

Abstract
Objective:
Deleterious copy number variants (CNVs) are identified in up to 20% of individuals with autism. However, levels of autism-risk conferred by most rare CNVs remain unknown. We recently developed statistical models to estimate the effect-size on IQ of all CNVs including undocumented ones. We aimed to extend this model to autism susceptibility.
Methods:
We identified CNVs in two autism (Simons Simplex Collection and MSSNG) and two unselected populations (IMAGEN and Saguenay Youth Study). Statistical models tested 9 quantitative variables associated with genes encompassed in CNVs to explain their effect on IQ, autism susceptibility and behavioural domains.
Results:
The “probability-of-being loss-of-function intolerant” (pLI) best explains the effect of CNVs on IQ and autism risk. Deleting 1 point of pLI decreases IQ by 2.6 points in autism and unselected populations. The effect of duplications on IQ is three-fold smaller. Autism susceptibility increases when deleting or duplicating any point of pLI. This is true for individuals with high or low IQ and after removing de novo and known recurrent neuropsychiatric CNVs. Once CNV effects on IQ are accounted for, autism susceptibility remains mostly unchanged for duplications but decreases for deletions. Model estimates for autism risk overlap with previously published observations. Deletions and duplications differentially affect social communication, behaviour, and phonological memory, whereas both equally affect motor skills.
Conclusions:
Autism risk conferred by duplications is less influenced by IQ compared to deletions. Our model, trained on CNVs encompassing >4,500 genes, suggests highly polygenic properties of gene dosage with respect to autism risk and IQ loss. These models will help to interpret CNVs identified in the clinic.
INTRODUCTION
Autism is a neurodevelopmental condition currently defined by atypical social communication and interaction, intense interests, and repetitive behaviour (1). Levels of general intelligence and language are not diagnostic criteria but are recognized as clinical specifiers which have been defined as important features of the heterogeneity of autism. (2) Neurodevelopmental and psychiatric comorbidities occur in up to 70% of children with autism. (3) The heritability of autism has been estimated between 50-80%. (4, 5) Deleterious Single Nucleotide Variants (SNV) and Copy Number Variants (CNVs) are identified in 15 to 20% of individuals with autism. (6–8) The largest rare variant autism case-control association studies to date have formally associated 102 genes and 16 CNVs at 13 genomic loci. (9–12) Many more genomic loci are likely implicated as suggested by the overall increase in CNV burden associated with autism. (9, 10, 13–15) Therefore, the susceptibility to autism conferred by most CNVs remains undocumented. This is particularly problematic in the neurodevelopmental clinic, where undocumented CNVs are routinely diagnosed in a large proportion of patients.
Even less is known about the effect-size of CNVs on the cognitive and behavioural dimensions related to autism, which have only been characterized for a handful of recurrent CNVs (e.g. 22q11.2, 16p11.2, 15q11.2, and 1q21.1 loci). These CNVs show reproducible effect-sizes on cognition, language, socio-communication, and brain structure, suggesting that these alterations drive their over-representation in autism or other neurodevelopmental and psychiatric conditions. (16–18)
Limited progress has been made in identifying phenotype-genotype relationships in autism. Studies have demonstrated that rare de novo variants are associated with lower intelligence quotient (IQ) and are over-represented in females. (15, 19–22) De novo variants have also been associated with an atypical autism profile characterized by less impairment in social communication and language, as well as greater likelihood of motor delay. (23, 24) Overall, the reasons underlying the overrepresentation of rare variants in autistic individuals remains unclear. It may be due to their effect on core symptoms of autism, or DSM-5-defined clinical specifiers of autism (intelligence, language, co-occurring conditions). Since CNVs have a strong influence on IQ and behavioural problems, including autism symptoms, it is of interest to examine the effect-size of CNVs on autism risk while accounting for their effect-size on IQ.
We previously reported that statistical models, trained on benign deletions in populations not selected for a clinical condition, can accurately estimate the effect-size of deleterious deletions on non-verbal IQ (NVIQ). (25) These results suggest that 1) the effect-size of deletions on NVIQ can be estimated using constraint scores, such as the “probability of being Loss-of-function Intolerant” (pLI, definition in textbox, Figure 1) (26), and 2) the effect of haploinsufficiency on NVIQ applies to a large proportion of the genome, consistent with a highly polygenic model. (27, 28) Using pLI as an explanatory variable, we estimated that one third of the coding genes affect NVIQ by >1 point, when deleted. (25) Previously, we were unable to establish the effect-size of duplications, likely due to inadequate power with the then-available sample size. Here, we propose to develop similar models to estimate autism susceptibility conferred by undocumented CNVs. We also aim to estimate their effects on cognitive and behavioural dimensions, which may underpin their overrepresentation in autism.
Figure 1. Methodological pipeline.
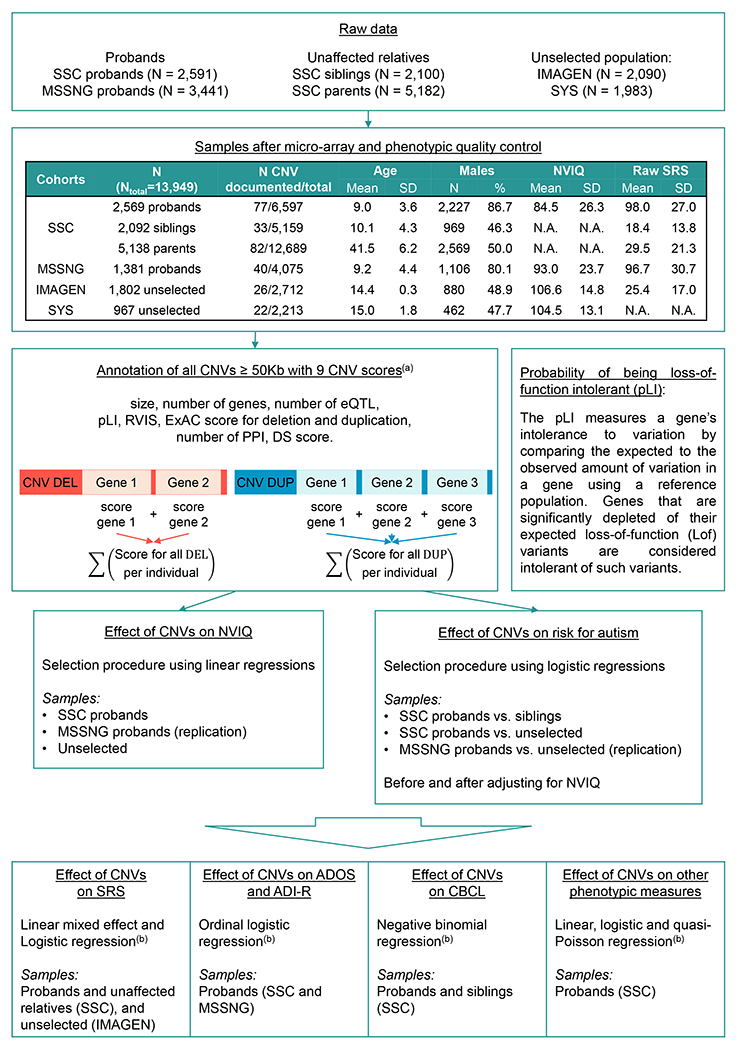
Probands from the SSC and MSSNG are defined as individuals recruited on the basis of a diagnosis of autism. Siblings and parents from the SSC did not meet diagnostic criteria for autism. 10 individuals from IMAGEN and none in SYS met criteria for ASD (as estimated by the Development and Well-Being Assessment, DAWBA). 1,490 unaffected siblings from the SSC, 3,660 unaffected parents from the SSC and 1,465 individuals from the general population carry at least one CNV ≥ 50kb (Table S1 in the online supplement). (a) Microarray quality control and CNV selection and annotation were performed as previously published (15) (Methods in the online supplement); (b) The model used and available data for each phenotype are detailed in Table 1 and Tables S7, S8, S9, and S10 in the online supplement. SSC: Simon Simplex Collection; SYS: Saguenay Youth Study; CNV: copy number variants; SD: standard deviation; N.A.: Not applicable; NVIQ: non-verbal intelligence quotient; SRS: Social Responsiveness Scale; ADOS: Autism Diagnostic Observation Schedule; ADI-R: Autism Diagnostic Interview-Revised; CBCL: Child behaviour Checklist.
We 1) tested whether the effect-size of gene dosage on NVIQ is the same across unselected populations and autism cohorts, 2) selected models that best explain the autism risk conferred by any deletions or duplications, while accurately adjusting for their effect on NVIQ established in step 1, and 3) investigated the cognitive, behavioural, and motor phenotypes that may explain the association between gene dosage and autism.
Models integrating genomic and functional scores of genes included in CNVs were trained on all CNVs ≥ 50 kb identified in two autism cohorts and two cohorts recruited from unselected populations. We provide a novel framework to model autism risk and the phenotypic profile of rare variants, regardless of effect-size and inheritance. This approach contrasts with previous genotype-phenotype studies restricted to small groups of individuals with de novo or recurrent variants.
METHODS
Cohorts
Autism cohorts
We studied two autism samples and intra-familial controls when available (Figure 1 and Table S1 in the online supplement). The Simons Simplex Collection (SSC) (29), a cohort of 2,569 simplex families: 2,074 quads (one autistic proband, unaffected parents, and one unaffected sibling) and 495 trios (one autistic proband and unaffected parents). The MSSNG database, used as an independent replication cohort, includes 1,381 probands with autism. (30)
Unselected cohorts
We included 2,769 individuals from two community-based cohorts that we previously studied (25): IMAGEN (N=1,802) (31) and the Saguenay Youth Study (SYS; N=967) (32) (Figure 1 and Table S1 in the online supplement).
CNV calling and annotation
We analyzed genotyping data from SSC, IMAGEN, and SYS and whole genome sequencing data from MSSNG. CNV detection, filtering, and annotation are detailed in Methods in the online supplement. We attributed 9 scores to deletions and duplications. These included size, number of genes, number of expression quantitative trait loci regulating genes expressed in the brain (33). Each coding gene with all isoforms fully encompassed in CNVs was annotated using 4 constraint scores which reflect genetic fitness. The pLI score (ExAC v1.0), which is available for 18,224 genes and ranges from 0 (the gene is tolerant to haploinsufficiency) to 1 (the gene is intolerant to haploinsufficiency with a 100% probability). (26) Genes with 80% or 90% probabilities of being intolerant are considered as intolerant (9, 26, 34). The 3 other constraint scores included the residual variation intolerance score (RVIS) (35), the deletion and duplication scores from ExAC (36). Coding genes were also scored using the number of protein-protein interactions (37) and the differential stability score (38). We computed the ancestry in the SSC, IMAGEN and SYS cohorts based on HapMap3 reference population. (39)
Clinical assessments
NVIQ data were available across all cohorts. (29–32) The assessment methods are detailed in Methods and Table S2 in the online supplement. All other cognitive, behavioural, and motor phenotypes are detailed in Table 1, Methods and Table S1 in the online supplement. Participants underwent age- and development-appropriate standardized cognitive and behavioural tests.
Table 1:
Effect-size of gene dosage measured by pLI on phenotypes in autistic probands from the SSC.
| Phenotypic measurements | N | CNV variable | No adjustment for NVIQ | Adjustment for NVIQ | ||||
|---|---|---|---|---|---|---|---|---|
| β or OR | SE or 95%CI | p | β or OR | SE or 95%CI | p | |||
| Autism related symptoms | ||||||||
| Regression | 2,568 | pLI DEL | 0.86 | 0.75-0.95 | 8.4×10−3 | 0.8 | 0.70-0.89 | 1.9×10−4 |
| pLI DUP | 0.99 | 0.92-1.04 | 0.65 | 0.96 | 0.90-1.02 | 0.19 | ||
| Language and phonology | ||||||||
| CTOPP(a) | 1,988 | pLI DEL | −0.08 | 0.02 | 5.5×10−4 | −0.02 | 0.02 | 0.24 |
| pLI DUP | −0.02 | 0.02 | 0.25 | 0.006 | 0.01 | 0.66 | ||
| Word delay | 2,567 | pLI DEL | 1.16 | 1.07-1.27 | 5.0×10−4 | 1.12 | 1.03-1.22 | 0.01 |
| pLI DUP | 1.03 | 0.98-1.09 | 0.24 | 1.02 | 0.96-1.08 | 0.6 | ||
| Phrase delay | 2,567 | pLI DEL | 1.04 | 0.98-1.09 | 0.42 | 0.95 | 0.87-1.04 | 0.25 |
| pLI DUP | 1.06 | 1.00-1.14 | 0.08 | 1.03 | 0.96-1.11 | 0.46 | ||
| Adaptive skills (VABS-II) | ||||||||
| Total score(a) | 2,569 | pLI DEL | −0.07 | 0.02 | 3.1×10−5 | −0.004 | 0.01 | 0.72 |
| pLI DUP | −0.03 | 0.01 | 2.6×10−3 | −0.01 | 0.01 | 0.4 | ||
| Daily living(a) | 2,569 | pLI DEL | −0.07 | 0.02 | 1.2×10−4 | −0.004 | 0.01 | 0.8 |
| pLI DUP | −0.04 | 0.01 | 3.4×10−3 | −0.01 | 0.01 | 0.38 | ||
| Communication(a) | 2,569 | pLI DEL | −0.07 | 0.02 | 3.4×10−4 | 0.01 | 0.01 | 0.54 |
| pLI DUP | −0.04 | 0.01 | 4.5×10−3 | −0.005 | 0.01 | 0.6 | ||
| Socialization(a) | 2,569 | pLI DEL | −0.06 | 0.02 | 4.8×10−4 | −0.01 | 0.01 | 0.67 |
| pLI DUP | −0.03 | 0.01 | 0.02 | −0.004 | 0.01 | 0.66 | ||
| Motor skills | ||||||||
| Motor VABS-II(a) | 919 | pLI DEL | −0.11 | 0.04 | 4.1×10−3 | −0.08 | 0.03 | 0.01 |
| pLI DUP | −0.07 | 0.02 | 1.3×10−3 | −0.04 | 0.02 | 0.02 | ||
| Gross motor VABS-II(a) | 926 | pLI DEL | −0.08 | 0.03 | 0.01 | −0.07 | 0.03 | 0.02 |
| pLI DUP | −0.05 | 0.02 | 4.6×10−3 | −0.04 | 0.02 | 0.03 | ||
| Fine motor | 923 | pLI DEL | −0.1 | 0.04 | 0.01 | −0.07 | 0.03 | 0.04 |
| VABS-II(a) | pLI DUP | −0.06 | 0.02 | 8.8×10−3 | −0.03 | 0.02 | 0.14 | |
| Onset for walking in months | 2,550 | pLI DEL | 1.03 | 1.02-1.04 | 2.2×10−11 | 1.03 | 1.02-1.04 | 4.6×10−9 |
| pLI DUP | 1.02 | 1.01-1.03 | 7.0×10−9 | 1.02 | 1.01-1.03 | 5.3×10−8 | ||
| Delayed onset for walking(a) | 2,564 | pLI DEL | 1.16 | 1.05-1.28 | 2.0×10−3 | 1.11 | 1.00-1.22 | 0.03 |
| pLI DUP | 1.2 | 1.11-1.30 | 6.1×10−6 | 1.19 | 1.09-1.29 | 4.2×10−5 | ||
| DCDQ score(a) | 2,209 | pLI DEL | −0.07 | 0.02 | 2.5×10−3 | −0.03 | 0.02 | 0.16 |
| pLI DUP | −0.03 | 0.01 | 0.04 | −0.01 | 0.01 | 0.33 | ||
| Associated neurological condition | ||||||||
| Non-febrile seizure | 2,566 | pLI DEL | 1.12 | 1.01-1.23 | 0.02 | 1.07 | 0.96-1.17 | 0.19 |
| pLI DUP | 1.04 | 0.95-1.11 | 0.3 | 1.02 | 0.94-1.09 | 0.63 | ||
phenotypic measures z-scored using normative data when available or computed using the full autistic proband group (Method in the online supplement). In bold: p-values significant above the statistical threshold (p ≤ 2.7×10−3). Effects in this table represent either the normalized β z-scores with their standard errors (SE) or an odds ratio (OR) with its 95% confidence interval (95%CI).
pLI: probability of being Loss-of-function Intolerant; pLI DEL or pLI DUP: deleted or duplicated point of pLI score; NVIQ: non-verbal intelligence quotient; CTOPP: Comprehensive Test of Phonological Processing; VABS-II: Vineland Adaptive behaviour Rating Scales - Second Edition; DCDQ: Developmental Coordination Disorder Questionnaire.
Statistical analyses
Effect-size of gene dosage on general intelligence in probands and the unselected populations
For each individual, we computed the sum of a given score for deletions and duplications separately (Figure 1, Methods in the online supplement). These deletion and duplication scores were used as two independent main effects in the model. We performed a stepwise variable selection procedure based on Bayesian information criteria to identify which score (among the 9 tested) best explain NVIQ for deletions and duplications. This was performed independently for the SSC probands, the unselected populations, and MSSNG as a replication dataset. To investigate the influence of the presence of lower IQ in the SSC, we assessed the effect-size of gene dosage on NVIQ in the SSC probands after performing 1:2 matching with MSSNG probands based on NVIQ (Methods and Figure S2 in the online supplement).Age, sex, ancestry and familial relatedness were used as covariates when applicable (Methods in the online supplement).
Effect-size of gene dosage on autism risk
We performed the same stepwise variable selection procedure to identify CNV scores that best explain the effect-size of deletions and duplications on autism risk. The dependent variable was the binary diagnosis (autism/control) and independent variables were the selected CNV scores. Conditional logistic regression was used when matching SSC probands with their unaffected siblings. Simple logistic regression was used when comparing SSC probands with the unselected populations. We assessed the effect-size of gene dosage on autism risk beyond its effect on NVIQ by adjusting for NVIQ or performing 1:1 matching of probands with individuals from the unselected populations based on NVIQ (Methods and Figure S1D in the online supplement). Replication analyses were performed using the MSSNG dataset. Sex, ancestry and familial relatedness were used as covariates when applicable (Methods in the online supplement).
To estimate the proportion of autism-risk potentially mediated by NVIQ for deletions and duplication, we performed a counterfactual-based mediation analysis on the pooled dataset.
Sensitivity analyses
For sensitivity analyses, we pooled all samples and excluded individuals with CNVs > 10 points of pLI (deletions with an effect > 2 standard deviations of NVIQ) or recurrent CNVs associated with neurodevelopmental disorders or rare de novo CNVs (Tables S3, S4 and S5 in the online supplement).
Estimating and validating the level of autism risk
We compared the autism risk estimated by our model to that previously published for recurrent CNVs. Our literature search identified 16 CNVs with available odds ratios (ORs) (9–11, 40) (Table S6 in the online supplement). The model was trained using a pooled dataset including SSC and MSSNG probands, unaffected siblings, and unselected populations, excluding these 16 CNVs.
To illustrate the output of our model, we computed the autism risk for each CNV called in both autism cohorts including at least one gene with a pLI annotation. We also computed autism risk for any 1MB CNV across the genome, generating a series of 1Mb deletions and duplications (Human Gene Nomenclature) by moving a sliding window in 50Kb steps across the genome. (41) We chose 1Mb CNVs based on thresholds for deleteriousness used in previous studies. (22, 42)
Effect-size of gene dosage on measures of core symptoms and specifiers of autism
We investigated the effect of the previously selected CNVs score on cognitive, behavioural, and motor phenotypes to understand why they increase susceptibility to autism. The choice of the statistical model depended on the distribution of the phenotypic measure (Methods and Table S7 in the online supplement). The Social Responsiveness Scale (SRS) was investigated using the entire SSC, MSSNG probands and IMAGEN cohorts (Methods and Table S8 in the online supplement). The Autism Diagnostic Observation Schedule (ADOS) and Autism Diagnostic Interview-Revised (ADI-R) were investigated using probands from SSC and MSSNG (Methods and Table S9 in the online supplements). The Child behaviour Checklist (CBCL) was investigated on probands and unaffected siblings from the SSC (Methods and Table S10 in the online supplements). All other phenotypic measurements were analysed using SSC probands alone. For all analyses, age, sex, ancestry and familial relatedness were used as covariates when applicable. Phenotypic measures were also tested with and without adjustment for NVIQ and/or autism diagnosis when available (Methods in the online supplement). Computation of the significance threshold is detailed in Methods in the online supplement.
RESULTS
Effect-size of gene dosage on general intelligence in probands and the unselected populations
As we previously observed in unselected populations (26), the variable selection procedure identified the sum of pLI scores as the variable that best explains the variance of NVIQ in the SSC for deletions (r2=0.014) and duplications (r2=0.004), compared to the 8 other scores. The sum of pLI scores per individual ranges from 0 to 18.92 and 35.71 for deletions and duplications respectively. As an example, a CNV scoring 2 points of pLI may include either 2 genes with a 100% probability of being intolerant or 3 genes with moderate to high probabilities (60 to 90%).
Deleting 1 point of pLI has the same effect-size on z-scored NVIQ in autism probands of both samples (SSC: β=−0.17, SE=0.03, p=8×10−10; MSSNG: β=−0.20, SE=0.07, p=3×10−3) and unselected populations (β=−0.19, SE=0.04, p=7×10−5). The pLI is also the score that best explains the impact of duplications on NVIQ, showing a three-fold smaller effect of pLI points on z-scored NVIQ in the SSC (β=−0.06, SE=0.02, p=1×10−3). No effect of duplications is detected in unselected populations or the MSSNG dataset (Table S11 in the online supplement, Figure 2A).
Figure 2. Effect of gene dosage on NVIQ and autism susceptibility.
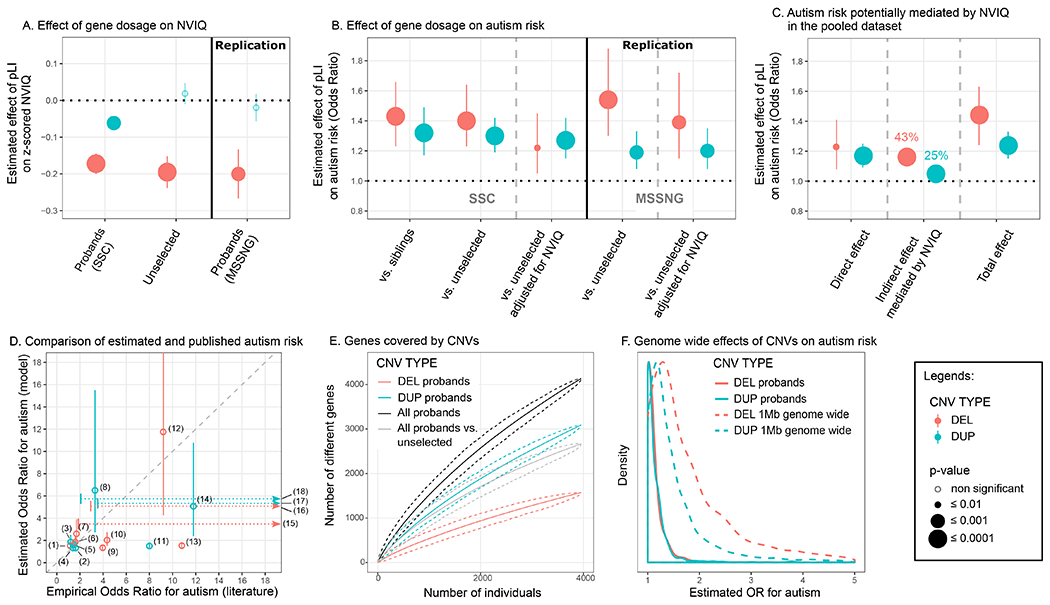
(A) Effect-size of a deleted (DEL, red) or duplicated (DUP, blue) point of pLI on NVIQ in autistic probands and unselected populations. Y axis values are z-scores for NVIQ (e.g. 0.2 z-score=3 points of NVIQ). (B) Autism risk conferred by a deleted (red) or duplicated (blue) point of pLI. Y axis values are odds ratios (OR) computed using a logistic regression to explain an autism diagnosis. Controls include unaffected siblings or unselected populations. Replication was performed using autistic probands from the MSSNG dataset and the unselected populations. (C) Autism risk potentially mediated by NVIQ on the pooled dataset. Y axis values are ORs computed using a counterfactual-based mediation analysis on a logistic regression. Direct effects of CNVs are those not mediated by NVIQ. Indirect are those potentially mediated by NVIQ. Percentage of effect mediated by NVIQ = (indirect / total effect) × 100. Total effects are those computed without adjusting for NVIQ. (D) We compared the risk of autism estimated by our model and the risk observed in previous published studies on 16 recurrent CNVs (4, 9, 22) (Table S3 in the online supplement). OR estimates from the model overlap with the 95% confidence interval (CI) of ORs from previous publications for 14 recurrent CNVs. For three CNVs, the horizontal dotted arrows represent the extreme variability for ORs reported in previous publications. Values are detailed in Table S3 in the online supplement. (1) DEL 15q11.2, (2) DUP 15q11.2, (3) DUP 16p11.2 distal, (4) DUP 15q13.3, (5) DUP 16p13.11, (6) DEL 1q21.1, (7) DEL 16p11.2 distal, (8) DUP 22q11.2, (9) DEL 17p12, (10) DEL 16p13.11, (11) DUP 1q21.1, (12) DEL 16p11.2, (13) DEL 15q13.3, (14) DUP 16p11.2, (15) DEL 17q12, (16) DEL 3q29, (17) DUP 7q11.23 and (18) DUP 17p11.2. (E) Genes covered by deletions (red), duplications (blue), or both (black) in autistic probands. The grey line represents the excess of genes encompassed in CNVs in autistic probands relative to the unselected populations. The Y-axis represents the number of distinct genes encompassed in CNVs (each gene is only counted once). The X-axis represents the number of individuals. The mean and 95%CI confidence interval were obtained using 1,000 iterations (bootstrap procedure). (F) Distribution of the estimated effects of deletions (red) and duplications (blue). Full line: Estimated effects computed for all CNVs ≥ 50Kb identified in both autism cohorts. Dotted line: Autism risk computed for any CNV of 1MB across the genome including at least one gene with a pLI annotation.
Matching the SSC and MSSNG based on NVIQ, or removing ratio NVIQ from the SSC, does not influence these effect sizes (Figure S2 and Table S4 in the online supplement). In the pooled dataset, an autism diagnosis does not influence the effect of deleted or duplicated points of pLI on NVIQ. There is also no interaction with sex. Removing carriers of CNVs with a pLI sum > 10, with a known psychiatric association, or one occurring de novo, results in similar effect-sizes for deletions. For duplications, our limited power only allowed us to observe an effect when removing CNVs enriched in neurodevelopmental disorders (Table S4 in the online supplement).
Effect-size of gene dosage on autism risk
The variable selection procedure identified again the sum of pLI scores as the variable that best explains the diagnosis of autism for deletions (r2=0.004) and duplications (r2=0.004). Susceptibility to autism increases for each deleted point of pLI and the effect-size is identical when comparing autistic probands with their paired siblings or unselected populations (OR=1.43, 95%CI=1.23-1.66, p=4×10−6; OR=1.40, 95%CI=1.23-1.64, p=2×10−6, respectively). A duplicated point of pLI also increases autism susceptibility (comparing with siblings: OR=1.32, 95%CI=1.17-1.49, p=5×10−6; and the unselected populations: OR=1.30, 95%CI=1.19-1.42, p=2×10−8) (Figure 2B, Table S12 in the online supplement). Of note, there is no difference in pLI burden between intra- and extra-familial controls (unselected populations) (Table S5 in the online supplement).
The risk conferred by deletions measured by pLI decreases substantially but remains borderline significant when the model is adjusted for NVIQ (OR=1.22, 95%CI=1.05-1.45, p=0.01) or when both autism and unselected populations are matched for NVIQ. In contrast, the autism risk conferred by each duplicated point of pLI remains unchanged when adjusting (OR=1.27, 95%CI=1.15-1.42, p=5×10−6) or matching for NVIQ (Figure 2B and Table S12 in the online supplement).
The replication analysis with the MSSNG dataset shows the same effect of deleted or duplicated points of pLI on autism susceptibility. We also replicate the differential effect of NVIQ adjustment on autism risk conferred by deletions and duplications (Figure 2B and Table S12 in the online supplement).
In the pooled dataset mediation analysis suggested that 43% and 25% of the autism risk conferred by deletions and duplications are potentially influenced by NVIQ (Figure 2C and Table S13). However, the effect-size of autism risk for deletions and duplications measured by pLI is the same in both subgroups of individuals above and below median NVIQ (Figure S3 and Table S14). There is no interaction with sex. Autism susceptibility related to gene dosage is unaffected by removing carriers of CNVs with a pLI sum > 10, CNVs with a known association to neurodevelopmental disorder, occurring de novo, or individuals from the unselected populations with a suspected diagnosis of autism (n=10) as well as no diagnostic information from the Development and Well-Being Assessment (DAWBA) (N=124) (Table S5 in the online supplement).
Estimating and validating the level of autism risk
ORs have previously been computed for a few recurrent CNVs with broad confidence intervals. The autism risk estimated by our model overlaps with that previously published for 16 recurrent CNVs, except for the 15q13.3 BP4-BP5 deletion and the 1q21.2 duplication, which are discordant (9–11, 40) (Figure 2D, Table S6 in the online supplement). The results are similar whether we include or exclude the 16 CNVs from the training dataset (Figure S3 in the online supplement). Our model is trained on deletions and duplications covering over 4,500 different genes in the autism and unselected populations (Figure 2E). The sharply ascending slope of genes encompassed in the CNVs shows no asymptotic effects. Model estimates show that any 1Mb coding deletion or duplication across the genome should increase autism susceptibility, with a median OR of 1.6 and 1.3, respectively (Figure 2F and Table S15 in the online supplement).
Effect-size of gene dosage on measures of core symptoms and specifiers of autism
We assessed the cognitive and behavioural symptoms that underlie autism susceptibility conferred by gene dosage.
Autism related symptoms
The pLI increases the SRS, with a 2:1 effect-size ratio for deletions and duplications in the pooled SSC and IMAGEN dataset (deletions: β=3.72 points of raw SRS score per point of pLI,, SE=0.57, p=5×10−11; duplications: β=1.87 points of raw SRS score per point of pLI, SE=0.43, p=1×10−5). The effect-size of pLI on SRS remains the same after adding data from MSSNG (deletions: β=3.68, SE=0.56, p=4×10−11; duplications: β=1.63, SE=0.42, p=1×10−4). This effect of gene dosage is entirely explained by NVIQ and the autism diagnosis (Figure 3A, Figure S5, Table S8 in the online supplement).
Figure 3. Effect of gene dosage on phenotypic measurements.
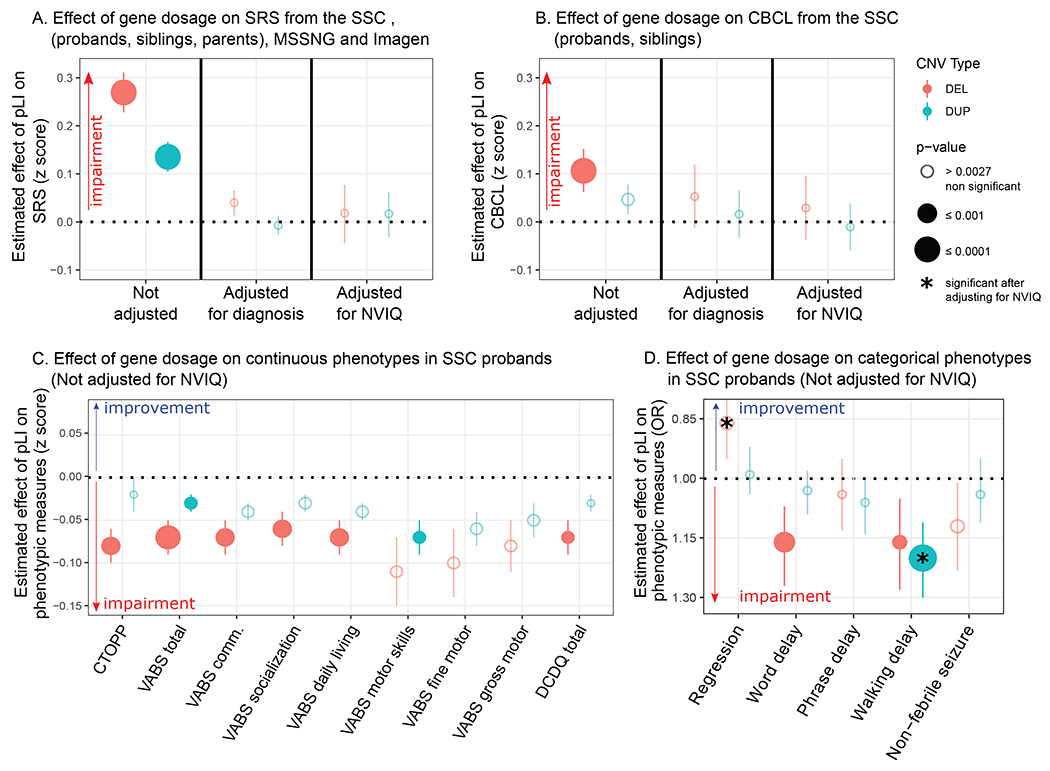
(A) Effect-size of a deleted (red) or duplicated (blue) point of pLI on the z-scored Social Responsiveness Scale (SRS) in autistic probands from the SSC pooled with their unaffected relatives (siblings and parents) and the unselected populations from the IMAGEN dataset. Effects were measured with and without adjustment for the diagnosis of autism, and NVIQ. The Y-axis represents the estimated effect of pLI on the SRS z-score computed using the mean and standard deviation of unaffected individuals (0.10 z-score = 1.28 SRS raw score point). The analysis adjusting for NVIQ only contains probands from the SSC and individuals from Imagen. (B) Effect-size of a deleted or duplicated point of pLI on the z-score of the Child behaviour Checklist (CBCL) in autistic probands from the SSC pooled with their unaffected siblings. Effects were measured with and without adjustment for the diagnosis of autism, and NVIQ. The estimates were originally computed as OR using a negative binomial regression. The Y-axis represents the estimated effect of pLI on z-scored CBCL computed using the mean and standard deviation of unaffected individuals (0.10 z-score = 1.52 CBCL raw score point). The analysis adjusting for NVIQ is only performed on probands from the SSC. (C, D) Effect-size of a deleted (red) or duplicated (blue) point of pLI on continuous (C) and categorical (D) phenotypes in autistic probands from the SSC unadjusted for NVIQ. Results adjusted for NVIQ are detailed in Figure S4. Y-axis values of panel (C) are measures z-scored using normative data (Table S7) except for DCDQ which was z-scored using the SSC autistic proband group. Y-axis values of panel (D) are odds ratios computed by logistic regression. The significance threshold was computed (51) to account for multiple testing: 0.0027. (✱) Effects significant after adjusting for NVIQ. CTOPP: Comprehensive Test of Phonological Processing; VABS-II: Vineland Adaptive behaviour Rating Scales - Second Edition; DCDQ: Developmental Coordination Disorder Questionnaire.
Deletions and duplications measured by pLI do not affect the ADOS or ADI-R scores in probands of the SSC and MSSNG datasets, pooled or separately (Table S9 in the online supplement). Moreover, deletions measured by pLI protect against regression in autism and this effect is enhanced after adjusting for NVIQ (OR=0.80, 95%CI=0.70-0.89, p=2×10−4) (Table 1, Figure 3D, Figure S6B in the online supplement).
Language and phonological memory
There is a clear dissociation between the effect of deletions and duplications on language. Deleted points of pLI are associated with a delay of first-words (OR=1.16, 95%CI=1.07-1.27, p=5×10−4) and negatively affects phonological memory, assessed by the non-word repetition of the Comprehensive Test of Phonological Processing (CTOPP) (β=0.08, SE=0.02, p=6×10−4). No effects are observed for deletions after adjusting for NVIQ and for duplications with or without adjusting for NVIQ (Table 1, Figure 3C and 3D, Figure S6A and 6B in the online supplement).
Behavioural and emotional symptoms
In the sample pooling probands and unaffected siblings, haploinsufficiency measured by pLI impacts the score of total problems from the CBCL (OR=1.05, 95%CI=1.03-1.08, p=2×10−6). The effect of duplications is weaker (OR=1.02, 95%CI=1.01-1.04, p=3×10−3) (Table S10, Figure 3B). This translates into an increase of 20.63 [95%CI=19.55-21.73] and 7.85 [95%CI=7.28-8.44] points for a deletion or a duplication encompassing 10 points of pLI, respectively. These effects are not observed within SSC probands or unaffected siblings samples.
Adaptive Skills
Adaptive skills measured by the second edition of the Vineland Adaptive behaviour Rating Scales (VABS-II) are negatively affected by the pLI, with a decrease of 2 and 1 point of VABS per deleted or duplicated point of pLI, respectively (p=3×10−5 and p=3×10−3). Total scores and all subscales are equally affected. NVIQ appears to account for most, if not all, of this effect (Table 1, Figure 3C, Figure S6A in the online supplement).
Motor skills and epilepsy
The relationship between the onset of walking measured in months and pLI (deletion: OR=1.03, 95%CI=1.02-1.04, p=2×10−11; duplication: OR=1.02, 95%CI=1.01-1.03, p=7×10−9) translates into a 5.46 [95%CI=5.27-5.65] or 3.58 [95%CI=3.45-3.72] month delay for a deletion or duplication encompassing 10 points of pLI, respectively (Figure S7 in the online supplement). This remains significant after adjusting for NVIQ for duplications only. The effect-size of gene dosage on motor skills, measured by the VABS-II and the Developmental Coordination Disorder Questionnaire (DCDQ), shows a 2:1 ratio for deletions and duplications with a similar effect for gross and fine motor skills. Gene dosage does not affect the risk of non-febrile seizures (Table 1, Figure 3C and 3D, Figure S6A and 6B in the online supplement).
Potential applications in the clinic
We developed a prediction tool available online (https://cnvprediction.urca.ca/) to estimate the effect-size of deletions and duplications on NVIQ, autism risk and the SRS score. As an illustration, our model estimates a decrease in NVIQ of 26.78 [95%CI=26.19-27.37] and 30.89 [95%CI=30.30-31.48] points, an increase in the SRS raw score of 36.93 [95%CI=35.82-38.04] and 42.59 [95%CI=41.48-43.70] points, and an increase in autism risk of 21.05 [95%CI=6.10-72.26] and 33.58 [95%CI=8.05-139.99] for the 16p11.2 and 22q11.2 deletion respectively. We detail the model output for 21 recurrent CNVs in Table S6. Briefly summarized, this tool should be viewed as a translation of gnomAD (34) information into phenotypic effect-sizes.
DISCUSSION
We propose a model to estimate the effect-size of gene dosage on autism susceptibility, core autism symptoms, general intelligence, and autism specifiers. Haploinsufficiency measured by pLI increases autism susceptibility across the genome but NVIQ drives a large proportion of this effect. Language, motor, social communication, and behavioural problems are also strongly affected by deletions. While these manifestations may increase the probability for deletion carriers of receiving an autism diagnosis, there is no evidence that core symptoms are affected (Figure 4). In contrast, duplicated points of pLI increase autism risk, genome-wide, and the influence of NVIQ is smaller. Increased risk measured by pLI is similar in subgroups of individuals with NVIQ below and above median.
Figure 4. Interpretation of the effect of gene dosage on autism risk.
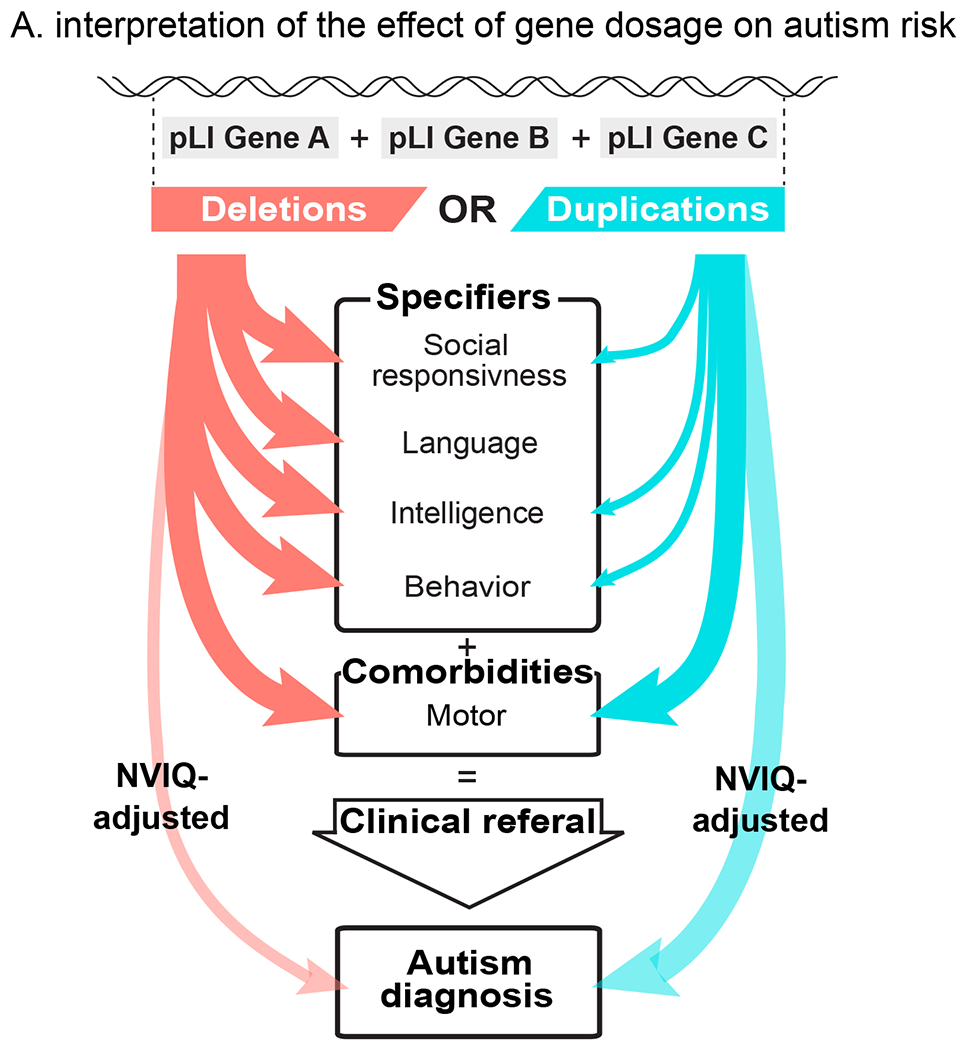
Summary interpretation of the differential effects of deletions and duplications on autism risk. Effect-sizes of deletion on most autism specifiers are larger than those of duplications. Duplications and to a lesser extent deletions increase the probability of an autism diagnosis after adjusting for their effect on NVIQ (transparent arrows).
Differential effects of deletions and duplications on autism core symptoms and specifiers
Model estimates show that any 1Mb coding deletion or duplication across the genome should increase autism susceptibility, with a median OR of 1.6 and 1.3, respectively (Figure 2F). GWAS conducted on common variants also showed that the bulk of the heritability for complex conditions (i.e. schizophrenia) is spread across the genome and largely driven by genes with no clear relevance to disease. (28, 43) Gene dosage affects NVIQ, social communication, and adaptive behaviour, with a deletion:duplication effect-size ratio of 2-3:1. Although both CNVs equally affect motor skills, phonological memory may be predominantly affected by haploinsufficiency. Similar differential profiles have been reported for 16p11.2 CNVs with phonological memory deficits in deletion but not duplication carriers. (44) We posit that general phenotypic profiles may be associated with deletions and duplications irrespective of the genomic loci. Genes included in the CNVs may mostly influence the effect-size but not the profile of symptoms. Consistent with this interpretation, the phenotypic profile of haploinsufficiency delineated by our model has been similarly reported in patients with de novo loss of function variants (23, 24). In addition, excluding large effect-size de novo variants from our analyses does not modify the effect-size of gene dosage, measured by pLI, on NVIQ and autism risk. Therefore, molecular functional networks enriched in genes with an excess of de novo mutations (chromatin remodelling, synaptic function) (14, 45, 46) may be related to large effect-sizes rather than specific effects on autism risk. Interestingly, although previous studies have shown lower NVIQ and a higher burden of deleterious CNVs in females from the SSC (22), we did not identify any interaction between the effect of pLI and sex. This suggests that deleting or duplicating one point of pLI affects NVIQ and increases autism risk similarly in both sexes.
Potential clinical applications
Our models are implemented in a prediction tool (https://cnvprediction.urca.ca/), which is designed to predict the effect-size of CNVs, not the symptoms of the individual who carries the CNV. If symptoms are discordant, the clinician may conclude that additional factors should be investigated. Discordance may be defined when the estimated effect-size of the CNV is 1 SD (15 IQ points) lower than the IQ loss observed in the carrier (compared to the population mean = 100). If a CNV with an effect-size of −10 IQ points is identified in a carrier with mild intellectual disabilities and an IQ of 60 (−40 compared to population mean) the majority of the cognitive deficits are caused by additional factors. The estimates of autism risk provided by models in this study overlap with risk computed in previous studies. As an example, our model estimates for 16p11.2 and 22q11.2 deletions are similar to the previously published effect for NVIQ, (loss of 25 (47) and 29 (48) points), autism risk (OR of 11.8 (10) and 32.37 (9)) and SRS (gain of 44 (47) and 49 (48) points). Overall, the output of these models can help interpret CNVs in the clinic, but estimates should be interpreted with caution.
Limitations
Discordance between autism risk estimated by the model and literature observations allows for the identification of CNVs, which may encompass genes with specific properties. For example, autism susceptibility and deficits associated with the 15q13.3 (CHRNA7) deletion appear to be underestimated by our model. This CNV may include genes for which the assigned pLI score does not capture the effects on psychiatric traits (e.g. gene dosage of CHRNA7, which has a pLI=0 may affect psychopathology without altering genetic fitness). The pLI was not developed to measure intolerance to duplications and results should, therefore, be interpreted with caution. Our findings suggest, however that pLI may be a general measure of dosage sensitivity, in line with recent data from gnomAD-SV. (49) Since gene dosage is not comparable between sex-linked and autosomal CNVs, we could not pool both types of CNVs. Sex-linked CNVs were excluded from this study because they were too rare in our samples to be studied separately. The effect of gene dosage on SRS was very robust but was mainly explained by the autism diagnosis. This suggests that the SRS may not measure a continuous dimension since this score is unable to provide additional granularity within the autism group or the controls despite large sample size. Some phenotypic measures such as phonological memory and motor skills were only available for autism probands and results may not be generalizable to non-autism samples. Larger samples, with additional intrafamilial controls, novel functional annotations, and more refined models are required to improve our estimates of CNV effect-sizes on cognitive dimensions.
Of note, although CNV with large effect-sizes have significant impacts on the development of an individual, they only explain a small fraction of the variance of general intelligence (1.4% and 0.4% for deletions and duplications) and liability for autism (0.4 and 0.4% for deletions and duplications) at the population level, which is concordant with previous reports. (5)
Conclusion
Our study highlights the extreme polygenicity of autism susceptibility conferred by gene dosage. It also delineates cognitive mechanisms which may explain in part the overrepresentation of CNVs in autism. Among mutations over-represented in autism, those truly related to core symptoms may be less common than previously thought. Future large-scale studies simultaneously investigating the effect of genomic variants on categorical diagnoses and continuous dimensions are warranted. This study represents a new framework to study rare variants and can help in the interpretation of the effect-size of undocumented CNVs identified in the neurodevelopmental clinic.
Supplementary Material
Funding/Support:
This research was enabled by support provided by Calcul Quebec (http://www.calculquebec.ca) and Compute Canada (http://www.computecanada.ca). Sebastien Jacquemont is a recipient of a Bursary Professor fellowship of the Swiss National Science Foundation, a Canada Research Chair in neurodevelopmental disorders, and a chair from the Jeanne et Jean Louis Levesque Foundation. Catherine Schramm is supported by an Institute for Data Valorization (IVADO) fellowship. Petra Tamer is supported by a Canadian Institute of Health Research (CIHR) Scholarship Program. Guillaume Huguet is supported by the Sainte-Justine Foundation, the Merit Scholarship Program for foreign students, and the Network of Applied Genetic Medicine fellowships. Eva Loth is supported by European Autism Interventions, which receives support from the Innovative Medicines Initiative Joint Undertaking under grant agreement 115300, the resources of which are composed of financial contributions from grant FP7/2007-2013 from the European Union’s Seventh Framework Programme, the European Federation of Pharmaceutical Industries and Associations companies’ in-kind contributions, and Autism Speaks. Thomas Bourgeron is a recipient of a chair of the Bettencourt-Schueler foundation. Laurent Mottron is a recipient of the Marcel & Rolande Gosselin research chair. This work is supported by a grant from the Brain Canada Multi-Investigator initiative and CIHR grant 159734 (Sebastien Jacquemont, Celia Greenwood, Tomas Paus). The Canadian Institutes of Health Research and the Heart and Stroke Foundation of Canada fund the Saguenay Youth Study (SYS). SYS was funded by the Canadian Institutes of Health Research (Tomas Paus, Zdenka Pausova) and the Heart and Stroke Foundation of Canada (Zdenka Pausova). Funding for the project was provided by the Wellcome Trust. This work was also supported by an NIH award U01 MH119690 granted to Laura Almasy, Sebastien Jacquemont and David Glahn and U01 MH119739. The authors gratefully acknowledge the resources provided by the Autism Speaks MSSNG project and the Autism Genetic Resource Exchange Consortium, as well as the participating families.
We are grateful to all the families who participated in the Simons Variation in Individuals Project (VIP) and the Simons VIP Consortium (data from Simons VIP are available through SFARI Base). We thank the coordinators and staff at the Simons VIP and SCC sites. We are grateful to all of the families at the participating SSC sites and the principal investigators (A. Beaudet, M.D., R. Bernier, Ph.D., J. Constantino, M.D., E. Cook, M.D., E. Fombonne, M.D., D. Geschwind, M.D., Ph.D., R. Goin-Kochel, Ph.D., E. Hanson, Ph.D., D. Grice, M.D., A. Klin, Ph.D., D. Ledbetter, Ph.D., C. Lord, Ph.D., C. Martin, Ph.D., D. Martin, M.D., Ph.D., R. Maxim, M.D., J. Miles, M.D., Ph.D., O. Ousley, Ph.D., K. Pelphrey, Ph.D., B. Peterson, M.D., J. Piggot, M.D., C. Saulnier, Ph.D., M. State, M.D., Ph.D., W. Stone, Ph.D., J. Sutcliffe, Ph.D., C. Walsh, M.D., Ph.D., Z. Warren, Ph.D., and E. Wijsman, Ph.D.). We appreciate obtaining access to phenotypic data on SFARI base.
Role of the Funder/Sponsor:
The funder had no role in the design and conduct of the study; collection, management, analysis, or interpretation of the data; preparation, review, or approval of the manuscript; or decision to submit the manuscript for publication.
Footnotes
Previous presentation
WCPG (World Congress of Psychiatry Genetics) 2019, Los Angeles, United-States – oral presentation. October the 27th 2019, 3:45 PM - 4:00 PM
INSAR (International Society for Autism Research) 2019, Montreal, Canada – Poster presentation. May the 3th 2019, 5:30 PM - 7:00 PM
REFERENCES
- 1.American Psychiatric Association: Diagnostic and Statistical Manual of Mental Disorders (DSM-5®). American Psychiatric Pub, 2013 [Google Scholar]
- 2.Ousley O, Cermak T: Autism Spectrum Disorder: Defining Dimensions and Subgroups. Curr Dev Disord Rep 2014; 1:20–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simonoff E, Pickles A, Charman T, et al. : Psychiatric disorders in children with autism spectrum disorders: prevalence, comorbidity, and associated factors in a population-derived sample. J Am Acad Child Adolesc Psychiatry 2008; 47:921–929 [DOI] [PubMed] [Google Scholar]
- 4.Sandin S, Lichtenstein P, Kuja-Halkola R, et al. : The Heritability of Autism Spectrum Disorder. JAMA 2017; 318:1182–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gaugler T, Klei L, Sanders SJ, et al. : Most genetic risk for autism resides with common variation. Nat Genet 2014; 46:881–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanders SJ, He X, Willsey AJ, et al. : Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015; 87:1215–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang Y, Yuen RKC, Jin X, et al. : Detection of Clinically Relevant Genetic Variants in Autism Spectrum Disorder by Whole-Genome Sequencing. Am J Hum Genet 2013; 93:249–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tammimies K, Marshall CR, Walker S, et al. : Molecular Diagnostic Yield of Chromosomal Microarray Analysis and Whole-Exome Sequencing in Children With Autism Spectrum Disorder. JAMA 2015; 314:895–903 [DOI] [PubMed] [Google Scholar]
- 9.Sanders SJ, Sahin M, Hostyk J, et al. : A framework for the investigation of rare genetic disorders in neuropsychiatry. Nat Med 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malhotra D, Sebat J: CNVs: Harbingers of a Rare Variant Revolution in Psychiatric Genetics. Cell 2012; 148:1223–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moreno-De-Luca D, Sanders SJ, Willsey AJ, et al. : Using large clinical data sets to infer pathogenicity for rare copy number variants in autism cohorts. Mol Psychiatry 2013; 18:1090–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Satterstrom FK, Kosmicki JA, Wang J, et al. : Novel genes for autism implicate both excitatory and inhibitory cell lineages in risk. bioRxiv 2018; 484113 [Google Scholar]
- 13.Marshall CR, Howrigan DP, Merico D, et al. : Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet 2017; 49:27–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krumm N, Turner TN, Baker C, et al. : Excess of rare, inherited truncating mutations in autism. Nat Genet 2015; 47:582–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Girirajan S, Brkanac Z, Coe BP, et al. : Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet 2011; 7:e1002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.D’Angelo D, Lebon S, Chen Q, et al. : Defining the Effect of the 16p11.2 Duplication on Cognition, Behavior, and Medical Comorbidities. JAMA Psychiatry 2016; 73:20–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernier R, Steinman KJ, Reilly B, et al. : Clinical phenotype of the recurrent 1q21.1 copy-number variant. Genet Med Off J Am Coll Med Genet 2016; 18:341–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Butcher NJ, Chow EWC, Costain G, et al. : Functional outcomes of adults with 22q11.2 deletion syndrome. Genet Med 2012; 14:836–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Girirajan S, Dennis MY, Baker C, et al. : Refinement and Discovery of New Hotspots of Copy-Number Variation Associated with Autism Spectrum Disorder. Am J Hum Genet 2013; 92:221–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iossifov I, O’Roak BJ, Sanders SJ, et al. : The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014; 515:216–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mottron L, Duret P, Mueller S, et al. : Sex differences in brain plasticity: a new hypothesis for sex ratio bias in autism. Mol Autism 2015; 6:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacquemont S, Coe BP, Hersch M, et al. : A Higher Mutational Burden in Females Supports a “Female Protective Model” in Neurodevelopmental Disorders. Am J Hum Genet 2014; 94:415–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bishop SL, Farmer C, Bal V, et al. : Identification of Developmental and Behavioral Markers Associated With Genetic Abnormalities in Autism Spectrum Disorder. Am J Psychiatry 2017; 174:576–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buja A, Volfovsky N, Krieger AM, et al. : Damaging de novo mutations diminish motor skills in children on the autism spectrum. Proc Natl Acad Sci U S A 2018; 115:E1859–E1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huguet G, Schramm C, Douard E, et al. : Measuring and Estimating the Effect Sizes of Copy Number Variants on General Intelligence in Community-Based Samples. JAMA Psychiatry 2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lek M, Karczewski KJ, Minikel EV, et al. : Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536:285–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weiner DJ, Wigdor EM, Ripke S, et al. : Polygenic transmission disequilibrium confirms that common and rare variation act additively to create risk for autism spectrum disorders. Nat Genet 2017; 49:978–985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wray NR, Wijmenga C, Sullivan PF, et al. : Common Disease Is More Complex Than Implied by the Core Gene Omnigenic Model. Cell 2018; 173:1573–1580 [DOI] [PubMed] [Google Scholar]
- 29.Fischbach GD, Lord C: The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron 2010; 68:192–195 [DOI] [PubMed] [Google Scholar]
- 30.Yuen RKC, Merico D, Bookman M, et al. : Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat Neurosci 2017; 20:602–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schumann G, Loth E, Banaschewski T, et al. : The IMAGEN study: reinforcement-related behaviour in normal brain function and psychopathology. Mol Psychiatry 2010; 15:1128–1139 [DOI] [PubMed] [Google Scholar]
- 32.Pausova Z, Paus T, Abrahamowicz M, et al. : Cohort Profile: The Saguenay Youth Study (SYS). Int J Epidemiol 2017; 46:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramasamy A, Trabzuni D, Guelfi S, et al. : Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci 2014; 17:1418–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karczewski KJ, Francioli LC, Tiao G, et al. : Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019; 531210 [Google Scholar]
- 35.Petrovski S, Gussow AB, Wang Q, et al. : The Intolerance of Regulatory Sequence to Genetic Variation Predicts Gene Dosage Sensitivity. PLoS Genet 2015; 11:e1005492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruderfer DM, Hamamsy T, Lek M, et al. : Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nat Genet 2016; 48:1107–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szklarczyk D, Franceschini A, Wyder S, et al. : STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015; 43:D447–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hawrylycz M, Miller JA, Menon V, et al. : Canonical genetic signatures of the adult human brain. Nat Neurosci 2015; 18:1832–1844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.International HapMap Consortium: The International HapMap Project. Nature 2003; 426:789–796 [DOI] [PubMed] [Google Scholar]
- 40.Chaste P, Sanders SJ, Mohan KN, et al. : Modest impact on risk for autism spectrum disorder of rare copy number variants at 15q11.2, specifically breakpoints 1 to 2. Autism Res Off J Int Soc Autism Res 2014; 7:355–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wain HM, Bruford EA, Lovering RC, et al. : Guidelines for human gene nomenclature. Genomics 2002; 79:464–470 [DOI] [PubMed] [Google Scholar]
- 42.Cooper GM, Coe BP, Girirajan S, et al. : A copy number variation morbidity map of developmental delay. Nat Genet 2011; 43:838–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyle EA, Li YI, Pritchard JK: An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017; 169:1177–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hippolyte L, Maillard AM, Rodriguez-Herreros B, et al. : The Number of Genomic Copies at the 16p11.2 Locus Modulates Language, Verbal Memory, and Inhibition. Biol Psychiatry 2016; 80:129–139 [DOI] [PubMed] [Google Scholar]
- 45.Pinto D, Delaby E, Merico D, et al. : Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet 2014; 94:677–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huguet G, Ey E, Bourgeron T: The genetic landscapes of autism spectrum disorders. Annu Rev Genomics Hum Genet 2013; 14:191–213 [DOI] [PubMed] [Google Scholar]
- 47.Moreno-De-Luca A, Evans DW, Boomer KB, et al. : The role of parental cognitive, behavioral, and motor profiles in clinical variability in individuals with chromosome 16p11.2 deletions. JAMA Psychiatry 2015; 72:119–126 [DOI] [PubMed] [Google Scholar]
- 48.Vangkilde A, Jepsen JRM, Schmock H, et al. : Associations between social cognition, skills, and function and subclinical negative and positive symptoms in 22q11.2 deletion syndrome. J Neurodev Disord 2016; 8:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Collins RL, Brand H, Karczewski KJ, et al. : An open resource of structural variation for medical and population genetics. bioRxiv 2019; 578674 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.