

VEGF receptors are essential regulators of blood and lymphatic vessel growth and maintenance. Karaman et al. used single and compound mutants of VEGFR1, 2, and 3 to reveal organ-specific roles for each of these receptors and interactions between them.
Abstract
Karaman et al. (2022. J. Exp. Med. https://doi.org/10.1084/jem.20210565) examined the differential effects of the conditional deletion of genes encoding each VEGF receptor, VEGFR1, VEGFR2 and VEGFR 3, as well as combinations thereof in mice. The results highlight the crosstalk between receptors in different organs and emphasize the importance of VEGF receptor expression and interplay in vascular heterogeneity.
Blood vessels play essential roles in health and disease. They not only allow delivery of nutrients to all organs, but also perform highly specialized functions in an organ-specific manner. For example, a portal system that connects hypothalamic and pituitary capillaries enables regulation of pituitary hormone release by hypothalamic factors, bypassing the systemic circulation; or a barrier that restricts entry of a variety of molecules in brain and retina. Indeed, the organ-specific structural characteristics of normal blood vessels has been long recognized (Aird, 2007). Such specificity is not limited to normal vessels. Almost a century ago, W.H. Lewis observed that the density and morphological characteristics of blood vessels in transplanted tumors were markedly different depending on the tumor type, leading to the far-reaching hypothesis that the tumor environment determines the vascular pattern of tumors (Lewis, 1927).

Insights from Pin Li and Napoleone Ferrara.
Over the last few decades, a variety of reciprocal interactions between vascular endothelium and local microenvironment has been uncovered, and investigation of the mechanisms of organ specificity of the vasculature is currently very active (Augustin and Koh, 2017; Red-Horse et al., 2007; Rafii et al., 2016). Elucidating the mechanisms of such diversity at the molecular level has important implications for embryonic development, adult homeostasis, pathological conditions, and regenerative medicine as well.
As illustrated in the figure, there is potentially a great deal of complexity in such mechanisms (Augustin and Koh, 2017; Red-Horse et al., 2007; Rafii et al., 2016). These include unique transcriptional programs that result in artery–vein specification; metabolic heterogeneity in endothelial cells (ECs) based on the surrounding tissues; instructive cues from the embryonic endothelium resulting in differentiation of organs like pancreas and liver; vascular bed–specific release of growth factors and cytokines, which can stimulate organ-specific growth and regeneration; and EC mitogens with a tissue-specific expression pattern and with action restricted to certain EC types (reviewed in Augustin and Koh, 2017; Red-Horse et al., 2007; Rafii et al., 2016). Also, a very recent study describes an unexpected facet of such diversity: EC type-specific signaling determines whether certain growth factors may stimulate or inhibit EC growth (Li et al., 2022). These features make it possible to regulate organ-specific growth and regeneration precisely according to unique needs and requirements in different organs.
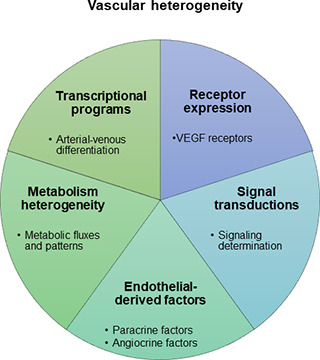
Heterogeneity in the vasculature allows different organs to fulfil distinct functions. There are several characteristics of organotypic vasculature, including specific transcriptional programs in differentiated EC; heterogeneity in EC based on the metabolic pattern of the surrounding tissues; unique endothelial-derived factors that regulate organ development; and unique signal transduction mechanisms for precise regulation.
In this issue, Karaman and colleagues have examined an important aspect of vascular heterogeneity, the organ-specific roles of key angiogenesis-related receptors vascular endothelial growth factor receptor 1 (VEGFR1), VEGFR2, and VEGFR3 (Karaman et al., 2022). These highly related receptor tyrosine kinases (RTKs) are key regulators of hemangiogenesis and lymphangiogenesis and work in conjunction with multiple coreceptors (Simons et al., 2016).
Interestingly, numerous studies have pointed to key functional differences among these receptors. It is now well established that VEGFR2 is the key signaling receptor for VEGF-A, while VEGFR3 mediates lymphangiogenesis and developmental angiogenesis stimulated by VEGF-C and VEGF-D (Adams and Alitalo, 2007; Tammela et al., 2008). These two receptors have some unique features, but their signaling properties are generally consistent with other RTKs (Simons et al., 2016). In contrast, VEGFR1 (Flt-1) does not fulfill the criteria of a conventional RTK, and even today some of its functions are object of debate. In the study that originally reported the identification of Flt-1 as a VEGFR, this role was almost missed because of lack of detectable tyrosine phosphorylation in response to VEGF stimulation (de Vries et al., 1992); the only evidence that at that time established this orphan RTK as a VEGFR was high-affinity VEGF binding (de Vries et al., 1992). This was very surprising and led to the hypothesis that VEGFR1 may be primarily a decoy receptor that regulates angiogenesis by sequestering VEGF rather than as a signaling molecule (Hiratsuka et al., 1998). However, this is only part of the story, as the study by Karaman et al. points out. In 2003, it was reported that VEGFR1 selective agonists, although unable to induce angiogenesis, were nearly as effective at protecting the liver from hepatotoxic damage as the pro-angiogenic VEGFR2 agonists (LeCouter et al., 2003). The protective effects of the VEGFR1 agonists were shown to be mediated by the release of hepatocyte growth factor and possibly other growth factors from quiescent EC.
Karaman et al. (2022) examined the organ-specific effects of VEGFR functions in EC by conditional gene targeting in mice. They found that single deletion of each VEGFR or combinations leads to organ-specific phenotypes in which vascular development and vessel maintenance are differentially affected. In the case of VEGFR2 deletion, there was a substantial vascular regression, which had some organ-specific features, since the most pronounced regression was observed in trachea and kidney glomeruli and the least pronounced was in the intestine. Interestingly, while VEGFR1 deletion resulted in increased vascular density in most tissues, it led to an increase in the density of retinal vasculature and no changes in liver vasculature. The absence of VEGFR3 induced hypersprouting in the retinal vasculature, in agreement with an earlier study (Tammela et al., 2011).
Previous understanding of EC heterogeneity was largely based on in vitro studies, using a limited number of EC model systems. The study by Karaman and colleagues provides an overall view of vascular changes throughout the body. The transgenic models with both newborn and adult mice represent a more physiological setting and might uncover novel perspectives on the mechanisms of vascular development and maintenance.
An especially significant finding in the study by Karaman et al. is the unexpected and complex interplay among VEGFRs. Vegfr2 deletion resulted in a significant decrease in VEGFR3 protein levels. Vascular densities of seven different types of genetic deleted mice, single or compound deletions of Vegfr1, Vegfr2, and/or Vegfr3, were analyzed and compared. The authors report for the first time that additional deletions of Vegfr1 and Vegfr3, in the context of Vegfr2 deletion, leads to further reduction in the vasculature. This is in apparent contrast with the observation that the single Vegfr3 deletion results in higher vascular density and increased permeability. These findings suggest that VEGFR1 and VEGFR3 can support mature vascular maintenance in the absence of VEGFR2 via crosstalk among different VEGFRs. At least for VEGFR1, this may be due to the aforementioned ability to mediate release of growth factors from EC.
Moreover, transcriptional changes were examined in response to VEGFR deletion. Single-cell transcriptomic analysis of ECs from cardiac and pulmonary ECs was performed before and after VEGFR deletions. A novel RNA velocity analysis was performed to summarize and predict the transcriptional changes and possible new trajectories after VEGFR deletions. Notably, the strong directionality of velocity vectors from EC III to EC IV arterial cluster was lost after VEGFR deletion, suggesting the possibility that VEGFR deletions alter arterial differentiation. Several other changes were also observed in EC resident in different organs. This supports the conclusion that both VEGFR deletion–sensitive and –resistant ECs undergo profound transcriptional changes after each gene deletion. The different transcriptional changes may indicate how vessels reciprocally instruct tissue differentiation and function, as well as the mechanisms that underlie vascular bed–specific diseases.
The thorough analysis by Karaman et al. (2022) illustrates how VEGFRs in EC are engaged in crosstalk with each other to maintain the morphology and function of vasculature, providing paradigms for vessel type- and organ-specific endothelial functions.
Current anti-angiogenic therapies rely primarily on targeting VEGF-A or VEGFR2 (Apte et al., 2019). Targeting of VEGFR1 or VEGFR3 so far has been less investigated. The study by Karaman and colleagues makes the case for considering the targeting of all three receptors in order to achieve a more complete inhibition of tumor angiogenesis that may avoid angiogenic escape.
Anti-angiogenic therapies are employed for treatment of tumors and ophthalmical vascular disorders such as age-related macular degeneration. However, the multitude of pathways implicated in vascular growth and maintenance may lead to resistance. The information from single-cell transcriptomics provided by the authors enhances our understanding of vascular specificity, generating new ideas for future research in the organotypic vasculature as a therapeutic target for vascular and organ diseases.
However, further studies are required to fully appreciate the translational implications of these findings, since the consequences of genetic deletion may be different from those of pharmacological inhibition achieved with antibodies or other agents. In an earlier study, Tammela and colleagues reported increased number of tip cells, vascular hypersprouting, and increased vascular leakage following Vegfr3 deletion (Tammela et al., 2011). These effects resembled loss of Notch signaling and were not observed using antibodies. In contrast, loss of Vegfc led to disruption of tip cell fusion points and inefficient angiogenesis and made the case for a bimodal role of VEGFR3 in the regulation of angiogenesis. It is important to point out here such dichotomy, considering that clinical trials with a VEGFR3 trap, which sequesters VEGF-C and VEGF-D, in combination with anti-VEGF, are ongoing in patients with intraocular neovascular disorders and show early evidence of clinical benefit. Interestingly, a discordance between genetic and pharmacological inhibition has been observed also with VEGF-A. Conditional Vegfa deletion in the retinal pigment epithelium or in podocytes of adult mice predicted dramatic retinal and kidney damage, effects that were very rarely observed in humans, using specific VEGF inhibitors (Quaggin, 2012).
In conclusion, the study by Karaman is thought-provoking and will undoubtedly stimulate research aiming to further elucidate the mechanisms underlying the organ-specific roles described in the study. Analysis of VEGFR interactions at the genetic level had not been previously reported and provides insights that are not possible using pharmacological approaches.
References
- Adams, R.H., and Alitalo K.. 2007. Nat. Rev. Mol. Cell Biol. 10.1038/nrm2183 [DOI] [PubMed] [Google Scholar]
- Aird, W.C. 2007. Circ. Res. 10.1161/01.RES.0000255691.76142.4a [DOI] [Google Scholar]
- Apte, R.S., et al. 2019. Cell. 10.1016/j.cell.2019.01.021 [DOI] [Google Scholar]
- Augustin, H.G., and Koh G.Y.. 2017. Science. 10.1126/science.aal2379 [DOI] [PubMed] [Google Scholar]
- de Vries, C., et al. 1992. Science. 10.1126/science.1312256 [DOI] [Google Scholar]
- Hiratsuka, S., et al. 1998. Proc. Natl. Acad. Sci. USA. 10.1073/pnas.95.16.9349 [DOI] [Google Scholar]
- Karaman, S., et al. 2022. J. Exp. Med. 10.1084/jem.20210565 [DOI] [Google Scholar]
- LeCouter, J., et al. 2003. Science. 10.1126/science.1079562 [DOI] [Google Scholar]
- Lewis, W.H. 1927. Bull. Johns Hopkins Hosp. 41:156–162. [Google Scholar]
- Li, P., et al. 2022. EMBO Mol. Med. 10.15252/emmm.202114511 [DOI] [Google Scholar]
- Quaggin, S.E. 2012. J. Clin. Invest. 10.1172/JCI65509 [DOI] [Google Scholar]
- Rafii, S., et al. 2016. Nature. 10.1038/nature17040 [DOI] [Google Scholar]
- Red-Horse, K., et al. 2007. Dev. Cell. 10.1016/j.devcel.2007.01.013 [DOI] [PubMed] [Google Scholar]
- Simons, M., et al. 2016. Nat. Rev. Mol. Cell Biol. 10.1038/nrm.2016.87 [DOI] [PubMed] [Google Scholar]
- Tammela, T., et al. 2008. Nature. 10.1038/nature07083 [DOI] [Google Scholar]
- Tammela, T., et al. 2011. Nat. Cell Biol. 10.1038/ncb2331 [DOI] [PMC free article] [PubMed] [Google Scholar]