

Abstract
Factor XII (FXII) is a serine protease involved in multiple cascades, including the kallikrein–kinin system. It may play a role in diseases in which the downstream cascades are dysregulated, such as hereditary angioedema. Garadacimab (CSL312) is a first‐in‐class, fully human, monoclonal antibody targeting activated FXII (FXIIa). We describe how translational pharmacokinetic (PK) and pharmacodynamic (PD) modeling enabled dose selection for the phase I, first‐in‐human trial of garadacimab. The PK/PD data used for modeling were derived from preclinical PK/PD and safety studies. Garadacimab plasma concentrations rose with increasing dose, and clear dose‐related PD effects were observed (e.g., a mechanism‐based prolongation of activated partial thromboplastin time). The PK/PD profile from cynomolgus monkeys was used to generate minimal physiologically‐based pharmacokinetic (mPBPK) models with target‐mediated drug disposition (TMDD) for data prediction in cynomolgus monkeys. These models were later adapted for prediction of human data to establish dose selection. Based on the final mPBPK model with TMDD and assuming a weight of 70 kg for an adult human, a minimal inhibition (<10%) of FXIIa with a starting dose of 0.1 mg/kg garadacimab and a near maximal inhibition (>95%) at 10 mg/kg garadacimab were predicted. The phase I study is complete, and data on exposure profiles and inhibition of FXIIa‐mediated kallikrein activity observed in the trial support and validate these simulations. This emphasizes the utility and relevance of translational modeling and simulation in drug development.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Models based on physiology and mechanisms of action can be highly useful in translating pharmacokinetic/pharmacodynamic (PK/PD) data from animal studies to expectations in humans.
WHAT QUESTION DID THIS STUDY ADDRESS?
This analysis sought to select doses for investigation in a phase I, first‐in‐human trial of garadacimab (CSL312), a first‐in‐class, fully human, immunoglobulin G4 monoclonal antibody that targets FXIIa.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
This analysis demonstrated how a mechanism‐based PK/PD model describing the relationship between drug administration and pharmacologic response observed in cynomolgus monkeys was generated and extrapolated to select doses for investigation in a phase I, first‐in‐human trial of garadacimab. The detailed explanation of the modeling and extrapolation methodology used in this study provides guidance to future researchers selecting doses for phase I, first‐in‐human trials of monoclonal antibodies.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
This study highlights the importance and utility of insightful, physiologic, and mechanistic mathematical modeling in conjunction with robust animal data for translation of PK/PD and accurate prediction of first‐in‐human dosing for clinical trials.
INTRODUCTION
Factor XII (FXII) is a serine protease involved in multiple cascades, including the coagulation cascade, and the kallikrein–kinin, fibrinolytic, and complement systems. 1 , 2 When activated, FXII contributes to the initiation of these pathways, and thus may play a role in diseases in which the downstream cascades are dysregulated, such as hereditary angioedema (HAE). 1 , 2 , 3 , 4 HAE is a rare and potentially life‐threatening genetic disorder caused by deficient or dysfunctional C1‐esterase inhibitor (C1‐INH), a serine protease inhibitor belonging to the serpin superfamily. 5 , 6
Activation of the kallikrein–kinin system by activated FXII (FXIIa) initiates a cascade leading to the production of bradykinin, a key mediator of angioedema. 6 , 7 C1‐INH inhibits the kallikrein–kinin system and thus the downstream production of bradykinin. 5 , 6 , 7 , 8 Therefore, in patients with HAE with deficient or dysfunctional C1‐INH, bradykinin is overproduced, which results in vascular permeability, extravasation, and ultimately edema. 6 , 7 As FXIIa is a key initiator of the kallikrein–kinin system, it has been speculated that pharmacological inhibition of FXIIa could reduce the production of bradykinin, decreasing the likelihood of an attack of angioedema in patients with HAE.
Garadacimab (CSL312) is a first‐in‐class, fully human, immunoglobulin G4 monoclonal antibody targeting FXIIa. 9 Garadacimab is an affinity‐matured, recombinant variant of the parental antibody 3F7, which was isolated from a Fab‐based phage display library following screening against a fragment of FXIIa. 10 Garadacimab has been shown to prevent bradykinin formation in samples of plasma from patients with HAE. 9 These preclinical data support the investigation of garadacimab in a clinical program.
Translational pharmacokinetic (PK) and pharmacodynamic (PD) models and simulations are used to aid target prioritization and selection, steer drug optimization, provide insight into a drug’s mechanism of action, support identification of mechanistic biomarkers, and enable selection of dosing regimens that balance efficacy against safety, thereby improving the quality of clinical studies. 11 , 12 This report demonstrates how a mechanism‐based PK/PD model describing the relationship between drug administration and pharmacologic response in cynomolgus monkeys was generated and extrapolated to select doses for investigation in the phase I, first‐in‐human trial of garadacimab.
METHODS
Animal studies
PK/PD measurements over time and the initial safety profile of garadacimab (or a research variant of garadacimab) were established in four studies using cynomolgus monkeys: one short‐term and one long‐term toxicity study exploring the intravenous (i.v.) and subcutaneous (s.c.) routes, one PK study using the i.v. and s.c. routes, and one PK/PD study applying the i.v. route only. Some aspects of the PK/PD study have been discussed previously, 9 but relevant sections have been highlighted here for modeling purposes.
Cynomolgus monkeys are an adequate model species for this study based on the FXII amino‐acid sequence similarity with humans, as well as data from functional in vitro assays using plasma from healthy monkeys/humans and patients with HAE. 9
Animals
Male and female cynomolgus monkeys (aged 27–77 months) were studied. In the short‐term and long‐term toxicity studies and the PK study, routine screening tests were performed prior to allocating the animals to the study. In all studies, the health status of the animals was reviewed by a veterinary officer and confirmed acceptable prior to the start of dose administration. The studies were approved by the Institutional Animal Care and Use Committee (IACUC) or a comparable local institutional animal ethics review committee prior to the start of the experimental phase.
Dosing
The short‐term toxicity study investigated garadacimab doses of 3, 10, 30, and 100 mg/kg for i.v. administration and 60 and 200 mg/kg for s.c. administration, over 8 days, applying single doses on days 1 and 8 only, to assess the systemic toxic potential and toxicokinetics of garadacimab. The control group received 0.9% isotonic saline on days 1 and 8.
The long‐term toxicity study investigated garadacimab at doses of 3, 10, 30, and 100 mg/kg i.v. and 60 and 200 mg/kg s.c. every 7 days for 5 weeks (single doses on days 1, 8, 15, 22, 29, and 36). The control group received 0.9% isotonic saline on the same occasions.
The single‐dose PK study investigated single doses of garadacimab 0.5, 1, and 3 mg/kg i.v. and 6 and 20 mg/kg s.c. over 8 weeks. The PK/PD study investigated single i.v. doses of 3, 9, and 27 mg/kg garadacimab administered over 36 days to investigate the PK profile of the anti‐FXIIa monoclonal antibodies.
PK assessment
Blood samples were collected at baseline and throughout the studies to analyze the PK profile of garadacimab. Full details of the blood‐sampling schedule for each study are provided in the Supplementary Material.
PD and no observed adverse effect level assessment
The PD markers included activated partial thromboplastin time (aPTT) and/or measurement of FXIIa‐mediated kallikrein activity. In the short‐term and long‐term toxicity studies, the no observed adverse effect level (NOAEL) was also determined. The aPTT was measured by an ACL series analyzer at the study sites, and inhibition of FXIIa‐mediated kallikrein activity by garadacimab was measured using a chromogenic assay (full methodology provided in the Supplementary Material). For the toxicity studies, safety assessments were performed to determine the NOAEL. Additional assessments were performed during the studies, and these will be described in detail within a separate manuscript.
PK/PD model development
The minimal physiologically‐based pharmacokinetic (mPBPK) modeling approach with target‐mediated drug disposition (TMDD) was utilized to characterize the PK and the FXIIa target binding kinetics of garadacimab. A stepwise modeling approach was adopted (Figure 1). First, the second‐generation mPBPK model was applied to describe garadacimab PKs using plasma concentration measurements from garadacimab in cynomolgus monkeys (Figure 1a). 13 A TMDD feature was then incorporated in the plasma compartment to interrogate the binding and suppression of FXIIa by garadacimab (Figure 1b). In the FXIIa‐mediated kallikrein activity assay, FXII in the plasma is activated by the negatively charged dextran sulfate to form FXIIa. As in attacks of angioedema, the FXIIa cleaves prekallikrein, producing enzymatic kallikrein, which is the predominant enzymatic activity in the assay. This aligns with the proposed pathophysiological pathway leading to bradykinin liberation in HAE and can be used as a surrogate for FXIIa target binding and suppression by garadacimab. It is assumed that inhibition of FXIIa‐mediated kallikrein activity reflects FXIIa suppression by garadacimab. Finally, the relationship between aPTT and FXIIa‐mediated kallikrein activity was described with a simple log‐log linear correlation, similar to that used in previous studies. 14 , 15 Parameters estimated in the prior step were fixed for model fitting in the subsequent step.
FIGURE 1.
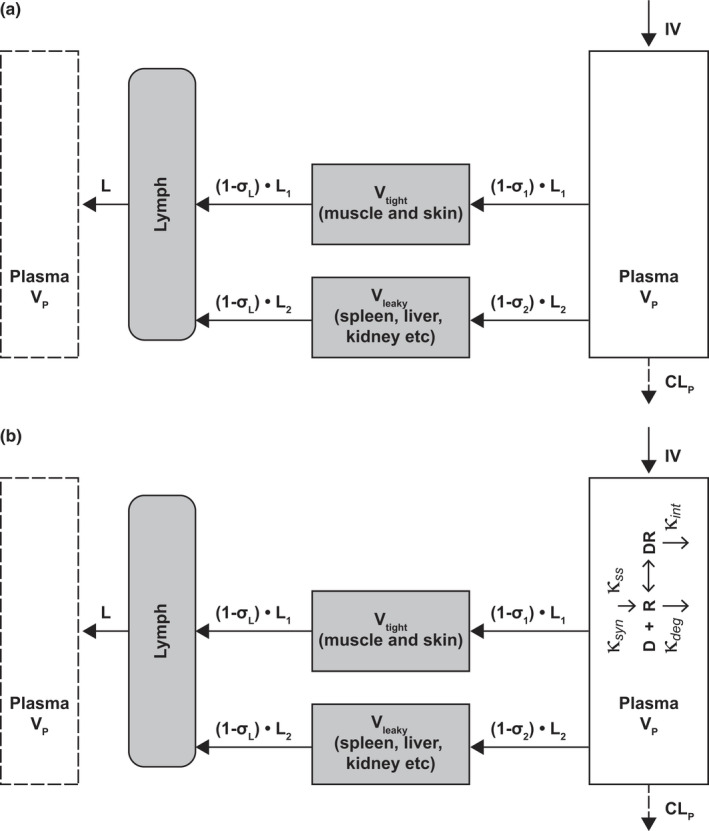
Scheme of stepwise mechanistic PK/PD model development: (a) second‐generation mPBPK model and (b) second‐generation mPBPK model with incorporated TMDD feature. IV, intravenous; mPBPK, minimal physiologically based pharmacokinetic; PD, pharmacodynamic; PK, pharmacokinetic; TMDD, target‐mediated drug disposition
Step I: Plasma PKs of garadacimab
The second‐generation mPBPK model was applied to characterize garadacimab plasma PKs (Figure 1a). Plasma concentration profiles from the three cynomolgus monkey studies (the short‐term toxicity study, the PK study, and the PK/PD study) across a wide dose range (0.5–100 mg/kg garadacimab i.v. and 6–200 mg/kg s.c.) were included for model development. The model includes a plasma compartment, lymph compartment, and two lumped tissue compartments based on vascular endothelium structures. Compartments are interconnected in an anatomic manner assuming convection and lymph drainage as the major pathways for garadacimab tissue distribution and nonspecific catabolism‐driven linear clearance from plasma. The model and initial conditions were described by:
| (1) |
| (2) |
| (3) |
| (4) |
where Cp is the total concentration of garadacimab in plasma, C tight and C leaky represent garadacimab concentrations in interstitial spaces in two types of lumped tissue (tight and leaky), and C lymph is the concentration of garadacimab in lymph. V tight (0.65 • ISF • Kp , where ISF represents total tissue interstitial fluid volume, and Kp is the fraction of ISF available for antibody distribution), and V leaky (0.35 • ISF • Kp ) are the ISFs of the two lumped tissues. Vlymph is lymph volume, which is assumed to be equal to blood volume. L represents total lymph flow rate, and L1 and L2 are one third and two thirds of total lymph flow rate, respectively. The σ1 and σ2 are vascular reflection coefficients for leaky and tight tissues. The σL is the lymphatic capillary reflection coefficient and is assumed to be 0.2. Vp and CLp represent the plasma volume and the linear clearance of garadacimab from plasma, respectively. For s.c. administration, a first‐order absorption rate constant and bioavailability were used to describe drug input into the blood circulation. Fixed parameters were sourced from Cao et al. 2013, 13 and the physiological parameter values are listed in Table 1, assuming the typical body weight of a monkey to be 3.5 kg. 16
TABLE 1.
Summary of model parameter estimates for garadacimab and its interaction with FXIIa in monkeys
| Parameter | Description | Estimate | (RSE %) |
|---|---|---|---|
| Step I | |||
| F | s.c. bioavailability | 0.529 | (17) |
| ka (h−1) | First‐order s.c. absorption rate constant | 0.0227 | (23) |
| σ 1 | Vascular permeability coefficient for tight tissue | 0.98 | (Fixed) |
| σ 2 | Vascular permeability coefficient for leaky tissue | 0.426 | (14) |
| CLp (ml/h/kg) | Plasma clearance of garadacimab | 0.215 | (14) |
| Step II | |||
| k deg (1/h) | Degradation rate constant of FXII | 0.0058 | (20) |
| k int (1/h) | Elimination rate constant of garadacimab‐FXII complex | 0.004 | (17) |
| α | Exponent factor of FXIIa‐mediated kallikrein activity assay | 1.59 | (11) |
| FXIIaB (nmol/ml) | Baseline concentration of FXIIa | 0.3 32 | (Fixed) |
| Step III | |||
| AAT | Power factor of aPTT‐FXIIa‐mediated kallikrein activity | −0.259 | (8) |
| AAI | Intercept factor of aPTT‐FXIIa‐mediated kallikrein activity | 18.9 | (2) |
| Physiological parameter values 16 | |||
| ISF (ml/kg) | Total tissue interstitial fluid volume | 210 | |
| Kp | Available fraction of ISF for antibody distribution | 0.4 | |
| VP (ml/kg) | Plasma volume | 44.9 | |
| VL (ml/kg) | Lymph volume | 89.7 | |
| L (ml/h/kg) | Total lymph flow rate | 3.51 | |
Abbreviations: FXII(a), (activated) factor XII; RSE, relative standard error; SC, subcutaneous.
Step II: Built‐in TMDD for FXIIa binding by garadacimab in the peripheral circulation
The binding of FXIIa to garadacimab was characterized using the TMDD kinetics incorporated in the plasma compartment of the second‐generation mPBPK model (Figure 1b). Plasma concentrations of garadacimab and FXIIa activity measured as inhibition of FXIIa‐mediated kallikrein activity by garadacimab were included. Parameters estimated from the previous step were fixed for model fitting. The model was described by:
| (5) |
| (6) |
| (7) |
| (8) |
| (9) |
where C free is the free garadacimab concentration in plasma, R total and AR represent the total FXIIa and the binding complex of FXIIa and garadacimab, respectively, k syn and k deg are the rate constants for synthesis and degradation for FXIIa homeostasis, and k int represents the elimination rate constant of the FXIIa–garadacimab binding complex. C tight and C leaky represent concentrations of free drug in the different tissue compartments.
The model‐assumed binding quasi‐equilibrium, C free, is described as:
| (10) |
Kss is the steady‐state constant defined as:
| (11) |
where k on and k off are the association and dissociation binding rate constants.
AR for the FXIIa‐garadacimab binding complex is described as:
| (12) |
FXIIa activity, measured as inhibition of FXIIa‐mediated kallikrein activity by garadacimab, which reflects suppression of free FXIIa, is described as:
| (13) |
where R free refers to free FXIIa concentration and R baseline is FXIIa concentration at baseline prior to garadacimab treatment. α is an exponential factor.
R free can be described as:
| (14) |
Step III: Relationship between aPTT and FXIIa‐mediated kallikrein activity
Prolongation of aPTT and inhibition of FXIIa‐mediated kallikrein activity results from changes in the regulation of the intrinsic coagulation pathway and the kallikrein–kinin pathway, respectively. The mathematical relationship between aPTT and FXIIa was explored. Observed data of aPTT and FXIIa‐mediated kallikrein activity from the PK study applying the i.v. and s.c. routes were evaluated. A linear correlation between the log‐transformed aPTT and FXIIa was used to describe aPTT with FXIIa‐mediated kallikrein activity:
| (15) |
Model prediction in humans
The mechanistic PK/PD models described above were utilized translationally to predict garadacimab concentration, FXIIa‐mediated kallikrein activity, and aPTT in humans. Physiological flows and volumes of humans used for garadacimab PK prediction are listed in Table 2, assuming a typical human body weight of 70 kg. Allometric scaling was applied for projection of garadacimab clearance and absorption rate constant. Binding kinetics of garadacimab and FXIIa (kon and koff ) were determined from in vitro experiments (Table 2, full methodology provided in the Supplementary Material). Values for baseline FXII concentration and in vivo degradation rate of FXII in humans were derived based on literature‐reported values. 17 , 18 For aPTT prediction, the power and intercept factors are based on parameters estimated in monkeys as well as previous publications. 19 , 20
TABLE 2.
Summary of parameter values used for prediction of garadacimab PK and interaction with FXIIa in humans
| Parameter | Description | Value | Source |
|---|---|---|---|
| Garadacimab pharmacokinetics | |||
| σ 1 | Vascular permeability coefficient for tight tissue | 0.98 | Same value as monkey |
| σ 2 | Vascular permeability coefficient for leaky tissue | 0.426 | Same value as monkey |
| CLp (ml/h/kg) | Plasma clearance of garadacimab | 0.137 | Allometric scaling with exponent 0.85 |
| F | s.c. bioavailability | 0.529 | Same value as monkey |
| ka (h−1) | First‐order s.c. absorption rate constant | 0.0107 | Allometric scaling with exponent −0.25 |
| FXIIa dynamics | |||
| FXIIaB (nmol/ml) | Baseline concentration of FXIIa | 0.395 | 17 |
| kon_FXII (ml/nmol/h) | Binding association constant | 3600 | Table S2 |
| k off_FXII (1/h) | Binding dissociation constant | 205 | Table S2 |
| kdeg_FXII (1/h) | Degradation rate constant | 0.017 | 17, 18 |
| k int (1/h) | Elimination rate constant of garadacimab‐FXII complex | 0.0031 | Allometric scaling with exponent 0.85, assuming elimination rate is the same as that of garadacimab |
| α | Exponent factor of FXIIa‐mediated kallikrein activity assay | 1.59 | Assuming the same assay‐specific parameter |
| aPTT assay | |||
| AAT | Slope | −0.259 | Assuming the same |
| AAI | Intercept | 28 | 19, 20 |
| Physiological parameter values 16 | |||
| ISF (ml/kg) | Total tissue interstitial fluid volume | 223 | |
| Kp | Available fraction of ISF for antibody distribution | 0.4 | |
| VP (ml/kg) | Plasma volume | 37.1 | |
| VL (ml/kg) | Lymph volume | 74.3 | |
| L (ml/h/kg) | Total lymph flow rate | 1.73 | |
Abbreviations: FXII(a), (activated) factor XII; PK, pharmacokinetic; SC, subcutaneous.
First‐in‐human dose selection and exposure margins
A minimum anticipated biological effect level (MABEL) 21 approach was adopted for dose selection for the first‐in‐human study, based on the predicted inhibition of FXIIa‐mediated kallikrein activity and the expectation that garadacimab exposure would not exceed exposures at the i.v. NOAEL. The exposure margins were calculated based on the observed garadacimab maximum concentration (Cmax; short‐term toxicity study: 2043 μg/ml after the first dose; long‐term toxicity study: 1903 μg/ml after the first dose) and area under the concentration–time curve (AUC; long‐term toxicity study: 96,896 h μg/ml after the first dose) values in cynomolgus monkeys at the i.v. NOAEL of 100 mg/kg divided by the model‐predicted human Cmax and AUC values.
Comparison of predicted versus observed garadacimab PK in humans
The predicted garadacimab Cmax and AUC values in humans were compared with the garadacimab PK parameters observed in the first‐in‐human study, which used doses ranging from 0.1 to 10 mg/kg. Data from the first‐in‐human study will be reported separately.
Software
Data management was performed using R (version 3.5.3). Graphical evaluations were performed using R or GraphPad Prism (version 9.0.0). Modeling of the monkey data and simulations of human PK/PD profiles were performed using nonlinear mixed effects modeling using NONMEM (version 7.3) 22 and Perl‐speaks‐NONMEM (version 3.4.2). 23 Pirana (version 2.9.0) was used as the graphical user interface. 24 All models were estimated using the first‐order conditional estimation method with interaction.
RESULTS
The PK/PD and initial safety profile of garadacimab were established in cynomolgus monkeys (Figure 2 gives an example for PK/PD assessment in the PK study applying i.v. and s.c. administration). In all cynomolgus studies, garadacimab plasma concentrations increased in a dose‐dependent manner. All doses of garadacimab had an inhibitory effect on FXII‐mediated kallikrein activity in the PK study applying the i.v. and s.c. routes. Differences in the maximal PD effect and sustainment of this effect between i.v. and s.c. routes of administration were demonstrated. Across all studies, administration of garadacimab, both i.v. and s.c., caused a dose‐dependent prolongation of aPTT, with no associated effects on the prothrombin time. In the short‐term and long‐term toxicity studies, which applied once‐weekly dosing, the NOAEL was determined to be 100 mg/kg for i.v. doses and 200 mg/kg for s.c. doses; these were the highest doses explored in these studies.
FIGURE 2.
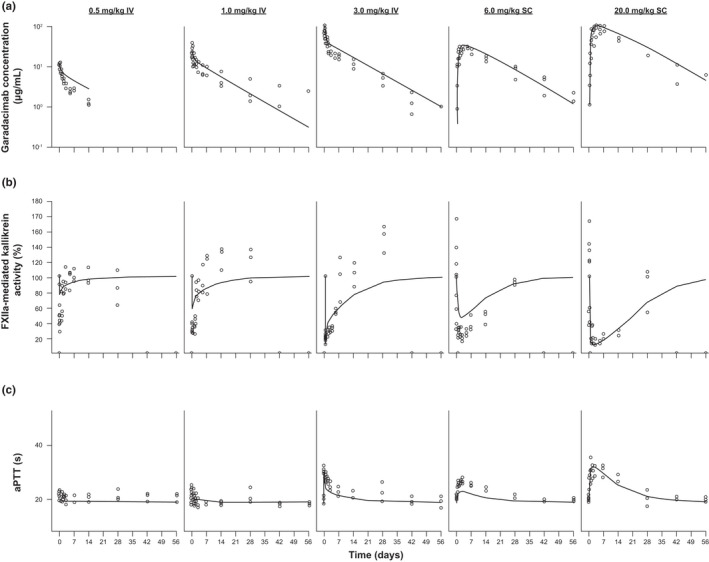
Model fitting of representative individual PK/PD data from the PK study applying the IV and SC routes in cynomolgus monkeys: (a) PK/PD data based on garadacimab exposure, (b) FXIIa‐mediated kallikrein activity, and (c) aPTT data. Symbols represent the individual PK/PD observations, and solid lines represent the model fitted PK/PD profiles. aPTT, activated partial thromboplastin time; FXIIa, activated factor XII; IV, intravenous; PD, pharmacodynamic; PK, pharmacokinetic; SC, subcutaneous
The final mPBPK model with TMDD (Figure 1) was developed to characterize garadacimab PK, inhibition of FXIIa‐mediated kallikrein activity, and aPTT values at various doses in cynomolgus monkeys, based on the observed data (Figure 2). The mPBPK model with the added TMDD component reasonably characterized garadacimab PK and PD profiles from multiple studies in cynomolgus monkeys across doses in the range of 0.5 to 200 mg/kg i.v. or s.c.; representative examples from a single study are shown in Figure 2a,b. The extent and duration of FXIIa‐mediated kallikrein activity inhibition was directly dependent on the concentration of garadacimab in the system, with activity returning to baseline as garadacimab was eliminated. The aPTT prolongation was also reasonably characterized (Figure 2c), returning to initial values as garadacimab was eliminated.
Physiological parameters in the model were fixed: their values are listed in Table 1, along with the parameter estimates at each step of model development. Overall, the model parameters were generally well estimated with relative standard errors of less than 30%.
In addition, a log‐log relationship between observed aPTT values and FXIIa‐mediated kallikrein activity was well‐described using linear regression (Figure 3). Applying Equation 15, the slope of the power factor of aPTT‐FXIIa‐mediated kallikrein activity was −0.259, and the intercept was 18.9.
FIGURE 3.
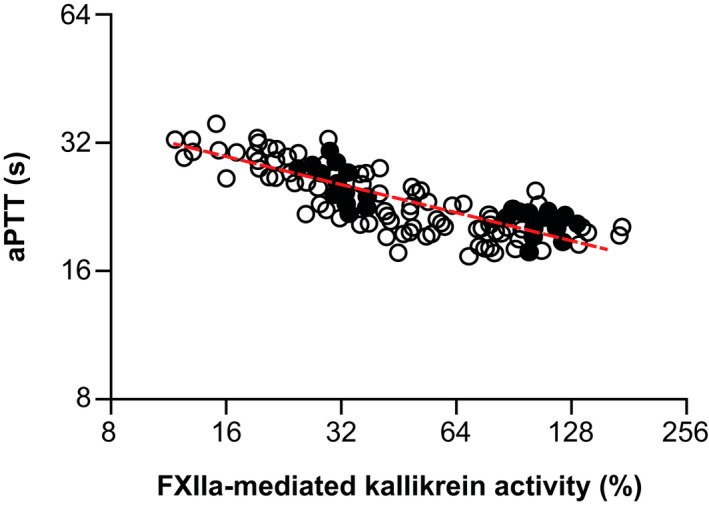
Relationship between aPTT and FXIIa‐mediated kallikrein activity data in cynomolgus monkeys, fit using linear regression. The dashed red line represents the linear regression, as determined by Equation 15. aPTT, activated partial thromboplastin time; FXIIa, activated factor XII
Garadacimab concentrations, FXIIa‐mediated kallikrein activity, and aPTT time profiles in healthy individuals following administration of various i.v. and s.c. doses were simulated based on the final PK/PD model (Table 2) and allometric scaling assuming an adult human weight of 70 kg (Figure 4). A dose of 0.1 mg/kg i.v. was predicted to cause slight (<10%) inhibition of FXIIa‐mediated kallikrein activity; 10 mg/kg i.v. was predicted to cause near maximal (>95%) inhibition of FXIIa‐mediated kallikrein activity (Figure 4). First‐in‐human doses were selected to target minimal inhibition of FXIIa‐mediated kallikrein activity at the starting dose and maximal inhibition at the highest dose (derived from the aforementioned PK/PD model), in accordance with the MABEL approach.
FIGURE 4.
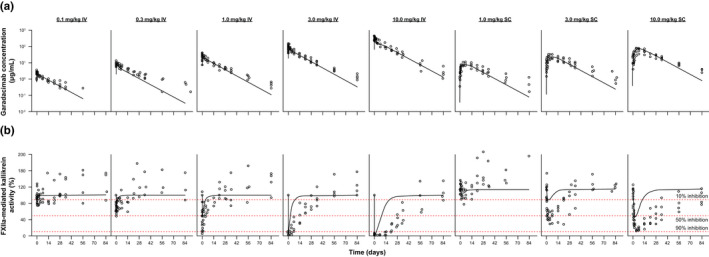
(a) Model‐predicted and observed human PK and (b) FXIIa‐mediated kallikrein activity from first‐in‐human study. The black symbols depict the individual PK/PD observations in phase I volunteers. Solid black lines reflect the simulated PK/PD profiles in humans. Red dotted lines represent the percentage inhibition. FXIIa, activated factor XII; IV, intravenous; PD, pharmacodynamic; PK, pharmacokinetic; SC, subcutaneous
At the starting dose of 0.1 mg/kg i.v., the model predicted a garadacimab Cmax and AUC in humans of 3 μg/ml and 1527 h μg/ml, respectively, based on the simulated PK profiles. Relative to the i.v. NOAEL, this represents ~ 681‐fold and 634‐fold lower values when compared with the i.v. NOAEL Cmax (short‐term toxicity study: 2043 µg/ml; long‐term toxicity study: 1903 μg/ml, each after the first dose) and 63‐fold lower values when compared with the i.v. NOAEL AUC (long‐term toxicity study: 96,896 h μg/ml after the first dose) at the human i.v. starting dose (Table S1). Exposures of garadacimab up to 10 mg/kg s.c. and i.v. were lower than that observed at i.v. NOAEL, thus providing a robust safety margin, considering the predicted safety margins were over 1 (Table S1).
Selected, single escalating doses were investigated as part of a phase I, single‐center study. Volunteers were randomized to one of eight cohorts: i.v. doses of 0.1, 0.3, 1, 3, and 10 mg/kg, and s.c. doses of 1, 3, and 10 mg/kg. Six volunteers in each cohort received garadacimab or placebo in a ratio of 2:1. Follow‐up for safety lasted until 85 days after dosing. Blood samples were collected throughout for PK/PD analysis. The 48 volunteers were aged 19–44 years with a mean body mass index of 24 kg/m2 (unpublished data).
The garadacimab concentration profiles observed in the first‐in‐human study (unpublished data) were well‐predicted based on the scaling of the PK/PD model, with the exception of the terminal phase in which the model’s predicted decline was faster than observed (Figure 4a). Inhibition of FXIIa‐mediated kallikrein activity in the first‐in‐human study was predicted reasonably well, with the exception of the lower doses where the model underpredicted the inhibition. The extent of inhibition is also underpredicted by the model (Figure 4b). Finally, garadacimab Cmax and AUC0–inf at all doses were well‐predicted based on scaling of the mPBPK model (Figure 5). The predicted PK parameters were within two‐fold of the observed values, which is the industry standard. 25 , 26 , 27
FIGURE 5.
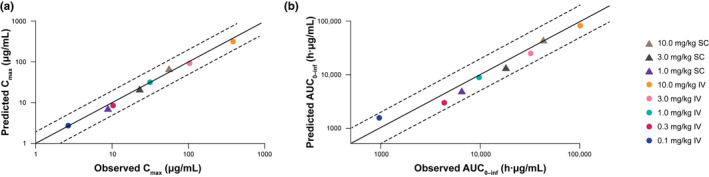
Model‐predicted and observed human (a) Cmax and (b) AUC0–inf from first‐in‐human study. The solid middle lines are the line of unity, while the upper and lower dashed lines represent two‐fold under‐ or overprediction. AUC0–inf, area under the plasma concentration–time curve extrapolated to infinity; Cmax, maximum concentration; IV, intravenous; PK, pharmacokinetic; SC, subcutaneous
DISCUSSION
Garadacimab, a recombinant monoclonal antibody, specifically and potently inhibits the catalytic domain of FXIIa. The in vitro pharmacological activity and in vivo PK/PD of garadacimab have been assessed in multiple preclinical studies, 9 and the data produced have enabled the translational modeling used to suggest well‐tolerated doses for investigation in the phase I, first‐in‐human trial of garadacimab. This report presents a quantitative translational approach for the selection of well‐tolerated starting doses and a pharmacologically relevant single‐dose escalation scheme for a phase I clinical trial in healthy volunteers.
Data collected from studies in cynomolgus monkeys have shown that garadacimab plasma concentrations increase in a dose‐dependent manner and have a concentration‐dependent inhibitory effect on FXII‐mediated kallikrein activity. Furthermore, administration of garadacimab caused a clear dose‐dependent prolongation of aPTT with no associated effect on the prothrombin time, which was consistent with the expected pharmacological response. There were no other findings of toxicological significance at any dose tested (up to 100 mg/kg i.v. and 200 mg/kg s.c. in the toxicity studies). Thus, it can be concluded that garadacimab has an expected PK/PD profile and a favorable safety profile at the tested doses.
Initially, both the mPBPK model and a two‐compartmental model were explored to characterize the PK of garadacimab (model building and results of the two‐compartment model not shown). The two models were able to characterize the observed data equally well. The mPBPK model lumps tissues with similar permeability to drug (i.e., with “tight” and “leaky” vasculature) while describing tissue distribution as a function of lymph flow and a vascular reflection coefficient. 13 The mPBPK model was chosen because it offers the advantage of translating preclinical data to predict the clinical PK of monoclonal antibodies by using the actual physiological values in humans and monkeys. The mPBPK model is structured in an anatomic and physiological manner and has been demonstrated to be advantageous for numerous antibodies. 24 Compared with the compartmental model, the mPBPK model offers advantages for interpreting parameter estimates in the context of physiological or disease pathological conditions without requiring additional numbers of parameters to be estimated. Based on this model, the predicted half‐life for garadacimab PK in the clinic is in line with the expected half‐life of similar monoclonal antibodies. 28 , 29
Inhibition of FXIIa‐mediated kallikrein activity provides a PD marker to assess the effect of garadacimab on the biological process of interest. The mechanistic mPBPK model with TMDD characterizes FXIIa‐mediated kallikrein activity that is reflective of the inhibition of FXIIa by garadacimab.
When determining the first‐in‐human dose and developing the necessary models, complex tissue distribution was also considered. An mPBPK model with additional complexities to incorporate the distribution of the FXIIa‐garadacimab binding complex in peripheral circulation was explored. Mechanistically, the complex, which comprises a free target FXIIa and garadacimab and is similar in size to garadacimab alone, is expected to follow the same mechanisms of tissue distribution as garadacimab. When the target expression is low relative to the antibody drug concentration, the concentration of the complex is considered negligible compared with the total antibody drug concentration. Therefore, not accounting for complex tissue distribution in the PK/PD model will not lead to a significant difference in total antibody drug concentration prediction. However, in our case, the baseline FXII target concentration is within the same order of magnitude as garadacimab concentrations at garadacimab doses less than 1 mg/kg. Modeling results suggest that the formed complex is minimal compared with the total antibody drug concentration; therefore, the mPBPK model with distribution of FXIIa‐garadacimab complex in peripheral circulation was not chosen as the final model.
PKs and FXIIa‐mediated kallikrein activity data predicted by the model were validated by comparison with the observed data from the phase I trial of garadacimab (Figure 4; unpublished data) and the clinical PK data aligned with predictions for PK (Figure 5). The predictions for Cmax and AUC were within two‐fold of the observed values, which is well‐accepted as the industry standard for large molecules and indicates that the model is suitable for translation to human predictions. 25 , 26 , 27 As observed in other studies, 30 the bioavailability data predicted in monkeys were similar to those in humans. Based on monkey data, the bioavailability in humans was predicted to be 53%. In the phase I study, the estimated plasma bioavailability of garadacimab s.c. versus i.v. administration was 63.3% for the 1 mg/kg dose, 54.3% for 3 mg/kg, and 42.4% for 10 mg/kg. Overall, the absolute bioavailability of garadacimab after s.c. injection was observed to be 49.7%. The similar values of bioavailability in humans and in monkeys justifies the use of the monkey values to predict human PKs of garadacimab. Furthermore, the predicted concentration‐dependent inhibition of FXIIa‐mediated kallikrein activity and aPTT prolongation was observed in the phase I study after single ascending doses of i.v. and s.c. garadacimab (unpublished data). This model will be regularly updated throughout the drug development process, based on available clinical data, to optimize predictability.
The translation accounted for between‐species differences in target expression and binding by utilizing data generated by in vitro experiments, thus improving the estimation of PKs and inhibition of FXIIa‐mediated kallikrein due to garadacimab. 25 There are multiple challenges to accurate estimation. The PK/PD data used for model development were limited to single‐dose studies. Combined FXIIa‐mediated kallikrein activity and aPTT measurements were limited to data from one study; there was no placebo group and thus it was not possible to assess the daily variation in FXIIa and aPTT baselines. Furthermore, the FXIIa data in Figure 2a were normalized with a set starting value of 100%, which presumes an exact known value 31 and does not take baseline variability into account. Nevertheless, the effects of garadacimab clearly stand out from the variability in the data, lending confidence to the estimated parameters.
Both FXIIa‐mediated kallikrein activity and aPTT assays rely on ex vivo activation of the plasma sample for their readout. They are thus not exact in vivo readouts but reflect the potential for activation of the respective biochemical pathways, namely FXII‐driven activation of the kallikrein–kinin pathway and FXII‐driven intrinsic coagulation in the case of aPTT. The activation of FXIIa‐mediated kallikrein activity reflects the triggering of this pathway that is thought to occur in attacks of angioedema.
A MABEL‐based approach, using translational PK/PD modeling, was adopted to select the first‐in‐human starting dose. The i.v. doses of 0.1, 0.3, 1, 3, and 10 mg/kg and s.c. doses of 1, 3, and 10 mg/kg were selected for inclusion in the phase I clinical study in healthy volunteers. Because these predictions were established, the first‐in‐human, phase I study has been completed: garadacimab PK and FXIIa‐mediated kallikrein activity data observed support and validate the simulations described in this manuscript. This again emphasizes the utility and relevance of modeling and simulations in drug development.
CONFLICT OF INTEREST
D.P., A.M., F.G., and M.T. are employees and shareholders of CSL Limited. A.R. and M.B. are employees of CSL Limited. F.M. and M.W.N. are employees of CSL Behring Innovation GmbH and shareholders of CSL Limited. W.J.J. received support from CSL Behring. All other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
D.P., X.C., F.G., F.M., A.R., M.B., W.J.J., A.M., M.W.N., and M.T. wrote the manuscript. F.M., A.R., and M.W.N. designed the research. D.P., X.C., F.G., F.M., A.R., M.B., A.M., M.W.N., and M.T. performed the research. D.P., X.C., F.G., F.M., A.R., M.B., A.M., M.W.N., and M.T. analyzed the data.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
The study was sponsored by CSL Behring. Writing support was provided by Hannah Brazier DPhil of OPEN Health Medical Communications and was funded by CSL Behring.
Pawaskar D, Chen X, Glassman F, et al. Pharmacokinetic/pharmacodynamic modeling for dose selection for the first‐in‐human trial of the activated Factor XII inhibitor garadacimab (CSL312). Clin Transl Sci. 2022;15:709–720. doi: 10.1111/cts.13192
Funding information
This study was funded by CSL Behring Innovation GmbH and CSL Limited.
REFERENCES
- 1. Didiasova M, Wujak L, Schaefer L, Wygrecka M. Factor XII in coagulation, inflammation and beyond. Cell Signal. 2018;51:257‐265. [DOI] [PubMed] [Google Scholar]
- 2. Weidmann H, Heikaus L, Long AT, Naudin C, Schlüter H, Renné T. The plasma contact system, a protease cascade at the nexus of inflammation, coagulation and immunity. Biochim Biophys Acta Mol Cell Res. 2017;1864:2118‐2127. [DOI] [PubMed] [Google Scholar]
- 3. Schmaier AH, Stavrou EX. Factor XII ‐ What’s important but not commonly thought about. Res Pract Thromb Haemost. 2019;3:599‐606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stavrou E, Schmaier AH. Factor XII: what does it contribute to our understanding of the physiology and pathophysiology of hemostasis & thrombosis. Thromb Res. 2010;125:210‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Donaldson VH, Evans RR. A biochemical abnormality in hereditary angioneurotic edema: absence of serum inhibitor of C’ 1‐esterase. Am J Med. 1963;35:37‐44. [DOI] [PubMed] [Google Scholar]
- 6. Hofman ZL, Relan A, Zeerleder S, Drouet C, Zuraw B, Hack CE. Angioedema attacks in patients with hereditary angioedema: Local manifestations of a systemic activation process. J Allergy Clin Immunol. 2016;138:359‐366. [DOI] [PubMed] [Google Scholar]
- 7. Schmaier AH. The elusive physiologic role of Factor XII. J Clin Invest. 2008;118:3006‐3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Davis AE 3rd. The pathogenesis of hereditary angioedema. Transfus Apher Sci. 2003;29:195‐203. [DOI] [PubMed] [Google Scholar]
- 9. Cao H, Biondo M, Lioe H, et al. Antibody‐mediated inhibition of FXIIa blocks downstream bradykinin generation. J Allergy Clin Immunol. 2018;142:1355‐1358. [DOI] [PubMed] [Google Scholar]
- 10. Larsson M, Rayzman V, Nolte MW, et al. A factor XIIa inhibitory antibody provides thromboprotection in extracorporeal circulation without increasing bleeding risk. Sci Transl Med. 2014;6:222ra17. [DOI] [PubMed] [Google Scholar]
- 11. Tiwari A, Bhattacharya I, Chan PLS, Harnisch L. Comparing model performance in characterizing the PK/PD of the anti‐myostatin antibody domagrozumab. Clin Transl Sci. 2020;13:125‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Visser SA, Aurell M, Jones RD, et al. Model‐based drug discovery: implementation and impact. Drug Discov Today. 2013;18:764‐775. [DOI] [PubMed] [Google Scholar]
- 13. Cao Y, Balthasar JP, Jusko WJ. Second‐generation minimal physiologically‐based pharmacokinetic model for monoclonal antibodies. J Pharmacokinet Pharmacodyn. 2013;40:597‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Benincosa LJ, Chow FS, Tobia LP, Kwok DC, Davis CB, Jusko WJ. Pharmacokinetics and pharmacodynamics of a humanized monoclonal antibody to factor IX in cynomolgus monkeys. J Pharmacol Exp Ther. 2000;292:810‐816. [PubMed] [Google Scholar]
- 15. Tilley D, Levit I, Samis JA. Measurement of factor v activity in human plasma using a microplate coagulation assay. J Vis Exp. 2012;9:3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhao J, Cao Y, Jusko WJ. Across‐species scaling of monoclonal antibody pharmacokinetics using a minimal PBPK model. Pharm Res. 2015;32:3269‐3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Björkqvist J, Nickel K, Stavrou E, Renné T. In vivo activation and functions of the protease factor XII. Thromb Haemost. 2014;112:868‐875. [DOI] [PubMed] [Google Scholar]
- 18. Haemochrom Diagnostica . Factor XII. 2021. https://www.haemochrom.de/en/produkte/general‐haemostasis/faktor‐xiii?key=1‐2&cHash=484972e2e674f1680fbbca70fd0040a7). Accessed September 9, 2021.
- 19. Cannon CP, Dingemanse J, Kleinbloesem CH, Jannett T, Curry KM, Valcke CP. Automated heparin‐delivery system to control activated partial thromboplastin time: evaluation in normal volunteers. Circulation. 1999;99:751‐756. [DOI] [PubMed] [Google Scholar]
- 20. Chow FS, Benincosa LJ, Sheth SB, et al. Pharmacokinetic and pharmacodynamic modeling of humanized anti‐factor IX antibody (SB 249417) in humans. Clin Pharmacol Ther. 2002;71:235‐245. [DOI] [PubMed] [Google Scholar]
- 21. Milton MN, Horvath CJ. The EMEA guideline on first‐in‐human clinical trials and its impact on pharmaceutical development. Toxicol Pathol. 2009;37:363‐371. [DOI] [PubMed] [Google Scholar]
- 22. Beal SL, Sheiner LB, Boeckmann AJ, Bauer RJ (Eds). NONMEM 7.4 user guides. 1989–2018.
- 23. Mats K, Rikard N, Svetlana F, Sebastian U, Gunnar Y. Perl speaks NONMEM version 4.8.1. 2018.
- 24. Keizer RJ, Karlsson MO, Hooker A. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst Pharmacol. 2013;2:e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dong JQ, Salinger DH, Endres CJ, et al. Quantitative prediction of human pharmacokinetics for monoclonal antibodies: retrospective analysis of monkey as a single species for first‐in‐human prediction. Clin Pharmacokinet. 2011;50:131‐142. [DOI] [PubMed] [Google Scholar]
- 26. Li H, Kock K, Wisler JA, et al. Prediction of clinical pharmacokinetics of AMG 181, a human anti‐alpha 4 beta 7 monoclonal antibody for treating inflammatory bowel diseases. Pharmacol Res Perspect. 2015;3:e00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Benet LZ, Sodhi JK. Investigating the theoretical basis for In Vitro‐In Vivo Extrapolation (IVIVE) in predicting drug metabolic clearance and proposing future experimental pathways. AAPS J. 2020;22:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Temrikar ZH, Suryawanshi S, Meibohm B. Pharmacokinetics and clinical pharmacology of monoclonal antibodies in pediatric patients. Paediatr Drugs. 2020;22:199‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ovacik M, Lin K. Tutorial on monoclonal antibody pharmacokinetics and its considerations in early development. Clin Transl Sci. 2018;11:540‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Musther H, Olivares‐Morales A, Hatley OJ, Liu B, Rostami HA. Animal versus human oral drug bioavailability: do they correlate? Eur J Pharm Sci. 2014;57:280‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Woo S, Pawaskar D, Jusko WJ. Methods of utilizing baseline values for indirect response models. J Pharmacokinet Pharmacodyn. 2009;36:381‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kogan AE, Kardakov DV, Khanin MA. Analysis of the activated partial thromboplastin time test using mathematical modeling. Thromb Res. 2001;101:299‐310. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material