

Abstract
Asymmetric catalytic azidation has increased in importance to access enantioenriched nitrogen containing molecules, but methods that employ inexpensive sodium azide remain scarce. This encouraged us to undertake a detailed study on the application of hydrogen bonding phase-transfer catalysis (HB-PTC) to enantioselective azidation with sodium azide. So far, this phase-transfer manifold has been applied exclusively to insoluble metal alkali fluorides for carbon–fluorine bond formation. Herein, we disclose the asymmetric ring opening of meso aziridinium electrophiles derived from β-chloroamines with sodium azide in the presence of a chiral bisurea catalyst. The structure of novel hydrogen bonded azide complexes was analyzed computationally, in the solid state by X-ray diffraction, and in solution phase by 1H and 14N/15N NMR spectroscopy. With N-isopropylated BINAM-derived bisurea, end-on binding of azide in a tripodal fashion to all three NH bonds is energetically favorable, an arrangement reminiscent of the corresponding dynamically more rigid trifurcated hydrogen-bonded fluoride complex. Computational analysis informs that the most stable transition state leading to the major enantiomer displays attack from the hydrogen-bonded end of the azide anion. All three H-bonds are retained in the transition state; however, as seen in asymmetric HB-PTC fluorination, the H-bond between the nucleophile and the monodentate urea lengthens most noticeably along the reaction coordinate. Kinetic studies corroborate with the turnover rate limiting event resulting in a chiral ion pair containing an aziridinium cation and a catalyst-bound azide anion, along with catalyst inhibition incurred by accumulation of NaCl. This study demonstrates that HB-PTC can serve as an activation mode for inorganic salts other than metal alkali fluorides for applications in asymmetric synthesis.
Introduction
Griess, Curtius, and Tiemann were the first to investigate the chemistry of metal and organic azides at the end of the 19th century,1 with greater interest in azidation chemistry emerging in the 1960s.2 Today, organic azides have established themselves as highly versatile intermediates for synthetic, material, and biological applications because they participate in diverse transformations including 1,3-dipolar cycloadditions, aza-Wittig reactions, Staudinger reductions and ligations, as well as C–H bond aminations.3 Various protocols for asymmetric azidation have been developed, often requiring toxic and volatile reagents such as hydrazoic acid or azidotrimethylsilane.4 Crystalline 1-azido-1,2-benziodoxol-3(1H)-one has also been used, but this azide source is of poor atom economy, and is prepared from azidotrimethylsilane.5 In contrast, only a few enantioenriched organic azides are obtainable directly from sodium azide, an inexpensive reagent compared to all aforementioned azide sources. Phase-transfer catalysis with chiral ammonium salts is the most successful approach for azidation with sodium azide, although details on how the azide ion interacts with the catalyst-substrate complex are scarce.6
In biology, the azide ion serves as an inhibitor of many enzymes, including cytochrome oxidases involved in the electron transport chain, and formate dehydrogenase for CO2 fixation or nicotinamide recycling (Figure 1A).7 Enzyme inhibition results from coordination of the azide ion to the metal or through H-bonding interactions as observed in the azide-bound NAD-dependent dehydrogenase complex (PDB ID 2NAD). The terminal nitrogen atoms of the azide anion can engage in H-bond contacts with several residues of the enzyme, a binding profile that enables azide to serve as a bridging ion between molecular fragments. As well as acting as an inhibitor, the azide ion has been used as a nucleophile in biocatalytic azidations. Janssen and co-workers reported the kinetic resolution of racemic epoxides by azidolysis with NaN3 in the presence of the halohydrin dehalogenase from Agrobacterium radiobacter AD1, an enzyme class that typically promotes (de)halogenation, with the exception of fluoride, in epoxide chemistry.8 This enzyme displays nucleophile promiscuity for a range of monovalent, linear anions other than N3- including cyanide, cyanate, and isocyanate ions. More recently, C–H azidation at aliphatic carbons enabled by an iron-dependent halogenase was disclosed, a process highlighting coordination of the azide anion to the enzyme’s Fe(II) cofactor.9
Figure 1.

Molecular principles of catalysis and inhibition featuring hydrogen bonding interactions in natural enzymes; translational design strategies for synthetic organocatalysts. (A) Azide binding in natural enzymes. (B) Azidation with sodium azide under hydrogen bonding phase-transfer catalysis (this work).
The metal-coordinating ability of the azide anion has been amply exploited for catalytic azidation.10 For example, Groves and co-workers have reported a manganese-catalyzed aliphatic C–H azidation reaction featuring a Mn-bound intermediate in the azido transfer step.11 In contrast, the ability of the azide anion to engage in H-bonding interactions has not been harnessed for catalytic azidation with NaN3. We however note that theoretical studies have suggested that H-bonds in methylpentynol-azide clusters may influence the regiochemical outcome of 1,3-dipolar cycloaddition reactions.12
This state of play encouraged an in-depth investigation into the coordination chemistry of the azide ion with hydrogen bond donors (HBD) for applications in asymmetric catalysis (Figure 1B). The study detailed herein focuses on the H-bonding of azide with ureas, a class of HBD widely used in catalysis for a wide range of (asymmetric) transformations other than azidations. Mechanistically, we envisaged a scenario based on anion binding catalysis whereby the urea-bound azide ion would intercept a cationic electrophile (E+) in the enantiodetermining step. In this approach, H-bonding interactions to the azide anion would enable the HBD urea to function as a phase-transfer catalyst and bring NaN3 into solution. These interactions would attenuate the nucleophilicity of the azide. In our previous work applying this mechanistic scenario for enantioselective fluorination, background reactivity was suppressed by using an insoluble metal alkali fluoride with the urea HBD serving as phase-transfer catalyst (hydrogen bonding phase-transfer catalysis, HB-PTC).13 This manuscript addresses whether HB-PTC is viable for enantioselective azidation with NaN3. Specifically, we demonstrate C(sp3)–N3 bond formation under catalytic conditions and report the successful application of a chiral BINAM-derived bisurea catalyst to promote asymmetric azidation with sodium azide for the synthesis of enantioenriched β-amino azides. Detailed information is provided on the structure and characterization of a diverse range of (a)chiral urea-azide complexes in the solid state and in solution. Moreover, X-ray diffraction analysis, quantum chemical calculations, and NMR spectroscopy provide insight on the coordination chemistry of the azide ion to a chiral BINAM-derived bisurea catalyst. The catalytic cycle has been interrogated using a combination of kinetics and computational studies.
Results and Discussion
1. Catalytic Azidation under HB-PTC
We started our investigation by employing β-chloroamines as substrates as these were previously found to be reactive under HB-PTC conditions with alkali metal fluorides.13 When treated with NaN3 in 1,2-difluorobenzene in the absence of a hydrogen bond donor, model substrate (±)-2a afforded (±)-3a in less than 10% yield after 1 h (Scheme 1). When 10 mol % of Schreiner’s urea 1a(14) was added to the reaction mixture and under otherwise identical conditions, the yield of this reaction increased to 90%.
Scheme 1. Azidation of β-Chloroamine (±)-2a Using NaN3 in 1,2-Difluorobenzene with(out) Schreiner’s Urea 1a.

The demonstration that the hydrogen bond donor urea 1a catalyzes azidation of (±)-2a prompted the development of an enantioselective variant of this reaction. We selected the chiral BINAM-derived bisurea catalyst (S)-1k that was highly successful for fluorination, well aware that the strength and binding mode of azide anion with (S)-1k may differ significantly from fluoride (Table 1). The reaction of (±)-2a with NaN3 in 1,2-difluorobenzene in the presence of (S)-1k (5 mol %) at room temperature provided 3a in 80% yield and 85:15 e.r., thereby demonstrating the feasibility of enantioselective azidation under HB-PTC (Table 1, entry 1). The nonalkylated (S)-BINAM catalyst (S)-1l with four instead of three sites for hydrogen bonding with azide afforded β-aminoazide 3a in 74% yield, but with significantly decreased e.r., a result highlighting the crucial effect of N-alkylation on enantiocontrol (Table 1, entry 2).13N-Methylated catalyst (S)-1m (Table 1, entry 3) or a switch to other solvents (Table 1, entries 4 and 5) led to decreased enantiocontrol. Enantioselectivity was improved by reducing the temperature, although this required increasing both the azide and catalyst loading to achieve full conversion. Use of catalyst (S)-1k (10 mol %) with 2.4 equiv NaN3 at −20 °C for 72 h yielded β-amino azide (S,S)-3a in 74% yield and 93.5:6.5 e.r. (Table 4, entry 6).15 The reaction of soluble tetrabutylammonium azide in the presence of (S)-1k (5 mol %) resulted in racemic product, suggesting that phase-transfer is essential for enantioinduction (Table 1, entry 7).
Table 1. Optimization of the Reaction Conditions.

| entry | cat. | azide source | solvent | yielda (%) | e.r.b |
|---|---|---|---|---|---|
| 1 | (S)-1k | NaN3 (1.2 equiv) | 1,2-DFB | 80 | 85:15 |
| 2 | (S)-1l | NaN3 (1.2 equiv) | 1,2-DFB | 74 | 58:42 |
| 3 | (S)-1m | NaN3 (1.2 equiv) | 1,2-DFB | 80 | 83:17 |
| 4 | (S)-1k | NaN3 (1.2 equiv) | CHCl3 | 86 | 77:23 |
| 5 | (S)-1k | NaN3 (1.2 equiv) | CH2Cl2 | 91 | 81:19 |
| 6c | (S)-1k | NaN3 (2.4 equiv) | 1,2-DFB | 74 | 93.5:6.5 |
| 7 | (S)-1k | Bu4N·N3 (1.2 equiv) | 1,2-DFB | 90 | 50:50 |
Yield determined by 1H NMR with Ph3CH (0.5 equiv) internal standard.
e.r. determined after prep TLC.
(S)-1k (10 mol %), 72 h, −20 °C. 1,2-DFB = 1,2-difluorobenzene.
Table 4. Donor–Acceptor N(H)···X– Bond Distances22a of 1k·TBAN3.
| NH···X– | X-ray (±)-1k·N3–d N···N3– (Å) | X-ray (S)-1k·F–d N···F– (Å)13a | DFT (S)-1k·N3–d N···N3– (Å) |
|---|---|---|---|
| 1 | 2.81(2) (S), 2.81(2) (R) | 2.667(2) | 2.90(4) |
| 2 | 2.94(2) (S), 2.91(2) (R) | 2.690(2) | 2.95(2) |
| 3 | 2.98(2) (S), 3.02(2) (R) | 2.662(2) | 3.03(2) |
The optimized conditions were successfully applied to a range of meso-aziridinium precursors (Scheme 2). Products containing pharmaceutically relevant saturated heterocycles ,16 including substituted piperidines 3a–c, pyrrolidine 3d, morpholine 3e, piperazine 3f–g, and tetrahydroisoquinoline 3h motifs were all formed with high enantioselectivity. Unsymmetrically substituted amine derivatives also performed well in this reaction to give 3h–i, despite the possibility for formation of two diastereomeric meso-aziridinium intermediates. The reaction was found to be tolerant of meta- and para-halogen substituents (3l, 3m, 3o), trifluoromethyl groups (3n, 3q), and larger alkyl groups (3p). The absolute configuration of 3o was determined by single-crystal X-ray diffraction and was used to assign the absolute configuration of 3a–n and 3p–r by analogy. The bis-pyridyl azide 3r was obtained in good yield and enantioselectivity. A cycloalkyl amino chloride successfully furnished product 3s in high yield but with no enantiocontrol. The model reaction was carried out on gram scale, yielding 1.23 g of (S,S)-3a in 80% yield and 93.5:6.5 e.r. No measures were taken to avoid moisture or oxygen, emphasizing the operational simplicity of reactions performed under HB-PTC. Reduction of azide (S,S)-3a by hydrogenation and subsequent bis-alkylation with 1,5-dibromopentane afforded 1.1 g of enantioenriched Kv1.5 blocker 4 from (±)-2a.17
Scheme 2. Substrate Scope, Scale-up, and Derivatization.

1.09 mmol scale.
Absolute configuration of major enantiomer not determined.
The successful application of HB-PTC to NaN3 encouraged a detailed analysis of how the azide ion interacts with hydrogen bond donors such as ureas in the solid state and in solution, and further mechanistic investigation based on kinetics combined with computational studies.
2. Insight on the Structure of Urea-Azide Complexes from Experimental and Computational Studies
Studies on H-bonded azide complexes have been reported,18 with a single example of azide anion encapsulated in a urea receptor.18l This limited knowledge on the binding modes of azide with urea motifs prompted us to prepare and characterize H-bonded azide complexes derived, at first instance, from a range of achiral hydrogen bond donors.19 A set of variously substituted 1,3-diarylureas was selected to examine how steric and electronic effects may influence the structures of azide complexes in the solid-state, well aware that hydrogen bond directionality and/or packing effects may be at play. All complexes were synthesized from either tetrabutylammonium (TBA) azide or a combination of sodium azide and 15-crown-5 (>95% yield). The azide salt was stirred overnight with an equimolar amount of hydrogen bond donor in acetonitrile (0.1 M), followed by evaporation of solvent to dryness. The resulting complexes were characterized by 1H NMR, 13C NMR, and IR spectroscopy and subsequently recrystallized to obtain samples suitable for X-ray analysis.15 A diverse set of structures was obtained, revealing distinct binding modes which were categorized depending on (i) the type of donor–acceptor interaction (side-on or end-on),20 (ii) whether the complex exists as a bridged or nonbridged structure—in the end-on binding mode, the azide could act as a bridging ion between two hydrogen bond donors, and (iii) the stoichiometry of the complex with a HBD:azide ratio of either 1:1 or 2:1. Figure 2 illustrates the coordination diversity of the urea-azide complexes that were successfully characterized by single-crystal X-ray diffraction analysis and distinguishes between 1:1 nonbridged, side-on (type I, Figure 2A), 1:1 nonbridged, end-on (type II, Figure 2B), and 2:1 bridged, end-on (type III, Figure 2C) complexes. Figure 3 and Table 2 highlight some key parameters for these complexes, such as donor–acceptor (D–A) distances and the angles θ and Φ, which indicate the extent to which the azide lies outside of the urea NC(=O)N plane in end-on and side-on complexes.19
Figure 2.

Coordination diversity of achiral urea-azide complexes. M+ = tetrabutylammonium or Na[15-crown-5].
Figure 3.

Achiral hydrogen bonded donor-azide complexes. Counter cations and crown ethers are omitted for clarity.
Table 2. Main Structural Features of Achiral Hydrogen-Bonded Urea Complexes.
| entry | HBD | complex type | complex | D–A distance22 | θ or Φ22 |
|---|---|---|---|---|---|
| 1a,b | 1a, R1 = R2 = 3,5-CF3 | 1:1 Non-bridged (side-on) | [1a·N3]·TBA | 2.966(4), 2.974(4), 2.962(4), 3.069(4) | 1.15(14), 15.79(7) |
| 2c | 1a, R1 = R2 = 3,5-CF3 | 1:1 Non-bridged (end-on) | [1a·N3]·[Na(15-crown-5)] | 2.780(9), 3.332(9) | 62.40(30) |
| 3 | 1b, R1 = R2 = 3-Cl | 1:1 Non-bridged (end-on) | [1b·N3]·TBA | 2.876(4), 3.000(4) | 51.90(14) |
| 4 | 1c, R1 = 3,5-CF3; R2 = H | 1:1 Non-bridged (end-on) | [1c·N3]·TBA | 2.852(9), 2.946(9), 2.834(9), 2.940(9)c | 27.60(19), 34.21(12)c |
| 5 | 1d, R1 = R2 = 4-CF3 | 1:1 Non-bridged (end-on) | [1d·N3]·TBA | 2.838(5), 2.905(5) | 46.53(8) |
| 6 | 1e, R1 = R2 = H | 1:1 Non-bridged (end-on) | [1e·N3]·TBA | 2.870(2), 2.996(2) | 7.43(14) |
| 7 | 1f, R1 = R2 = 4-Br | 1:1 Non-bridged (end-on) | [1f·N3]·TBA | 2.909(8), 2.923(8) | 24.90(3) |
| 8 | 1g, R1 = R2 = 4-F | 1:1 Non-bridged (end-on) | [1g·N3]·TBA | 2.833(3), 2.912(3) | 37.13(6) |
| 9d | 1h, R1 = R2 = 4-CN | 2:1 Bridged (end-on) | {[1h]2·N3·2H2O}·TBA | 2.860(2), 2.976(2) | nd |
| 10e | 1i, R1 = R2 = H | 1:1 Non-bridged (end-on) | [1i·N3]·TBA | 2.842(2), 3.202(2) | nd |
| 11f | 1j, R1 = R2 = 3,5-CF3 | 1:1 Bridged (end-on) | {[1j]2·N3}·TBA | 2.923(3)d | nd |
Calculated for the two crystallographically distinct motifs.
This complex is side-on. The values correspond to N(1) to N(2) and N(1′) to N(4) distances, respectively.
Under slightly different crystallization conditions, a 2:1 complex was also obtained (see Supporting Information for details).
The second HB donor involves a single N–H in binding (N–H–N3 = 2.144(2) Å).
The hydrogen bond donor is a guanidine;
The hydrogen bond donor is an oxalyl amide, and the terminal nitrogen of the azide anion is bound to each HB donor via a single N–H. TBA = tetrabutylammonium.
The [1a·N3]·TBA complex derived from Schreiner’s urea 1a (entry 1, Table 2) features two crystallographically distinct motifs (see Supporting Information for details) and is a 1:1 complex with the azide bound side-on. This arrangement is likely favored due to the electron-deficient 3,5-bis(trifluoromethyl)phenyl groups which allow for an additional interaction between the two terminal nitrogens of the azide and the weakly acidic aryl ortho C–H bonds (D–A distance: 3.364(4) Å). An additional point of interest resides in weak long-range interactions between the azide and α-C–H bonds of the tetrabutylammonium countercation.17 In both crystallographic motifs, the azide lies in the plane of the NC(=O)N motif of the urea (1.15(14)°; 15.79(7)°). When the same urea was bound to azide but featured Na+(15-crown-5) as the countercation, an end-on 1:1 complex of type II was obtained with the azide out of the urea plane (θ = 62.40(30)°, ([1a·azide]·[Na(15-crown-5)], entry 2, Table 2). This result underlines the role of the cation in influencing the coordination mode of the azide in the solid-state. Complex [1a·azide]·[Na(15-crown-5)] features the shortest and longest D–A distances observed among all complexes examined in this study (2.780(9) Å and 3.332(9) Å). Similar coordination modes were observed for [1b·N3]·TBA and [1c·N3]·TBA (entries 3 and 4, Table 2) derived from symmetrical urea 1b featuring 3-Cl substituents, and unsymmetrical urea 1c substituted with 3,5-bis(trifluoromethyl) group on a single aryl ring, respectively. Complexes formed with 1d–1g presented two different packing arrangements, a dimeric structure whereby the urea N–H bonds point toward each other with two linking azides (entries 5 and 6, Table 2, [1d·N3]·TBA and [1e·N3]·TBA), and a structure in which the NHs of each urea point in the same direction thus forming an extended chain (entries 7 and 8, Table 2, [1f·N3]·TBA and [1g· N3]·TBA). Interestingly, [1f·N3]·TBA revealed the possibility of halogen bond interactions (3.140(2) and 3.131(2) Å) between the C–Br and the terminal nitrogen of the azide.21 Both urea 1f and 1g led to the formation of symmetry-related interdigitated antiparallel chains. For complexes derived from 1d–1g, the azide is out of the NC(=O)N plane of the urea with angles in the range of θ = 24–62°, but to a lesser extend for [1e· N3]·TBA (θ = 7.43(14)). A single 2:1 urea-azide complex was obtained, which included water of crystallization (type III, entry 9, {[1h]2· N3·2H2O}·TBA). In this complex, each urea with one of its NH binds the azide while the other NH is coordinated to water, which presumably originated from TBAF·3H2O.
Given the range of structures that are accessible within a narrow family of urea-derived complexes, the question of whether the urea unit could be replaced by another hydrogen bonding entity arose. These queries encouraged the synthesis of additional azide complexes, two of which successfully crystallized. The guanidine-based complex [1i·N3]·TBA (type II, entry 10) crystallized as the amino rather than imino tautomer whereby both NH2 and NH interact with N3– with NH2 binding significantly more weakly than NH (D–A distance: 3.202(2) Å versus 2.842(2) Å). The C=NPh forces the phenyl ring to bend, thus giving an angle between the two aryls of ∼63° (average values for ureas in this set: ∼6–30°). Finally, a near symmetrical complex {[1j]·N3}·TBA (entry 11) was obtained with diphenyloxalamide as HB donor. In this structure, the presence of two carbonyl groups sets the two aryl units of diphenyloxalamide in plane with the two N–H bonds that are oriented anti to each other. This generates a 1:1 bridged complex which is distinct from all others and in which each oxalamide unit binds a different terminal nitrogen of the azide anion (D–A distance: 2.923(3) Å).
The binding properties of 1a–i with N3– in solution were also investigated by 1H NMR spectroscopy. 1H NMR titrations were carried out by adding increasing amounts of tetrabutylammonium azide (TBA·N3) to a solution of HBD (CH3CN/CD3CN 8:2, at 2 mM concentration). Deshielding and broadening of 1H resonances ascribed to the NH groups was observed, an indicator of H-bonding interactions between azide and urea. The chemical shift variation of the aromatic signals was plotted against the concentration of added TBA·N3, and association constants extrapolated from nonlinear least-squares regression using Bindfit.23 Titration data were fitted to 1:1 and 2:1 binding isotherms; for 1a and 1d, the fitting was optimal when accounting for the formation of a 2:1 complex, while the 1:1 binding mode resulted in a better fit for 1c, 1g, 1e, and 1i.15 The association constants (Ka(1:1)) for 1:1 urea-azide complexes ranged between 102–103 M–1 with the more electron-deficient diarylureas resulting in stronger binding to azide, which is consistent with the enhanced acidity of the NH groups (Table 3). The two most acidic ureas 1a and 1d featuring 3,5-bis(trifluoromethyl) or 4-trifluoromethyl substituents gave binding constants Ka(1:1) = 1.57 ± 0.06 × 103 M–1, Ka(2:1) = 7 ± 3 × 101 M–1 and Ka(1:1) = 1.25 ± 0.12 × 103, Ka(2:1) = 1.3 ± 0.5 × 102, respectively. Ureas 1c (R1 = 3,5-CF3; R2 = H), 1g (R1 = R2 = 4-F), and 1e (R1 = R2 = H) presented progressively reduced binding affinity, as expected from their electronic properties. Diphenyl guanidine 1i displayed the weakest binding among all receptors studied.
Table 3. Association Constants for the Formation of 1:1, Ka(1:1), and 2:1, Ka(2:1), Complexes between Receptor 1a–i and TBA·N3 (CH3CN/CD3CN, 2 mM), Ordered by Decreasing Strength.
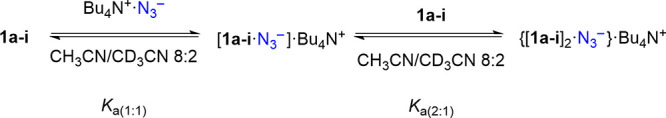
| entry | HBD | Ka(1:1) (M–1) | Ka(2:1) (M–1) |
|---|---|---|---|
| 1 | 1a | 1.57 ± 0.06 × 103 | 7 ± 3 × 101 |
| 2 | 1d | 1.25 ± 0.12 × 103 | 1.3 ± 0.5 × 102 |
| 3 | 1c | 9.4 ± 1.7 × 102 | – |
| 4 | 1g | 4.82 ± 0.05 × 102 | – |
| 5 | 1e | 3.14 ± 0.03 × 102 | – |
| 6 | 1i | 1.4 ± 0.3 × 102 | – |
These data confirm the ability of azide to engage in hydrogen bonding interactions with dual HBD donors in solution. The predominant binding mode is 1:1, with an additional weaker 2:1 binding mode observed only for the strongest donors 1a and 1d. Compared with complexes derived from fluoride,24 the binding affinity is substantially reduced, a measure of the lower propensity of azide to engage in hydrogen bonding interaction (cf. 1d·F– ∼ 105 M–1, 1d·N3– ∼ 103 M–1). The weaker binding of azide compared to fluoride has implications in the development of a catalytic method aimed at bringing insoluble azide salts into solution via complexation with hydrogen bond donors; while the phase-transfer of azide salts may differ from that of fluoride salts, the nucleophilicity of soluble bound urea-azide complex is expected to be less attenuated as a consequence of weaker binding to the HBD catalyst. Also, urea 1a binds chloride (Ka(1:1) = 4.7 ± 1.6 × 104 M–1; Ka(2:1) = 1.7 ± 0.9 × 102 M–1) more effectively than azide (Ka(1:1) = 1.57 ± 0.06 × 103 M–1; Ka(2:1) = 7 ± 3 × 101 M–1), raising awareness of a possible inhibition pathway.
Next, we focused on the characterization of the chiral BINAM-derived bisurea-azide complex that led to successful enantioselective azidation with sodium azide. The proposed hydrogen-bonded association between azide and (S)-1k was investigated computationally and experimentally. Conformational analysis of the solution-phase structure of a 1:1 complex formed between (S)-1k and azide was performed computationally. While our previous studies on fluoride complexation focused on the use of explicitly solvated classical molecular dynamics,25 here we used semiempirical GFN2-xTB calculations and the iMTD-GC workflow implemented in Grimme’s CREST for sampling including implicit solvation for dichloromethane,26 followed by DFT optimizations of the low energy conformers.27 Low-lying conformers were obtained with three NH-azide H-bonding interactions, which can be further categorized into three distinct catalyst-azide binding-modes: (i) Type A conformers show side-on binding in which the azide termini form H-bonds with proximal N–H groups in the catalyst; (ii) Type B conformers show side-on binding in which the azide termini form H-bonds to distal N–H groups; (iii) Type C conformers show end-on binding in a tripodal fashion to all three N–H bonds in 1k (Figure 4). The syn,anti-conformation with respect to the catalyst N-isopropylated urea is found in these low-lying conformers.
Figure 4.

DFT computed conformers and relative Gibbs energies (kJ·mol–1) of the [(S)-1k·N3]− complex. N–H distances (Å) and natural charges on azide N atoms (au) also shown.
The end-on binding mode in Type C conformers is energetically most favorable by over 10 kJ·mol–1, displaying the shortest average N–H distance (2.02 Å). Additionally, the quadrupole moment of the azide anion is polarized upon binding, with the terminus coordinated to the highest possible number of N–H bonds (3 for end-on; 2 for side-on), and NA, bearing the largest residual negative charge. In the Type C conformation, this effect is largest.
Next, 1H NMR titrations were conducted using TBA·N3 (CDCl3 at 2 mM concentration). Similar to achiral ureas 1a and 1d, a 1:1 binding model was insufficient to provide an accurate description of the system. The inclusion of a 2:1 complex ([(S)-1k]2·N3–) resulted in improved fits, leading to a Ka(1:1) of 9.14 ± 0.9 × 103 M–1 and a Ka(2:1) of 1.0 ± 0.6 × 102 M–1 (Figure 5A). This finding is analogous to fluoride, where 2:1 urea-fluoride complexes were also observed in solution.13d The Ka(1:1) and Ka(2:1) for the complexes of (S)-1k with fluoride are 1.43 ± 0.04 × 106 M–1 and 3.1 ± 0.9 × 103 M–1 in CH2Cl2, approximately 2 orders of magnitude higher than azide. 14N NMR spectroscopy provided further insight. The highly symmetric environment of unbound TBAN3 (CDCl3, 25 mM) gives three signals in 14N NMR spectrum corresponding to the tetrabutylammonium cation (66 ppm), to the central azide nitrogen (251 ppm), and to the terminal one (102 ppm). In an equimolar mixture of (S)-1k and TBAN3 (CDCl3, 25 mM), the central azide nitrogen appears significantly broader and the signal of the terminal azide nitrogen is broadened beyond detection; negligible change is observed for the tetrabutylammonium cation (Figure 5B). 14N is a quadrupolar nucleus which shows sharp signals only in symmetric environments;28 the extreme line broadening observed for (S)-1k·TBA·N3 is thus consistent with a lack of symmetry of the azide anion likely resulting from an interaction with (S)-1k. Further analysis was performed using isotopically enriched tetrabutylammonium [1-15N]azide (TBA·[1-15N]N3). At room temperature, a sample prepared by mixing TBA·[1-15N]N3 and 1 equiv of (S)-1k (CDCl3, 25 mM) exhibited broad lines in 1H NMR supporting the existence of multiple equilibrating species present in solution, ascribed to the unbound ligand, the 1:1 [(S)-1k]·N3– and 2:1 [(S)-1k]2· complexes. 15N NMR at room temperature shows one resonance at 101.8 ppm. Steady-state heteronuclear 1H–15N NOE experiments provided evidence of azide binding to the NH groups of (S)-1k. Selective irradiation of the urea NH protons resulted in a decrease of 15N signal intensities, whereas irradiation of the CH protons gave no evidence of heteronuclear NOEs, supporting the proposal that the urea NH protons are responsible for azide coordination.15 At 213 K a sample prepared using (S)-1k (CDCl3, 25 mM) and two equivalents of TBA[1-15N]N3 shows sharp resonances in 1H NMR, with a major species assigned to the 1:1 complex 1k·[1-15N]azide and a minor species which is consistent with the 2:1 complex [(S)-1k]2·azide, as noted in the course of the 1H NMR titrations. At this temperature, applying a 1H–15N HMBC sequence, it was possible to detect couplings across hydrogen bonding (1hJNH) between the three NH groups and [1-15N]N3– (Figure 5C). The magnitude of 1hJNH, measured using 1D 1H15N HMBC, was found to be around 5 Hz, although accurate measurement was hindered by the line width of the cross-peaks. This value is consistent with those reported for 15N-labeled DNA duplex, which are typically in the range of 1–4 Hz.29 The data obtained by 1H–15N NOE and HMBC experiments unambiguously indicate coordination of the azide with all three NH hydrogen bond donors in solution.
Figure 5.

(A) Ka(1:1) and Ka(2:1) for the complexes of TBA·N3 with (S)-1k in CDCl3 (2 mM). (B) 14N spectra of and TBA·N3 complexed to 1 equiv of (S)-1k in CDCl3 (25 mM). (C) 1D and 2D 1H–15N HMBC spectra of (S)-1k·[1-15N]N3 (CDCl3, 25 mM, 213 K).
Further insight on the nature of the complexation of (±)-1k with azide was obtained in the solid-state. A sample of (±)-1k complexed to TBAN3 (1:1 ratio) was prepared by stirring both components in MeCN (0.1 M) and subsequently evaporating the mixture to dryness. Crystals of (±)-1k·TBAN3 suitable for single crystal X-ray diffraction were successfully grown by slow evaporation of a saturated solution of the amorphous solid in hot hexane and EtOAc. In a single asymmetric unit cell, both enantiomers (R)- and (S)-1k were observed, each complexed to azide. In both enantiomeric complexes, the azide anion is coordinated at one terminus by three hydrogen bonds from the NH groups of one catalyst unit. As previously observed for the corresponding fluoride complex,13a−13d the N-isopropylated urea adopts a syn-anti conformation with the iPr group pointing away from the chiral pocket, thus allowing for azide to interact with the three NHs (Figure 6A). Comparison with (S)-1k·tetrabutylammonium fluoride revealed similar geometries, although the donor–acceptor distances22a for the azide complex were consistently longer (by ∼0.2–0.4 Å) than observed for fluoride (Figure 6B, Table 4). In (S)-1k·TBAF, the relative N(H)···F donor–acceptor distances were NH(3)···F ∼ NH(1)···F < NH(2)···F; a reversal of these distances is found in (S)-1k·TBAN3, with NH(1)···N3 < NH(2)···N3 < NH(3)···N3 (Table 4). The crystal structure is analogous to the computed Type C coordination mode, which shows end-on binding of the azide (Figure 6C, Table 4).
Figure 6.
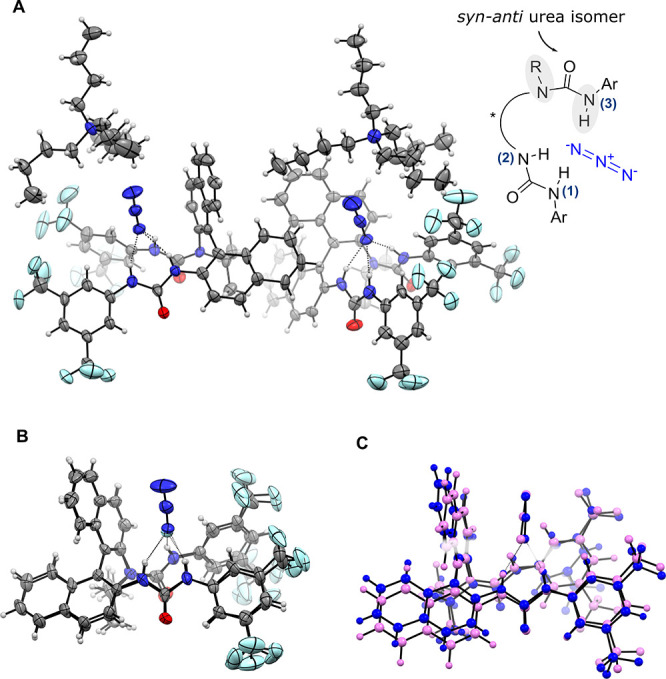
(A) Asymmetric unit of a Z′ = 2 crystal structure consisting of both (R)-1k and (S)-1k complexed to tetrabutylammonium azide. (B) View of (S)-1k complexed to azide. Distances provided in Ångstroms, displacement ellipsoids drawn at 50% probability level. (C) Overlay of [1k·N3]− (DFT vs X-ray).
3. Mechanistic Insight from Kinetic and Computational Studies
To shed light on the reaction mechanism, we explored the kinetics of the reaction of β-chloroamine (±)-2a with NaN3 in 1,2-difluorobenzene, in the presence and absence of catalyst (S)-1k at ambient temperature. The growth of the substitution product (3a) was monitored by in situ ATR-FT-IR, analyzing the absolute intensity of the signal arising from organoazide stretching band at 2100 cm–1. After some initial optimization of conditions,15 the reactions gave kinetics that were sufficiently reproducible for further analysis (Figure 7). The temporal concentration profiles for product 3a obtained at a series of different initial concentrations of 2a and 1k were investigated using a series of simple models that included the net enantioselectivity.15 Detailed kinetic analysis was precluded by the absence of information on catalyst speciation from the in situ FT-IR spectra, and by the solid-phase form of the sodium azide reactant and sodium chloride coproduct; {NaN3}s and {NaCl}s, from the overall reaction, eq 1. Nonetheless, three key features that govern the reaction evolution emerged: (i) the rate of turnover has a first-order dependency on the initial concentration of catalyst, [(S)-1k]0; (ii) the rate of turnover has a fractional order (∼0.5) dependency on the temporal concentration of the substrate, [2a]t; and (iii) as the reactions proceed, the rate of turnover is attenuated to a greater degree than dictated by the progressive reduction in the quantities of the reactants (2a and {NaN3}s). The latter is consistent with inhibition by accumulation of {NaCl}s.15
| 1 |
| 2 |
Figure 7.

In situ ATR-FTIR analysis of the reaction of 2a with {NaN3}s in 1,2-difluorobenzene, catalyzed by (S)-1k. Data, open circles. Kinetic model (eq 2), solid red lines/crosses. (A) Temporal growth of [3a] from [2a]0 = 0.25, 0.18, and 0.13 M, at [(S)-1k]0 = 0.025M. (B) Temporal growth of [3a] from [2a]0 = 0.25 M, at catalyst (S)-1k loadings (mol %) indicated. (C) Net enantiomeric excess of (S,S)-3a at catalyst (S)-1k loadings (0.6, 1.3, 2.6, 7.7, 10, and 20 mol %). Constants used for fitting eq 2: a = 0.081(±0.018) M–0.5 s–1; b = 2.1(±0.9); c = 1.8(±0.5) × 10–5 M0.5 s–1; r = {NaN3}s/{NaCl}s. Enantioselectivity employed in all fits as aS,S/aR,R = 7.94 (e.r. = 88.8:11.2) and cS,S/cR,R = 1.00 (e.r. = 50:50).
The temporal concentration profiles for [3a] can be satisfactorily correlated (Figure 7A and B) using the simple empirical relationship shown in eq 2. When aS,S/aR,R = 7.94 (e.r. = 88.8:11.2) and cS,S/cR,R = 1.00 (e.r. = 50:50), eq 2 also correctly predicts the net enantioselectivity for (S,S)-3a as a function of catalyst loading (Figure 7C). eq 2 is consistent with two processes operating in parallel: one enantioselective, and one a background racemic reaction. Their relative flux, and thus the net enantioselectivity, is governed by the initial concentrations of substrate 2a and catalyst 1k, the proportions of reactants, the extent of conversion, and the magnitude of constants a, b, and c. Two kinetically equivalent processes that are consistent with the empirical eq 2 are shown in Figure 8. Both processes involve competing complexation (KX; X = N3 or Cl) of catalyst 1k with either azide or chloride ion, and a pre-equilibrium (KiKIPD) involving 2a that generates ion-pair separated aziridinium ([A]+) cation and chloride anion. The two pathways diverge in the sequence of their reaction of {[1k·X]−[Na+]} where X = N3, with [A]+ and [Cl]−. But in both cases, they lead to the same key species: an aziridinium/catalyst-bound azide ion pair {[1k·N3][A+]}. This rapidly and irreversibly generates (kee) the product 3a in 89:11 e.r. The fitting parameter “a” reflects the series of equilibria and reactions that lead to {[1k·N3][A+]}. The fitting parameter “b” reports the differential binding of chloride over azide to the catalyst, in an equilibrium that is limited by the common-ion Na+. The term “r” reflects the evolving stoichiometry ratio {NaN3}s/{NaCl}s. The fitting parameter “c” reports on the rate of the competing background racemic (e.r. = 50:50) process involving direct reaction (krac) of the aziridinium ([A]+) cation with azide.
Figure 8.

Two pathways for generation of 3a from NaN3 and 2a catalyzed by 1k, in competition with a racemic background reaction. The pathways are kinetically equivalent in the context of eq 2.15xN3 is the mole fraction of {[1k·X]−[Na+]}in which X is azide.
Alternative approaches involving more complex models and holistic simulations were also effective, but did not prove advantageous, or allow elucidation of any discrete kinetic constants. Conducting reactions in the presence of exogenous NaCl led to the expected changes in rate and enantioselectivity.15 Other general mechanisms, where catalyst 1k interacts first with the substrate 2a, in its neutral or ionized forms, are inconsistent with eq 2.15 Overall, the kinetics support a process where the turnover rate limiting event directly, or indirectly, results in the generation of an ion pair {[1k·N3][A+]} containing an aziridinium cation and a catalyst-bound azide anion. The substitution product (S,S)-3a is then generated in excess over (R,R)-3a through enantiocontrol by ligand ((S)-1k) that is coordinated to the azide being delivered to the aziridinium cation as the ion-pair collapses.
Computationally, we studied several elementary steps in the proposed azidation mechanism. First, we considered the achiral transformation with catalyst 1a (Figure 9) and subsequently the enantioselective reaction with 1k (Figure 10).
Figure 9.
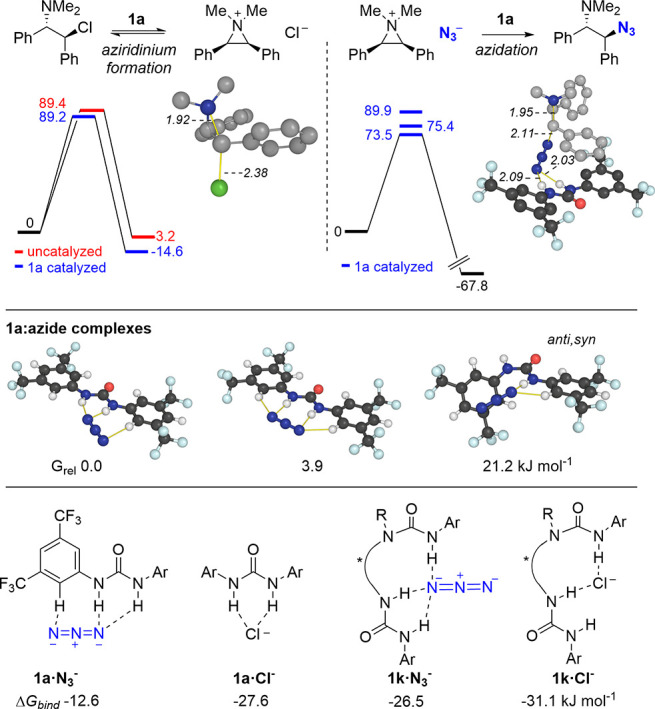
DFT computed transition structures for aziridinium formation, azidation with 1a·N3–, and binding of azide vs chloride to catalysts 1a and 1k.30 Distances in Å and energies in kJ·mol–1.
Figure 10.

Low-lying enantiodetermining azidation TSs with (S)-1k. Relative distortion energies of aziridinium and [(S)-1k·N3]− fragment in each TS shown. The reduced density gradient isosurface (RDG = 0.3) around the substrate is shown to indicate qualitatively the extent of substrate-catalyst noncovalent interactions in each TS.
Transition structures (TSs) were located for aziridinium formation in the absence and presence of the achiral urea catalyst 1a. Little energetic difference (0.2 kJ·mol–1) was found between these pathways, which corroborates previous computational studies.13 Consistent with the kinetic model above, aziridinium formation by autoionization is computed to be feasible and reversible, with ion-pair formation endergonic by 3 kJ·mol–1. Azidation TSs were also located for the addition of the urea-bound azide anion. Barrier heights are lower than for aziridinium formation by >15 kJ·mol–1, and the addition of azide is computed to occur irreversibly, with product formation exergonic by 68 kJ·mol–1. We also considered the relative stabilities of chloride and azide bound urea catalyst. For 1a, we located three distinct azide binding modes, of which the end-on structure is most stable. Chloride binding, however, is computed to be more favorable by 15 kJ·mol–1 (−27.6 vs −12.6 kJ·mol–1). Interestingly, the differential binding of chloride over azide for catalyst 1k is reduced to 4 kJ·mol–1, since azide anion is able to form three N–H bonds. This value is consistent with the kinetic model developed for 1k.
In order to understand the origins of asymmetric induction in azidation promoted by (S)-1k, we computed the competing TSs for the enantiodetermining step. We manually located a TS for the formation of major and minor enantiomer products, followed by a constrained conformational search with CREST. This produced 168 major and 219 minor structures,15 from which we finally obtained eight DFT-optimized (using the same methodology described above) energetically low-lying TSs within 14 kJ·mol–1 of the most stable structure (Figure 10). The two most stable competing TSs (TS-A and TS-B) involve attack from the H-bonded end of the azide anion (NA), while the remaining six higher energy structures are characterized by the distal nitrogen NC as the reactive center of the nucleophile, of which TS-C is the most stable example. Attack from NC results in a bridged structure in which there is comparatively little geometric distortion of the catalyst and substrate in the TS (relative distortion energies are shown in Figure 10). However, in the two most stable structures, attack from NA results in more significant noncovalent interactions between the substrate and catalyst—as can be seen qualitatively from the extent of the RDG isosurface produced by NCI plot in each of the TSs. These arise from several dispersive interactions between aromatic rings (both face-to-face and edge-to-face are evident) as well as CH(substrate)-π(catalyst) interactions. All three H-bonds are retained in the TS, however, as seen in asymmetric HB-PTC fluorination, the H-bond between nucleophile and the monodentate urea most noticeably lengthens along the reaction coordinate. The Boltzmann-averaged enantioselectivity arising from these low-lying structures is 69:31 in favor of the major enantiomer observed experimentally (Table 1, entry 5).
Conclusion
In this work, we have described the expansion of hydrogen-bonding phase-transfer catalysis and the privileged BINAM-derived bisurea catalyst scaffolds to the recognition of an anion other than fluoride for applications in catalysis. By employing a linear anion such as azide rather than the spherical charge-dense fluoride anion, we have demonstrated that the bisurea catalyst acts as an azide receptor and enables enantioselective azidation of β-chloroamine-derived meso aziridinium electrophiles using sodium azide. Kinetic studies support a process with the turnover rate limiting event that directly or indirectly generates an ion pair containing an aziridinium cation and a catalyst-bound azide anion, with catalyst inhibition incurred by accumulation of NaCl. Structural data in the solid state and in solution of a range of hydrogen bonded azide complexes inform that azide end-on binding is more often observed. For the chiral monoalkylated bisurea catalyst, 1H–15N NOE and HMBC experiments in solution as well as data in the solid state arising from single crystal X-ray diffraction analysis indicate coordination of the azide with all three NH hydrogen bond donors. Computationally, azide end-on bound to all three NH bonds of the BINAM urea in a tripodal fashion is found to be energetically most favorable. This binding mode induces polarization of the azide ion with the bound nitrogen bearing the largest negative charge, which indicates that this nitrogen is amenable to electrophilic attack. This analysis corroborates with the features of the most stable transition state leading to the major enantiomer. More generally, this study highlights the potential of hydrogen bonding phase transfer (HB-PTC) catalysis beyond fluorination and as a general activation mode for abundant alkali metal salts as reagents in asymmetric synthesis.
Acknowledgments
This work was supported by the EU Horizon 2020 Research and Innovation Programme (Marie Skłodowska-Curie Agreement 675071 and 789553), the Engineering and Physical Sciences Research Council (EP/R010064; Centre for Doctoral Training in Synthesis for Biology and Medicine EP/L015838/1), and the European Research Council (Agreement 832994). R.S.P. acknowledges support from the National Science Foundation (CHE 1955876), the RMACC Summit supercomputer, which is supported by the National Science Foundation (ACI-1532235 and ACI-1532236), the University of Colorado Boulder and Colorado State University, and the Extreme Science and Engineering Discovery Environment (XSEDE) through allocation TG-CHE180056. Y.G. thanks Edinburgh University for an Edinburgh Global Researcher Scholarship. We thank Mr. Songshi Jing for preliminary experiments, Dr. John M. Brown FRS for insightful discussion during the course of this project, Prof. Paul Beer for a helpful comment, and Dr. Amber L. Thompson for proofreading the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c13434.
Accession Codes
CCDC 2129236–2129248 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Author Contributions
# J.W., M.A.H., and A.B.D. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- a Griess P. On a New Class of Compounds in Which Nitrogen Is Substituted for Hydrogen. Proc. R. Soc. London 1864, 13, 375–384. 10.1098/rspl.1863.0082. [DOI] [Google Scholar]; b Curtius T. Ueber Stickstoffwasserstoffsäure (Azoimid) N3H. Berichte der Dtsch. Chem. Gesellschaft 1890, 23 (2), 3023–3033. 10.1002/cber.189002302232. [DOI] [Google Scholar]; c Curtius T. Hydrazide Und Azide Organischer Säuren I. Abhandlung. J. für Prakt. Chemie 1894, 50 (1), 275–294. 10.1002/prac.18940500125. [DOI] [Google Scholar]; d Tiemann F. Ueber Die Einwirkung von Benzolsulfonsäurechlorid Auf Amidoxime. Berichte der Dtsch. Chem. Gesellschaft 1891, 24 (2), 4162–4167. 10.1002/cber.189102402316. [DOI] [Google Scholar]
- The Azido Group (1971); Patai S., Ed.; John Wiley & Sons, Ltd.: Chichester, UK, 1971. [Google Scholar]
- Bräse S.; Gil C.; Knepper K.; Zimmermann V. Organic Azides: An Exploding Diversity of a Unique Class of Compounds. Angew. Chemie Int. Ed. 2005, 44 (33), 5188–5240. 10.1002/anie.200400657. [DOI] [PubMed] [Google Scholar]
- For a recent review:; a Ding P.-G.; Hu X.-S.; Zhou F.; Zhou J. Catalytic Enantioselective Synthesis of α-Chiral Azides. Org. Chem. Front. 2018, 5 (9), 1542–1559. 10.1039/C8QO00138C. [DOI] [Google Scholar]; Selected examples:; b Nugent W. A. Chiral Lewis Acid Catalysis. Enantioselective Addition of Azide to Meso Epoxides. J. Am. Chem. Soc. 1992, 114 (7), 2768–2769. 10.1021/ja00033a090. [DOI] [Google Scholar]; c Martínez L. E.; Leighton J. L.; Carsten D. H.; Jacobsen E. N. Highly Enantioselective Ring Opening of Epoxides Catalyzed by (Salen)Cr(III) Complexes. J. Am. Chem. Soc. 1995, 117 (21), 5897–5898. 10.1021/ja00126a048. [DOI] [Google Scholar]; d Hansen K. B.; Leighton J. L.; Jacobsen E. N. On the Mechanism of Asymmetric Nucleophilic Ring-Opening of Epoxides Catalyzed by (Salen)Cr(III) Complexes. J. Am. Chem. Soc. 1996, 118 (44), 10924–10925. 10.1021/ja962600x. [DOI] [Google Scholar]; e Schaus S. E.; Larrow J. F.; Jacobsen E. N. Practical Synthesis of Enantiopure Cyclic 1,2-Amino Alcohols via Catalytic Asymmetric Ring Opening of Meso Epoxides. J. Org. Chem. 1997, 62 (12), 4197–4199. 10.1021/jo970146p. [DOI] [Google Scholar]; f Konsler R. G.; Karl J.; Jacobsen E. N. Cooperative Asymmetric Catalysis with Dimeric Salen Complexes. J. Am. Chem. Soc. 1998, 120 (41), 10780–10781. 10.1021/ja982683c. [DOI] [Google Scholar]; g Annis D. A.; Helluin O.; Jacobsen E. N. Stereochemistry as a Diversity Element: Solid-Phase Synthesis of Cyclic RGD Peptide Derivatives by Asymmetric Catalysis. Angew. Chemie Int. Ed. 1998, 37 (13–14), 1907–1909. . [DOI] [Google Scholar]; h Myers J. K.; Jacobsen E. N. Asymmetric Synthesis of β-Amino Acid Derivatives via Catalytic Conjugate Addition of Hydrazoic Acid to Unsaturated Imides. J. Am. Chem. Soc. 1999, 121 (38), 8959–8960. 10.1021/ja991621z. [DOI] [Google Scholar]; i Bellavista T.; Meninno S.; Lattanzi A.; Della Sala G. Asymmetric Hydroazidation of Nitroalkenes Promoted by a Secondary Amine-Thiourea Catalyst. Adv. Synth. Catal. 2015, 357 (14–15), 3365–3373. 10.1002/adsc.201500403. [DOI] [Google Scholar]; j Nielsen M.; Zhuang W.; Jørgensen K. A. Asymmetric Conjugate Addition of Azide to α,β-Unsaturated Nitro Compounds Catalyzed by Cinchona Alkaloids. Tetrahedron 2007, 63 (26), 5849–5854. 10.1016/j.tet.2007.02.047. [DOI] [Google Scholar]; k Rowland E. B.; Rowland G. B.; Rivera-Otero E.; Antilla J. C. Brønsted Acid-Catalyzed Desymmetrization of Meso-Aziridines. J. Am. Chem. Soc. 2007, 129 (40), 12084–12085. 10.1021/ja0751779. [DOI] [PubMed] [Google Scholar]; l Guerin D. J.; Miller S. J. Asymmetric Azidation–Cycloaddition with Open-Chain Peptide-Based Catalysts. A Sequential Enantioselective Route to Triazoles. J. Am. Chem. Soc. 2002, 124 (10), 2134–2136. 10.1021/ja0177814. [DOI] [PubMed] [Google Scholar]; m Horstmann T. E.; Guerin D. J.; Miller S. J. Asymmetric Conjugate Addition of Azide to α,β-Unsaturated Carbonyl Compounds Catalyzed by Simple Peptides. Angew. Chemie Int. Ed. 2000, 39 (20), 3635–3638. . [DOI] [PubMed] [Google Scholar]; n Tiffner M.; Stockhammer L.; Schörgenhumer J.; Röser K.; Waser M. Towards an Asymmetric Organocatalytic α-Azidation of β-Ketoesters. Molecules 2018, 23 (5), 1142. 10.3390/molecules23051142. [DOI] [PMC free article] [PubMed] [Google Scholar]; o Humbrías-Martín J.; Pérez-Aguilar M. C.; Mas-Ballesté R.; Dentoni Litta A.; Lattanzi A.; Della Sala G.; Fernández-Salas J. A.; Alemán J. Enantioselective Conjugate Azidation of α,β-Unsaturated Ketones under Bifunctional Organocatalysis by Direct Activation of TMSN3. Adv. Synth. Catal. 2019, 361 (20), 4790–4796. 10.1002/adsc.201900831. [DOI] [Google Scholar]; p Liang Y.; Zhao X. Enantioselective Construction of Chiral Sulfides via Catalytic Electrophilic Azidothiolation and Oxythiolation of N -Allyl Sulfonamides. ACS Catal. 2019, 9 (8), 6896–6902. 10.1021/acscatal.9b01900. [DOI] [Google Scholar]
- Zhdankin V. V.; Krasutsky A. P.; Kuehl C. J.; Simonsen A. J.; Woodward J. K.; Mismash B.; Bolz J. T. Preparation, X-Ray Crystal Structure, and Chemistry of Stable Azidoiodinanes-Derivatives of Benziodoxole. J. Am. Chem. Soc. 1996, 118 (22), 5192–5197. 10.1021/ja954119x. [DOI] [Google Scholar]
- a da Silva Gomes R.; Corey E. J. A Method for the Catalytic Enantioselective Synthesis of Chiral α-Azido and α-Amino Ketones from Racemic α-Bromo Ketones, and Its Generalization to the Formation of Bonds to C, O, and S. J. Am. Chem. Soc. 2019, 141 (51), 20058–20061. 10.1021/jacs.9b12315. [DOI] [PubMed] [Google Scholar]; b Zhang X.; Ren J.; Tan S. M.; Tan D.; Lee R.; Tan C. An Enantioconvergent Halogenophilic Nucleophilic Substitution (SN2X) Reaction. Science 2019, 363 (6425), 400–404. 10.1126/science.aau7797. [DOI] [PubMed] [Google Scholar]; c Uyanik M.; Sahara N.; Tsukahara M.; Hattori Y.; Ishihara K. Chemo- and Enantioselective Oxidative α-Azidation of Carbonyl Compounds. Angew. Chemie Int. Ed. 2020, 59 (39), 17110–17117. 10.1002/anie.202007552. [DOI] [PubMed] [Google Scholar]
- a Yoshikawa S.; Shinzawa-Itoh K.; Nakashima R.; Yaono R.; Yamashita E.; Inoue N.; Yao M.; Fei M. J.; Libeu C. P.; Mizushima T.; Yamaguchi H.; Tomizaki T.; Tsukihara T. Redox-Coupled Crystal Structural Changes in Bovine Heart Cytochrome c Oxidase. Science 1998, 280 (5370), 1723–1729. 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]; b Fei M. J.; Yamashita E.; Inoue N.; Yao M.; Yamaguchi H.; Tsukihara T.; Shinzawa-Itoh K.; Nakashima R.; Yoshikawa S. X-Ray Structure of Azide-Bound Fully Oxidized Cytochrome c Oxidase from Bovine Heart at 2.9 Å Resolution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2000, 56 (5), 529–535. 10.1107/S0907444900002213. [DOI] [PubMed] [Google Scholar]; c Guo Q.; Gakhar L.; Wickersham K.; Francis K.; Vardi-Kilshtain A.; Major D. T.; Cheatum C. M.; Kohen A. Structural and Kinetic Studies of Formate Dehydrogenase from Candida Boidinii. Biochemistry 2016, 55 (19), 2760–2771. 10.1021/acs.biochem.6b00181. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Bowler M. W.; Montgomery M. G.; Leslie A. G. W.; Walker J. E. How Azide Inhibits ATP Hydrolysis by the F-ATPases. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (23), 8646–8649. 10.1073/pnas.0602915103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Hasnaoui-Dijoux G.; Majerić Elenkov M.; Lutje Spelberg J. H.; Hauer B.; Janssen D. B. Catalytic Promiscuity of Halohydrin Dehalogenase and Its Application in Enantioselective Epoxide Ring Opening. ChemBioChem. 2008, 9 (7), 1048–1051. 10.1002/cbic.200700734. [DOI] [PubMed] [Google Scholar]; b Majerić Elenkov M.; Primožič I.; Hrenar T.; Smolko A.; Dokli I.; Salopek-Sondi B.; Tang L. Catalytic Activity of Halohydrin Dehalogenases towards Spiroepoxides. Org. Biomol. Chem. 2012, 10 (26), 5063. 10.1039/c2ob25470k. [DOI] [PubMed] [Google Scholar]
- Matthews M. L.; Chang W.; Layne A. P.; Miles L. A.; Krebs C.; Bollinger J. M. Direct Nitration and Azidation of Aliphatic Carbons by an Iron-Dependent Halogenase. Nat. Chem. Biol. 2014, 10 (3), 209–215. 10.1038/nchembio.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews M. L.; Chang W.; Layne A. P.; Miles L. A.; Krebs C.; Bollinger J. M. Direct Nitration and Azidation of Aliphatic Carbons by an Iron-Dependent Halogenase. Nat. Chem. Biol. 2014, 10 (3), 209–215. 10.1038/nchembio.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X.; Bergsten T. M.; Groves J. T. Mangananese-Catalyzed Late-Stage Aliphatic C–H Azidation. J. Am. Chem. Soc. 2015, 137 (16), 5300–5303. 10.1021/jacs.5b01983. [DOI] [PubMed] [Google Scholar]
- Hashemi M.; Taherpour A. A. Structural Assessment of Hydrogen Bonds on Methylpentynol–Azide Clusters to Achieve Regiochemical Outcome of 1,3-Dipolar Cycloaddition Reactions Using Density Functional Theory. ACS Omega 2020, 5 (11), 5964–5975. 10.1021/acsomega.9b04333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Pupo G.; Ibba F.; Ascough D. M. H.; Vicini A. C.; Ricci P.; Christensen K. E.; Pfeifer L.; Morphy J. R.; Brown J. M.; Paton R. S.; Gouverneur V. Asymmetric Nucleophilic Fluorination under Hydrogen Bonding Phase-Transfer Catalysis. Science 2018, 360 (6389), 638–642. 10.1126/science.aar7941. [DOI] [PubMed] [Google Scholar]; b Pupo G.; Vicini A. C.; Ascough D. M. H.; Ibba F.; Christensen K. E.; Thompson A. L.; Brown J. M.; Paton R. S.; Gouverneur V. Hydrogen Bonding Phase-Transfer Catalysis with Potassium Fluoride: Enantioselective Synthesis of β-Fluoroamines. J. Am. Chem. Soc. 2019, 141 (7), 2878–2883. 10.1021/jacs.8b12568. [DOI] [PubMed] [Google Scholar]; c Roagna G.; Ascough D. M. H.; Ibba F.; Vicini A. C.; Fontana A.; Christensen K. E.; Peschiulli A.; Oehlrich D.; Misale A.; Trabanco A. A.; Paton R. S.; Pupo G.; Gouverneur V. Hydrogen Bonding Phase-Transfer Catalysis with Ionic Reactants: Enantioselective Synthesis of γ-Fluoroamines. J. Am. Chem. Soc. 2020, 142 (33), 14045–14051. 10.1021/jacs.0c05131. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ibba F.; Pupo G.; Thompson A. L.; Brown J. M.; Claridge T. D. W.; Gouverneur V. Impact of Multiple Hydrogen Bonds with Fluoride on Catalysis: Insight from NMR Spectroscopy. J. Am. Chem. Soc. 2020, 142 (46), 19731–19744. 10.1021/jacs.0c09832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Schreiner P. R.; Wittkopp A. H-Bonding Additives Act Like Lewis Acid Catalysts. Org. Lett. 2002, 4, 217–220. 10.1021/ol017117s. [DOI] [PubMed] [Google Scholar]; b Schreiner P. R. Metal-free organocatalysis through explicit hydrogen bonding interactions. Chem. Soc. Rev. 2003, 32, 289–296. 10.1039/b107298f. [DOI] [PubMed] [Google Scholar]
- See the Supporting Information for full details, including full optimization.
- Vitaku E.; Smith D. T.; Njardarson J. T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57 (24), 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- Boström J. Symmetric Kv1.5 Blockers Discovered by Focused Screening. ACS Med. Chem. Lett. 2012, 3 (9), 769–773. 10.1021/ml3001787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected examples of hydrogen bonded azide crystal structures:; a Kimura E.; Anan H.; Koike T.; Shiro M. A Convenient Synthesis of a Macrocyclic Dioxo Pentaamine and X-Ray Crystal Structure of Its Monohydrazoic Acid Salt. J. Org. Chem. 1989, 54, 3998–4000. 10.1021/jo00277a051. [DOI] [Google Scholar]; b Lex J.; Linke K. Beiträge Zur Chemie Des Hydrazins Und Seiner Derivate, LI: Kristall- Und Molekülstruktur von N, N, N′,N′-Tetraaminopiperazindiium-Bis(Azid). Chem. Ber. 1976, 109, 2684–2690. 10.1002/cber.19761090804. [DOI] [Google Scholar]; c Bushmarinov I. S.; Nabiev O. G.; Kostyanovsky R. G.; Antipin M. Y.; Lyssenko K. A. The Azide Anion as a Building Block in Crystal Engineering from a Charge Density Point of View. CrystEngComm 2011, 13, 2930. 10.1039/c0ce00859a. [DOI] [Google Scholar]; d Serpell C. J.; Cookson J.; Thompson A. L.; Beer P. D. A Dual-Functional Tetrakis-Imidazoliummacrocycle for Supramolecular Assembly. Chem. Sci. 2011, 2, 494–500. 10.1039/C0SC00511H. [DOI] [Google Scholar]; e Christe K. O.; Wilson W. W.; Bau R.; Bunte S. W. New Synthesis, Crystal Structure, and Vibrational Spectra of Tetramethylammonium Azide and Reactions of the Fluoride Anion with Hydrazoic Acid and of the Azide Anion with Hydrogen Fluoride. J. Am. Chem. Soc. 1992, 114, 3411–3414. 10.1021/ja00035a037. [DOI] [Google Scholar]; f Kang S. O.; Day V. W.; Bowman-James K. Tricyclic Host for Linear Anions. Inorg. Chem. 2010, 49, 8629–8636. 10.1021/ic1013297. [DOI] [PubMed] [Google Scholar]; g Escuer A.; Esteban J.; Font-Bardia M. Anion Coordination by Metallamacrocycles: A Cryptand-like Cavity. Chem. Commun. 2012, 48, 9777. 10.1039/c2cc34061e. [DOI] [PubMed] [Google Scholar]; h Esteban J.; Font-Bardia M.; Escuer A. Anionic Guests in Prismatic Cavities Generated by Enneanuclear Nickel Metallacycles. Inorg. Chem. 2014, 53, 1113–1121. 10.1021/ic402651k. [DOI] [PubMed] [Google Scholar]; i Tuo D.-H.; Ao Y.-F.; Wang Q.-Q.; Wang D.-X. Benzene Triimide Cage as a Selective Container of Azide. Org. Lett. 2019, 21, 7158–7162. 10.1021/acs.orglett.9b02782. [DOI] [PubMed] [Google Scholar]; j Ray S. K.; Homberg A.; Vishe M.; Besnard C.; Lacour J. Efficient Synthesis of Ditopic Polyamide Receptors for Cooperative Ion Pair Recognition in Solution and Solid States. Chem. Eur. J. 2018, 24, 2944–2951. 10.1002/chem.201704895. [DOI] [PubMed] [Google Scholar]; k Evers J.; Göbel M.; Krumm B.; Martin F.; Medvedyev S.; Oehlinger G.; Steemann F. X.; Troyan I.; Klapötke T. M.; Eremets M. I. Molecular Structure of Hydrazoic Acid with Hydrogen-Bonded Tetramers in Nearly Planar Layers. J. Am. Chem. Soc. 2011, 133 (31), 12100–12105. 10.1021/ja2027053. [DOI] [PubMed] [Google Scholar]; For a single example of a crystal structure of a urea-azide complex, see:; l Wang X.; Jia C.; Huang X.; Wu B. Azide Anion Encapsulation in a Tetraurea Receptor. Inorg. Chem. Commun. 2011, 14 (9), 1508–1510. 10.1016/j.inoche.2011.06.004. [DOI] [Google Scholar]
- Low temperature single-crystal X-ray diffraction data were collected using a Rigaku Oxford Diffraction SuperNova diffractometer. Raw frame data were reduced using CrysAlisPro, and the structures were solved using “Superflip”:; a Palatinus L.; Chapuis G. SUPERFLIP - A Computer Program for the Solution of Crystal Structures by Charge Flipping in Arbitrary Dimensions. J. Appl. Crystallogr. 2007, 40, 786. 10.1107/S0021889807029238. [DOI] [Google Scholar]; Successive refinement was performed with CRYSTALS:; b Parois P.; Cooper R. I.; Thompson A. L. Crystal Structures of Increasingly Large Molecules: Meeting the Challenges with CRYSTALS Software. Chem. Cent. J. 2015, 9, 30. 10.1186/s13065-015-0105-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cooper R. I.; Thompson A. L.; Watkin D. J. CRYSTALS Enhancements: Dealing with Hydrogen Atoms in Refinement. J. Appl. Crystallogr. 2010, 43, 1100. 10.1107/S0021889810025598. [DOI] [Google Scholar]; Full refinement details are given in the Supporting Information. Crystallographic data have been deposited with the Cambridge Crystallographic Data Centre (CCDC 2129236–2129248) and can be obtained via www.ccdc.cam.ac.uk/data_request/cif.
- End-on binding implies hydrogen bonds from the hydrogen bond donor moieties are formed to the same terminal nitrogen of the azide, whereas side-on binding indicates that one or more H-bonds are formed with both terminal nitrogens of the azide.
- For an example of halogen-bonding to azide in solution, see:; a Lim J. Y. C.; Beer P. D. A Halogen Bonding 1,3-Disubstituted Ferrocene Receptor for Recognition and Redox Sensing of Azide. Eur. J. Inorg. Chem. 2017, 2017, 220. 10.1002/ejic.201600805. [DOI] [Google Scholar]; And in the solid-state:; b Rosokha S. V.; Stern C. L.; Swartz A.; Stewart R. Halogen Bonding of Electrophilic Bromocarbons with Pseudohalide Anions. Phys. Chem. Chem. Phys. 2014, 16, 12968–12979. 10.1039/C4CP00976B. [DOI] [PubMed] [Google Scholar]
- D–A distance relates to the distance expressed in Å between the nitrogen atom of the donor NH and the proximal nitrogen of the azide. θ for end-on complexes is defined as the angle between the vector defined by the carbonyl of the urea and the vector defined by the terminal nitrogen atoms of the azide; ϕ for side-on complexes is defined as the angle between the N–N urea vector and the vector defined by the terminal nitrogen atoms of the azide. Both angles were calculated using PLATON:; a Spek A. L.PLATON, A Multipurpose Crystallographic Tool; Utrecht, the Netherlands, 1998.; b Spek A. L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. 10.1107/S0021889802022112. [DOI] [Google Scholar]
- a Supramolecular.org. http://supramolecular.org (accessed September 18, 2019).; b Brynn Hibbert D.; Thordarson P. The Death of the Job Plot, Transparency, Open Science and Online Tools, Uncertainty Estimation Methods and Other Developments in Supramolecular Chemistry Data Analysis. Chem. Commun. 2016, 52 (87), 12792–12805. 10.1039/C6CC03888C. [DOI] [PubMed] [Google Scholar]; c Thordarson P.Binding Constants and Their Measurement. In Supramolecular Chemistry; John Wiley & Sons, Ltd.: Chichester, UK, 2012. [Google Scholar]; d Howe E. N. W.; Bhadbhade M.; Thordarson P. Cooperativity and Complexity in the Binding of Anions and Cations to a Tetratopic Ion-Pair Host. J. Am. Chem. Soc. 2014, 136 (20), 7505–7516. 10.1021/ja503383e. [DOI] [PubMed] [Google Scholar]; e Thordarson P. Determining Association Constants from Titration Experiments in Supramolecular Chemistry. Chem. Soc. Rev. 2011, 40 (3), 1305–1323. 10.1039/C0CS00062K. [DOI] [PubMed] [Google Scholar]
- Pfeifer L.; Engle K. M.; Pidgeon G. W.; Sparkes H. A.; Thompson A. L.; Brown J. M.; Gouverneur V. Hydrogen-Bonded Homoleptic Fluoride–Diarylurea Complexes: Structure, Reactivity, and Coordinating Power. J. Am. Chem. Soc. 2016, 138 (40), 13314–13325. 10.1021/jacs.6b07501. [DOI] [PubMed] [Google Scholar]
- Bickelhaupt F. M.; Houk K. N. Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chemie Int. Ed. 2017, 56 (34), 10070–10086. 10.1002/anie.201701486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pracht P.; Bohle F.; Grimme S. Automated Exploration of the Low-Energy Chemical Space with Fast Quantum Chemical Methods. Phys. Chem. Chem. Phys. 2020, 22 (14), 7169–7192. 10.1039/C9CP06869D. [DOI] [PubMed] [Google Scholar]
- Energetics in the text refer to ωB97XD3/(ma)-def2-TZVPP single point calculations on M06-2X geometries using a mixed def2-SVP (C, H), def2-TZVP (F) and def2-TZVPD (N, O) basis set with CPCM solvation (see Supporting Information for details).
- Mason J.Nitrogen. In Multinuclear NMR; Mason J., Ed.; Springer US: Boston, MA, 1987; pp 335–367. [Google Scholar]
- a Yan X.; Kong X.; Xia Y.; Sze K. H.; Zhu G. Determination of Internucleotide h J HN Couplings by the Modified 2D J NN -Correlated [15N, 1H] TROSY. J. Magn. Reson. 2000, 147 (2), 357–360. 10.1006/jmre.2000.2221. [DOI] [PubMed] [Google Scholar]; b Pervushin K.; Ono A.; Fernandez C.; Szyperski T.; Kainosho M.; Wuthrich K. NMR Scalar Couplings across Watson-Crick Base Pair Hydrogen Bonds in DNA Observed by Transverse Relaxation-Optimized Spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 1998, 95 (24), 14147–14151. 10.1073/pnas.95.24.14147. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dingley A. J.; Masse J. E.; Peterson R. D.; Barfield M.; Feigon J.; Grzesiek S. Internucleotide Scalar Couplings Across Hydrogen Bonds in Watson–Crick and Hoogsteen Base Pairs of a DNA Triplex. J. Am. Chem. Soc. 1999, 121 (25), 6019–6027. 10.1021/ja9908321. [DOI] [Google Scholar]
- The three barrier values correspond to different binding modes of the urea to the azide anion: end-on azide reacting at the distal N atom, shown in graphics (73.5 kJ·mol–1), end-on azide reacting at the proximal N atom (75.4 kJ·mol–1), side on bound azide (89.9 kJ·mol–1).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.