

Abstract
Phylogenomics via ultraconserved elements (UCEs) has led to improved phylogenetic reconstructions across the tree of life. However, inadvertently incorporating non‐targeted DNA into the UCE marker design will lead to misinformation being incorporated into subsequent analyses. To date, the effectiveness of basic metagenomic filtering strategies has not been assessed in arthropods. Designing markers from museum specimens requires careful consideration of methods due to the high levels of microbial contamination typically found in such specimens. We investigate if contaminant sequences are carried forward into a UCE marker set we developed from insect museum specimens using a standard bioinformatics pipeline. We find that the methods currently employed by most researchers do not exclude contamination from the final set of targets. Lastly, we highlight several paths forward for reducing contamination in UCE marker design.
Keywords: contamination, insects, museum specimens, ultraconserved elements
We investigate if genomic bacterial and laboratory contamination is introduced into ultraconserved element (UCE) probe design from five novel Coleoptera museum specimens. Our findings indicate that even very clean samples have contamination that is carried into the UCE probe design. These results indicate that metagenomic filtering should be a consideration in UCE probe design.

1. INTRODUCTION
Phylogenomic methods, including improvements to both sequencing and analytical techniques, have facilitated the resolution of long‐recalcitrant phylogenetic relationships across the tree of life (Blaimer et al., 2015; Brady et al., 2015; Brewer & Bond, 2013; Faircloth, 2016, 2017; Haddad et al., 2018; Lemmon et al., 2012; Locke et al., 2018; McCormack et al., 2012, 2016; McKenna et al., 2019; Misof et al., 2014; Van Dam et al., 2017). Ultraconserved elements (UCEs), conserved orthologous nuclear protein‐coding genes, and other phylogenomic markers have traditionally been developed from a wide variety of starting material, including fresh tissue for DNA or RNA marker development, ethanol‐preserved tissues, and museum specimens more than 100‐years‐old (Blaimer et al., 2016; Branstetter et al., 2017; Brewer & Bond, 2013; Derkarabetian et al., 2019; Locke et al., 2018; McGuire et al., 2018; Shin et al., 2018; Van Dam et al., 2017). Even formalin‐fixed specimens have shown promise for use in phylogenomic analyses (Hykin et al., 2015; Peacock et al., 2017; Ruane & Austin, 2017). It is generally assumed that using either newer tissues or published genomes, the amount of contamination from microbes and parasites will be so small as to be inconsequential and will therefore have little or no negative impact on phylogenomic marker development. Consequently, when freshly sourced tissue or genomes from NCBI are used, no significant metagenomic filtering is generally carried out (Faircloth, 2017; Gustafson et al., 2019).
However, such contaminants are common in older specimens, laboratory reagents, and the environment. Since phylogenetic analyses are likely to be affected by microbial contaminants, it is unsettling that this issue is not typically addressed in phylogenomic studies (Glassing et al., 2016; Hadfield & Eldridge, 2014; Sangiovanni et al., 2019). For larger taxa, it may be possible to simply use a single tissue type not known to host symbiotic microbes as the source of DNA (Łukasik et al., 2017; Seutin et al., 1991), but in smaller, soft‐bodied organisms, this may not be possible (e.g., Acari, micro‐Hymenoptera, Coccoidea, Nematoda, protists). Additionally, some taxa, including many insects, contain symbionts whose bacteriome may be nearly impossible to physically remove (McKenna, 2020).
Although filtering of metagenomic contaminants from UCE bait‐capture experiments has been performed (Bossert & Danforth, 2018), this has only been done after the target loci have been designed. Moreover, simply using a more stringent target cutoff will not remove baits that have inadvertently been designed from contaminants. Coverage‐based approaches work well with clean starting material, but when samples are old, coverage tends to be lower or uneven (Blaimer et al., 2016; McCormack et al., 2016; Van Dam et al., 2017). Other authors have developed pipelines to harvest UCE’s from exons (Van Dam et al., 2021), or simply blast scaffolds to exon accessions on NCBI. While both of these approaches are likely to reduce bacterial contamination, they will not reduce contamination from microbial eukaryotes in the tissue, and worse still, they rely on the assumption that loci containing exons retrieved by blast to the database of choice are not underlying mis‐annotations contributed by algorithm‐based gene annotation programs, as opposed to experimentally proven annotations. Presently, no standard metagenomic filtering strategy has been proposed as part of the UCE marker development workflow. This can be problematic when designing UCE markers from museum specimens or organisms whose tissues are routinely contaminated by symbionts or other organisms from the same environment.
Recent metagenomic filtering methods, such as canopy clustering and machine learning, rely on a reference database and multiple samples to create metagenomic bins (Alneberg et al., 2014; Eren et al., 2015; Nielsen et al., 2014; Nissen et al., 2018). While these methods are ideal, they are cost‐prohibitive for UCE marker pilot studies because they require many tens of specimens to deliver enough statistical power (Kang et al., 2015; Nielsen et al., 2014). Instead, three main strategies of metagenomic filtering are most commonly used with single specimen assemblies. These are (1) Metagenomic filtering of small specimens where symbionts are present. This method includes aligning reads to a reference genome of a model organism, for example, nematodes to Caenorhabditis elegans, or in ancient DNA studies on humans and many other animals, aligning reads back to the reference genome (Kumar et al., 2013; Rasmussen et al., 2015); (2) Examining nucleotide base composition bias, along with scaffold coverage, can also help cluster scaffolds (Kumar et al., 2013; Teeling et al., 2004); (3) Binning of scaffolds by taxonomic proximity and presence of exons in the hope that results provide the best guess as to taxonomic identity (Huson et al., 2018; Wood et al., 2019).
Binning scaffolds into taxonomic groups by genetic distance, base composition, and coverage should also have some merit for work with museum specimens; however, the degree of relatedness between non‐model organisms and their model anchor is a limiting factor. Even eukaryotic model organisms require metagenomic filtering to remove bacterial contamination (Fierst & Murdock, 2017). Throughout this paper, we refer to museum specimens as those specimens that are long preserved, poorly preserved, or dried and otherwise not from single tissue sources stored at or below −80°C. Because contamination may even exist in sequences from a reference genome (e.g., Koutsovoulos et al., 2016), the genomes derived from museum specimens of non‐model organisms will almost certainly require some metagenomic filtering. This is critical for phylogenomic marker development to prevent contamination from being represented in the probe set. To circumvent this problem, systematists, particularly those that study arthropods, typically try to anchor non‐model groups to a “clean” reference genome that is carefully extracted from fresh muscle tissue (Faircloth, 2017). However, “clean” material may not always be available in rare or small‐bodied arthropod groups. Thus, there is a need to establish best practices for developing phylogenomic markers from tissues that may be contaminated.
We explored the taxonomic composition of low‐coverage genome assemblies from insect museum specimens to produce a UCE probe set for the jewel weevils, tribe Pachyrhynchini (Coleoptera: Curculionidae: Entiminae). We then used these data to assess how metagenomic filtering affected UCE probe design. UCE alignments were constructed to determine whether anchoring UCEs to a base draft genome obtained from muscle tissue, as is standard practice, significantly reduces or eliminates contamination in the resulting probe design. If it does, the resulting UCEs should only contain endogenous insect DNA with few to no scaffolds originating from contaminant taxa. We describe our methods and findings below to investigate contamination levels resulting from using a clean draft genome to anchor museum specimens to develop a custom UCE marker set.
2. MATERIALS AND METHODS
2.1. Taxon sampling
Pachyrhynchini weevils are a charismatic group of beetles containing more than 155 species, with their center of diversity in the Philippines and Australasia (Rukmane, 2018). They are large and flightless, have aposematic coloration, and are ideal for investigating the evolution of island‐endemism and biogeography of the Philippines’ endemic fauna because of the large number of closely related species showing intriguing patterns of genetic and morphological divergence and (in contrast) convergent morphological evolution in allopatry and parapatry (Wang et al., 2018). While there are other phylogenomic markers for beetles (Faircloth, 2017; Haddad et al., 2018; Johnson et al., 2018; Shin et al., 2018), a phylogenomic marker set specifically tailored for optimal loci capture in the Pachyrhynchini does not yet exist. Here, we use three weevil species from the Philippines, one from Papua New Guinea (PNG), and a fourth species as an outgroup. The outgroup, Diaprepes abbreviatus, is an entimine weevil native to the Caribbean island of Puerto Rico. It is an economically important pest of a variety of fruit trees in the Caribbean region and the southern United States (Simpson et al., 1996). The addition of this outgroup taxon will ensure that the marker set is universal across the ingroup (Faircloth, 2017), and will likely also ensure utility across many other weevils in the extremely large subfamily Entiminae (more than 12,000 described species).
2.2. Collection localities
Oribius sp.: Papua New Guinea, Mt. Wilhelm, −5.714639 145.275 1680 m 7–8‐XI‐2014 leg. M Van Dam: Pachyrhynchus spp. and Metapocyrtus sp.: Makiling Forest Reserve Laguna, Calamba, Laguna, Philippines 2011. Pachyrhynchini samples were initially preserved in 95% EtOH upon collection in 2011, stored at room temperature, then extracted in 2017. The Oribius sp. Marshall, 1956 (placed in the tribe Celeuthetini) sample was preserved in 95% EtOH in 2014 and kept at ambient temperature while in the field for 5 weeks, and then preserved at −20°C until DNA extraction in 2017. Diaprepes abbreviatus (DDM2014024) was obtained by DDM from a laboratory research colony maintained by Dr. S. L. Lapointe in 2013. USDA/ARS, Horticultural Research Laboratory. 2001 South Rock Road. December 2013.
2.3. DNA extraction
For all DNA extractions only one sample was used to build downstream libraries and sequencing. Except for D. abbreviatus, DNA was extracted using DNeasy Tissue kits (Qiagen; Macherey‐Nagel) following the methods of Van Dam et al. (2017). Briefly, using sterilized forceps (soaked in 10% bleach and flame sterilized between use). Muscle tissue was removed from the pronotum, mesothorax, and abdomen and placed in a 1.5–2.0 ml centrifuge tube for tissue lysis and extraction (Van Dam et al., 2017). A single adult D. abbreviatus (DDM2938) was placed live in cold RNAlater and stored at −80 °C until extraction. Total genomic DNA was extracted from the prothorax and one hindleg using the G‐Biosciences Omniprep gDNA extraction kit/protocol and treated with RNAse A before genomic DNA library preparation following the methods of Shin et al. (2018).
2.4. Illumina library preparation and sequencing
2.4.1. Diaprepes abbreviates
Following visualization on a 1× agarose gel, samples were sonicated to a size of approximately 400–700 bp on a Covaris M220 ultrasonicator (Covaris). Dual‐indexed shotgun genomic libraries were prepared by the University of Illinois Roy J. Carver Biotechnology Center/W.M. Keck Center (Champaign‐Urbana, Illinois) using the Hyper Library construction kit from Kapa Biosystems following manufacturer's instructions. All libraries were constructed using six cycles of PCR and size selected for fragments 400 to 500 bp in length. Paired‐end (PE) sequencing was conducted on a MiSeq v2 (250 bp PE reads) at the University of Illinois and using a HiSeq 2500 (150 bp PE reads) at Florida State University (Tallahassee, FL). Paired‐end SRA reads in FASTQ format were trimmed to remove low‐quality bases and adapter sequences and used as the input file for de novo assembly using the CLC Genomics Workbench version 4.9 (www.qiagenbioinformatics.com/products/clc‐genomics‐workbench/). Statistical parameters were maintained at their defaults.
2.4.2. Other taxa
Following visualization on a 1× agarose gel, samples were sonicated to a size of approximately 400 bp on a Covaris M220 ultrasonicator (Covaris). Library preparation procedures followed Van Dam et al. (2017). Sequencing was performed via an Illumina HiSeq4000 at GeneWiz Next Generation Sequencing Center (South Plainfield, New Jersey).
2.4.3. Illumina data quality filtering
Data were first inspected via a fastqc (Andrews, 2010) quality report. Data were then quality filtered with Trimmomatic v. 0.36 (Bolger et al., 2014) using the settings “PE: ILLUMINACLIP: TruSeq3‐PE.fa:2:30:10:2:keepBothReads LEADING:3 TRAILING:3 SLIDINGWINDOW:4:20 MINLEN:36.” A summary of the quality‐filtered reads, organized by sample, can be found in Table S1.
2.4.4. Nanopore sequencing
Because the Oribius sp. sample was the most recently preserved, an attempt was made to add additional long reads to improve the assembly. This sample was not fragmented and went directly to library construction following the protocols in Oxford Nanopore Technologies (ONT) 1D PCR barcoding genomic DNA (SQK‐LSK108) for version R9 chemistry to construct the libraries. We followed the ONT protocol for the SpotOn Flow Cell version R9 chemistry (ONT cat No. FLO‐MIN 107 R9). The Library Loading Bead kit (ONT cat No. EXP‐LLB001) was used to help load samples onto the flow cell. The flow cell was loaded onto an ONT MinION sequencer and ran for 48 hours using ONT MinKNOW software.
2.4.5. Nanopore data quality filtering
We used the albacore basecaller v2.1.3 (Oxford Nanopore Technologies, 2017a) to convert the fast5 data to FASTQ format (Oxford Nanopore Technologies, 2017b). Quality filtering was executed in NanoFilt 2.6.0 as part of NanoPack (De Coster et al., 2018). Read length, Phred quality scores, and other summary statistics were calculated using Pauvre (Schultz, 2018) and NanoPlot. A total of 171,816 and 87,639 reads met albacore quality standards on two flow cells. These data were combined with the Illumina data (Table S1) to generate a hybrid assembly. The ONT reads were then used in the gap closer process with the SPAdes assembler (Bankevich et al., 2012).
2.4.6. Draft genome assembly
Kmergenie (Chikhi & Medvedev, 2014) was used to find the optimal k‐mer size for assembly. The resulting optimal k‐mer size was then added to k values of 21,33,55 and 77 in the SPAdes‐3.11.1 (Bankevich et al., 2012) genome assembly pipeline. BUSCO v1.22 (Seppey et al., 2019; Simão et al., 2015) was used to assess draft genome completeness of capturing conserved single‐copy genes using the Arthropoda Odb10 database. For summary statistics on assembly quality bbmap stat.sh (Bushnell, 2015) script was used to assess the quality of draft genome assemblies.
2.5. Metagenomic clustering strategy to explore sources of contamination
2.5.1. Metagenomic filtering
Metagenomic filtering from a single library per sample necessitates using methods relying on scaffold coverage cutoff, GC content, and annotating the scaffolds via blast. The Blobology pipeline used here employs read coverage, blast+ (Kent, 2012) to the nt database for taxonomic annotation (blast v.2.7.1, nt database March 2019), and GC content to inform manual metagenomic clustering of assembly taxonomic annotation (Kumar et al., 2013). Blast+ settings were blastn megablast with an e‐value of 1e‐5 cutoff. Reads that passed the quality trimming step from Trimmomatic were aligned to their respective genome using the mem algorithm in bwa version 0.7.3a, with default settings used to get an estimate of contig coverage and generate coverage plots (Li & Durbin, 2009). Plots produced via Blobology (Kumar et al., 2013) or BlobTools (Laetsch & Blaxter, 2017) assisted with annotation. This plotting method has proven successful in a wide variety of Eukaryotic genome sequencing projects for exploring potential sources of contamination and host‐symbiont relationships (Dentinger et al., 2016; Husnik & McCutcheon, 2016; Szitenberg et al., 2017). The blob graphs allow for the visualization of the relative degree of contamination in each draft genome, where scaffolds clustered based on taxonomy, coverage, and GC content (Figure 2).
FIGURE 2.
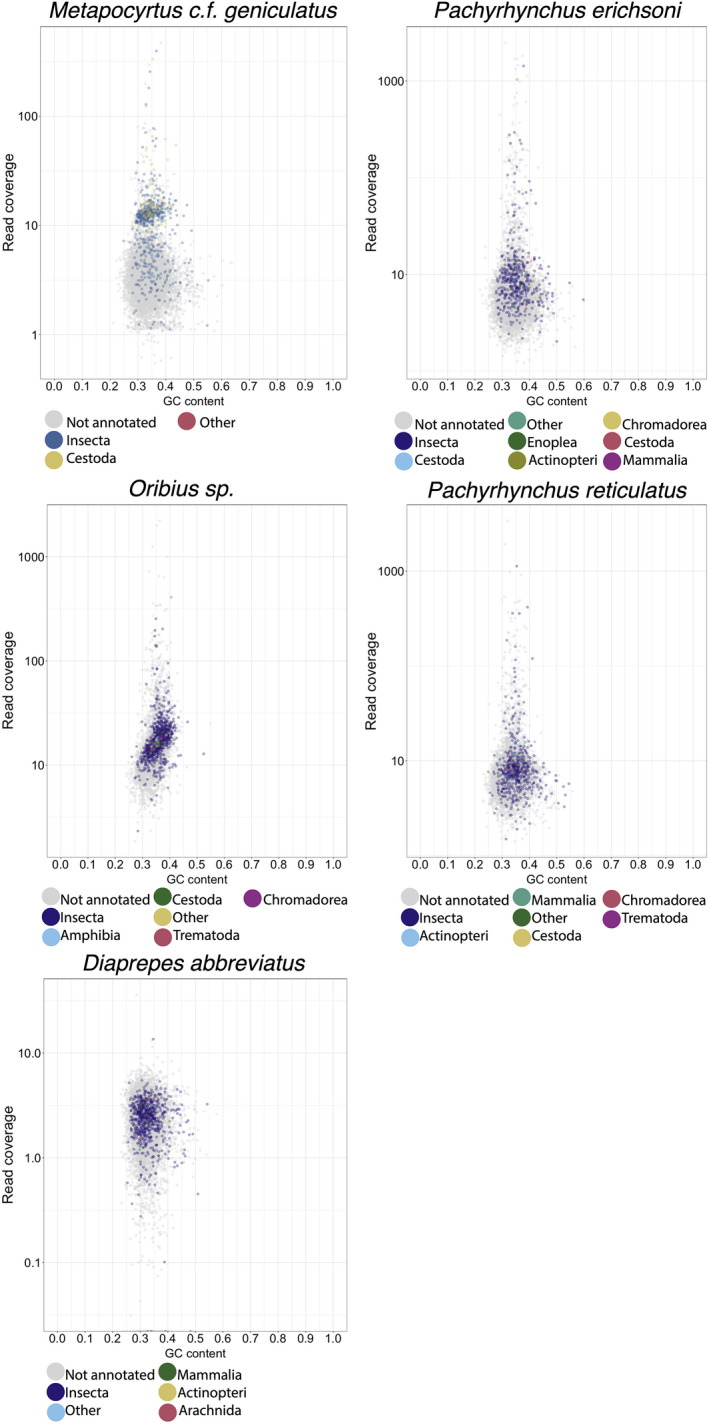
Blob plots of UCE‐bearing scaffolds from the master UCE probe set. The Y‐axis is read coverage via the bwa‐mem algorithm, and the X‐axis is the GC content of individual scaffolds. Color is coded by taxonomic class
2.5.2. UCE marker development
We used the PHYLUCE v 1.5.0 pipeline (Faircloth, 2016) to develop UCEs from our five weevil genome assemblies. We used the D. abbreviatus draft genome as the base genome for this analysis. The D. abbreviatus genome was repeat masked using RepeatMasker 4.0.6 (Smit et al., 2015). A UCE combined probe length of 160 bp was used for probe development. UCE probes were culled to have an ≥80% match across the ingroup taxa. Duplicated probes mapped via Lastz (Harris, 2007) to multiple regions in their host genomes were removed (Faircloth, 2016). This resulted in 9217 UCEs and 86,079 master UCE probes. Probes were then mapped back to all five draft genomes via PHYLUCE and Lastz (Harris, 2007). The final data matrix was composed of genomic regions matching the master‐UCE probes with 80% coverage and 80% match across the five taxa with 500 bp flanking regions.
2.5.3. UCE taxonomic annotation and metagenomic binning
The taxonomic mapping from the initial blast+ mapping was used to provide an initial putative taxonomic assignment to UCE‐based scaffolds (Figure 2). Blatq (Henderson, 2018; Kent, 2002, 2012) was used to rapidly align the UCEs to each genome (Figure 2). This initial taxonomic annotation revealed many scaffolds that remained unannotated.
2.5.4. UCE probe parent scaffold annotation
Briefly, three methods were chosen as binning strategies for UCE parent scaffolds using this custom database (1) blast+ alignment‐based mapping (2) Kraken2 k‐mer‐based annotation (3) Lastz nucleotide similarity‐based cutoff mapping. In more detail, the UCE loci's parent scaffolds were identified via the Lastz PHYLUCE results.
Using the three methods described above, scaffolds were assigned a taxonomic identity using a custom reference database. The custom references included the Rice Weevil genome v‐2.0 (Sitophilus oryzae, NCBI RefSeq:13876818) and the Kraken2 (Wood et al., 2019) bacterial and fungal databases, as well as additional representative genomes from likely contaminant clades that were identified by the initial blast+ nt analysis. These clades were Acari, Nematoda, Platyhelminthes, Chordata, and Viridiplantae. A complete list of contaminating taxa used in the custom database is provided in Appendix S1: Table S4.
Using the custom reference set, the UCE‐based scaffolds were binned via Kraken2 using default settings (Wood et al., 2019). Using the Blast v 2.7.1 tool, “blastn megablast” was used to report only the annotation with the highest bit‐score E‐5 cut‐off. Lastz alignment tool was used with default settings and an 85% alignment cut‐off (as in Bossert & Danforth, 2018).
Finally, a linear regression model was used to see if k‐mer coverage was predicted by UCE parent scaffold length and contaminant taxa via R 3.3.3 (Cran, 2010). As short scaffolds are expected to contain more contaminants, the linear regression was used to help visualize contaminants by parent UCE scaffold size and coverage.
3. RESULTS
3.1. Illumina and nanopore sequencing and data quality filtering
After sequencing the Pachyrhynchini species, there were between 109 M and 122 M paired‐end reads (post Trimmomatic quality filtering) for each sample. The Nanopore sequencing produced 171,816 and 87,639 reads with a Q5 quality score or better on the two flow cells. See Appendix S1: Tables S1 and S2 and Figure S1 for a full summary of sequencing results.
3.2. Draft genome assembly
The Pachyrhynchini assemblies contained between 1.5 M and 0.5 M contigs, with an N50 ranging from 370–2189 bp, revealing that the assemblies are generally low quality (see Appendix S1: Table S3 for a full summary). BUSCO scores were also low ranging from 60 to 732 complete single‐copy genes out of 1367 single copy genes from the Arthropoda Odb10 database (see Appendix S1: Figure S2 for full BUSCO score summary). The relatively low quality is acceptable for building UCEs and is expected from museum specimens (Locke et al., 2018; Van Dam et al., 2017, 2019). The D. abbreviatus draft genome was highly fragmented, with 3.8 M contigs and an N50 of 437 (Appendix S1: Table S3), the BUSCO score of 164 was also low (Appendix S1: Figure S2).
3.3. Metagenomic clustering
The Blobology profile results revealed that every sample was heavily contaminated with bacterial contigs (Figure 1). There were two to three major clusters of Bacteria clades in each sample (Figure 1). The most common clades of Bacteria in the samples were as follows: Acidobacteria, Actinobacteria, α‐proteobacteria, β‐proteobacteria, and γ‐proteobacteria. Based on the Blast+ annotation in the Blob plots, it is clear that Blast+ retrieves a variety of spurious annotations (e.g., tropical amphibian lineages; see Figure 1). It is less clear whether some of the other eukaryotic annotations are spurious, such as Acari (mites) and Cestoda (parasitic flatworms) (Figure 1), because both mites and cestodes are common symbionts of beetles (Baumann, 2018; Shostak, 2014). Some scaffolds were annotated to Mammalia and could be spurious annotations and/or laboratory contamination (Figure 1). The D. abbreviates draft genome was quite clean compared to the other four taxa with very few contigs assigned to Bacteria, and no Bacteria contigs forming noticeable blob‐like clusters (Figure 1).
FIGURE 1.
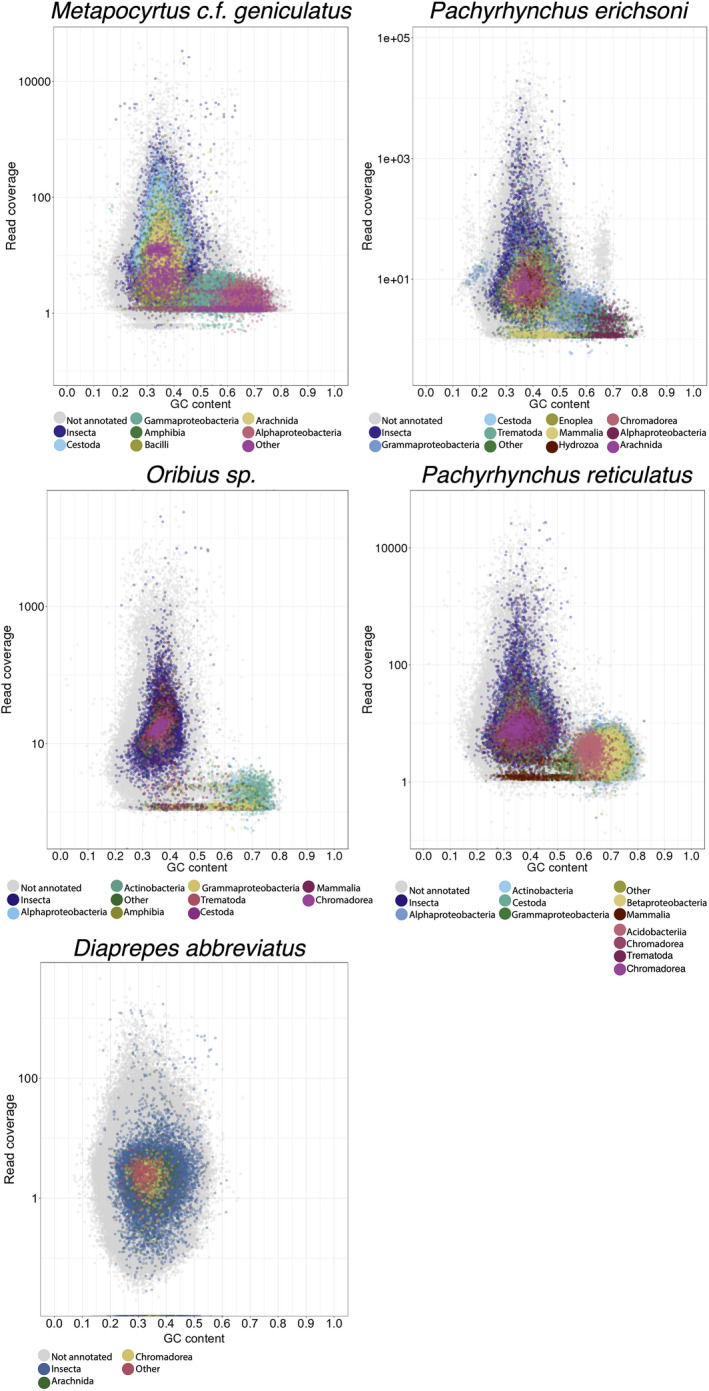
Blob plots of draft genome assembly scaffolds, Y‐axis is read coverage via the bwa‐mem algorithm, X‐axis is GC content of individual scaffolds. Color is coded by taxonomic class
The bacterial contaminants are unlikely to be spurious since these formed distinct blobs that tended to have higher GC content than the insect contigs and less than 100x coverage. There is also a scattering of both insect and contaminant reads with high coverage and lower GC content (Figure 1). Also, there is a significant overlap of low coverage contigs between the eukaryote/insect blob and the bacteria blobs (Figure 1).
3.4. UCE markers and metagenomic UCE loci binning
PHYLUCE produced 9217 UCE loci that were conserved across the five taxa. From this master‐probe list, there were 8688 UCE loci in the final matrix. The count of UCE loci that is comprised of the final matrix by taxon are as follows: 8404 Metapocyrtus (Artapocyrtus) c.f. geniculatus, 8073 Pachyrhynchus erichsoni, 7970 Pachyrhynchus reticulatus, 7780 Oribius sp., and 7732 Diaprepes abbreviatus. The rapid UCE scaffold annotation via blatq revealed that the final UCE dataset consistently mapped to various contaminating sequences, including Bacteria, Acari, and Cestoda, across all five weevil taxa (Figures 2 and 3).
FIGURE 3.
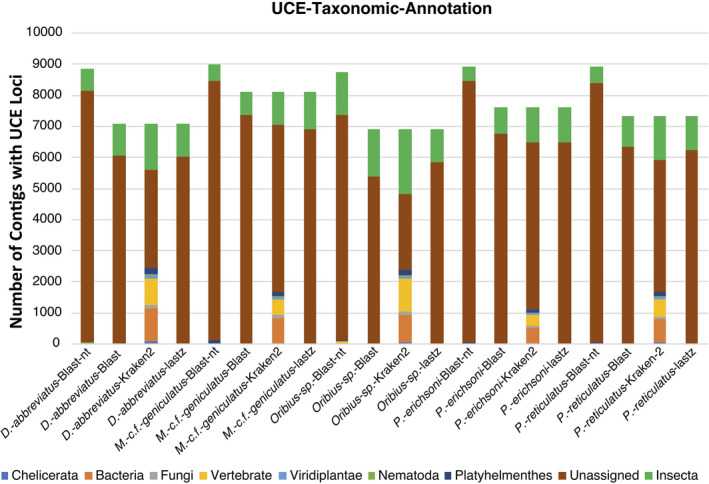
UCE bar chart. Y‐axis is the total number of UCEs, and the X‐axis is the method used to annotate associated species. Color is coded by taxonomic annotation
The metagenomic binning via Kraken2 of the UCE loci in the final data matrix also had significant bacterial contamination (Figure 3). The Kraken2 annotation retrieved >160 UCE loci of Insecta for each of the five species compared to the Blast+ annotation to the same database (Figure 3). The Kraken2 binning revealed that all taxa had significant bacterial contamination (4%–10%; range 341–966 Bacteria UCE contigs Figure 3). The blast+ annotation with both the nt and custom database revealed that the UCE parent scaffolds had bacterial and/or fungal contamination across all five weevil species (see Figure 3). Similar results were obtained via the Kraken2 annotation but with more scaffolds identified as contaminants than Blast+ (Figure 3). While the lastz methods produced a consistent set of UCE scaffolds across the species, many were dubious because they overlapped with contaminant scaffolds using blast+and Kraken2 (Figures 3 and 4).
FIGURE 4.
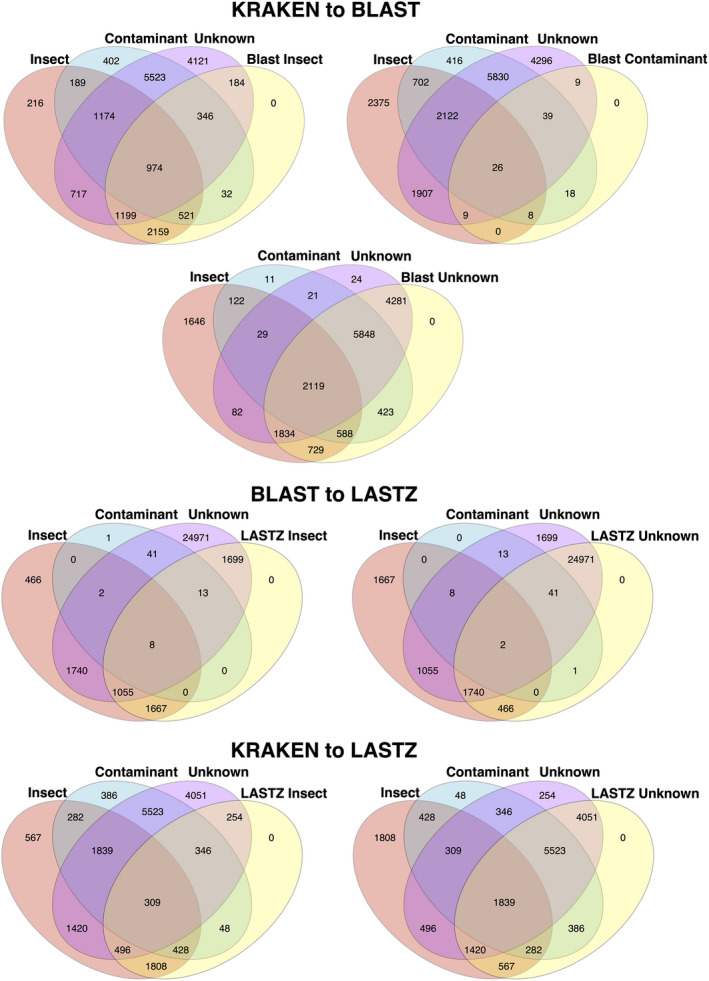
Venn diagrams of UCE bearing scaffolds annotated by taxonomic groups. The groupings are the same ones used in the final data matrix
The linear regression model of UCE parent scaffolds length and k‐mer coverage did not show any significant breaks in coverage demarcating potential contaminants (Figure 5). Scaffolds of both contaminant UCEs and Insecta UCEs had lengths over 10 kb and over 100× coverage (Figure 5), making it impossible to discriminate between insect and non‐insect scaffolds by coverage/length alone. K‐mer coverage and scaffold length were also significantly correlated (p‐value: <2.2e−16).
FIGURE 5.
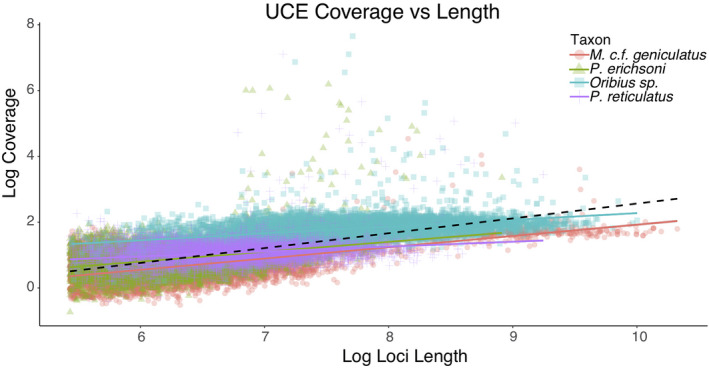
Linear regression of log UCE loci length versus the log of coverage for Pachyrhynchini taxa in the final data matrix
4. DISCUSSION
In all five taxa, <50% of the UCE parent scaffolds had their origins definitively binned by either blast+, lastz, or Kraken2 (Figure 2). We have demonstrated that despite careful extraction to avoid contamination, museum specimens of weevils consistently contain high levels of bacterial and non‐target contamination. This contamination was carried over to the UCE alignment matrix in the form of UCEs that reside in scaffolds of bacterial origin. The level of contamination varies by reference database (nt vs. custom) and the method of metagenomic binning (blast+, Kraken2, lastz). Contamination is consistent across all methods and databases via cross‐checking the methods against each other. The UCE contamination does not appear to be restricted to low coverage (<10×), short scaffolds (~1 kb), but appears to populate long scaffolds (>10 kb) with high coverage (>50×). In fact, many of the contaminant scaffolds that were incorporated into the UCE data matrix had high coverage and were relatively long. An artifact of de Bruijn graph assembly programs is that smaller genomes tend to have higher coverage (Kumar et al., 2013). While coverage may be an excellent method to eliminate low‐level laboratory contamination (Douglass et al., 2019), it is doubtful that contamination in museum specimens would be eliminated by coverage and length alone because of near‐complete coverage‐depth overlap in these specimens (Figures 1, 2 and 5). Simply running blast+ to an exemplar bacterial genome (e.g., Wolbachia or E. coli) or an exemplar eukaryote model organism (e.g., Sitophilus orizae or Drosophila) will produce hundreds to thousands of ambiguous un‐annotated scaffolds. Most of the systematics community works on organisms distantly related to model organisms, suggesting that this metagenomic binning approach will continue to be problematic across taxa. Because of the costly nature of phylogenomic studies, a reduction in loci is not ideal. Most studies that benchmark the accuracy and recall of metagenomic mapping/binning methods fail to consider the genetic distance between non‐model organisms and the data in the NCBI nt and RefSeq databases (Sarmashghi et al., 2019). This problem is manifested here, as the majority of contigs remained unannotated.
4.1. Paths forward for museum‐based insect phylogenomics
With the advent of truly high‐throughput Illumina sequencers (e.g., NovaSeq), the cost of producing draft genomes is greatly reduced, so it is possible to create relatively decent draft genomes and extract loci from a single museum specimen (Cotoras et al., 2017; Derkarabetian et al., 2019; McGuire et al., 2018; McKenna et al., 2019). The lower cost of sequencing will also make it possible to use more statistically robust methods, such as canopy clustering‐based methods, for example, CONCOCT or MetaBAT, assuming you have a close reference genome (Alneberg et al., 2014; Kang et al., 2015). Machine deep learning metagenomic binning methods (e.g., VAMB) should also be explored and could be a way to rescue these types of heterogeneous multi‐taxa data (Nissen et al., 2018). These methods could potentially work for studies entirely composed of museum specimens but remain untested. Another potential path is reference assemblies that incorporate Hi‐C data (Dudchenko et al., 2017). These data types group scaffolds into ordered chromosomes based on their Hi‐C mapping interactions. Consequently, genomes that do not map to the other chromosomes, such as bacteria, can easily be identified and removed.
Another possible way to reduce the chances of contamination is using Anchored Hybrid Enrichment (AHE) probe sets or other exon‐based markers (Bi et al., 2012; Haddad et al., 2018; Lemmon et al., 2012; Shin et al., 2018). With these methods, the initial probe set is anchored to single‐copy nuclear genes, ideally derived from transcriptomes. Anchoring marker sets to poly‐A tailed RNA‐seq data would certainly eliminate most if not all bacterial contamination but may still require screening for microbial eukaryotes. However, these methods typically require an annotated genome and/or transcriptomes for their design (Bi et al., 2012; Lemmon et al., 2012), limiting their use to clades where freshly collected material is available (i.e., not preserved museum specimens). To capture a diverse set of genomic categories, using UCEs for their intergenic and intronic markers (Van Dam et al., 2021) coupled with AHE seems to be a beneficial combination (Wood et al., 2018).
Several other aspects of contamination on UCE datasets remain unresolved. DNA degradation in museum specimens may further confound the accuracy of binning because longer scaffolds may be more accurately binned than shorter scaffolds (Leidenfrost et al., 2020). Examining the effects of UCE parent scaffold or contig length on the accuracy of metagenomic binning should also be explored in the future to potentially improve the accuracy of novel UCE marker set development. While we demonstrate that contaminants are incorporated into a UCE dataset, the effects of contamination on the subsequent phylogeny remain unexplored.
The potential contamination of UCE data used in phylogenomic papers involving museum specimens is not limited to insects. Similar levels of contamination are likely prevalent in other invertebrates and in herbaria or archeological specimens. The methods used by the systematics community have advanced rapidly in the era of phylogenomics. However, relatively little attention has been given to potential contamination in these datasets. Future studies should incorporate a robust metagenomic binning method (beyond just a blast+filtering step) to eliminate sources of contamination from downstream phylogenomic analyses.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
AUTHOR CONTRIBUTIONS
Alex R. Van Dam: Conceptualization (lead); Data curation (equal); Formal analysis (equal); Funding acquisition (equal); Investigation (equal); Methodology (equal); Project administration (equal); Resources (equal); Software (equal); Supervision (equal); Validation (equal); Visualization (equal); Writing – original draft (equal); Writing – review & editing (equal). Javier O. Covas Orizondo: Data curation (equal); Formal analysis (equal); Investigation (equal). Athena W. Lam: Data curation (equal); Investigation (equal); Methodology (equal); Project administration (equal); Resources (equal); Validation (equal); Writing – original draft (equal); Writing – review & editing (equal). Duane D. McKenna: Data curation (equal); Formal analysis (equal); Funding acquisition (equal); Resources (equal); Writing – original draft (equal); Writing – review & editing (equal). Matthew H. Van Dam: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Funding acquisition (equal); Investigation (equal); Methodology (equal); Project administration (equal); Resources (equal); Software (equal); Supervision (equal); Validation (equal); Visualization (equal); Writing – original draft (equal); Writing – review & editing (equal).
Supporting information
Appendix S1
ACKNOWLEDGMENTS
Funding has been provided by the NSF‐DEB‐1856402 to MHVD and NSF‐DEB‐1355169 to DDM, the USDA‐NIFA‐HSI‐12600558 to ARVD for funding the publication charges, NIH‐PR‐INBRE‐5P20GM103475 sub‐award to ARVD for funding the ONT sequencing. We also thank Sarah Crews (CAS) for their help editing the manuscript and Seunggwan Shin (University of Memphis) for assistance with data management.
Van Dam, A. R. , Covas Orizondo, J. O. , Lam, A. W. , McKenna, D. D. , & Van Dam, M. H. (2022). Metagenomic clustering reveals microbial contamination as an essential consideration in ultraconserved element design for phylogenomics with insect museum specimens. Ecology and Evolution, 12, e8625. 10.1002/ece3.8625
DATA AVAILABILITY STATEMENT
Raw data associated with this work can be accessed via NCBI BioProject ID PRJNA800670.
REFERENCES
- Alneberg, J. , Bjarnason, B. S. , de Bruijn, I. , Schirmer, M. , Quick, J. , Ijaz, U. Z. , Lahti, L. , Loman, N. J. , Andersson, A. F. , & Quince, C. (2014). Binning metagenomic contigs by coverage and composition. Nature Methods, 11(11), 1144–1146. 10.1038/nmeth.3103 [DOI] [PubMed] [Google Scholar]
- Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Google Scholar]
- Bankevich, A. , Nurk, S. , Antipov, D. , Gurevich, A. A. , Dvorkin, M. , Kulikov, A. S. , Lesin, V. M. , Nikolenko, S. I. , Pham, S. , Prjibelski, A. D. , Pyshkin, A. V. , Sirotkin, A. V. , Vyahhi, N. , Tesler, G. , Alekseyev, M. A. , & Pevzner, P. A. (2012). SPAdes: A new genome assembly algorithm and its applications to single‐cell sequencing. Journal of Computational Biology, 19(5), 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann, J. (2018). Tiny mites on a great journey – a review on scutacarid mites as phoronts and inquilines (Heterostigmatina, Pygmephoroidea, Scutacaridae). Acarologia, 58(1), 192–251. 10.24349/acarologia/20184238 [DOI] [Google Scholar]
- Bi, K. , Vanderpool, D. , Singhal, S. , Linderoth, T. , Moritz, C. , & Good, J. M. (2012). Transcriptome‐based exon capture enables highly cost‐effective comparative genomic data collection at moderate evolutionary scales. BMC Genomics, 13(403). 10.1186/1471-2164-13-403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaimer, B. B. , Brady, S. G. , Schultz, T. R. , Lloyd, M. W. , Fisher, B. L. , & Ward, P. S. (2015). Phylogenomic methods outperform traditional multi‐locus approaches in resolving deep evolutionary history: A case study of formicine ants. BMC Evolutionary Biology, 15, 271. 10.1186/s12862-015-0552-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaimer, B. B. , Lloyd, M. W. , Guillory, W. X. , & Brady, S. G. (2016). Sequence capture and phylogenetic utility of genomic ultraconserved elements obtained from pinned insect specimens. PLoS One, 11(8), e0161531. 10.1371/journal.pone.0161531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossert, S. , & Danforth, B. N. (2018). On the universality of target‐enrichment baits for phylogenomic research. Methods in Ecology and Evolution, 9(6), 1453–1460. 10.1111/2041-210X.12988 [DOI] [Google Scholar]
- Brady, G. , Faircloth, B. C. , Branstetter, M. G. , White, N. D. , & Brady, S. G. (2015). Target enrichment of ultraconserved elements from arthropods provides a genomic perspective on relationships among Hymenoptera. Molecular Ecology Resources, 15, 489–501. 10.1111/1755-0998.12328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branstetter, M. G. , Danforth, B. N. , Pitts, J. P. , Faircloth, B. C. , Ward, P. S. , Buffington, M. L. , Gates, M. W. , Kula, R. R. , & Brady, S. G. (2017). Phylogenomic Insights into the Evolution of Stinging Wasps and the Origins of Ants and Bees. Current Biology, 27(7), 1019–1025. 10.1016/j.cub.2017.03.027 [DOI] [PubMed] [Google Scholar]
- Brewer, M. S. , & Bond, J. E. (2013). Ordinal‐level phylogenomics of the arthropod class Diplopoda (millipedes) based on an analysis of 221 nuclear protein‐coding loci generated using next‐generation sequence analyses. PLoS One, 8(11), 1–15. 10.1371/journal.pone.0079935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell, B. (2015). BBMap short‐read aligner, and other bioinformatics tools. https://sourceforge.net/projects/bbmap/ [Google Scholar]
- Chikhi, R. , & Medvedev, P. (2014). Informed and automated k‐mer size selection for genome assembly. Bioinformatics, 20, 31–37. 10.1093/bioinformatics/btt310 [DOI] [PubMed] [Google Scholar]
- Cotoras, D. , Murray, G. , Kapp, J. , Gillespie, R. , Griswold, C. , Simison, W. , Green, R. , & Shapiro, B. (2017). Ancient DNA resolves the History of Tetragnatha (Araneae, Tetragnathidae) spiders on Rapa Nui. Genes, 8(12), 403. 10.3390/genes8120403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cran (2010). The comprehensive R Archive Network. 10.1002/wics.1212 [DOI] [Google Scholar]
- De Coster, W. , D’Hert, S. , Schultz, D. T. , Cruts, M. , & Van Broeckhoven, C. (2018). NanoPack: Visualizing and processing long‐read sequencing data. Bioinformatics, 34(15), 2666–2669. 10.1093/bioinformatics/bty149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dentinger, B. T. M. , Gaya, E. , O'Brien, H. , Suz, L. M. , Lachlan, R. , Díaz‐Valderrama, J. R. , Koch, R. A. , & Aime, M. C. (2016). Tales from the crypt: genome mining from fungarium specimens improves resolution of the mushroom tree of life. Biological Journal of the Linnean Society, 117(1), 11–32. 10.1111/bij.12553 [DOI] [Google Scholar]
- Derkarabetian, S. , Benavides, L. R. , & Giribet, G. (2019). Sequence capture phylogenomics of historical ethanol‐preserved museum specimens: Unlocking the rest of the vault. Molecular Ecology Resources, 19(6), 1531–1544. 10.1111/1755-0998.13072 [DOI] [PubMed] [Google Scholar]
- Douglass, A. P. , O’Brien, C. E. , Offei, B. , Coughlan, A. Y. , Ortiz‐Merino, R. A. , Butler, G. , & Wolfe, K. H. (2019). Coverage‐versus‐length plots, a simple quality control step for de Novo Yeast Genome sequence assemblies. G3: Genes, Genomes, Genetics, 9(3), 879–887. 10.1534/g3.118.200745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudchenko, O. , Batra, S. S. , Omer, A. D. , Nyquist, S. K. , Hoeger, M. , Durand, N. C. , Shamim, M. S. , Machol, I. , Lander, E. S. , Aiden, A. P. , & Aiden, E. L. (2017). De novo assembly of the Aedes aegypti genome using Hi‐C yields chromosome‐length scaffolds. Science, 356(6333), 92–95. 10.1126/science.aal3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eren, A. M. , Esen, Ö. C. , Quince, C. , Vineis, J. H. , Morrison, H. G. , Sogin, M. L. , & Delmont, T. O. (2015). Anvi’o: An advanced analysis and visualization platform for ‘omics data. PeerJ, 3, e1319. 10.7717/peerj.1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faircloth, B. C. (2016). PHYLUCE is a software package for the analysis of conserved genomic loci. Bioinformatics, 32, 786–788. 10.1093/bioinformatics/btv646 [DOI] [PubMed] [Google Scholar]
- Faircloth, B. C. (2017). Identifying conserved genomic elements and designing universal bait sets to enrich them. Methods in Ecology and Evolution, 8(9), 1103–1112. 10.1111/2041-210X.12754 [DOI] [Google Scholar]
- Fierst, J. L. , & Murdock, D. A. (2017). Decontaminating eukaryotic genome assemblies with machine learning. BMC Bioinformatics, 18(1), 533. 10.1186/s12859-017-1941-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glassing, A. , Dowd, S. E. , Galandiuk, S. , Davis, B. , & Chiodini, R. J. (2016). Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathogens, 8(1), 24. 10.1186/s13099-016-0103-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafson, G. T. , Baca, S. M. , Alexander, A. M. , & Short, A. E. Z. (2019). Phylogenomic analysis of the beetle suborder Adephaga with comparison of tailored and generalized ultraconserved element probe performance. Systematic Entomology, 45(3), 552–570. 10.1111/syen.12413 [DOI] [Google Scholar]
- Haddad, S. , Shin, S. , Lemmon, A. R. , Lemmon, E. M. , Svacha, P. , Farrell, B. , Ślipiński, A. , Windsor, D. , & Mckenna, D. D. (2018). Anchored hybrid enrichment provides new insights into the phylogeny and evolution of longhorned beetles (Cerambycidae). Systematic Entomology, 43(1), 68–89. 10.1111/syen.12257 [DOI] [Google Scholar]
- Hadfield, J. , & Eldridge, M. D. (2014). Multi‐genome alignment for quality control and contamination screening of next‐generation sequencing data. Frontiers in Genetics, 5, 31. 10.3389/fgene.2014.00031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, R. S. (2007). Improved pairwise alignment of genomic DNA. Ph.D. thesis, The Pennsylvania State University. http://www.bx.psu.edu/~rsharris/rsharris_phd_thesis_2007.pdf [Google Scholar]
- Henderson, J. B. (2018). BLATq v. 1.0.2. https://github.com/calacademy‐research/BLATq/blob/master/README.md [Google Scholar]
- Husnik, F. , & McCutcheon, J. P. (2016). Repeated replacement of an intrabacterial symbiont in the tripartite nested mealybug symbiosis. Proceedings of the National Academy of Sciences of the United States of America, 113(37), E5416–E5424. 10.1073/pnas.1603910113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson, D. H. , Albrecht, B. , Bağcı, C. , Bessarab, I. , Górska, A. , Jolic, D. , & Williams, R. B. H. (2018). MEGAN‐LR: new algorithms allow accurate binning and easy interactive exploration of metagenomic long reads and contigs. Biology Direct, 13, 6. 10.1186/s13062-018-0208-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hykin, S. M. , Bi, K. , & McGuire, J. A. (2015). Fixing formalin: A method to recover genomic‐scale DNA sequence data from formalin‐fixed museum specimens using high‐throughput sequencing. PLoS One, 10(10), e0141579. 10.1371/journal.pone.0141579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, A. J. , McKenna, D. D. , Jordal, B. H. , Cognato, A. I. , Smith, S. M. , Lemmon, A. R. , Lemmon, E. M. , & Hulcr, J. (2018). Phylogenomics clarifies repeated evolutionary origins of inbreeding and fungus farming in bark beetles (Curculionidae, Scolytinae). Molecular Phylogenetics and Evolution, 127, 229–238. 10.1016/j.ympev.2018.05.028 [DOI] [PubMed] [Google Scholar]
- Kang, D. D. , Froula, J. , Egan, R. , & Wang, Z. (2015). MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ, 3, e1165. 10.7717/peerj.1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W. J. (2002). BLAT – The BLAST‐Like Alignment Tool. Genome Research, 12(4), 656–664. 10.1101/gr.229202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent, W. J. (2012). BLAT—The BLAST‐Like Alignment Tool. Version 35. https://users.soe.ucsc.edu/~kent [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutsovoulos, G. , Kumar, S. , Laetsch, D. R. , Stevens, L. , Dau, J. , Conlon, C. , Maroon, H. , Thomas, F. , Aboobaker, A. A. , & Blaxter, M. (2016). No evidence for extensive horizontal gene transfer in the genome of the tardigrade Hypsibius dujardini . Proceedings of the National Academy of Sciences of the United States of America, 113(18), 5053–5058. 10.1073/pnas.1600338113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Jones, M. , Koutsovoulos, G. , Clarke, M. , & Blaxter, M. (2013). Blobology: Exploring raw genome data for contaminants, symbionts and parasites using taxon‐annotated GC‐coverage plots. Frontiers in Genetics, 4, 237. 10.3389/fgene.2013.00237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laetsch, D. R. , & Blaxter, M. L. (2017). BlobTools: Interrogation of genome assemblies [version 1; peer review: 2 approved with reservations]. F1000Research, 6, 1287. 10.12688/f1000research.12232.1 [DOI] [Google Scholar]
- Leidenfrost, R. M. , Pöther, D.‐C. , Jäckel, U. , & Wünschiers, R. (2020). Benchmarking the MinION: Evaluating long reads for microbial profiling. Scientific Reports, 10(1), 5125. 10.1038/s41598-020-61989-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon, A. R. , Emme, S. A. , & Lemmon, E. M. (2012). Anchored hybrid enrichment for massively high‐throughput phylogenomics. Systematic Biology, 61(5), 727–744. 10.1093/sysbio/sys049 [DOI] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke, S. A. , Van Dam, A. , Caffara, M. , Pinto, H. A. , López‐Hernández, D. , & Blanar, C. A. (2018). Validity of the Diplostomoidea and Diplostomida (Digenea, Platyhelminthes) upheld in phylogenomic analysis. International Journal for Parasitology, 48(13), 1043–1059. 10.1016/j.ijpara.2018.07.001 [DOI] [PubMed] [Google Scholar]
- Łukasik, P. , Nazario, K. , Van Leuven, J. T. , Campbell, M. A. , Meyer, M. , Michalik, A. , Pessacq, P. , Simon, C. , Veloso, C. , & McCutcheon, J. P. (2017). Multiple origins of interdependent endosymbiotic complexes in a genus of cicadas. Proceedings of the National Academy of Sciences of the United States of America, 115(2), E226–E235. 10.1073/pnas.1712321115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack, J. E. , Faircloth, B. C. , Crawford, N. G. , Gowaty, P. A. , Brumfield, R. T. , & Glenn, T. C. (2012). Ultraconserved elements are novel phylogenomic markers that resolve placental mammal phylogeny when combined with species‐tree analysis. Genome Research, 22, 746–754. 10.1101/gr.125864.111.746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack, J. E. , Tsai, W. L. E. , & Faircloth, B. C. (2016). Sequence capture of ultraconserved elements from bird museum specimens. Molecular Ecology Resources, 16(5), 1189–1203. 10.1111/1755-0998.12466 [DOI] [PubMed] [Google Scholar]
- McGuire, J. A. , Cotoras, D. D. , O’Connell, B. , Lawalata, S. Z. S. , Wang‐Claypool, C. Y. , Stubbs, A. , Huang, X. , Wogan, G. O. U. , Hykin, S. M. , Reilly, S. B. , Bi, K. E. , Riyanto, A. , Arida, E. , Smith, L. L. , Milne, H. , Streicher, J. W. , & Iskandar, D. T. (2018). Squeezing water from a stone: high‐throughput sequencing from a 145‐year old holotype resolves (barely) a cryptic species problem in flying lizards. PeerJ, 6, e4470. 10.7717/peerj.4470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, D. (2020). Evolution: Symbiotic microbes mediate host range of herbivorous beetles. Current Biology, 30(15), R893–R896. 10.1016/j.cub.2020.05.089 [DOI] [PubMed] [Google Scholar]
- McKenna, D. D. , Shin, S. , Ahrens, D. , Balke, M. , Beza‐Beza, C. , Clarke, D. J. , Donath, A. , Escalona, H. E. , Friedrich, F. , Letsch, H. , Liu, S. , Maddison, D. , Mayer, C. , Misof, B. , Murin, P. J. , Niehuis, O. , Peters, R. S. , Podsiadlowski, L. , Pohl, H. , … Beutel, R. G. (2019). The evolution and genomic basis of beetle diversity. Proceedings of the National Academy of Sciences of the United States of America, 116(49), 24729–24737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misof, B. , Liu, S. , Meusemann, K. , Peters, R. S. , Donath, A. , Mayer, C. , Frandsen, P. B. , Ware, J. , Flouri, T. , Beutel, R. G. , Niehuis, O. , Petersen, M. , Izquierdo‐Carrasco, F. , Wappler, T. , Rust, J. , Aberer, A. J. , Aspöck, U. , Aspöck, H. , Bartel, D. , … Zhou, X. (2014). Phylogenomics resolves the timing and pattern of insect evolution. Science, 346(6210), 763–767. [DOI] [PubMed] [Google Scholar]
- Nielsen, H. B. , Almeida, M. , Juncker, A. S. , Rasmussen, S. , Li, J. , Sunagawa, S. , Plichta, D. R. , Gautier, L. , Pedersen, A. G. , Le Chatelier, E. , Pelletier, E. , Bonde, I. , Nielsen, T. , Manichanh, C. , Arumugam, M. , Batto, J.‐M. , Quintanilha dos Santos, M. B. , Blom, N. , Borruel, N. , … Ehrlich, S. D. (2014). Identification and assembly of genomes and genetic elements in complex metagenomic samples without using reference genomes. Nature Biotechnology, 32(8), 822–828. 10.1038/nbt.2939 [DOI] [PubMed] [Google Scholar]
- Nissen, N. J. , Sønderby, C. K. , Almagro Armenteros, J. J. , Grønbech, C. H. , Nielsen, H. B. , Petersen, T. N. , & Rasmussen, S. (2018). Binning microbial genomes using deep learning. 10.1101/490078 [DOI] [Google Scholar]
- Oxford Nanopore Technologies (2017a). Albacore basecaller from Oxford Nanopore. https://mirror.oxfordnanoportal.com/software/analysis/ont_albacore [Google Scholar]
- Oxford Nanopre Technologies (2017b). New basecaller now performs ‘raw basecalling’, for improved sequencing accuracy. https://nanoporetech.com/about‐us/news/new‐basecaller‐now‐performs‐raw‐basecalling‐improved‐sequencing‐accuracy [Google Scholar]
- Peacock, M. M. , Hekkala, E. R. , Kirchoff, V. S. , & Heki, L. G. (2017). Return of a giant: DNA from archival museum samples helps to identify a unique cutthroat trout lineage formerly thought to be extinct. Royal Society Open Science, 4(11), 171253. 10.1098/rsos.171253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruane, S. , & Austin, C. C. (2017). Phylogenomics using formalin‐fixed and 100+ year‐old intractable natural history specimens. Molecular Ecology Resources, 17(5), 1003–1008. 10.1111/1755-0998.12655 [DOI] [PubMed] [Google Scholar]
- Rasmussen, S. , Allentoft, M. E. , Nielsen, K. , Orlando, L. , Sikora, M. , Sjögren, K.‐G. , Pedersen, A. G. , Schubert, M. , Van Dam, A. , Kapel, C. M. O. , Nielsen, H. B. , Brunak, S. , Avetisyan, P. , Epimakhov, A. , Khalyapin, M. V. , Gnuni, A. , Kriiska, A. , Lasak, I. , Metspalu, M. , … Willerslev, E. (2015). Early divergent strains of Yersinia pestis in Eurasia 5,000 years ago. Cell, 163(3), 571–582. 10.1016/j.cell.2015.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rukmane, A. (2018). An annotated checklist of genus Pachyrhynchus (Coleoptera: Curculionidae: Pachyrhynchini). Acta Biologica Universitatis Daugavpiliensis, 18, 63–68. [Google Scholar]
- Sangiovanni, M. , Granata, I. , Thind, A. S. , & Guarracino, M. R. (2019). From trash to treasure: detecting unexpected contamination in unmapped NGS data. BMC Bioinformatics, 20(S4), 168. 10.1186/s12859-019-2684-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmashghi, S. , Bohmann, K. , Gilbert, P. M. T. , Bafna, V. , & Mirarab, S. (2019). Skmer: Assembly‐free and alignment‐free sample identification using genome skims. Genome Biology, 20(1), 34. 10.1186/s13059-019-1632-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz, D. n. d. (2018). Pauvre: QC and genome browser plotting Oxford Nanopore and PacBio long reads. https://github.com/conchoecia/pauvre [Google Scholar]
- Seppey, M. , Manni, M. , & Zdobnov, E. M. (2019). BUSCO: Assessing genome assembly and annotation completeness. Methods in Molecular Biology, 1962, 227–245. 10.1007/978-1-4939-9173-0_14 [DOI] [PubMed] [Google Scholar]
- Seutin, G. , White, B. D. , & Boag, P. T. (1991). Preservation of avian blood and tissue samples for DNA analyses. Canadian Journal of Zoology, 69(1), 82–90. 10.1139/z91-013 [DOI] [Google Scholar]
- Shin, S. , Clarke, D. J. , Lemmon, A. R. , Moriarty Lemmon, E. , Aitken, A. L. , Haddad, S. , Farrell, B. D. , Marvaldi, A. E. , Oberprieler, R. G. , & McKenna, D. D. (2018). Phylogenomic data yield new and robust insights into the phylogeny and evolution of weevils. Molecular Biology and Evolution, 35(4), 823–836. 10.1093/molbev/msx324 [DOI] [PubMed] [Google Scholar]
- Shostak, A. W. (2014). Hymenolepis diminuta infections in tenebrionid beetles as a model system for ecological interactions between helminth parasites and terrestrial intermediate hosts: A review and meta‐analysis. Journal of Parasitology, 100(1), 46–58. 10.1645/13-347.1 [DOI] [PubMed] [Google Scholar]
- Simão, F. A. , Waterhouse, R. M. , Ioannidis, P. , Kriventseva, E. V. , & Zdobnov, E. M. (2015). BUSCO: Assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics, 31(19), 3210–3212. 10.1093/bioinformatics/btv351 [DOI] [PubMed] [Google Scholar]
- Simpson, S. E. , Nigg, H. N. , Coile, N. C. , & Adair, R. A. (1996). Diaprepes abbreviatus (Coleoptera: Curculionidae): Host Plant Associations. Environmental Entomology, 25(2), 333–349. 10.1093/ee/25.2.333 [DOI] [Google Scholar]
- Smit, A. F. A. , Hubley, R. , & Green, P. (2015). RepeatMasker Open‐4.0.6 . http://www.repeatmasker.org/RepeatMasker/
- Szitenberg, A. , Salazar‐Jaramillo, L. , Blok, V. C. , Laetsch, D. R. , Joseph, S. , Williamson, V. M. , Blaxter, M. L. , & Lunt, D. H. (2017). Comparative genomics of apomictic root‐knot nematodes: hybridization, ploidy, and dynamic genome change. Genome Biology and Evolution, 9(10), 2844–2861. 10.1093/gbe/evx201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling, H. , Waldmann, J. , Lombardot, T. , Bauer, M. , & Glöckner, F. O. (2004). TETRA: A web‐service and a stand‐alone program for the analysis and comparison of tetranucleotide usage patterns in DNA sequences. BMC Bioinformatics, 5, 163. 10.1186/1471-2105-5-163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dam, M. H. , Henderson, J. B. , Esposito, L. , & Trautwein, M. (2021). Genomic characterization and curation of UCEs improves species tree reconstruction. Systematic Biology, 70(2), 307–321. 10.1093/sysbio/syaa063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dam, M. H. , Lam, A. W. , Sagata, K. , Gewa, B. , Laufa, R. , Balke, M. , Faircloth, B. C. , & Riedel, A. (2017). Ultraconserved elements (UCEs) resolve the phylogeny of Australasian smurf‐weevils. PLoS One, 12(11), e0188044. 10.1371/journal.pone.0188044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dam, M. H. , Trautwein, M. , Spicer, G. S. , & Esposito, L. (2019). Advancing mite phylogenomics: Designing ultraconserved elements for Acari phylogeny. Molecular Ecology Resources, 19(2), 465–475. 10.1111/1755-0998.12962 [DOI] [PubMed] [Google Scholar]
- Wang, L. Y. , Huang, W. S. , Tang, H. , Huang, L. C. , & Lin, C. P. (2018). Too hard to swallow: a secret secondary defence of an aposematic insect. Journal of Experimental Biology, 221, jeb172486. 10.1242/jeb.172486 [DOI] [PubMed] [Google Scholar]
- Wood, D. E. , Lu, J. , & Langmead, B. (2019). Improved metagenomic analysis with Kraken 2. Genome Biology, 20, 257. 10.1186/s13059-019-1891-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood, H. M. , González, V. L. , Lloyd, M. , Coddington, J. , & Scharff, N. (2018). Next‐generation museum genomics: Phylogenetic relationships among palpimanoid spiders using sequence capture techniques (Araneae: Palpimanoidea). Molecular Phylogenetics and Evolution, 127, 907–918. 10.1016/j.ympev.2018.06.038 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Data Availability Statement
Raw data associated with this work can be accessed via NCBI BioProject ID PRJNA800670.