

Abstract
Efficient quality control and export of procollagen from the cell is crucial for extracellular matrix homeostasis, yet it is still incompletely understood. One of the debated questions is the role of a collagen-specific ER chaperone HSP47 in these processes. Most ER chaperones preferentially bind to unfolded polypeptide chains, enabling selective export of natively folded proteins from the ER after chaperone release. In contrast, HSP47 preferentially binds to the natively folded procollagen and is believed to be released only in the ER-Golgi intermediate compartment (ERGIC) or cis-Golgi. HSP47 colocalization with procollagen in punctate structures observed by immunofluorescence imaging of fixed cells has thus been interpreted as evidence for HSP47 export from the ER together with procollagen in transport vesicles destined for ERGIC or Golgi. To understand the mechanism of this co-trafficking and its physiological significance, we imaged the dynamics of fluorescently tagged type I procollagen and HSP47 punctate structures in live MC3T3 murine osteoblasts with up to 120 nm spatial and 500 ms time resolution. Contrary to the prevailing model, we discovered that most bona fide carriers delivering procollagen from ER exit sites (ERESs) to Golgi contained no HSP47, unless the RDEL signal for ER retention in HSP47 was deleted or mutated. These transport intermediates exhibited characteristic rapid, directional motion along microtubules, while puncta with colocalized HSP47 and procollagen similar to the ones described before had only limited, stochastic motion. Live cell imaging and fluorescence recovery after photobleaching revealed that the latter puncta (including the ones induced by ARF1 inhibition) were dilated regions of ER lumen, ERESs, or autophagic structures surrounded by lysosomal membranes. Procollagen was colocalized with HSP47 and ERGIC53 at ERESs. It was colocalized with ERGIC53 but not HSP47 in Golgi-bound transport intermediates. Our results suggest that procollagen and HSP47 sorting occurs at ERES before procollagen is exported from the ER in Golgi-bound transport intermediates, providing new insights into mechanisms of procollagen trafficking.
Keywords: Collagen, HSP47, trafficking, ER exit sites, live cell imaging
Introduction
Procollagen trafficking and secretion has been a subject of numerous studies over the years, yet the mechanism of procollagen delivery from the Endoplasmic Reticulum (ER) to Golgi is still being debated [1–3]. Collagens are by far the most abundant vertebrate proteins distinguished by triple helical domains, which have the distinct sequence of repeating Gly-X-Y triplets with many Pro and Hyp in the X and Y positions. Collagens are essential for forming structural scaffolds of extracellular matrix in all tissues and organs. They are synthesized by cells as procollagen precursors, in which triple helical domains are flanked by globular N- and C-terminal domains. The most abundant and widely studied is type I collagen, which is the main structural protein in bones, tendons, and ligaments.
Procollagens are assembled and folded in the ER, transported through Golgi, and secreted. Like other secretory proteins, procollagen trafficking from the ER to Golgi involves COPII coat and TANGO1/cTAGE machinery of ER exit sites (ERESs) [1–3]. However, whether procollagen is exported from ERESs in enlarged COPII-coated vesicles [4–6], in ERGIC-membrane-derived megacarriers [7, 8], through “short-loop” ER-Golgi connections [9], or through a combination of these and other pathways is unclear. From the cell biology perspective, the membrane trafficking pathways followed by procollagen exemplify a general problem of poorly understood secretory pathways for large cargoes [1–3]. It is not even resolved whether ER-Golgi intermediate membrane structures represent a distinct subcellular compartment (ERGIC, aka IC) or just transport intermediates between the ER and Golgi [10]. From the matrix biology and pathophysiology perspective, procollagen trafficking defects likely underlie skeletal dysplasias caused by mutations in COPII proteins [11–17], CREB3L1 that regulates COPII protein expression [18–20], and TANGO1 [21]. In general, deficient trafficking of procollagens and other matrix molecules might be involved in a wide plethora of diseases since extracellular matrix remodeling is a crucial regulator of tissue homeostasis [22, 23].
An important part of the puzzle of ER-to-Golgi procollagen trafficking is the role of HSP47 in its regulation. HSP47 is a collagen-specific ER chaperone co-expressed with procollagen, which is required for proper folding of procollagen’s triple helix [24–26]. Other ER chaperones, such as BIP and HSP90B (GRP94), assist protein folding by preferentially binding to unfolded, mostly hydrophobic regions of polypeptide chains, thereby preventing non-native interactions and aggregation [27]. Once native conformation of a protein is attained, these chaperones are released, which is a signal for protein loading into ERESs and export from the ER. In contrast, HSP47 enables folding of intrinsically unstable procollagen by preferentially binding to the triple helical rather than unfolded conformation [28–31]. Unlike other ER chaperones, HSP47 is accrued at multiple binding sites along the triple helix instead of being released upon completion of procollagen folding. It might also serve as an adapter for procollagen loading into ERESs by TANGO1 [32]. Thus, it is widely thought that HSP47 is co-transported with procollagen from the ER to ERGIC or cis-Golgi, followed by its release at lower pH [33, 34], capture by KDEL/RDEL receptors, and retrograde traffic to the ER [25, 26].
Previously, HSP47 was observed by immunofluorescence in vesicle-like procollagen puncta after brefeldin A [35] or GTPγS [36] disruption of ER-Golgi trafficking. It was also reported to be colocalized with TANGO1 and COPII in large procollagen carriers by immunofluorescence and by fractionation in a cell-free vesicle budding reaction [6]. However, these techniques cannot unambiguously identify bona fide transport vesicles. In fixed cells imaged by immunofluorescence, procollagen/HSP47 puncta might instead represent dilated regions of the ER, ERESs, autophagic structures, other cell compartments, or non-physiological structures caused by the disruption of cellular function with brefeldin A or GTPγS. Cell-free assays might produce vesicles that do not form inside the cell under physiological conditions. Unambiguous identification of any procollagen puncta as transport intermediates requires a proof of their disconnection from the ER, directional (non-stochastic) motion, and entry into Golgi. Having developed an assay for live cell imaging capable of monitoring these processes [37], we therefore decided to revisit procollagen trafficking regulation by HSP47.
Here we describe dynamic imaging of puncta containing fluorescently tagged type I procollagen and HSP47 in live cells with ~120 nm spatial and up to 500 ms/frame time resolution, which reveals that procollagen is normally exported from osteoblast ER without HSP47. We created fluorescent constructs of type I procollagen and HSP47 and imaged them in a murine osteoblastic cell line MC3T3 together with protein markers of the ER lumen, COPII coat, ERGIC membranes, cis-Golgi, autophagic structures, and lysosomes. Osteoblasts, which are the cells responsible for making bone, are some of the most active secretory cells that produce and secrete massive amounts of type I collagen. Expression of the fluorescent procollagen and HSP47 constructs in these cells did not disrupt their function. The constructs faithfully reproduced the subcellular localization of the corresponding endogenous proteins observed by immunofluorescence as well as procollagen/HSP47 localization patterns previously reported in static snapshots of fixed cells. Upon live cell imaging, however, we found no evidence for HSP47 accompanying procollagen out of the ER in Golgi-destined transport intermediates. Rather, we observed HSP47 only in the ER and in stochastically moving vesicle-like structures, which were distinct from bona fide procollagen carriers that moved directionally toward the Golgi apparatus. Upon deleting or modifying the RDEL retention signal, HSP47 was co-trafficked with procollagen to Golgi in directionally moving vesicles. Our results support a revised trafficking mechanism in which HSP47 dissociates from procollagen at ERESs, before the latter is exported in ERGIC53-postive transport intermediates bound for the Golgi.
Results
Fluorescently tagged HSP47, proα1(I), and proα2(I) constructs for live cell imaging of procollagen and HSP47 trafficking
We built fluorescent protein (FP)-tagged mouse HSP47 constructs, in which different color FPs were added on the N-terminus of the molecule, between the ER signal peptide and the start site of HSP47. GFP-HSP47 transiently transfected into the MC3T3-E1 mouse osteoblast line [38] co-immunoprecipitated with procollagen, suggesting that its binding to collagen was similar to endogenous HSP47 (Kd ~ 1 μM, Fig. 1A). We observed complete colocalization of FP-tagged HSP47 molecules with each other and antibody-tagged HSP47, i.e. all spots positive for one of the tags were also positive for the other tags (Fig. 1B). Variation in the relative intensity of the tags at different spots appeared to be related primarily to antibody accessibility, since the largest difference was observed behind the cell nucleus. Localization of all FP-HSP47 constructs (TagBFP2, GFP, Venus, Apple, and Cherry) was similar to endogenous HSP47.
Fig. 1. Fluorescent protein-tagged HSP47 (FP-HSP47) interacts with procollagen and colocalizes with endogenous HSP47.

(A) Western blots of cell lysates before and after IP of GFP-HSP47 with Anti-GFP beads. Green arrow shows transfected GFP-HSP47; red arrow shows endogenous HSP47. Significant enrichment of procollagen relative to vinculin on the beads revealed that it co-immunoprecipitated with GFP-HSP47. Since the estimated procollagen concentration in cell lysates was lower than 1 μM and HSP47 binds procollagen with Kd ~ 1–10 μM [30], the co-immunoprecipitation indicated minimal or no disruption of HSP47-procollagen interaction by the fluorescent tag in GFP-HSP47. Because multiple endogenous HSP47 and GFP-HSP47 molecules would be bound to the same procollagen triple helix, a significant amount of endogenous HSP47 was found on the Anti-GFP beads. (B) Confocal imaging of colocalization between two FP-HSP47 constructs co-transfected into MC3T3 cells and HSP47 antibodies that label both transfected and endogenous HSP47. All spots labeled with anti-HSP47 were also positive for GFP-HSP47 and Cherry-HSP47, indicating complete colocalization of transfected and endogenous molecules in all subcellular compartments. Variations in relative fluorescence intensity of different tags are affected by antibody accessibility, uneven photobleaching during sample preparation and imaging, and other uncontrolled factors. Here and throughout the paper, different channels in multichannel images are merged using RGB pseudo color scheme and the corresponding color labels for individual channels. The scale bar is 10 μm; N=6 cells were examined.
We performed procollagen/HSP47 sorting experiments separately with FP-proα1(I) and FP- proα2(I) constructs because of the difference in cell adaptation mechanisms responding to overexpression of proα1(I) and proα2(I) chains. Excessive synthesis of proα2(I) due to the exogenous Col1a2 produces unassociated proα2(I) chains that must be removed from the ER and degraded (only one proα2(I) can be incorporated into trimeric procollagen). In contrast, excess of proα1(I) relative to proα2(I) causes formation of proα1(I) homotrimers, which appear to be secreted normally by osteoblasts and cause no significant bone pathology even in individuals who do not produce proα2(I) at all [39, 40].
The design and validation of the FP-proα1(I) and FP- proα2(I) constructs was described in more detail in our previous study [37]. Briefly, we found that both FP-proα1(I) and FP-proα2(I) chains assembled into procollagen heterotrimers, progressed through the normal secretory pathway, and after being secreted were incorporated into extracellular collagen fibers. In MC3T3 cells with low-to-moderate amounts of FP-procollagen (i.e., 12–24 h post transfection) we observed no overexpression artifacts and the results were the same for both constructs. In MC3T3 cells with excessive expression of FP-procollagen and after more than 24 h post transfection, we noticed the formation of large dilated ER regions filled by FP-procollagen chains (Supp. Fig. 1), which was probably due to irreversible aggregation of procollagen. The effects were more pronounced after FP-proα1(I) transfection, probably because of larger disruptions in procollagen folding and trafficking caused by incorporation of more than one FP-proα1(I) chain into the same molecule.
Since FP tags used for live cell imaging might perturb folding and trafficking of the corresponding proteins, we performed multiple control experiments. We utilized only the tags that have been well characterized before or additionally validated by colocalization with each other and endogenous protein antibodies (Fig. 1B, Supp. Fig. 2, and Experimental Procedures). To avoid ER disruption caused by slow accumulation of FP-tagged proteins, we did not use over-transfected cells with large regions of dilated ER and limited experiments to 18–24 h post transfection. We compared FP-proα1(I) and FP-proα2(I) transfections, exploiting the difference in the number of FP-tagged chains that can be incorporated into the same procollagen molecule. We performed key experiments with different combinations of FP tags, since different FPs might produce different artifacts. To emphasize the importance of such controls, most images in this manuscript are shown for both FP-proα1(I) and FP- proα2(I) transfections and different combinations of FP tags.
Additionally, we generated an MC3T3 cell line, in which one endogenous Col1a2 allele was modified with CRISPR/CAS9 to produce the same proα2(I) chain as the GFP-proα2(I) construct used here. The endogenous GFP-procollagen was trafficked similar to the transfected one. It also gradually accumulated in the ER after stimulation of procollagen synthesis with ascorbic acid, eventually causing ER disruption and suppression of collagen synthesis or cell demise. This cell line provided no added benefits for the present study and its generation, characterization, and possible utilization will be described in detail separately.
Dynamic imaging of FP-HSP47 co-localization with procollagen
As we reported previously [37], FP-proα1(I) and FP- proα2(I) were observed in the ER lumen marked by ssFP-KDEL (Fig. 2A,B) and in cis-Golgi marked by FP-GM130 (Fig. 2C,D). FP- HSP47 perfectly co-localized with FP-proα1(I) and FP-proα2(I) within the ER lumen. It was not observed in the cis-Golgi, unless the cells were subjected to excessive stress or over-transfected with FP-HSP47. The strict ER localization of FP-HSP47 under normal expression conditions suggested FP-HSP47 was either rapidly removed by retrograde transport back to the ER or never left the ER to enter the cis-Golgi. To distinguish between these two possibilities, we photobleached FP-HSP47 and FP-procollagen in the Golgi area to remove fluorescence in this area and then imaged transport vesicles moving into this region from distant ERESs.
Fig. 2. FP-HSP47 colocalizes with procollagen in the ER but does not colocalize with the cis-Golgi marker GM130.

(A,B) MC3T3 cells transfected with Cherry-HSP47, Venus-proα1(I) (A) or Venus-proα2(I) (B), and marker of ER lumen (ssCFP-KDEL). (C,D) MC3T3 cells transfected with Cherry-HSP47 (C) or TagBFP2-HSP47 (D), Venus-proα1(I) (C) or GFP-proα2(I) (D) and marker of cis-Golgi CFP-GM130 (C) or Cherry-GM130 (D). Switching of the fluorescent tag colors in different images illustrates that each of these and subsequent experiments were performed with different combinations of fluorescent tag colors, to eliminate potential tag-specific effects. Arrows mark Golgi structures containing procollagen. All images are Airyscan (A,B) or confocal (C,D) single slices; scale bars = 10 μm (full cell) and 1 μm (zoomed areas); N≥20 (≥4 experiments with different FP combinations) for each panel.
Time lapse imaging of fluorescence recovery after photobleaching (FRAP) revealed rapidly moving FP-proα1(I)/α2(I) puncta entering into the cis-Golgi, but these puncta did not contain any FP-HSP47 (Fig. 3; Movies 1 and 2). We also observed numerous vesicle-like puncta of FP-proα1(I)/α2(I) co-localized with FP-HSP47, but their restricted, stochastic-like movements were inconsistent with those of ER-to-Golgi transport intermediates.
Fig. 3, Movies 1,2. FP-HSP47 is not found in FP-procollagen-positive transport vesicles entering cis-Golgi.

(A, Movie 1) Individual frames and time-lapse video of a cell transfected with Cherry-HSP47, Venus-proα1(I) and CFP-GM130 before and after Cherry-HSP47 and Venus-proα1(I) were photobleached from Golgi region. (B, Movie 2) Frames and time-lapse video of a cell transfected with TagBFP2-HSP47, GFP-proα2(I) and Cherry-GM130 before and after TagBFP2-HSP47 and GFP-proα2(I) were photobleached from Golgi region. Arrows in post-FRAP frames (A, B) mark Golgi-destined procollagen transport vesicles identified by time-lapse imagining (white circles in Movies 1,2). Arrowheads in pre-FRAP whole-cell images point to some of the vesicle-like procollagen/HSP47 structures (magenta circles) that exhibit limited stochastic motion. The images are confocal single slices; scale bars = 10 μm (whole cell) and = 1 μm (zoom); N≥15 (3 experiments with different FP combinations) for each panel. (Movie 2) White circle outlines the ER-to-Golgi movement of 3 procollagen transport vesicles, none of which contain HSP47.
To better visualize procollagen and HSP47 dynamics in the cell and trace rapidly moving procollagen puncta, we performed time-lapse imaging without photobleaching at 2 s/frame (Fig. 4A; Movie 3), 1 s/frame (Fig. 4B; Movie 4), and 500 ms/frame (Supp. Fig. 3, Movie 5). Regardless of the imaging speed, we observed no directional movement of FP-HSP47-labeled structures. Rather, their limited movements appeared to reflect stochastic fluctuations caused by rearrangements of the ER network. We traced numerous procollagen transport intermediates distinguished by rapid directional movement, likely along microtubules, some moving toward the cis-Golgi and some toward the plasma membrane (Movies 3–5). We detected HSP47 in just one or two out of the hundreds of vesicles that appeared to be Golgi-bound (for more accurate quantification, see below).
Fig. 4, Movies 3,4. Dynamics of vesicle-like FP-HSP47 structures resembles stochastic fluctuations of the reticular ER network but not rapid directional movement of FP-procollagen transport vesicles.

(A, Movie 3) Confocal single slice frame and 2s/frame time-lapse video of a cell transfected with GFP-proα1(I) and Apple-HSP47. (B, Movie 4) Airyscan single slice frame and 1 s/frame video of a cell transfected with Venus-proα2(I) and Cherry-HSP47. Some of procollagen transport vesicles identified by rapid directional movement are outlined by white circles in the time-lapse videos and also marked by arrows in the still frames. These transport intermediates contain procollagen but not HSP47. Stochastically moving vesicle-like structures containing procollagen and HSP47 are outlined by magenta circles in the videos and also marked by arrowheads in the still frames. Scale bars = 10 μm (whole cell) and = 1 μm (zoom); N>70 (≥14 experiments with different FP combinations) for each panel.
Effects of the RDEL sequence on FP-HSP47 localization and trafficking
One possible explanation for the absence of FP- HSP47 in transport vesicles delivering procollagen to Golgi is that HSP47 is retained in the ER because of its ER localization sequence, RDEL. To test this hypothesis, we made FP constructs of HSP47 in which this sequence was deleted (ΔRDEL) or mutated to RNGL. FP-HSP47ΔRDEL colocalized with FP-proα1(I)/α2(I) in the ER and cis-Golgi (Fig. 5A), but its expression caused cis-Golgi disruption and formation of large FP-GM130 spheres within the nucleus similar to droplets previously observed upon GM130 overexpression [41]. FP-HSP47RNGL also co-localized with FP-proα1(I)/α2(I) in the ER and cis-Golgi, causing less pronounced yet noticeable Golgi disruption and nuclear FP-GM130 spheres (Fig. 5B). Time lapse imaging revealed that FP-HSP47RNGL was present in many FP-proα1(I)/α2(I) transport vesicles (Fig. 5C; Movie 6 and 7). Apparently, HSP47 release from the ER and trafficking to cis-Golgi was responsible for the Golgi disruption and mislocalization of FP-GM130 in the nucleus. This effect might be important for understanding how HSP47 mutations cause osteogenesis imperfecta [42, 43], but its detailed analysis is beyond the scope of the present paper and will be reported elsewhere.
Fig. 5, Movies 6,7. Deletion of RDEL and RDEL→RNGL mutation release HSP47 from the ER to cis-Golgi, causing Golgi disruption.
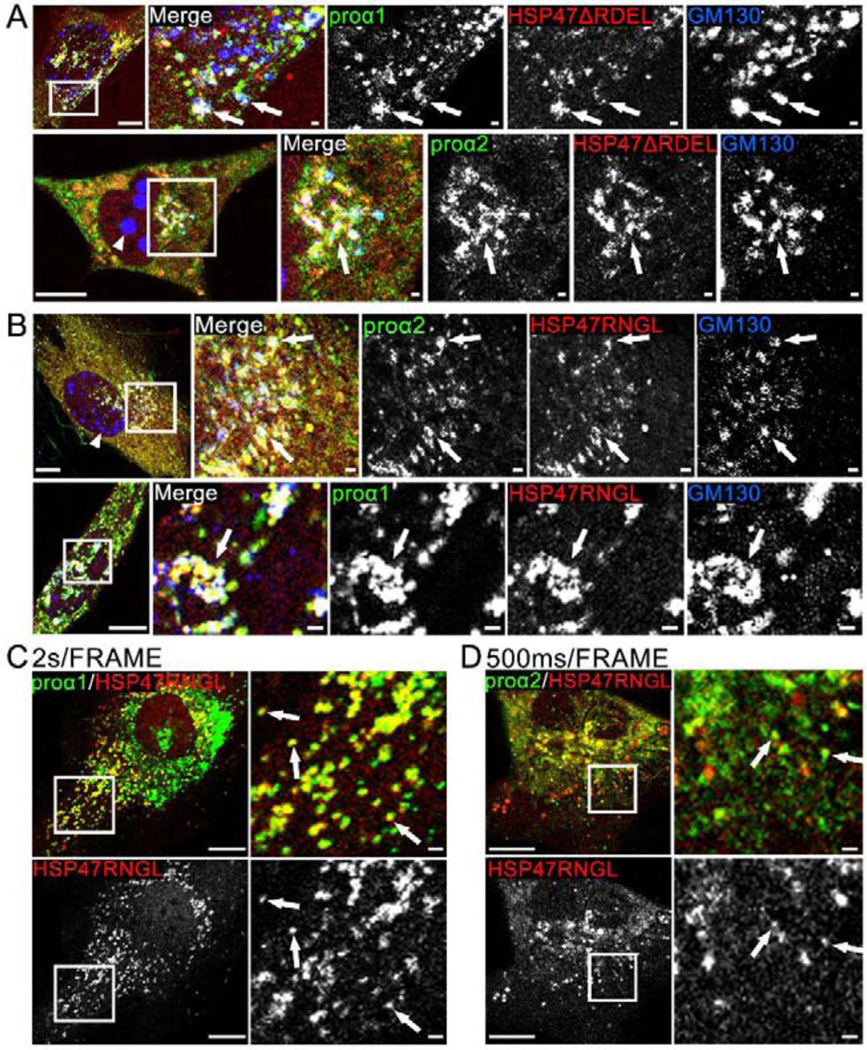
(A) Airyscan colocalization of Cherry-HSP47ΔRDEL with Venus-proα1(I)/α2(I) and cis-Golgi marker CFP-GM130 (white arrows); N=4 (proα1(I)) and N=4 (proα2(I)). Based on imaging of several z-slices, large CFP-GM130 spheres (arrowhead) are located inside the nuclei. At least some Golgi fragmentation was noticeable in all cells transfected with Cherry-HSP47ΔRDEL. (B) Airyscan colocalization of Cherry-HSP47RNGL with Venus-proα1(I)/α2(I) and cis-Golgi marker CFP-GM130 (white arrows); N=19 (proα1(I)) and N=7 (proα2(I)). In these cells, Golgi fragmentation was less pronounced. The nuclear CFP-GM130 spheres (arrowhead) were smaller and absent from some of the cells. (C, Movie 6) Confocal single slice frame and time-lapse video of Apple-HSP47RNGL colocalization with GFP-proα1(I)in transport vesicles marked by arrows in the still frame; N=63 (14 experiments). (D, Movie 7) Airyscan single slice frame and video of similar colocalization of Cherry-HSP47RNGL with Venus-proα2(I); N=18 (3 experiments). Scale bars = 10 μm (whole cell) and = 1 μm (zoom).
Identification of vesicle-like structures containing FP-HSP47 and FP-proα1(I)/α2(I)
The observation of FP-HSP47ΔRDEL and FP-HSP47RNGL but not FP-HSP47 in procollagen transport vesicles was consistent with HSP47 retention in the ER. If true, this interpretation would also mean that vesicle-like structures containing both procollagen and HSP47 were either regions of the ER or autophagic structures derived from the ER, as suggested by their stochastic, ER-like dynamics.
To validate this deduction, we co-transfected the cells with ER and autophagy markers (Fig. 6, Movie 8). Some vesicle-like puncta containing FP-proα1(I)/α2(I) and FP-HSP47 were indeed colocalized with the ER lumen marker ssFP-KDEL (Fig. 6A, Movie 8) and surrounded by ER membrane (Supp. Fig. 1). Almost instantaneous recovery of ssFP-KDEL fluorescence after photobleaching in these puncta indicated that they were vesicle-like regions of ER lumen connected to the ER network rather than actual vesicles (Fig. 6A, Movie 8). Slower fluorescence recovery of FP-HSP47 and FP-procollagen was consistent with larger size and therefore slower diffusion of these molecules (Fig. 7C). Other procollagen/HSP47 puncta were autophagic structures colocalized with and/or surrounded by the lysosome and late endosome membrane marker FP-LAMP1 (Fig. 6B, Supp. Fig. 4), which could be formed by lysosomal engulfment of ER exit sites (ERES) [37], lysosomal engulfment of ER lumen [44], or ER-phagy [45]. Correlative light and electron microscopy supported the existence of ERESs engulfed by lysosomal membranes (Supp. Fig. 4 and Fig. 7 in Ref. [37]) as well as vesicle-like ER lumen regions containing procollagen (Supp. Fig. 4). These structures were not procollagen transport vesicles, yet static snapshots of such structures could be misinterpreted as transport vesicles.
Fig. 6, Movie 8. Vesicle-like structures containing procollagen and HSP47 inside ER lumen and lysosomal membranes.
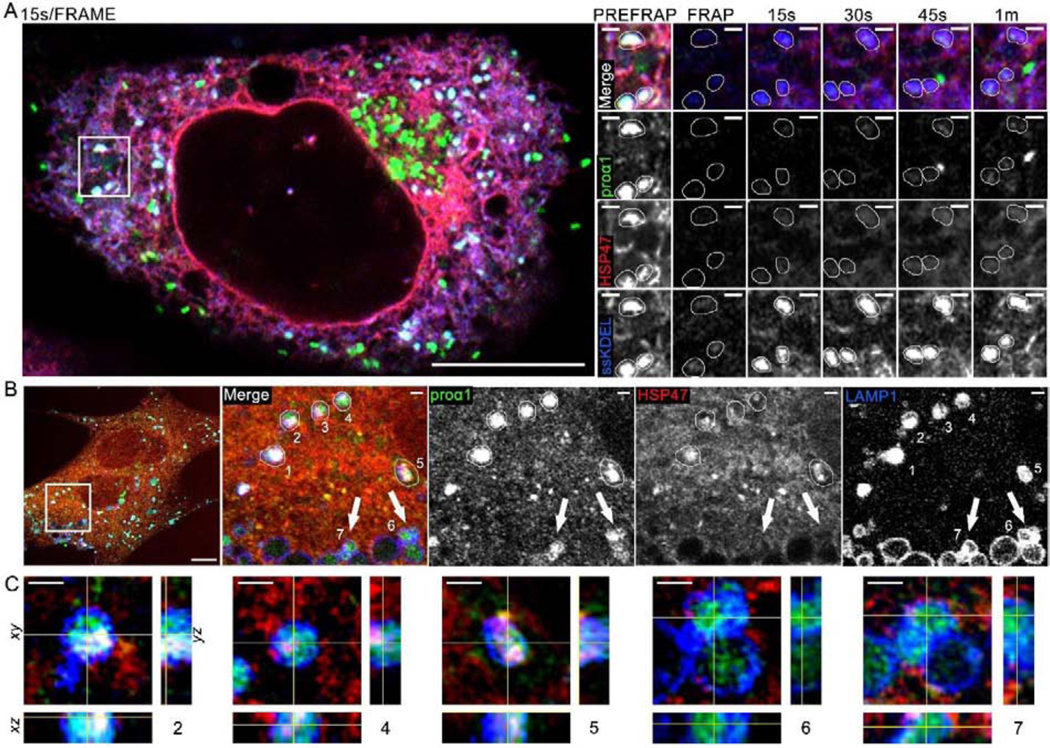
(A, Movie 8) Airyscan single slice frames and time-lapse video of colocalization of Venus-proα1(I), Cherry-HSP47, and ssCFP-KDEL in vesicle-like structures inside ER lumen and fluorescence recovery of these markers after photobleaching (FRAP); N=18 (3 experiments). Zoomed still frames show the same area as the inset in Movie 8. In these zoomed frames, 3 vesicle-like structures marked by Venus-proα1(I), Cherry-HSP47, and ssCFP-KDEL are traced by thin white lines. One of these structures is also highlighted by a white circle in the movie inset. The bright green structure in 45 s and 1 min frames is likely a procollagen transport vesicle containing no Cherry-HSP47 and no ssCFP-KDEL, which entered the field of view between 30 and 45 s post-bleaching (the 15s/frame imaging rate in this experiment was not fast enough for more definitive identification). Quantitative analysis of FRAP kinetics in shown in Fig. 7C with and without BFA treatment. (B, C) 3D Airyscan imaging of Venus-proα1(I) and Cherry-HSP47 colocalization in lysosomes/late endosomes marked by membrane protein LAMP1-CFP; N=7. (B) A slice from the 3D z-stack shows whole cell and zoomed images of vesicle-like structures 1–7, in which Venus-proα1(I) is colocalized with LAMP1-CFP. (C) Orthogonal cross-sections of structures 2, 4, 5, 6, and 7. Structures 2, 4, and 5 were likely formed by direct lysosomal engulfment of ERES [37] or ER lumen [44], since they are surrounded by LAMP1-positive lysosomal membrane and contain internalized procollagen, HSP47, and LAMP1. Lysosomes 6 and 7 contain little or no internalized HSP47 and thereby might have a different origin, e.g. endocytosis of secreted procollagen molecules. Scale bars = 10 μm (whole cell) and = 1 μm (zoom).
Fig. 7, Movie 9. Golgi disruption by brefeldin A (BFA) causes accumulation of vesicle-like regions of ER lumen filled with procollagen and HSP47, which were previously misinterpreted as procollagen transport vesicles.
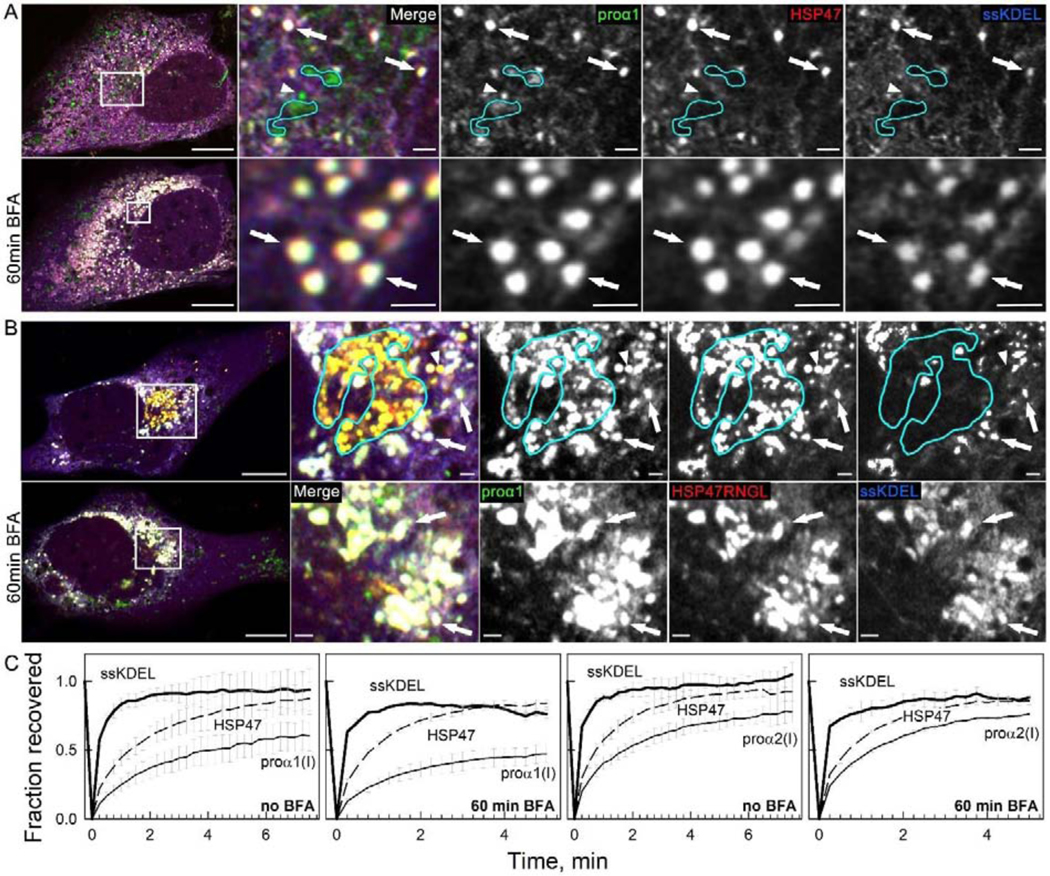
(A) Colocalization of Venus-proα1(I), Cherry-HSP47, and ssCFP-KDEL in vesicle-like structures inside ER lumen (white puncta in merged images marked by arrows) and accumulation of these structures after 60 min treatment of the same cell with 5 μg/ml BFA (bottom panels); N=9. Golgi cisternae (outlined by cyan lines) and likely transport vesicle (arrowhead) contain Venus-proα1(I) but not Cherry-HSP47 or ssCFP-KDEL. They disappear rather than accumulate after BFA treatment. (Movie 9) Time-lapse video of similar Venus-proα1(I)/Cherry-HSP47/ssCFP-KDEL puncta in a different cell after BFA treatment shows nearly instantaneous ssCFP-KDEL fluorescence recovery after photobleaching, demonstrating that these puncta are integrated within the ER lumen network. (B) Similar accumulation of Venus-proα1(I), Cherry-HSP47RNGL, and ssCFP-KDEL in vesicle-like white puncta (arrows) inside ER lumen after 60 min treatment with 5 μg/ml BFA; N=8. Golgi cisternae (outlined in cyan) and likely transport vesicle (arrowhead) contain Venus-proα1(I) and Cherry-HSP47RNGL but not ssCFP-KDEL. They disappear after BFA treatment. (C) FRAP kinetics in vesicle-like structures containing colocalized ssCFP-KDEL, Venus-proα1(I)/proα2(I), and Cherry-HSP47; N=3. The images in (A,B) are Airyscan single slices; scale bars = 10 μm (whole cell) and = 1 μm (zoom).
We next revisited a frequently cited observation of accumulation of vesicle-like procollagen/HSP47 structures after Golgi disruption by brefeldin A (BFA), which was interpreted as accumulation of transport vesicles that had nowhere to go [35]. We similarly observed accumulation of spherical, vesicle-like puncta containing FP-HSP47 and FP-proα1(I)/α2(I) after 1 h treatment with 5 μg/ml BFA (Fig. 7A, Supp. Fig. 5). However, these puncta were colocalized with ssFP-KDEL. Nearly instantaneous recovery of ssFP-KDEL fluorescence after their photobleaching revealed that they were vesicle-like ER regions rather than transport vesicles (Fig. 7C, Movie 9). Instead of accumulation, BFA caused disappearance of transport vesicles (Fig. 7A). Consistently, in cells expressing FP-HSP47RNGL, BFA caused accumulation of vesicle-like ER regions containing ssFP-KDEL and disappearance of Golgi and transport vesicles containing just FP-proα1(I)/α2(I) and FP-HSP47RNGL (Fig. 7B).
In addition to HSP47 retention in the ER, these effects suggested dependence of procollagen export from the ER on activation of ARF1 GTPase by GDP→GTP exchange, since BFA is an inhibitor of this exchange [46, 47]. Given that GDP→GTP exchange activates membrane attachment of ARF1 and COPI coatomer recruitment, we examined ARF1 localization in cells co-transfected with FP-ARF1, FP-proα2(I), and FP-SEC23 (Supp. Fig. 6). Outside of the Golgi area, we found numerous punctate ARF1 structures, ~ 50±8% of which were colocalized with procollagen and most of which appeared to be associated with ERESs.
FP-HSP47 and FP-proα1(I)/α2(I) co-localization with ERGIC53
How could we reconcile the evidence pointing to HSP47 retention in the ER with the preferential HSP47 binding to natively folded procollagen in the ER? One hint is the putative role of HSP47 as an adapter for procollagen loading into the ERES by TANGO1 [32], which suggests that HSP47 might be loaded into ERESs as well. Indeed, colocalization of FP-HSP47, FP-proα1(I)/α2(I), and COPII coat marker FP-SEC23 in punctate structures supported the entry of HSP47 into ERESs together with procollagen (Fig. 8A, Movie 10). Subsequent formation of transport intermediates at ERES could then be preceded by HSP47 release and retrieval back to the ER (enabled by distinct ERES environment).
Fig. 8, Movies 10–12. HSP47 entry into ERES and release at ERES regions marked by ERGIC53.
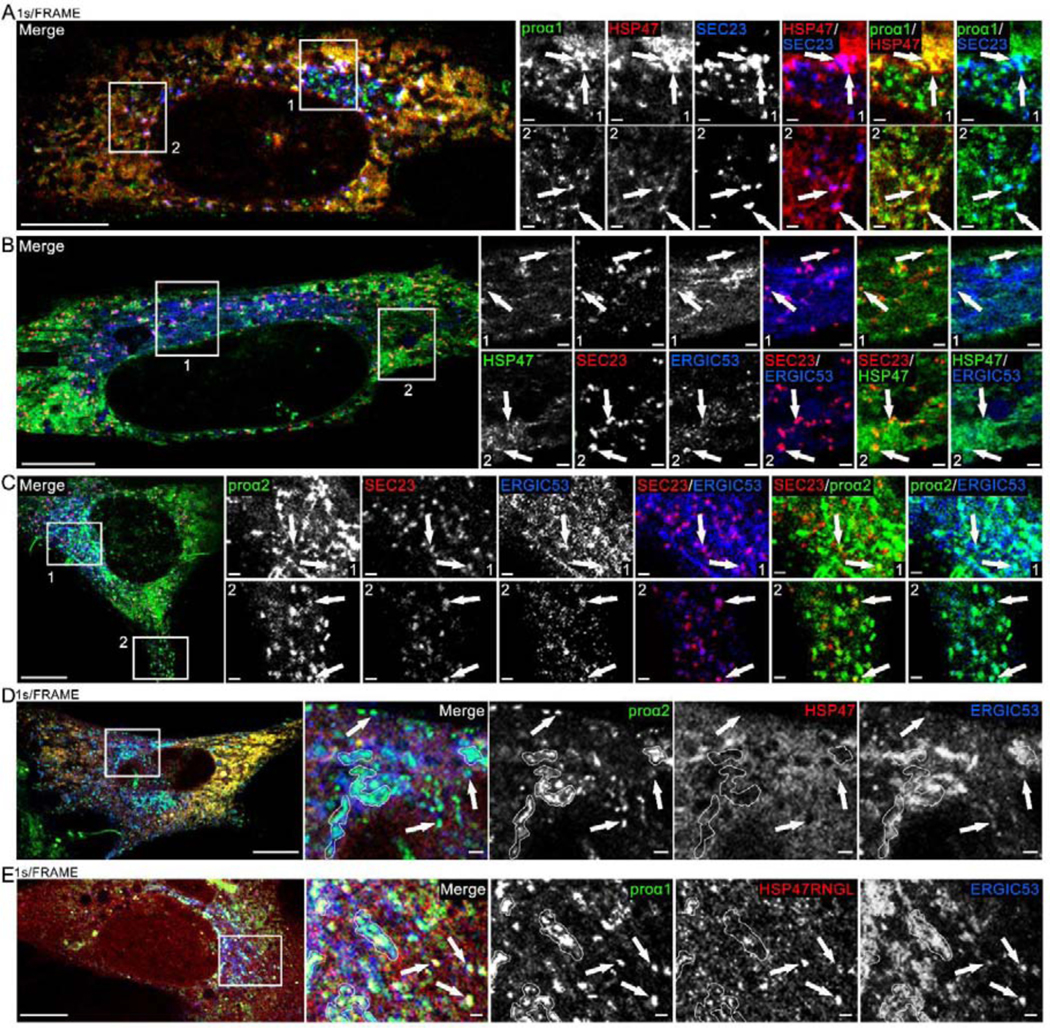
(A, Movie 10) Airyscan single slice frame and time-lapse video of GFP-proα1(I), Cherry-HSP47, and TagBFP2-SEC23 colocalization in punctate structures representing ERES (arrows) in the Golgi region (Movie inset and zoomed still frame in top panels) and away from the Golgi region (zoomed still frame in bottom panels). Based on larger size and slower motion, procollagen carrier appearing at 00:28 s frame in the movie inset is more likely a secretory vesicle than ER-Golgi transport intermediate. N=20 (3 experiments). (B) Airyscan slice of similar colocalization of Venus-HSP47, Halo-SEC23 and Cerulean-ERGIC53 at ERES (arrows); N=6. (C) Airyscan slice of similar colocalization of Venus-proα2(I), Halo-SEC23, and Cerulean-ERGIC53 at ERES (arrows); N=22 (5 experiments). (D, Movie 11) Airyscan single slice frame and time-lapse video of Venus-proα2(I) and Cerulean-ERGIC53 colocalization without Cherry-HSP47 in procollagen transport vesicles (arrows) and Golgi (white outlines); N=4. (E, Movie 12) Airyscan single slice frame and time-lapse video of Venus-proα1(I), Cherry-HSP47RNGL, and Cerulean-ERGIC53 colocalization in procollagen transport vesicles (arrows) and Golgi (white outlines); N=17 (3 experiments). Scale bars = 10 μm (whole cell) and = 1 μm (zoom).
To test the latter hypothesis, we created, validated, and imaged the intracellular localization and dynamics of a fluorescently tagged membrane marker FP-ERGIC53 (Supp. Fig. 2A). This membrane protein is known to cycle between the ER and Golgi in transport intermediates believed to have reduced pH [48]. Live cell imaging showed FP-ERGIC53 was colocalized with procollagen in the area of cis-Golgi, at ERESs (including ones located far from the Golgi, Fig. 8B,C), and in procollagen transport intermediates destined for Golgi that exhibited rapid directional motion (Fig. 8D, Movie 11, Supp. Fig. 7A). FP-ERGIC53 labeling identified ER-Golgi transport intermediates that could not be traced to their origin or destination, distinguishing them from secretory vesicles. FP-HSP47 was detected in fewer than 1±3% of these intermediates and only in cells with higher FP-HSP47 expression (Supp. Fig. 7C). In contrast, FP-HSP47RNGL was detected in over 90±12% of procollagen transport intermediates regardless of FP-HSP47RNGL expression level (Fig. 8E, Movie 12, Supp. Fig. 7B). Therefore, HSP47 appeared to be only leaking rather than co-transported with procollagen to Golgi.
Discussion
Because HSP47 was shown to be a collagen-specific ER chaperone and its mutations were found to cause severe osteogenesis imperfecta with pronounced bone fragility and deformities, its interactions with procollagen have been extensively studied. Multiple observations have suggested co-trafficking of procollagen with bound HSP47, release of HSP47 at reduced pH in cis-Golgi (or ERGIC), and return to the ER by retrograde transport [25, 26].
We tested this hypothesis by imaging trafficking of fluorescently tagged type I procollagen and HSP47 in live MC3T3 osteoblasts with up to ~120 nm spatial and 500 ms/frame time resolution. Unlike previous snapshots of fixed cells, time lapse imaging of live cells unequivocally distinguished procollagen transport intermediates from other vesicle-like punctate structures based on their directional vs. stochastic motion. Co-transfection of the cells with fluorescently tagged protein markers of different subcellular compartments, FRAP experiments, and tracking the origin and destination of transport intermediates revealed the nature and composition of different vesicle-like structures. Many post-Golgi procollagen carriers appeared to be larger and slower than ER-Golgi transport intermediates. Nonetheless, to avoid misidentification, we examined the composition of only those procollagen transport intermediates that could be traced to their origin, destination, or both. To quantify HSP47 content, we identified ER-Golgi procollagen carriers based on the presence of FP-ERGIC53 (observed in most ER-Golgi transport intermediates; Fig. 8, Supp. Fig. 7, Movies 11, 12, S2, S3). Multiple control experiments revealed no artifacts or off-target effects of low-to-moderate expression of the FP-tagged constructs in MC3T3 cells within 24 h of transfection.
Our observations suggest that HSP47 is released from procollagen before rather than after procollagen transport intermediate fission from ERESs (Fig. 9) and that the RDEL sequence in HSP47 prevents the protein from being exported from the ER in such intermediates. The bona fide transport intermediates contained only procollagen and ERGIC53, emerged from ERESs, exhibited characteristic directional motion consistent with movement along microtubules, and merged into the cis-Golgi (Figs. 3 and 8A–C; Movies 1–5, 10, and 11). Their composition and dynamics were consistent with the previously described ERGIC-like carriers transporting proteins to the cis-Golgi [49, 50]. With very few exceptions (< 1±3%), we observed no detectable HSP47 in these or any other (Fig. 4, Supp. Fig. 3, Movies 1–5) procollagen transport intermediates in the secretory pathway, unless the ER localization sequence RDEL of HSP47 was either deleted or mutated to RNGL (Figs. 5, 8E; Supp. Fig. 7B; Movies 6,7, and 12). We observed colocalization of procollagen, HSP47, and COPII proteins in a variety of vesicle-like structures, some of which had similar appearance to structures interpreted by other studies as procollagen transport vesicles in various human cell lines [6, 51]. In MC3T3 cells, all these structures exhibited slow, limited, stochastic motion and none of them merged into the cis-Golgi (Fig. 8A and Movie 10). These structures were either LAMP1-negative ER exit sites (Fig. 8A), larger dilated ER regions (some decorated with COPII-coated ERES), or LAMP1-positive autophagic structures (Fig. 6B), as illustrated in Fig. 9. We previously reported that all procollagen/COPII puncta that also contained ubiquitin, CUL3, or KLHL12 were positive for LC3, LAMP1, or both, indicating that they were degradative autophagic structures rather than secretory transport vesicles [37]. In this earlier study we found no evidence of directionally moving procollagen transport vesicles destined for Golgi that were marked by COPII, CUL3, or KLHL12.
Fig. 9. Model of HSP47 and procollagen sorting at ER exit sites (ERES).
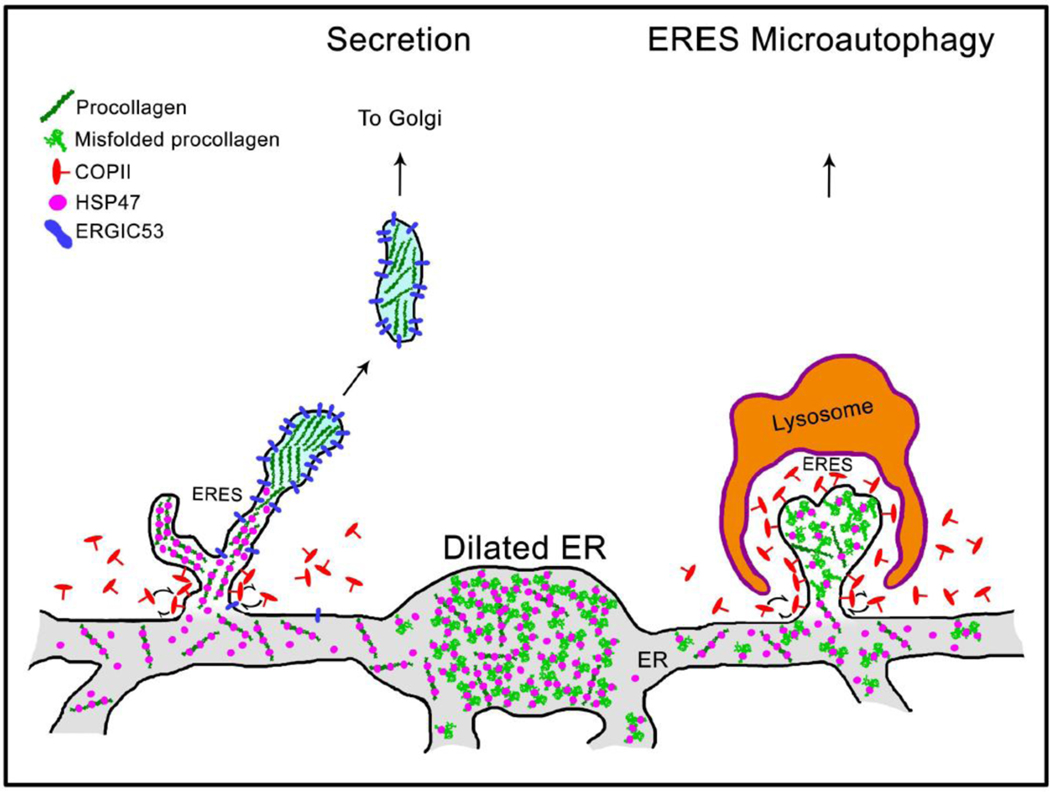
Grey structure at the bottom represents interconnected network of rough ER cisternae. Procollagen is loaded into ERES together with HSP47, as indicated by colocalization of procollagen, HSP47, and SEC23 (Fig. 8A, Movie 10). Maturation of procollagen transport intermediate precursors at distal ERES is accompanied by HSP47 release and accumulation of ERGIC53, since both ERGIC53 and HSP47 colocalize with procollagen at ERES and only ERGIC53 colocalizes with procollagen in Golgi-bound transport intermediates (Fig. 8, Movies 10–12). Unless its RDEL sequence is deleted or mutated, HSP47 is found in < 1±3% ERGIC53/procollagen transport intermediates, indicating that HSP47 is primarily returned to the ER from ERES (Supp. Fig. 7C). Only few escaping HSP47 molecules are likely to be captured by cis-Golgi KDEL/RDEL receptors and returned to the ER by retrograde trafficking. As reported before, ERESs containing misfolded procollagen and HSP47 might be engulfed by lysosomes and degraded (ERES microautophagy pathway) [37]. Under a microscope, procollagen ERESs (left), dilated ER regions (middle), and ERES microautophagy intermediates (right) appear as either small puncta or larger vesicle-like structures containing procollagen and HSP47 (Figs. 6 and 7, see also Figs. 1–7 in Ref. [37]). They accumulate upon inhibition of procollagen export from the ER by brefeldin A and are easy to confuse with transport vesicles, unless their motion and/or composition is imaged.
We have also found the export of procollagen transport intermediates from the ER to be ARF1 GTPase dependent. In immunofluorescence studies, vesicle-like procollagen/HSP47 [35, 36] or TANGO1/HSP47 [6] structures formed after SAR1 and ARF1 inhibition with GTPγS [36], ARF1 inhibition with brefeldin A (BFA) [35], COPI knockdown [6], or ARF1 Q71L transfection [6] were interpreted as transport intermediates containing HSP47. We reproduced the accumulation of such structures after BFA treatment (Fig. 7), which prevents GDP→GTP exchange at ARF1 and thereby ARF1 localization at membranes and COPI coatomer recruitment [52–54]. However, analysis of their composition, dynamics, and FRAP suggested that BFA blocked the secretory export of procollagen and HSP47 from the ER and that these structures were not transport intermediates but mostly dilated ER regions plus some ERESs and lysosomes (Figs. 6 and 7, Movies 8 and 9). At the same time, we previously found that BFA did not impede procollagen delivery from the ER to lysosomes for degradation (Fig. S3 in Ref. [37]). In the absence of BFA, we observed ARF1 colocalization with procollagen within elongated structures adjacent to COPII at ERES, which are consistent with transport intermediate precursors (Supp. Fig. 6). From quantitative analysis of N=9 cells in 3 such experiments with different FP combinations, we estimate that ~ 50±8 % of punctate ARF1 structures outside Golgi contain procollagen.
Taken together, these observations suggest that HSP47 release is accompanied by ARF1 and COPI dependent formation of procollagen transport intermediates at distal ERES regions (Fig. 9). This hypothesis is consistent with recent reports of COPII and TANGO1 rings at the ER-ERES interface [55, 56], suggesting that only procollagen entry into the ERES but not its exit might be COPII and TANGO1 dependent [1]. It provides an alternative to ERGIC intermediates believed to form from COPII vesicles that bud from ERESs and lose their COPII coats [10, 57], consistent with a recent observation of long tubular ERES membranes interacting with COPI proteins [58]. Another possible interpretation of ARF1-dependent procollagen transport intermediates that contain ERGIC53 but not HSP47 is that these intermediates form upon fusion of ERGIC membranes with ERES [7]. Our findings are consistent with the latter study, yet we propose a somewhat different point of view. We do see ARF1 structures that do not contain procollagen adjacent to SEC23 at ERES (Supp. Fig. 6). They might be arriving retrograde transport intermediates [59, 60] or ERGIC membranes fusing with ERES [7]. We cannot distinguish the two interpretations and we are not sure that there is a substantive difference between them. None of the transport intermediates we tracked in this study stopped over at a distinct ERGIC compartment between ERES and cis-Golgi and none appeared to change their size or intensity, consistent with previously reported VSVG trafficking [61, 62]. Therefore, we lean toward viewing transport intermediates as having all the attributes typically associated with ERGIC [61, 62]. However, our data can be interpreted in either way.
Some of the questions raised by our study will have to be answered by future work that might require additional development of live cell imaging and other technologies. Indeed, it is presently unclear whether HSP47 dissociation from procollagen at ERES is mediated by reduced pH or other features of the ERES environment. We also do not know whether the procollagen transport intermediates we observed are rapidly moving vesicles like the megacarriers proposed by Santos et al [7], short tubular connections between the cis-Golgi and proximal ER like the short-loop connections proposed by McCaughey et al [9], long tubular regions of ERES membranes proposed by Weigel et al. [58], or all of the above. The existing technology might not be able to clearly answer these questions. On one hand, the time resolution required for visualizing highly dynamic transport intermediates necessitates imaging a single z-slice, in which tubular intermediates could be easily confused with transport vesicles (given that procollagen flow could occur in discrete bursts). On the other hand, the spatial resolution required for visualizing such structures in 3D necessitates imaging of fixed/frozen specimens, in which the dynamics can be only deduced but not shown. Nonetheless, we believe that a consistent picture of secretory procollagen trafficking is beginning to emerge.
Overall, our observations provide the following mechanistic insights into ER-Golgi procollagen trafficking and the role of HSP47 in this process. Type I procollagen and HSP47 appear to be separated at ERESs followed by ARF1-dependent formation of Golgi-destined procollagen transport intermediates that contain ERGIC53 but no COPII coat and no HSP47. Perhaps the release of HSP47 enables entry of procollagen from ERES into transport intermediates, like the release of general ER chaperones enables entry of folded proteins from the ER lumen into ERES. This hypothesis is consistent with HSP47 retention at ERES by RDEL receptors and degradative targeting of procollagen together with HSP47 to lysosomes from ERESs. However, it has not been validated yet and its experimental testing is ongoing, e.g. the identity of the RDEL receptors for HSP47 and their relationship to cis-Golgi KDEL/RDEL receptors are presently unknown. In MC3T3 cells, HSP47 is exported from ERESs into the degradative but not secretory pathway, although some molecules likely escape this ERES sorting process and travel to the cis-Golgi, particularly under conditions of increased HSP47 expression or cell stress. Like general ER chaperones escaping from the ER lumen, the latter HSP47 molecules would then be captured by cis-Golgi KDEL/RDEL receptors and returned to the ER by retrograde transport. In the absence of cell stress or RDEL mutations, HSP47 leakage to cis-Golgi was detectable only in <1±3% of procollagen transport intermediates in a few cells with higher levels of FP-HSP47 expression. We do not know whether cells other than MC3T3 have a different mechanism for HSP47 release and procollagen export from ERES and whether other collagen types are trafficked similar to type I procollagen. Osteoblasts could be utilizing Golgi for HSP47 retrieval only as a backup (to capture molecules missed by the ERES sorting), because they produce more procollagen than other cells. However, from an evolutionary perspective, the ERES procollagen/HSP47 sorting and export mechanisms are unlikely to be collagen type or cell specific.
Experimental procedures
Cell lines and primary cell culture
MC3T3-E1 Subclone 4 osteoblastic cell lines, acquired from ATCC (ATCC CRL-2594), were cultured in αMEM + Glutamax (GIBCO 32571–036) and supplemented with 10% fetal bovine serum (FBS, Sigma Aldrich) and 1% Penn/Strep (Corning). To stimulate procollagen synthesis and secretion, 100 μM ascorbic acid 2-phospate (Sigma Aldrich) was supplemented during transfection, 12–24 h before imaging.
Constructs.
Constructs for fluorescently tagged proteins FP-proα2(I), FP-proα1(I), GM130-FP, Ii33-FP, LAMP1-FP, and FP-SEC23 and ssFP-KDEL have been previously described in [37]. Additional validation of LAMP1-FP and FP-SEC23 markers is illustrated in Supp. Fig. 2. ER lumen marker ssFP-KDEL constructs (FPs with N-terminal signal sequence (ss) for ER targeting and translocation plus C-terminal KDEL for ER retention) were modified from mEmerald-ER-3, a gift from Michael Davidson, Florida State University, Tallahassee, FL (Addgene 54082). The FP-HSP47 constructs were generated by placing the FP cDNA after the ER signal sequence, before the first amino acid of HSP47. In FP-HSP47ΔRDEL and FP-HSP47RNGL, the C-terminal RDEL sequence was either deleted (ΔRDEL) or mutated to RNGL using Quickchange-XL site-directed mutagenesis kit (Agilent). The fluorescently tagged FP-ERGIC53 constructs were modified from pMXs-IP spGFP-ERGIC53 (a gift from Noburu Mizushima, Addgene plasmid # 38270). For generation of these constructs, the following plasmids encoding fluorescent proteins (FPs) were utilized: eGFP-N1/C1 (GFP), mCherry-N1/C1 (Cherry), Halo-N1/C1 (Halo), mApple-N1/C1 (Apple), mVenus-N1/C1 (Venus), mCerulean-N1/C1 (Cerulean), mTagBFP2-N1/C1 (TagBFP2). Janelia Fluor Dye 646 was utilized for marking Halo-tagged molecules as described in [63, 64].
GFP or Venus-tagged FP-proα2(I) and FP-proα1(I) displayed a low transfection efficiency but were otherwise normally trafficked through the cell and secreted. Specifically, low-to-moderate expression of FP-procollagen appeared to have minimal or no impact on procollagen synthesis, localization inside the cell, trafficking, or cell function within 24 h of transfection. Over-transfected cells with high expression of FP-procollagen appeared to accumulate large procollagen pools within the ER lumen, causing severe ER dilation (Supp. Fig. 1). In cells with low-to-moderate expression of FP-procollagen, such procollagen pools usually did not accumulate during the first 24 h after transfection. Therefore, imaging experiments were performed within 12–24 h after transfection only with the latter cells. TagBFP2, Cerulean or Apple-procollagen constructs had similar features but lower transfection efficiencies. Cherry-procollagen was not used for this study, because it exhibited abnormal localization patterns inside the cell (likely caused by Cherry dimerization).
Transfection and treatments.
MC3T3 cells were transiently transfected with Fugene 6 (Promega) using the manufacturer’s protocols. The DNA:Fugene ratio was optimized for MC3T3 cells, as was the total DNA:Fugene concentration in the cell culture media. Due to rapid MC3T3 proliferation, the cell density at the time of transfection was optimized as well, with 50–60% confluence being an ideal timepoint for 12–24 h transfection without cells becoming overconfluent. During transfection, cells were incubated with αMEM + Glutamax media supplemented with 100 μM ascorbic acid 2-phoshate (Sigma Aldrich) and 10% FBS from Valley Biomedical (Lot #2C0550 tested for supporting osteoblast differentiation). Cells were then imaged 12–24 h after the transfection. Brefeldin A (Cell Signaling) treatment was performed by adding an equal volume of 2x solution (in the same media with ascorbic acid 2-phoshate) to chamber slides at the timepoints indicated in the text, resulting in 5 μg/ml final concentration.
Immunofluorescence
Cells were fixed for 10–15 min in freshly prepared methanol-free 2% formaldehyde (Thermo Fisher Scientific) solution in phosphate buffered saline, pH 7.4 (PBS), washed in PBS, permeabilized in 0.4% Triton X in PBS for 10min, and returned to PBS. After 30 min blocking in 3% BSA in PBS, cells were incubated overnight at 4 °C with primary antibody diluted in the same blocking buffer, then washed, and incubated for 30–60 min at room temperature with secondary Alexa-Fluor-labeled antibodies (Thermo Fisher Scientific) diluted with 1.5% BSA in PBS. After the final PBS wash, cells were either imaged immediately or mounted with Prolong Diamond Antifade with DAPI (Thermo Fisher Scientific) for subsequent imaging. The following primary antibodies were utilized (1:100–1:200): anti-HSP47 - Enzo LifeSciences (M16.10A1); anti-LMAN1 (ERGIC53) – Abcam (ab125006); anti-SEC31 – BD BioSciences (612351).
Imaging and image processing.
Live or fixed cell imaging and correlative light and electron microscopy (CLEM) were performed as previously described in [37]. Live cell imaging was performed using temperature-controlled incubation stage set to maintain 37°C and 5% CO2. Photobleaching experiments were performed with 10–30% laser power, with 405, 488, 514, or 561 nm lasers, as needed. Except for FRAP data, all time lapse sequences and videos were corrected for photobleaching using a Bleach Correction plugin with histogram matching within the FIJI image processing package [65]. Images were enhanced using linear brightness and contrast controls without any additional modification.
Immunoprecipitation
Immunoprecipitation of GFP-HSP47 was performed using anti-GFP bead system (μMACS, Miltenyi Biotec) according to manufacturer’s protocols with 25–30 μl magnetic beads per 1ml lysate. Degassed lysis buffer containing 150 mM NaCl, 50 mM Tris, 1% NP-40, 5 mM EDTA, protease inhibitors (5mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 5 mM benzamidine, 10 mM N-ethylmaleimide), pH 7.5 was used instead of the manufacturer-supplied lysis buffer. All steps were performed at room temperature rather than 4 °C, to prevent gelation of incompletely folded procollagen. Western Blots were performed as previously described in [34, 53] with the following antibodies: anti-HSP47, 1:2000, Enzo LifeSciences (M16.10A1); anti-proα1(I), 1:1000 (LF-42, a gift from L. Fisher, Kerafast ENH017-FP); anti-GFP, 1:1000 – 1:2000 (Alexa Fluor 647 Conjugate), Invitrogen (A-31852); anti-Vinculin, 1:1000, Sigma (V284).
Supplementary Material
Highlights.
120 nm, 500 ms/frame imaging of live osteoblasts identifies ER-Golgi procollagen carriers
ER-Golgi procollagen carriers contain ERGIC protein ERGIC53 but not HSP47
HSP47 is released from procollagen at ER exit sites rather than ERGIC or cis-Golgi
Procollagen carrier formation requires GDP to GTP exchange at ARF1 GTPase
Acknowledgements
We thank Michael Davidson, Noboru Mizushima and David Stephens for sharing their constructs; and Daniel Feliciano, Vivek Malhotra, Edward Mertz, Lynn Mirigian, Chris Obara, Carolyn Ott, Ishier Raote, Anna Roberts-Pilgrim, Antonio Santos, Prasanna Satpute-Krishnan, Aubrey Weigel, Chad Williamson, and particularly Prabuddha Sengupta, for plasmids, assistance, discussions, and advice.
Financial Support
This work was funded in part by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Abbreviations used
- ER
Endoplasmic Reticulum
- ERES
ER exit site
- ERGIC
ER Golgi Intermediate Compartment
- BFA
Brefeldin A
- FP
fluorescent protein
- FRAP
Fluorescence Recovery After Photobleaching
Footnotes
Declaration of Competing Interests
The authors have no competing interests.
Open access article is available from the Publisher: http://dx.doi.org/10.1016/j.matbio.2020.06.002
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Raote I, Malhotra V, Protein transport by vesicles and tunnels, J Cell Biol 218(3) (2019) 737–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].McCaughey J, Stephens DJ, ER-to-Golgi Transport: A Sizeable Problem, Trends Cell Biol 29(12) (2019) 940–953. [DOI] [PubMed] [Google Scholar]
- [3].Hutchings J, Zanetti G, Coat flexibility in the secretory pathway: a role in transport of bulky cargoes, Curr Opin Cell Biol 59 (2019) 104–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].McGourty CA, Akopian D, Walsh C, Gorur A, Werner A, Schekman R, Bautista D, Rape M, Regulation of the CUL3 Ubiquitin Ligase by a Calcium-Dependent Co-adaptor, Cell 167(2) (2016) 525–538 e14. [DOI] [PubMed] [Google Scholar]
- [5].Jin L, Pahuja KB, Wickliffe KE, Gorur A, Baumgartel C, Schekman R, Rape M, Ubiquitin-dependent regulation of COPII coat size and function, Nature 482(7386) (2012) 495–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yuan L, Kenny SJ, Hemmati J, Xu K, Schekman R, TANGO1 and SEC12 are copackaged with procollagen I to facilitate the generation of large COPII carriers, Proc Natl Acad Sci U S A 115(52) (2018) E12255-E12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Santos AJ, Raote I, Scarpa M, Brouwers N, Malhotra V, TANGO1 recruits ERGIC membranes to the endoplasmic reticulum for procollagen export, Elife 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nogueira C, Erlmann P, Villeneuve J, Santos AJ, Martinez-Alonso E, Martinez-Menarguez JA, Malhotra V, SLY1 and Syntaxin 18 specify a distinct pathway for procollagen VII export from the endoplasmic reticulum, Elife 3 (2014) e02784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].McCaughey J, Stevenson NL, Cross S, Stephens DJ, ER-to-Golgi trafficking of procollagen in the absence of large carriers, J Cell Biol 218(3) (2019) 929–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Saraste J, Marie M, Intermediate compartment (IC): from pre-Golgi vacuoles to a semi-autonomous membrane system, Histochem Cell Biol 150(5) (2018) 407–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Takeyari S, et al. , Japanese patient with Cole-carpenter syndrome with compound heterozygous variants of SEC24D, Am J Med Genet A 176(12) (2018) 2882–2886. [DOI] [PubMed] [Google Scholar]
- [12].Zhang H, Yue H, Wang C, Gu J, He J, Fu W, Hu W, Zhang Z, Novel mutations in the SEC24D gene in Chinese families with autosomal recessive osteogenesis imperfecta, Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA 28(4) (2017) 1473–1480. [DOI] [PubMed] [Google Scholar]
- [13].Garbes L, et al. , Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta, Am J Hum Genet 96(3) (2015) 432–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Boyadjiev SA, Kim SD, Hata A, Haldeman-Englert C, Zackai EH, Naydenov C, Hamamoto S, Schekman RW, Kim J, Cranio-lenticulo-sutural dysplasia associated with defects in collagen secretion, Clin Genet 80(2) (2011) 169–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fromme JC, Ravazzola M, Hamamoto S, Al-Balwi M, Eyaid W, Boyadjiev SA, Cosson P, Schekman R, Orci L, The genetic basis of a craniofacial disease provides insight into COPII coat assembly, Dev Cell 13(5) (2007) 623–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lang MR, Lapierre LA, Frotscher M, Goldenring JR, Knapik EW, Secretory COPII coat component Sec23a is essential for craniofacial chondrocyte maturation, Nat Genet 38(10) (2006) 1198–203. [DOI] [PubMed] [Google Scholar]
- [17].Boyadjiev SA, et al. , Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking, Nat Genet 38(10) (2006) 1192–7. [DOI] [PubMed] [Google Scholar]
- [18].Lindahl K, et al. , Homozygosity for CREB3L1 premature stop codon in first case of recessive osteogenesis imperfecta associated with OASIS-deficiency to survive infancy, Bone 114 (2018) 268–277. [DOI] [PubMed] [Google Scholar]
- [19].Keller RB, Tran TT, Pyott SM, Pepin MG, Savarirayan R, McGillivray G, Nickerson DA, Bamshad MJ, Byers PH, Monoallelic and biallelic CREB3L1 variant causes mild and severe osteogenesis imperfecta, respectively, Genet Med 20(4) (2018) 411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Guillemyn B, Kayserili H, Demuynck L, Sips P, De Paepe A, Syx D, Coucke PJ, Malfait F, Symoens S, A homozygous pathogenic missense variant broadens the phenotypic and mutational spectrum of CREB3L1-related osteogenesis imperfecta, Hum Mol Genet 28(11) (2019) 1801–1809. [DOI] [PubMed] [Google Scholar]
- [21].Lekszas C, et al. , Biallelic TANGO1 mutations cause a novel syndromal disease due to hampered cellular collagen secretion, Elife 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Iozzo RV, Gubbiotti MA, Extracellular matrix: The driving force of mammalian diseases, Matrix Biol 71–72 (2018) 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Karamanos NK, Theocharis AD, Neill T, Iozzo RV, Matrix modeling and remodeling: A biological interplay regulating tissue homeostasis and diseases, Matrix Biol 75–76 (2019) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ishikawa Y, Bachinger HP, A molecular ensemble in the rER for procollagen maturation, Biochim Biophys Acta 1833(11) (2013) 2479–91. [DOI] [PubMed] [Google Scholar]
- [25].Ito S, Nagata K, Biology of Hsp47 (Serpin H1), a collagen-specific molecular chaperone, Semin Cell Dev Biol 62 (2017) 142–151. [DOI] [PubMed] [Google Scholar]
- [26].Ito S, Nagata K, Roles of the endoplasmic reticulum-resident, collagen-specific molecular chaperone Hsp47 in vertebrate cells and human disease, J Biol Chem 294(6) (2019) 2133–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Braakman I, Bulleid NJ, Protein folding and modification in the mammalian endoplasmic reticulum, Annu Rev Biochem 80 (2011) 71–99. [DOI] [PubMed] [Google Scholar]
- [28].Fujii KK, Taga Y, Sakai T, Ito S, Hattori S, Nagata K, Koide T, Lowering the culture temperature corrects collagen abnormalities caused by HSP47 gene knockout, Sci Rep 9(1) (2019) 17433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Makareeva E, Leikin S, Procollagen triple helix assembly: an unconventional chaperone-assisted folding paradigm, PLoS One 2(10) (2007) e1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Koide T, et al. , Specific recognition of the collagen triple helix by chaperone HSP47. II. The HSP47-binding structural motif in collagens and related proteins, J Biol Chem 281(16) (2006) 11177–85. [DOI] [PubMed] [Google Scholar]
- [31].Koide T, Asada S, Takahara Y, Nishikawa Y, Nagata K, Kitagawa K, Specific recognition of the collagen triple helix by chaperone HSP47: minimal structural requirement and spatial molecular orientation, J Biol Chem 281(6) (2006) 3432–8. [DOI] [PubMed] [Google Scholar]
- [32].Ishikawa Y, Ito S, Nagata K, Sakai LY, Bachinger HP, Intracellular mechanisms of molecular recognition and sorting for transport of large extracellular matrix molecules, Proc Natl Acad Sci U S A 113(41) (2016) E6036–E6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Oecal S, Socher E, Uthoff M, Ernst C, Zaucke F, Sticht H, Baumann U, Gebauer JM, The pH-dependent Client Release from the Collagen-specific Chaperone HSP47 Is Triggered by a Tandem Histidine Pair, J Biol Chem 291(24) (2016) 12612–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Saga S, Nagata K, Chen WT, Yamada KM, pH-dependent function, purification, and intracellular location of a major collagen-binding glycoprotein, J Cell Biol 105(1) (1987) 517–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Satoh M, Hirayoshi K, Yokota S, Hosokawa N, Nagata K, Intracellular interaction of collagen-specific stress protein HSP47 with newly synthesized procollagen, J Cell Biol 133(2) (1996) 469–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Smith T, Ferreira LR, Hebert C, Norris K, Sauk JJ, Hsp47 and cyclophilin B traverse the endoplasmic reticulum with procollagen into pre-Golgi intermediate vesicles. A role for Hsp47 and cyclophilin B in the export of procollagen from the endoplasmic reticulum, J Biol Chem 270(31) (1995) 18323–8. [DOI] [PubMed] [Google Scholar]
- [37].Omari S, Makareeva E, Roberts-Pilgrim A, Mirigian L, Jarnik M, Ott C, Lippincott-Schwartz J, Leikin S, Noncanonical autophagy at ER exit sites regulates procollagen turnover, Proc Natl Acad Sci U S A 115(43) (2018) E10099-E10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sudo H, Kodama HA, Amagai Y, Yamamoto S, Kasai S, In vitro differentiation and calcification in a new clonal osteogenic cell line derived from newborn mouse calvaria, J Cell Biol 96(1) (1983) 191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Schwarze U, Hata RI, McKusick VA, Shinkai H, Hoyme HE, Pyeritz RE, Byers PH, Rare autosomal recessive cardiac valvular form of Ehlers-Danlos syndrome results from mutations in the COL1A2 gene that activate the nonsense-mediated RNA decay pathway, Am. J. Hum. Genet 74(5) (2004) 917–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Malfait F, Symoens S, Coucke P, Nunes L, De Almeida S, De Paepe A, Total absence of the alpha 2(I) chain of collagen type I causes a rare form of Ehlers-Danlos syndrome with hypermobility and propensity to cardiac valvular problems, J. Med. Genet 43(7) (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rebane AA, Ziltener P, LaMonica LC, Bauer AH, Zheng H, Lopez-Montero I, Pincet F, Rothman JE, Ernst AM, Liquid-liquid phase separation of the Golgi matrix protein GM130, FEBS Lett 594(7) (2020) 1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Christiansen HE, et al. , Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta, Am J Hum Genet 86(3) (2010) 389–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Schwarze U, Cundy T, Liu YJ, Hofman PL, Byers PH, Compound heterozygosity for a frameshift mutation and an upstream deletion that reduces expression of SERPINH1 in siblings with a moderate form of osteogenesis imperfecta, Am J Med Genet A 179(8) (2019) 1466–1475. [DOI] [PubMed] [Google Scholar]
- [44].Schafer JA, et al. , ESCRT machinery mediates selective microautophagy of endoplasmic reticulum in yeast, EMBO J 39(2) (2020) e102586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].De Leonibus C, Cinque L, Settembre C, Emerging lysosomal pathways for quality control at the endoplasmic reticulum, FEBS Lett 593(17) (2019) 2319–2329. [DOI] [PubMed] [Google Scholar]
- [46].Hsu VW, Lee SY, Yang JS, The evolving understanding of COPI vesicle formation, Nat Rev Mol Cell Biol 10(5) (2009) 360–4. [DOI] [PubMed] [Google Scholar]
- [47].Hsu VW, Yang JS, Mechanisms of COPI vesicle formation, FEBS Lett 583(23) (2009) 3758–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Appenzeller-Herzog C, Hauri HP, The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function, J Cell Sci 119(Pt 11) (2006) 2173–83. [DOI] [PubMed] [Google Scholar]
- [49].Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J, ER-to-Golgi transport visualized in living cells, Nature 389(6646) (1997) 81–5. [DOI] [PubMed] [Google Scholar]
- [50].Scales SJ, Pepperkok R, Kreis TE, Visualization of ER-to-Golgi transport in living cells reveals a sequential mode of action for COPII and COPI, Cell 90(6) (1997) 1137–48. [DOI] [PubMed] [Google Scholar]
- [51].Gorur A, Yuan L, Kenny SJ, Baba S, Xu K, Schekman R, COPII-coated membranes function as transport carriers of intracellular procollagen I, J Cell Biol 216(6) (2017) 1745–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Arakel EC, Schwappach B, Formation of COPI-coated vesicles at a glance, J Cell Sci 131(5) (2018). [DOI] [PubMed] [Google Scholar]
- [53].Jackson LP, Structure and mechanism of COPI vesicle biogenesis, Curr Opin Cell Biol 29 (2014) 67–73. [DOI] [PubMed] [Google Scholar]
- [54].Spang A, The life cycle of a transport vesicle, Cell Mol Life Sci 65(18) (2008) 2781–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Reynolds HM, Zhang L, Tran DT, Ten Hagen KG, Tango1 coordinates the formation of endoplasmic reticulum/Golgi docking sites to mediate secretory granule formation, J Biol Chem 294(51) (2019) 19498–19510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Raote I, Ortega-Bellido M, Santos AJ, Foresti O, Zhang C, Garcia-Parajo MF, Campelo F, Malhotra V, TANGO1 builds a machine for collagen export by recruiting and spatially organizing COPII, tethers and membranes, Elife 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Brandizzi F, Barlowe C, Organization of the ER-Golgi interface for membrane traffic control, Nat Rev Mol Cell Biol 14(6) (2013) 382–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Weigel A, et al. , COPI and COPII Cooperate At ER Exit Sites to Support ER-to-Golgi Protein Trafficking Revealed by 3D Ultrastructure Analyses and Live Cell Imaging, Mol. Biol. Cell 30 (2019) M42 (abstract#). [Google Scholar]
- [59].Roy Chowdhury S, Bhattacharjee C, Casler JC, Jain BK, Glick BS, Bhattacharyya D, ER arrival sites associate with ER exit sites to create bidirectional transport portals, J Cell Biol 219(4) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Schroter S, Beckmann S, Schmitt HD, ER arrival sites for COPI vesicles localize to hotspots of membrane trafficking, EMBO J 35(17) (2016) 1935–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Lippincott-Schwartz J, The secretory membrane system studied in real-time. Robert Feulgen Prize Lecture, 2001, Histochem Cell Biol 116(2) (2001) 97–107. [DOI] [PubMed] [Google Scholar]
- [62].Lippincott-Schwartz J, Roberts TH, Hirschberg K, Secretory protein trafficking and organelle dynamics in living cells, Annu Rev Cell Dev Biol 16 (2000) 557–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Grimm JB, et al. , A general method to improve fluorophores for live-cell and single-molecule microscopy, Nat Methods 12(3) (2015) 244–50, 3 p following 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Grimm JB, Brown TA, English BP, Lionnet T, Lavis LD, Synthesis of Janelia Fluor HaloTag and SNAP-Tag Ligands and Their Use in Cellular Imaging Experiments, Methods Mol Biol 1663 (2017) 179–188. [DOI] [PubMed] [Google Scholar]
- [65].Miura K, Rueden C, Hiner M, Schindelin J, Rietdorf J, ImageJ Plugin CorrectBleach V2.0.2, 2014. https://zenodo.org/record/30769#.XmI_j6hKiUl.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.