with cold water disease worldwide (Kumru et al., 2020). They can cause diseases, such as red‐skin disease and ulcers. Pseudomonadaceae can cause septicemia and rot in aquatic animals (Miklos et al., 1999). These bacteria act on target cells by secreting proteins that interfere with their normal function to cause pathogenicity (Abby et al., 2016). Studies have shown that amphibians exhibit poor resilience after infection from such pathogens (Jani et al., 2021). Whether these two pathogenic bacteria cause widespread infection of H. maoershanensis and population decline remains to be determined.
In summary, the structure and function of the intestinal microbiota of H. maoershanensis at different developmental stages are significantly different. Future studies will expand on the information gathered on the intestinal microbiota of H. maoershanensis, including whether gender and reproduction period affect the structure and diversity of intestinal microbiota. Since our research was significantly restricted, more behavioral observations and other studies will be carried out in later stages, such as the maintenance and improvement of population numbers through the study of activities, environment and social habits in H. maoershanensis during nonreproductive periods.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Bo Yang: Data curation (lead); Investigation (equal); Validation (equal); Writing – original draft (lead). Zhenzhen Cui: Investigation (equal). Meihong Ning: Investigation (equal). Yu Chen: Investigation (equal). Zhengjun Wu: Funding acquisition (equal); Methodology (equal); Resources (equal). Huayuan Huang: Conceptualization (equal); Funding acquisition (equal); Project administration (equal); Supervision (equal); Visualization (equal); Writing – review & editing (equal).
OPEN RESEARCH BADGES
All data are available in the Dryad repository, and the link to the data is https://doi.org/10.5061/dryad.zs7h44j9t.
Supporting information
Appendix S1‐S3
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (No. 31860609). We are very grateful to the Guangxi Maoershan National Nature Reserve for allowing us to conduct research at the site. We appreciate the assistance of Ye Jianping of the Maoershan National Nature Reserve in Guangxi. We also thank the members of the Bajiaotian Management Station of the Maoershan National Nature Reserve in Guangxi for their assistance in the field. We also thank the Shanghai Meiji Biomedical Technology Co., Ltd. for technical support. The authors would like to thank Enago (www.enago.cn) for providing English touch up.
Yang, B. , Cui, Z. , Ning, M. , Chen, Y. , Wu, Z. , & Huang, H. (2022). Variation in the intestinal microbiota at different developmental stages of Hynobius maoershanensis . Ecology and Evolution, 12, e8712. 10.1002/ece3.8712
Contributor Information
Zhengjun Wu, Email: wu_zhengjun@aliyun.com.
Huayuan Huang, Email: hhy-121@163.com.
DATA AVAILABILITY STATEMENT
The raw sequencing data from the current study are available in the Dryad repository at https://doi.org/10.5061/dryad.zs7h44j9t.
REFERENCES
- Abby, S. S. , Cury, J. , Guglielmini, J. , Néron, B. , Touchon, M. , & Rocha, E. P. (2016). Identification of protein secretion systems in bacterial genomes. Scientific Reports, 6, 23080. 10.1038/srep23080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alford, R. (2011). Bleak future for amphibians. Nature, 480, 461–462. 10.1038/480461a [DOI] [PubMed] [Google Scholar]
- Bennett, A. F. (1992). Environmental physiology of the amphibians. Trends in Ecology & Evolution, 7(12), 426–427. 10.1016/0169-5347(92)90032-7 [DOI] [Google Scholar]
- Besten, G. D. , Eunen, K. V. , Albert, K. G. , Venema, K. , Dirk‐Jan, R. , & Bakker, B. M. (2013). The role of short‐chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. Journal of Lipid Research, 54(9), 2325–2340. 10.1194/jlr.R036012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bletz, M. C. , Goedbloed, D. J. , Sanchez, E. , Reinhardt, T. , Tebbe, C. C. , Bhuju, S. , Geffers, R. , Jarek, M. , Vences, M. , & Steinfartz, S. (2016). Amphibian gut microbiota shifts differetially in community structure but converges on habitat‐specific predicted functions. Nature Communications, 7, 13699–13701. 10.1038/ncomms13699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonder, M. J. , Kurilshikov, A. , Tigchelaar, E. F. , Mujagic, Z. , Imhann, F. , Vila, A. V. , Deelen, P. , Vatanen, T. , Schirmer, M. , Smeekens, S. P. , Zhernakova, D. V. , Jankipersadsing, S. A. , Jaeger, M. , Oosting, M. , Cenit, M. C. , Masclee, A. A. , Swertz, M. A. , Li, Y. , Kumar, V. , … Zhernakova, A. (2016). The effect of host genetics on the gut microbiome. Nature Genetics, 48(11), 1407–1412. 10.1038/ng.3663 [DOI] [PubMed] [Google Scholar]
- Chai, L. , Dong, Z. , Chen, A. , & Wang, H. (2018). Changes in intestinal microbiota of Bufo gargarizans and its association with body weight during metamorphosis. Archives of Microbiol, 200, 1087–1099. 10.1007/s00203-018-1523-1 [DOI] [PubMed] [Google Scholar]
- Chen, L. , Li, S. , Xiao, Q. I. , Lin, Y. , Li, X. , Qu, Y. , Wu, G. , & Li, H. (2021). Composition and diversity of gut microbiota in Pomacea canaliculata in sexes and between developmental stages. BMC Microbiology, 21(1), 200. 10.1186/s12866-021-02259-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , Cui, Z. Z. , Yang, B. , Ning, M. H. , Wu, Z. J. , Ye, J. P. , & Huang, H. Y. (2021). Histology and distribution of 5‐hydroxytryptamine cells in the digestive tract of Hynobius maoershanensis and Pachytriton intexpectatus . Chinese Journal of Zoology, 56(4), 597–607. 10.13859/j.cjz.202104012 [DOI] [Google Scholar]
- Colman, D. R. , Toolson, E. C. , & Takacs‐Vesbach, C. D. (2012). Do diet and taxonomy influence insect gut bacterial communities? Molecular Ecology, 21(20), 5124–5137. 10.1111/j.1365-294X.2012.05752.x [DOI] [PubMed] [Google Scholar]
- Colston, T. J. , Noonan, B. P. , Jackson, C. R. , & Gabriel, M. H. (2015). Phylogenetic analysis of bacterial communities in different regions of the gastrointestinal tract of Agkistrodon piscivorus, the cottonmouth snake. PLoS One, 10(6), e0128793. 10.1371/journal.pone.0128793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch, C. E. , Stagaman, K. , Spaan, R. S. , Combrink, H. J. , Sharpton, T. J. , Beechler, B. R. , & Jolles, A. E. (2021). Diet and gut microbiome enterotype are associated at the population level in African buffalo. Nature Communications, 12, 2267. 10.1038/s41467-021-22510-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David, L. , Maurice, C. F. , Carmody, R. N. , Gootenberg, G. E. , Button, J. E. , Wolfe, B. E. , Ling, A. V. , Devlin, A. S. , Varma, Y. , Fischbach, M. A. , Biddinger, S. B. , Button, R. J. , & Turnbaugh, P. J. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature, 505, 559–563. 10.1038/nature12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippo, C. , Cavalieri, D. , Di Paola, M. , Ramazzotti, M. , Poullet, J. B. , Massart, S. , Collini, S. , Pieraccini, G. , & Lionetti, P. (2010). Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proceedings of the National Academy of Sciences of the United States of America, 107(33), 14691–14696. 10.1073/pnas.1005963107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint, H. J. , Scott, K. P. , Louis, P. , & Duncan, S. H. (2012). The role of the gut microbiota in nutrition and health. Nature Reviews Gastroenterology & Hepatology, 9, 577–589. 10.1038/nrgastro.2012.156 [DOI] [PubMed] [Google Scholar]
- Frese, S. A. , Parker, K. , Calvert, C. C. , & Mills, D. A. (2015). Diet shapes the gut microbiome of the pigs during nursing and weaning. Microbiome, 3, 28. 10.1186/s40168-015-0091-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillman, S. J. , McKenney, E. A. , & Lafferty, D. J. R. (2020). Wild black bears harbor simple gut microbial communities with little difference between the jejunum and colon. Scientific Reports, 10, 20779. 10.1038/s41598-020-77282-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green, S. , Michel, F. , Hadar, Y. , & Minz, D. (2007). Contrasting patterns of seed and root colonization by bacteria from the genus Chryseobacterium and from the family Oxalobacteraceae. The ISME Journal, 1(4), 291–299. 10.1038/ismej.2007.33 [DOI] [PubMed] [Google Scholar]
- Greener, M. S. , Verbrugghe, E. , Kelly, M. , Blooi, M. , Beukema, W. , Canessa, S. , Carranza, S. , Croubels, S. , Troyer, N. D. , Fernandez‐Giberteau, D. , Goethals, P. , Lens, L. , Li, Z. M. , Stegen, G. , Strubbe, D. , Leeuwenberg, R. V. , Praet, S. V. , Vila‐Escale, M. , Vervaeke, M. , … Martel, A. (2020). Presence of low virulence chytrid fungi could protect European amphibians from more deadly strains. Nature Commuciations, 11, 5393. 10.1038/s41467-020-19241-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang, I. , Park, Y. J. , Kim, Y. R. , Kim, Y. N. , Ka, S. , Lee, H. Y. , Seong, J. K. , Seok, Y. J. , & Kim, L. B. (2015). Alteration of gut microbiota by vancomycin and bacitracin improves insulin resistance via glucagon‐like peptide 1 in diet‐induced obesity. The FASEB Journal, 29(6), 2397–2411. 10.1096/fj.14-265983 [DOI] [PubMed] [Google Scholar]
- Jani, A. J. , Bushell, J. , Arisdakessian, C. G. , Belcaid, M. , Boiano, D. M. , Brown, C. , & Knapp, R. A. (2021). The amphibian microbiome exhibits poor resilience following pathogen‐induced disturbance. The ISME Journal, 15, 1628–1640. 10.1038/s41396-020-00875-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, A. W. , & Jiang, D. B. (2006). A new species of the genus Hynobius from Guangxi Zhuang autonomous region, China. (Caudata, Hynobiidae). Acta Zootaxonomica Sinica, 31(3), 670–674. 10.3969/j.issn.1000-0739.2006.03.049 [DOI] [Google Scholar]
- Karasov, W. H. (1992). Test of adaptive modulation hypothesis for dietary control of intestinal transport. The American Journal of Physiology, 263(3 pt2), R496–R502. 10.1152/ajpregu.1992.263.3.R496 [DOI] [PubMed] [Google Scholar]
- Kirsten, G. , Sandercock, B. K. , Ari, J. , & Zeglin, L. H. (2018). The avian gut microbiota: Community, physiology and function in wild birds. Journal of Avian Biology, 49(11), e01788. 10.1111/jav.01788 [DOI] [Google Scholar]
- Koeppel, M. , Garcia‐Alcalde, F. , Glowinski, F. , Schlaermann, P. , & Meyer, T. F. (2015). Helicobacter pylori infection causes characteristic DNA damage patterns in human cells. Cell Reports, 11(11), 1703–1713. 10.1016/j.celrep.2015.05.030 [DOI] [PubMed] [Google Scholar]
- Kohl, K. D. , Cary, T. L. , Karasov, W. H. , & Dearing, M. D. (2013). Restructuring of the amphibian gut microbiota through metamorphosis. Environmental Microbiology Reports, 5(6), 899–903. 10.1111/1758-2229.12092 [DOI] [PubMed] [Google Scholar]
- Kumru, S. , Tekedar, H. C. , Blom, J. , Lawrence, M. L. , & Karsi, A. (2020). Genomic diversity in Flavobacterial pathogens of aquatic origin. Microbial Pathogenesis, 142, 104053. 10.1016/j.micpath.2020.104053 [DOI] [PubMed] [Google Scholar]
- Lapébie, P. , Lombard, V. , Drula, E. , Terrapon, N. , & Henrissat, B. (2019). Bacteroidetes use thousands of enzyme combinations to break down glycans. Nature Communications, 10, 2043. 10.1038/s41467-019-10068-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley, R. E. , Hamady, M. , Lozupone, C. , Turnbaugh, P. J. , Ramey, R. R. , Bircher, J. S. , Schlegel, M. L. , Tucker, T. A. , Schrenzel, M. D. , Knight, R. , & Gordon, J. I. (2008). Evolution of mammals and their gut microbes. Science, 320(5883), 1647–1651. 10.1126/science.1155725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. H. , Chen, T. , Li, Y. B. , Tang, Y. , & Huang, Z. H. (2021). Gut microbiota are associated with sex and age of host: Evidence from semi‐provisioned rhesus macaques in southwest Guangxi, China. Ecology and Evolution, 11, 8096–8122. 10.1002/ece3.7643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miklos, S. M. , Tan, M. W. , Rahme, L. G. , & Ausubel, F. M. (1999). Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa– Caenorhabditis elegans Pathogenesis Model. Cell, 96(1), 47–56. 10.1016/S0092-8674(00)80958-7 [DOI] [PubMed] [Google Scholar]
- Moll, R. M. , Romoser, W. S. , Modrakowski, M. C. , Moncayo, A. C. , & Lerdthusnee, K. (2001). Meconial peritrophic membranes and the fate of midgut bacteria during mosquito (diptera: culicidae) metamorphosis. Journal of Medical Entomology, 38(1), 29–32. 10.1603/0022-2585-38.1.29 [DOI] [PubMed] [Google Scholar]
- Mori, H. , Maruyama, F. , Kato, H. , Toyoda, A. , Dozono, A. , Ohtsubo, Y. , Nagata, Y. , Fujiyama, A. , Tsuda, M. , & Kurokawa, K. (2014). Design and experimental application of a novel non‐degenerate universal primer set that amplifies prokaryotic 16S rRNA genes with a low possibility to amplify eukaryotic rRNA genes. DNA Research, 21(2), 217–227. 10.1093/dnares/dst052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya, A. , & Ferrer, M. (2016). Functional redundancy‐induced stability of gut microbiota subjected to disturbance. Trends in Microbiology, 24(5), 402–413. 10.1016/j.tim.2016.02.002 [DOI] [PubMed] [Google Scholar]
- Ning, M. H. , Chi, H. , Chen, Y. , Yang, B. , Wu, Z. J. , & Huang, H. Y. (2021). A preliminary study on the ontogenetic characteristics of captive‐bred Hynobius maoershanensis . Sichuan Journal of Zoology, 40(2), 196–202. 10.11984/j.issn.1000-7083.20200422 [DOI] [Google Scholar]
- Owens, L. A. , Colitti, B. , Hirji, I. , Pizarro, A. , Jaffe, J. E. , Moittié, S. , Bishop‐Lilly, K. A. , Estrella, L. A. , Voegtly, L. J. , Kuhn, J. H. , Suen, G. , Deblois, C. L. , Dunn, C. D. , Juan‐Sallés, C. , & Goldberg, T. L. (2021). A Sarcina bacterium linked to lethal disease in sanctuary chimpanzees in Sierra Leone. Nature Communications, 12, 763. 10.1038/s41467-021-21012-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman, A. S. , Gehrig, J. L. , Venkatesh, S. , Chang, H. W. , Hibberd, M. C. , Subramanian, S. , Kang, G. , Bessong, P. O. , Lima, A. A. M. , Kosek, M. N. , Petri Jr, W. A. , Rodionov, D. A. , Arzamasov, A. A. , Leyn, S. A. , Osterman, A. L. , Huq, S. , Mostafa, I. , Isiam, M. , Mahfuz, M. , … Gordon, J. I. (2019). A sparse covarying unit that describes healthy and impaired human gut microbiota development. Science, 365(6449). 10.1126/science.aau4735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimal, B. , & Patterson, A. D. (2021). Role of bile acids and gut bacteria in healthy ageing of centenarians. Nature, 599, 380–381. 10.1038/d41586-021-02196-0 [DOI] [PubMed] [Google Scholar]
- Schreiber, A. M. , Cai, L. Q. , & Brown, D. D. (2005). Remodeling of the intestine during metamorphosis of Xenopus laevis . PNAS, 102(10), 3720–3725. 10.1073/pnas.0409868102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, X. W. , Song, J. H. , Song, H. H. , Zeng, Q. , & Shi, K. K. (2018). A robust noninvasive approach to study gut microbiota structure of amphibian tadpoles by feces. Asian Herpetological Research, 9(01), 1–12. 10.16373/j.cnki.ahr.170062 [DOI] [Google Scholar]
- Sonnenburg, J. L. , Xu, J. , Leip, D. D. , Chen, C. H. , Westover, B. P. , Weatherford, J. , Buhler, J. D. , & Gordon, J. I. (2005). Glycan foraging in vivo by an intestine‐adapted bacterial symbiont. Science, 307(5717), 1955–1959. 10.1126/science.1109051 [DOI] [PubMed] [Google Scholar]
- Soverini, M. , Quercia, S. , Biancani, B. , Furlati, S. , Turroni, S. , Biagi, E. , Consolandi, C. , Peano, C. , Severgnini, M. , Rampelli, S. , Brigidi, P. , & Candela, M. (2016). The bottlenose dolphin (Tursiops truncatus) faecal microbiota. Fems Microbiology Ecology, 92(4), fiw055. 10.1093/femsec/fiw055 [DOI] [PubMed] [Google Scholar]
- Sullam, K. E. , Essinger, S. D. , Lozupone, C. A. , O’Connor, M. P. , Rosen, G. L. , Knight, R. , Kilham, S. S. , & Russell, J. A. (2012). Environmental and ecological factors that shape the gut bacterial communities of fish: A meta‐analysis. Molecular Ecology, 21(13), 3363–3378. 10.1111/j.1365-294X.2012.05552.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, F. , Hehemann, J. H. , Rebuffet, E. , Czjzek, M. , & Michel, G. (2011). Environmental and gut bacteroidetes: The food connection. Frontiers in Microbiology, 2(93), 93. 10.3389/fmicb.2011.00093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu, Y. , Zhu, W. Y. , & Lu, C. P. (2005). Bacterial 16S rDNA sequence analysis of Siberian tiger faecal flora. Acta Microbiologica Sinica, 45(05), 671–674. 10.13343/j.cnki.wsxb.2005.05.003 [DOI] [PubMed] [Google Scholar]
- Vangay, P. , Johnson, A. J. , Ward, T. L. , Al‐Ghalith, G. A. , Shields‐Cutler, R. R. , Hillmann, B. M. , Lucas, S. K. , Beura, L. K. , Thompson, E. A. , Till, L. M. , Batres, R. , Paw, B. , Pergament, S. L. , Saenyakul, P. , Xiong, M. , Kim, A. D. , Kim, G. , Masopust, D. , Martens, E. C. , … Knights, D. (2018). US immigration westernizes the human gut microbiome. Cell, 175(4), 10.1016/j.cell.2018.10.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vences, M. , Lyra, M. L. , Kueneman, J. G. , Bletz, M. C. , Archer, H. M. , Canitz, J. , Handreck, S. , Randrianiaina, R. , Struck, U. , Bhuju, S. , Jarek, M. , Geffers, R. , Mckenzie, V. J. , Tebbe, C. C. , Haddad, C. F. B. , & Glos, J. (2016). Gut bacterial communities across tadpole ecomorphs in two diverse tropical anuran faunas. Science of Nature, 103(3–4), 25. 10.1007/s00114-016-1348-1 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Smith, H. K. , Goossens, E. , Hertzog, L. , Bletz, M. C. , Bonte, D. , Verheyen, K. , Lens, L. , Vences, M. , Pasmans, F. , & Martel, A. (2021). Diet diversity and environment determine the intestinal microbiome and bacterial pathogen load of fire salamanders. Scientific Reports, 11, 20493. 10.1038/s41598-021-98995-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng, C. H. , Yang, Y. J. , & Wang, D. (2016). Functional analysis for gut microbes of the brown tree frog (polypedates megacephalus) in artificial hibernation. BMC Genomics, 17(S13), 31–42. 10.1186/s12864-016-3318-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, P. , He, X. M. , Baer, M. , Beirinckx, S. , Tian, T. , Moya, Y. A. T. , Zhang, X. C. , Deichmann, M. , Frey, F. P. , Bresgen, V. , Li, C. J. , Razavi, B. S. , Schaaf, G. , von Wirén, N. , Su, Z. , Bucher, M. , Tsuda, K. , Goormachtig, S. , Chen, X. P. , & Hochholdinger, F. (2021). Plant flavones enrich rhizosphere Oxalobacteraceae to improve maize performance under nitrogen deprivation. Nature Plants, 4, 481–499. 10.1038/s41477-021-00897-y [DOI] [PubMed] [Google Scholar]
- Zhang, M. J. , Chen, H. , Liu, L. S. , Xu, L. L. , Wang, X. G. , Chang, L. M. , Chang, Q. , Lu, G. Q. , Jiang, J. P. , & Zhu, L. F. (2020). The changes in the frog gut microbiome and its putative oxygen‐related phenotypes accompanying the development of gastrointestinal complexity and dietary shift. Frontiers in Microbiology, 11, 162. 10.3389/fmicb.2020.00162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z. , Hu, T. , Lu, G. Q. , & Zhu, L. F. (2019). Lessons from bamboo‐eating pandas and their gut microbiome: Gut microbiome flow and applications. Evolutionary Applications, 13, 615–619. 10.1111/eva.12915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M. J. , Sarah, G. , Chang, Q. , Chen, H. , Lu, G. Q. , Wang, X. G. , Xu, L. L. , Zhu, L. F. , & Jiang, J. P. (2018). Age‐related changes in the gut microbiota of the Chinese giant salamander (Andrias davidianus). MicrobiologyOpen, 8(7), 1–14. 10.1002/mbo3.778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Wen, B. , Meng, L. J. , Gao, J. Z. , & Chen, Z. Z. (2021). Dynamic changes of gut microbiota of discus fish (Symphysodon haraldi) at different feeding stages. Aquaculture, 531, 735912. 10.1016/j.aquaculture.2020.735912 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1‐S3
Data Availability Statement
The raw sequencing data from the current study are available in the Dryad repository at https://doi.org/10.5061/dryad.zs7h44j9t.