

Abstract
The purpose of this study was to investigate the potential clinical relevance of estimating the apparent clearance (CL/F) of atorvastatin through population pharmacokinetic (PopPK) modeling with samples collected in a real‐life setting in a cohort of ambulatory patients at risk of cardiovascular disease by using an opportunistic sampling strategy easily accessible in clinical routine. A total of 132 pharmacokinetic (PK) samples at a maximum of three visits were collected in the 70 included patients. The effects of demographic, genetic, and clinical covariates were also considered. With the collected data, we developed a two‐compartment PopPK model that allowed estimating atorvastatin CL/F relatively precisely and considering the genotype of the patient for SLCO1B1 c.521T>C single‐nucleotide polymorphism (SNP). Our results indicate that the estimation of the CL/F of atorvastatin through our PopPK model might help in identifying patients at risk of myalgia. Indeed, we showed that a patient presenting a CL/F lower than 414.67 L h−1 is at risk of suffering from muscle discomfort. We also observed that the CL/F was correlated with the efficacy outcomes, suggesting that a higher CL/F is associated with a better drug efficacy (i.e., a greater decrease in total and LDL‐cholesterol levels). In conclusion, our study demonstrates that PopPK modeling can be useful in daily clinics to estimate a patient’ atorvastatin clearance. Notifying the clinician with this information can help in identifying patients at risk of myalgia and gives indication about the potential responsiveness to atorvastatin therapy.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Atorvastatin is the major statin used at present for reducing cholesterol circulating levels. It is estimated that 10%–15% of statin users develop statin‐related muscle side effects. Interindividual variability also exists with regard to LDL‐cholesterol lowering response. Statin‐associated muscle‐related side effects and lipid‐lowering responses are dose‐dependent and related to the drug systemic exposure, which supports the importance of studying factors affecting atorvastatin pharmacokinetic (PK) behavior.
WHAT QUESTION DID THIS STUDY ADDRESS?
Our study aimed at constructing a population PK (PopPK) model with data prospectively collected in a cohort of ambulatory patients sparsely sampled and including relevant pharmacogenetic, clinical, and demographic information to estimate individual PK parameters and to relate PKs with drug response.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Based on sparse ambulatory PK data, the final validated model is able to provide an individual estimation of atorvastatin apparent clearance (CL/F) considering the genotype of the patient for SLCO1B1 c.521T>C single‐nucleotide polymorphism (SNP). The estimation of the atorvastatin CL/F helps in identifying patients at risk of myalgia and is correlated with drug efficacy: a patient presenting a CL/F lower than 414.67 L h−1 is at risk of suffering from muscle discomfort and a higher CL/F is associated with a better drug efficacy.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
PopPK modeling can be useful in daily clinics to estimates a patient’s atorvastatin clearance. Notifying the clinician with this information can help in identifying patients at risk of myalgia and gives indication about the potential responsiveness to atorvastatin therapy in complement to cholesterol measurements.
INTRODUCTION
Hypercholesterolemia remains one of the most important risk factors for cardiovascular disease (CVD), which is a leading cause of death worldwide. 1 HMG‐CoA reductase inhibitors, commercialized under the name of statins, constitute the first‐line medication used for primary and secondary CVD prevention because of their proven efficacy for lowering low‐density lipoprotein (LDL) cholesterol and triglyceride concentrations as well as reducing the risk of CVD. 2 , 3 Atorvastatin is one of the world’s bestselling drugs of all time and the major statin used at present. Although it is generally well‐tolerated, observational studies estimate that 10%–15% of statin users develop statin‐related muscle side effects ranging from mild myalgia (cramps, complaints, discomfort … ) to severe muscle symptoms (rare rhabdomyolysis). 4 On the other hand, interindividual variability also exists with regard to LDL‐cholesterol lowering response, as well as efficacy in reducing major CVD events. Statin‐associated muscle‐related side effects and lipid‐lowering responses are dose‐dependent. However, whereas atorvastatin systemic exposure has been constantly related to the drug‐related muscle toxicity, 5 , 6 , 7 cholesterol lowering efficacy better correlates with the dose than with systemic exposure markers, such as maximal concentration (Cmax) and area under the concentration curve (AUC) 5 , 6 , 7 , 8 probably because the liver is the site of HMG‐CoA reductase activity and atorvastatin undergoes extensive first‐pass metabolism. 6 Nevertheless, these dose‐response and pharmacokinetic‐pharmacodynamic (PK‐PD) relationships support the importance of studying factors affecting atorvastatin PK behavior to explain differential pharmacological responses.
Atorvastatin is administered orally as a calcium salt of the active acid form with a clinical dosage ranging commonly from 10 to 80 mg/day. It is rapidly and well‐absorbed but has a weak and variable oral bioavailability of ~ 14% due to substantial first‐pass metabolism. 5 The pharmacologically active atorvastatin is biotransformed to its corresponding inactive lactone form and both are further metabolized into OH‐metabolites by CYP3A isoenzymes. 9 The main metabolite, 2‐OH‐atorvastatin, is pharmacologically active and significantly contributes to the inhibitory activity on HMG–CoA reductase. By contrast, little is known about the role of these metabolites in the toxicity related to atorvastatin but in vitro data suggested that the myotoxic potency of atorvastatin lactone is higher than that of its acid form. 10 The lactone forms of atorvastatin and its metabolites can also be hydrolyzed back into their corresponding acid forms either nonenzymatically or by esterases and paraoxonases. 11 Atorvastatin is more than 98% bound to plasma proteins. Despite being highly protein‐bound, the mean volume of distribution of atorvastatin is reported to be 381 L after intravenous infusion of 5 mg, which denotes extensive distribution in peripheral tissues. 5 Atorvastatin is also substrate for transporter proteins, including active efflux transporters (ABCB1, 12 ABCC1, 13 ABCC2, 14 and ABCC4 13 ) and influx carriers (OATP1B1 [SLCO1B1], 13 , 15 , 16 OATP1B3 [SLCO1B3], 16 and OATP2B1 [SLCO2B1] 13 ). Genetic polymorphisms in the genes coding for these proteins can affect each step of the PK path covered by the absorption, distribution, metabolism, and excretion (ADME) processes. 17 Some of those transporters are expressed not only in excretory and absorptive organs, which might affect systemic exposure but also at the site of therapeutic action (i.e., the liver, and/or in the skeletal muscle tissue 13 where the toxic action is exerted), therefore impacting the active fraction of the drug reaching its target, which is more directly related to the clinical response. Atorvastatin and its metabolites are eliminated in the bile and less than 1% of the oral dose is excreted in urine. 5
Earlier studies have identified genetic variants in PK genes, such as ABCB1, ABCC2, ABCG2, CYP3A, POR, and SLCO genes associated with differential atorvastatin exposure, LDL‐cholesterol response, and/or muscle related side‐effects. 17 , 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 , 35 , 36 , 37 , 38 , 39 With few exceptions, the PK studies reported to date consist mainly in classical association studies using noncompartmental PK analysis sometimes performed on healthy volunteers after single dose administration 21 , 28 , 32 , 35 , 36 , 37 or physiologically‐based PK models. 40 , 41 , 42 , 43 , 44 Some population PK (PopPK) studies have been published but with strict, rich sampling and/or in a limited number of (healthy) individuals without considering genetic polymorphisms and, thus, are not likely to reflect the constraints of real‐life clinical settings. 29 , 45 , 46 , 47 , 48 In this context, with the support of rich datasets from former studies, 49 , 50 our study aimed at constructing a PopPK model with data prospectively collected in a cohort of ambulatory patients sparsely sampled and including relevant pharmacogenetic information. Next, we derived the apparent clearance (CL/F) of the drug in each individual and related it with indicators of statin‐related muscle toxicity and cholesterol‐lowering response.
MATERIALS AND METHODS
Datasets
Three datasets were used in the present study. The investigation dataset comprised newly collected data from patients with hypercholesterolemia who initiated atorvastatin‐based therapy or who were already treated with this drug at the time of their enrollment. Patients were prospectively recruited at the Cliniques Universitaires Saint‐Luc (Brussels, Belgium) from August 2017 until August 2019 and had a normal hepatic function, as evaluated with common hepatic markers (liver transaminases, gamma glutamyl transferase, and alkaline phosphatase). Patients provided written informed consent before any study‐related procedure and the study was approved by the local ethics committee (Comité d’Ethique Hospitalo‐Facultaire, approval number B403201732532, ClinicalTrials.gov registration number NCT03604471). Participants were sampled once at each study visit, for a total of one to four visits (depending on the individual), and the plasma obtained by centrifugation. Because patients were asked not to modify their drug‐taking habits and were allowed to come into the hospital at any time, the post‐intake delay was random. At the first study visit, an extra blood sample was also drawn for DNA extraction and genotyping (see below). All samples were frozen at −20°C until the day of analysis. In addition to the investigation dataset, PK data was obtained from two support datasets: a study in healthy volunteers 50 and a study in patients suffering from atorvastatin‐related myopathy and healthy volunteers. 49
Atorvastatin quantification
Atorvastatin acid plasma concentrations in the investigation cohort were determined using a liquid chromatography‐tandem mass spectrometry (LC‐MS/MS) method. Two hundred µl of plasma were spiked with d5‐atorvastatin (internal standard) and proteins were precipitated with 600 µl ice‐cold acetone. Samples were placed at −20°C for 2 h, then the supernatant was recovered by centrifugation and the solvents removed by evaporation at 30°C under a stream of nitrogen. The dry residue was reconstituted in methanol. For LC‐MS/MS analysis, 2 µl of each sample was injected on an Acquity UPLC class H system (Waters) coupled to a Xevo TQ‐S (Waters) mass spectrometer. A gradient between MeOH‐H2O‐acetic acid (75:24.9:0.1; v/v/v) and MeOH‐acetic acid (99.9:0.1; v/v) was used for analyte separation on an Ascentis Express C18 column (100 × 4.6 mm; 2.7 µm). The flow rate was set at 500 µl/min. For each compound, a quantification (Q) and a qualification (q) m/z transition were used: 559.2>440.2 (Q) and 559.2>249.9 (q) for atorvastatin; 575.2>440.2 (Q) and 575.2>249.9 (q) for ortho‐ and para‐hydroxyatorvastatin; 541.2>448.2 (Q) and 541.2>249.9 (q) for atorvastatin lactone; and 564.3>440.3 (Q) and 564.3>250.0 (q) for d5‐atorvastatin (internal standard). Calibration curves were obtained in the same conditions.
Genotyping
For the investigation cohort, DNA was extracted from whole blood using the QIAamp DNA mini‐kit according to the manufacturer’s instructions. Allelic discrimination was performed on a Step One Plus Real Time PCR system using Taqman genotyping assays for CYP3A4*22 (rs35599367); CYP3A5*3 (rs776746); ABCB1 c.1199G>A (rs2229109), and c.3435C>T (rs1045642); ABCC1 c.2012G>T; SLCO1B1 c.388A>G, and SLCO1B1*5 (c.521T>C); SLCO2B1 c.935G>A; and SLCO1B3 c.334T>G. Haldane’s exact test was used to check for deviations from Hardy‐Weinberg equilibrium.
Structural pharmacokinetic model and goodness‐of‐fit
Because of the limited amount of PK data in the investigation cohort, it would have been difficult to develop a model based solely on this data. Therefore, the datasets from Lemahieu et al. 50 and Hermann et al. 49 were used to support the development of the structural model. All three dataset were used to build a nonlinear mixed‐effects model in NONMEM (version 7.4.3). One‐ and two‐compartment disposition models were evaluated, whereas absorption was modeled as a first‐order process. Hierarchical structural and covariate models were compared with regard to the objective function value (OFV) in addition to parameter plausibility, precision (in terms of relative standard error [RSE]), and shrinkage. The final model was evaluated using graphical goodness‐of‐fit methods and a dose‐stratified, prediction‐corrected visual predictive check (pcVPC). Additionally, we verified the normality of NPDE computed from 1000 simulations.
Covariate analysis
The following covariates were investigated: race (Hispanic or White/other), sex, atorvastatin dosage, genotypes, drug‐drug interactions (DDIs), age, and body mass index (BMI). Genotypes were defined as wild‐type homozygous, variant homozygous, or heterozygous (see section 0 for a list of all single‐nucleotide polymorphisms [SNPs]). DDIs were defined by the presence of known inhibitors of CYP3A, ABCB1, OATP1B1, OATP1B3, or OATP2B1, or known inducers of CYP3A. List of inhibitors and inducers of CYP3A and ABCB1 were obtained from online sources, 51 , 52 whereas for OATP inhibitors, compounds from Karlgren et al. 53 showing more than 50% inhibition were selected. DDIs were ignored if the perpetrator drug was taken infrequently by the patient. Time‐varying covariates were summarized over the entire observation period because it was not possible to characterize interoccasion PK variability in any of the datasets. Missing covariate values in the investigation cohort were substituted by the most recent value, the population median or the most frequent category, as appropriate, whereas covariates that were unavailable in the other two cohorts were coded as missing. Stepwise covariate modeling was performed: covariates were added to the model in a univariate way (α = 0.05), all significant covariates were included in a multivariate model, then backward elimination of covariates was performed (α = 0.01), followed by assessment of the goodness‐of‐fit of the final model.
Pharmacodynamic outcomes
Toxicity outcomes were mean creatine kinase (CK) serum levels over the study period and occurrence of myalgia, which was defined as any patient‐reported muscle pain or cramps over the entire study period, regardless of serum CK level at the time of the complaint. Efficacy outcomes were evaluated through the changes in total and LDL cholesterol levels measured at the last patient visit from baseline expressed in percentages. Baseline was considered as the cholesterol levels measured at the date of either therapy initiation or switch to atorvastatin. For experienced patients, baseline cholesterol measurements were retrieved in the medical file through retrospective datamining.
Statistical analysis
PD outcomes were tested for univariate association with individual Bayesian estimates of atorvastatin apparent oral clearance using linear (continuous outcomes; i.e., CK levels or cholesterol lowering response) or logistic regression (binary outcome; i.e., occurrence of myalgia Yes/No) to derive the odds of presenting myalgia when considering the value of an explicative variable (CL/F). For the binary outcome, a receiver operating characteristic (ROC) curve plotting sensitivity (true positives) against 1‐specificity (false positives) for each level of the independent variable (CL/F) was then built to identify a cutoff value to discriminate cases from non‐cases. In all circumstances, a p value of less than 0.05 was considered as statistically significant.
RESULTS
Datasets
Three datasets were included for model development: the investigation dataset and the two support datasets (Table S1). The study by Lemahieu et al. 50 used a crossover design to evaluate the effect of calcineurin inhibitors on atorvastatin PK; only data from the control phase (atorvastatin alone) was used for modeling. Regarding the study by Hermann et al., 49 both the data from 13 patients with statin‐induced myopathy and data from the 15 healthy controls were used. Summary characteristics of the investigation cohort are presented in Table 1 whereas genotyping results are reported in Table 2. The investigation dataset included 70 patients and a total of 132 PK samples. The timing of the last dose was self‐reported by the patients. Patients were also asked to confirm whether they had followed a regular dosing schedule or if their schedule over the past few days included irregular or missed doses. Unless the timing of the last few doses was known, data obtained from patients with irregular dosing schedules were discarded. For patients who reported missed doses over the past few days but who otherwise followed a strict schedule, missed doses were coded as such in the database. In this dataset, the post‐intake delay ranged from 2.2 to 40 h (median: 14.6 h). 10 samples (7.6%) were excluded from the analysis because the post‐intake delay was unknown and 1 sample (0.8%) was excluded due to improper storage conditions. Two atorvastatin concentrations (1.5%) were below the limit of quantification and were set to half of that value. Six patients (8.6%) in the investigation cohort presented with myalgia at any point during follow‐up.
TABLE 1.
Summary characteristics of the investigation cohort
| n | % | |
|---|---|---|
| Atorvastatin dosage | ||
| 5 mg q24h | 4 | 5.7 |
| 10 mg q24h | 14 | 20.0 |
| 20 mg q24h | 22 | 31.4 |
| 30 mg q24h | 1 | 1.4 |
| 40 mg q24h | 19 | 27.1 |
| 80 mg q24h | 10 | 14.3 |
| Indication | ||
| Primary prevention | 64 | 91.4 |
| Secondary prevention | 6 | 8.6 |
| Type of patient | ||
| De novo therapy | 19 | 27.1 |
| Recently switched statin | 12 | 17.1 |
| Long‐term treatment | 39 | 55.7 |
| Age (years) | ||
| Median (IQR) | 53.8 (21.6) | |
| Sex | ||
| Female | 35 | 50.0 |
| Male | 35 | 50.0 |
| BMI | ||
| median (IQR) | 26.0 (5.4) | |
| Smoker | ||
| No | 59 | 84.4 |
| 1–5 cigarettes/day | 3 | 4.3 |
| >5 cigarettes/day | 8 | 11.4 |
| Race | ||
| White | 66 | 94.3 |
| Hispanic | 2 | 2.9 |
| Other | 2 | 2.9 |
| Drug‐drug interactions | ||
| CYP3A inhibitors | 2 | 2.9 |
| CYP3A inducers | 0 | 0 |
| ABCB1 inhibitors | 1 | 1.4 |
| OATP1B1 inhibitors | 20 | 28.6 |
| OATP1B3 inhibitors | 0 | 0 |
| OATP2B1 inhibitors | 7 | 10.0 |
| Myalgia at any time during follow‐up | 6 | 8.6 |
Data given at baseline, unless noted otherwise.
Abbreviations: BMI, body mass index; IQR, interquartile range.
TABLE 2.
Genotypes frequencies in the investigation cohort
| Gene | SNP | Genotype | N (%) | p value |
|---|---|---|---|---|
| ABCB1 | c.1199G>A | GG | 63 (90.0%) | 1 |
| GA | 2 (2.9%) | |||
| AA | 0 (0%) | |||
| Missing | 5 (7.1%) | |||
| c.3435C>T | CC | 14 (20.0%) | 0.619 | |
| CT | 30 (42.9%) | |||
| TT | 21 (30.0%) | |||
| Missing | 5 (7.1%) | |||
| ABCC1 | c.2012G>T | GG | 56 (80.0%) | 0.312 |
| GT | 8 (11.4%) | |||
| TT | 1 (1.4%) | |||
| Missing | 5 (7.1%) | |||
| CYP3A5 | g.6986A>G | *1/*1 | 0 (0%) | 1 |
| *1/*3 | 7 (10.0%) | |||
| *3/*3 | 58 (82.9%) | |||
| Missing | 5 (7.1%) | |||
| CYP3A4 | g.15389C>T | *1/*1 | 61 (87.1%) | 1 |
| *1/*22 | 4 (5.7%) | |||
| *22/*22 | 0 (0%) | |||
| Missing | 5 (7.1%) | |||
| SLCO1B1 | c.388A>G | AA | 25 (35.7%) | 0.799 |
| AG | 30 (42.9%) | |||
| GG | 10 (14.3%) | |||
| Missing | 5 (7.1%) | |||
| c.521T>C | TT | 44 (62.9%) | 0.109 | |
| TC | 16 (22.9%) | |||
| CC | 5 (7.1%) | |||
| Missing | 5 (7.1%) | |||
| SLCO1B3 | c.334T>G | TT | 46 (65.7%) | 0.661 |
| GT | 17 (24.3%) | |||
| GG | 2 (2.9%) | |||
| Missing | 5 (7.1%) | |||
| SLCO2B1 | c.935G>T | GG | 1 (1.4%) | 0.486 |
| GT | 11 (15.7%) | |||
| TT | 53 (75.7%) | |||
| Missing | 5 (7.1%) |
The p value is for Haldane’s exact test (*<0.05).
Abbreviation: SNP, single nucleotide polymorphism.
Structural model
A two‐compartment model fit the data best (ΔOFV compared to a one‐compartment model = −167). Due to difficulties in estimating this parameter with our design, the absorption rate constant ka was fixed to a literature value of 2.5 h−1 and its interindividual variability term was set to zero. Sensitivity analysis for values of ka ranging from 0.5 to 4.5 showed this had little impact on the other parameter estimates (data not shown). Residual error was modeled using an exponential function (a mixed additive/exponential model was also tested, ΔOFV compared to exponential model = −0.3). The use of a full omega matrix between random effects improved the OFV as well (ΔOFV = −47.5) but also caused numerical difficulties and made no appreciable differences on goodness‐of‐fit plots and thus was not used.
Covariate analysis
The effect of covariates was investigated on atorvastatin CL/F only as the other PK parameters were estimated with lower precision and higher shrinkage, which could have biased the results. Because only the investigation cohort contained covariate information, clearance was defined with two fixed and two random effects: one pair for the investigation cohort and one pair for the rest of the dataset, in such a way that covariate relationships could be included in the model without affecting parameter estimates for the subjects from the studies by Lemahieu et al. 50 and Hermann et al. 49 Model parameters and empirical Bayesian estimates of CL/F were not significantly affected by this split parameterization (data not shown). Covariates included in the forward search were OATP2B1 inhibitor (ΔOFV = −4.3), sex (ΔOFV = −4), and SLCO1B1 521T>C (ΔOFV = −20.7) (Figure 1a,b,c). Patients co‐medicated with an inhibitor of OATP2B1 (usually L‐thyroxine) had lower CL/F than the rest of the population, women had lower CL/F than men, and CL/F was reduced in carriers of the SLCO1B1 521C allele compared with carriers of the T allele, although strangely, this effect was more pronounced in heterozygotes (and the effect in CC homozygotes was estimated with very poor precision). The addition of this covariate lowered the unexplained interindividual variability of CL/F from 0.127 to 0.0677. Treating it in a binary manner (carrier of the C allele vs. non‐carrier) had a more moderate effect on the OFV. Of these three covariates, only SLCO1B1 521T>C was retained at the end of the backward elimination process.
FIGURE 1.
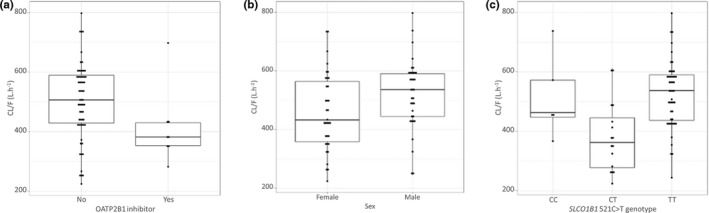
Box‐and‐whisker plots representing atorvastatin CL/F estimation according to (a) coadministration with OATP2B1 inhibitors, (b) sex, and (c) SLCO1B1 521C>T genotype. CL/F, apparent clearance
Final model evaluation
Final PopPK model estimates are reported in Table 3. Population predictions for the final model were biased for concentration values above 10 ng ml−1 (which mostly originate from the dataset by Lemahieu et al. 50 ) whereas individual predictions correlated well with observed concentrations (Figure 2; Spearman’s rho = 0.84 and 0.96 for population and individual predictions, respectively). Further, there was no apparent trend in the residuals (Figure 2). The pcVPC (Figure 3a), was satisfactory as well, with most observed concentrations lying in the 95% prediction interval. The pcVPC stratified by the dose are presented in the Supplementary Data S2 (5 and 30 mg q24h dose regimens are not shown on the plot due to the low number of subjects). NPDE were normally distributed, with mean 0 and variance 1 (Figure 3b–e).
TABLE 3.
Final population pharmacokinetic model estimates
| Parameter | Estimate | RSE (%) | 95% CI | Shrinkage (%) |
|---|---|---|---|---|
| Structural model | ||||
| θCL investigation (L h−1) | 535 | 6.2 | [470–600] | |
| θCL support (L h−1) | 400 | 7.4 | [342–458] | |
| ωCL investigation (SD) | 0.068 | 16.5 | [0.176–0.344] | 44.3 |
| ωCL support (SD) | 0.195 | 9.8 | [0.357–0.527] | 40.1 |
| θQ (L h−1) | 1690 | 20.3 | [1018–2362] | |
| ωQ (SD) | 0.715 | 19.3 | [0.526–1.164] | 52.4 |
| θVc (L) | 1960 | 21.7 | [1125–2795] | |
| ωVc (SD) | 1.18 | 16.1 | [0.745–1.435] | 40.3 |
| θVp (L) | 3900 | 15.7 | [2700–5100] | |
| ωVp (SD) | 0.508 | 19.5 | [0.441–0.985] | 38.8 |
| θka (h−1) | 2.5 (fixed) | |||
| σ (SD) | 0.085 | 5.5 | [0.067–0.103] | 13.9 |
| Covariate model | ||||
| θSLCO1B1 521TC | −0.402 | 15.2 | [−0.522–−0.282] | |
| θSLCO1B1 521CC | −0.041 | 469.7 | [−0.422–0.339] |
Clearances and volumes of distribution are apparent parameters.
Abbreviations: CI, confidence interval; CL, clearance; ka, absorption rate constant; Q, inter‐compartmental clearance; RSE, relative standard error; VC, central volume of distribution; VP, peripheral volume of distribution; θ, fixed effect; σ, random effect (residual variability); ω, random effect (between‐subject variability).
In the investigation cohort: CL/F = θCL × (1 + θSLCO1B1), where θSLCO1B1 is 0 for 521TT individuals.
FIGURE 2.
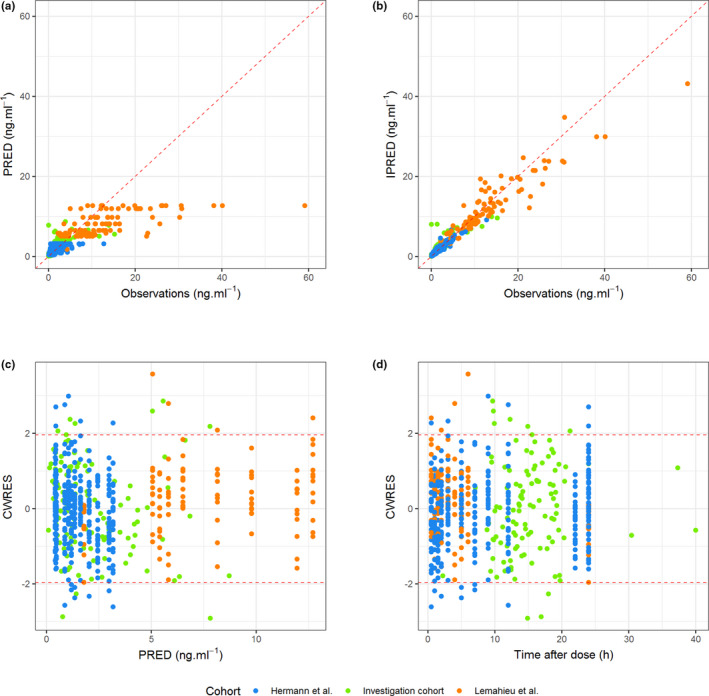
Goodness‐of‐fit plots. (a) Population predicted concentrations (PRED) versus observations (OBS), (b) individual predicted concentrations (IPRED) versus OBS, (c) conditional weighted residuals (CWRES) versus PRED, and (d) CWRES versus time after dose
FIGURE 3.
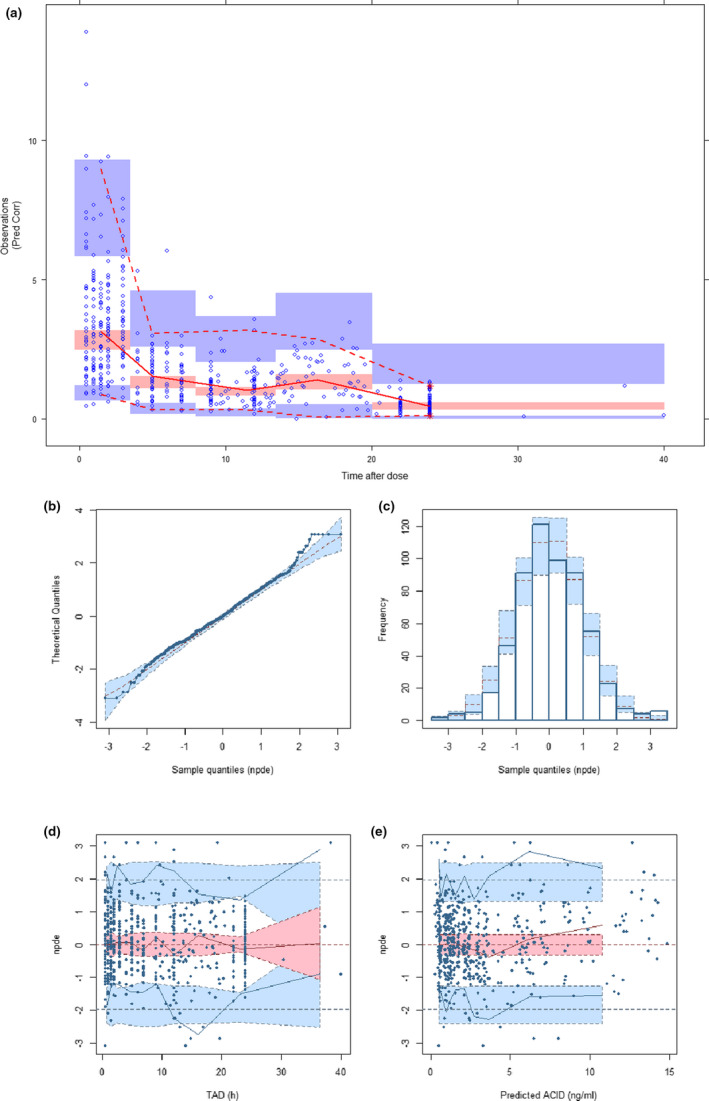
(a) Prediction‐corrected visual predictive check. (b to e) Normalized distribution prediction error (NPDE), (b) Q‐Q plot of NPDE, (c) Histogram of NPDE. Shaded area represents theoretical distribution, (d) NPDE versus time after dose (TAD). Shaded areas represent the prediction intervals associated with the 5th, 50th, and 95th percentiles. (e) NPDE versus predicted concentrations
PK/PD relationships
In univariate analysis, the CL/F and CK levels were negatively correlated (r = −0.26, p = 0.042; Figure 4a); for each increment of 1 L h−1 in the CL/F, CK levels decreased by 0.20 units. In line with this, the odds of myalgia, another indicator of statin‐related muscle toxicity, were correlated with atorvastatin CL/F, with an odds ratio (OR) of 0.68 IC95% (0.46–0.99) for each 50 L h−1 increase in CL/F (p = 0.031), indicating that a higher clearance protects against the occurrence of myalgia with an averaged 1.5‐fold lower chance of developing myalgia for every ~10% (i.e., 50 L/h) increase in CL/F. The ROC AUC, reflecting the classification performance of the test, was 0.77, which indicates a good discriminant power (Figure 4b). Optimal sensitivity (83.3%) and specificity (73.0%) were obtained with a CL/F threshold of 414.67 L h−1. Only one patient out of six patients presenting myalgia had a CL/F above this threshold (528.82 L h−1). When comparing patients with a so‐defined low CL/F to patients with a high CL/F, the OR of suffering from myalgia was 13.43 IC95% (2.47–124.31), confirming that low clearance patients are at higher risk of developing myalgia.
FIGURE 4.
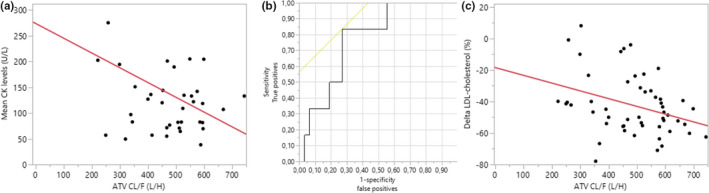
PK‐PD analysis. (a) Correlation between atorvastatin CL/F and CK levels. (b) Receiver operating characteristic (ROC) curve plotting sensitivity (true positives) against 1‐specificity (false positives) for each level of CL/F as cutoff point. Yellow line represents the 45‐degree angle tangent to the ROC curve indicating the best cutoff point. (c) Correlation between atorvastatin CL/F and LDL‐cholesterol reduction in percentages from baseline (% Delta LDL‐cholesterol). CK, creatine kinase; CL/F, apparent clearance; PD, pharmacodynamic; PK, pharmacokinetic
Considering treatment efficacy outcomes, CL/F and decreases in total and LDL cholesterol levels (%) were positively correlated (Figure 4c); for any increment in the CL/F, the decrease in total and LDL cholesterol from baseline was even more important (r = −0.34 and −0.36; p = 0.005 and 0.014, respectively). Adjusting the regression models for the administered dose, slightly increased the relationships between changes in either total or LDL cholesterol levels and atorvastatin CL/F (r = −0.44 and −0.49 vs. −0.34 and −0.36).
DISCUSSION
We developed a two‐compartment PopPK model for estimating atorvastatin CL/F in a cohort of ambulatory patients at risk of CVD by using an opportunistic sampling strategy easily accessible in clinical practice. This model is able to provide an estimation of atorvastatin CL/F considering the genotype of the patient for SLCO1B1 c.521T>C SNP. In addition, our results indicate that the estimation of the CL/F of atorvastatin through our PopPK model might help in identifying patients at risk of myalgia. Indeed, we show that a patient presenting a CL/F lower than 414.67 L h−1 is at risk of suffering from muscle discomfort. We also observed that the CL/F was correlated with the efficacy outcomes, suggesting that a higher CL/F is associated with a better drug efficacy (i.e., a greater decrease in total and LDL cholesterol levels).
Few atorvastatin PopPK models have been reported to date. The majority of existing PK studies used noncompartmental methods for analyzing the data. Yet, as in other PopPK studies, our final model consists of a two‐compartment model and the CL/F estimations obtained are similar to those derived in other PopPK studies, even if the populations were not comparable. In the study of Knebel et al., 45 the population consisted of 39 pediatric patients treated with low dose‐atorvastatin, whereas in the population included in the study of Narwal et al., 46 intensive 24 h sampling was performed on a mix of healthy volunteers (n = 10) and patients (n = 13) after receiving a single similar atorvastatin dose (10 mg). Both studies reported that a two‐compartment model fitted the data the best and estimated a mean CL/F of 652 and 504 L h−1, respectively. Here, we showed that, even with very sparsely sampled data in an adult ambulatory setting, we can still obtain a meaningful estimation of the atorvastatin CL/F with our model (535 L h−1). This is supported by the results obtained by Schwartz et al. 47 who used a PopPK model built based on PK samples collected in 143 patients in nursing homes and in the community and reported a typical CL/F value of 596.4 L h−1 (considering a bodyweight of 70 kg). However, whereas, like ours, this latter study investigated the effect of demographic and clinical covariates on the CL/F of atorvastatin, the population study comprised a majority of elderly patients and did not consider any genetic covariates in the model. The patients included in their study were, on average, 10 years older than the ambulatory patients of our clinical trial. Consequently, their patients had more than nine co‐medications recorded on average. Furthermore, the authors only assessed the impact of CYP3A inducers and inhibitors without considering perpetrator drugs having an impact on OATP or other transporters activities. In the study of Dostalek et al., 48 a sparse sampling design with a total of 312 PK points fitted to a two‐compartment model was also conducted but the population study consisted of 20 nondiabetic and 32 diabetic renal transplant recipients as it was specifically designed for investigating the effect of diabetes on atorvastatin conversion to its lactone form and no genetic or co‐medication covariates were considered. Their model was simplified to derive a metabolic CL/F of atorvastatin to its lactone form and no global CL/F was estimated.
Considering the PK‐PD associations, in the above‐mentioned PopPK studies, generally, no toxicity outcomes were assessed for correlation with PK parameters. However, in the study of Narwal et al., 46 which was based on the same rich dataset of Herman et al., 49 used to support the development of our structural model, no significant difference in the PK parameters of the acid and lactone forms of atorvastatin was found between healthy subjects and patients with myopathy. This can potentially be explained by the fact that, contrarily to our model, none of the genetic covariates were retained in their final model, including the SLCO1B1 c.521T>C, which was probably due to the low number of subjects included in their study (n = 26). Considering the cholesterol lowering effect of atorvastatin, in the study of Shwartz et al., 47 the authors did not detect any relationships between atorvastatin concentrations and either low‐density lipoprotein cholesterol or total cholesterol concentrations. By contrast, in our study, we observed a correlation between changes in total and LDL cholesterol levels from baseline and atorvastatin CL/F, suggesting that a higher CL/F is associated with a better efficacy. This observation is not surprising as the liver is the main site of HMG‐CoA reductase action, which represents also the main place of drug elimination. Our results suggest that a higher clearance would be associated with higher atorvastatin hepatic extraction from the blood to the hepatocyte thereby promoting the interaction between atorvastatin and its target in the hepatic tissue. This is supported by a preclinical study that linked the hepatic uptake of statins to their cholesterol‐lowering efficacy in a mouse model of hyperlipidemia. 54 A higher atorvastatin CL/F can thus potentially lead to a stronger HMG‐CoA reductase inhibition and, consequently, a larger reduction of cholesterol levels. This is in line with the fact that several studies associated the SLCO1B1 c.521T>C SNP with lower atorvastatin efficacy. 18 , 19 , 22 Furthermore, a higher atorvastatin CL/F might be also associated with a higher active metabolite formation, which accounts for about 70% of the circulating HMG‐CoA reductase inhibitory activity. 55 Accordingly, we observed a positive correlation between the mean metabolite ratio of atorvastatin hydroxy‐metabolites among patients with a higher atorvastatin clearance (r = 0.30, p = 0.015, data not shown).
The SLCO1B1 c521T>C SNP (rs4149056, p.V174A) constitutes a functional SNP that decreases the intrinsic transport activity of the encoded protein, 56 OATP1B1, a hepatic xenobiotic influx transporter participating in the extraction of statins from the portal vein into the hepatocyte, 56 the first step of statin hepatic elimination. In healthy volunteers and in patients, carrying one or two variant alleles has been consistently associated with an increase in atorvastatin AUC. 32 , 35 , 36 , 37 , 38 , 39 The influence of this variant on atorvastatin PKs has been ultimately confirmed in a comprehensive genomewide association study. 57 Furthermore, despite controverted, it has been linked with atorvastatin‐related muscle toxicity, with the variant allele predisposing to toxicity in a meta‐analysis. 18 The fact that the SLCO1B1 c.521T>C SNP was retained in our final PopPK model and was associated with lower CL/F that in turn correlates with toxicity and efficacy outcomes is thus in line with data reported in the literature. All in all, it suggests that carriers of the variant allele are characterized by a lower clearance, putting them at risk of myalgia and lower cholesterol‐lowering effect. The former association might potentially be explained by a higher blood exposure in patients with lower CL/F and thus a higher risk of muscle side effects while the latter observation might reflect a lower HMG‐CoA reductase inhibitory effect in patients with low CL/F values due to a lower drug hepatic extraction. Concerning myotoxicity, as mentioned in the introduction, it should be kept in mind that atorvastatin lactone is suspected to be more muscle toxic than the acid form. 10 , 58 In the present data set, the collected PK data were too sparse to allow modeling the metabolite PKs. However, we did observe a relatively good correlation between atorvastatin acid and lactone concentrations (r = 0.56, p < 0.0001, data not shown) whatever the time post‐intake or the atorvastatin dose, indicating that the relationship linking atorvastatin CL/F and toxicity outcomes might be a reflect of this association. In line with this, patients with a low atorvastatin CL/F (i.e., ≤414.67 L h−1) had on average higher atorvastatin‐lactone concentrations (17.19 nM ±3.00 vs. 6.82 ± 2.05, p = 0.0058) considering the whole follow‐up period.
Even if not retained in our final model, the clinician should also consider the impact of co‐administration of OATP2B1 inhibitors in atorvastatin‐treated patients. OATP2B1 is an uptake transporter located in the luminal membrane of enterocytes and in the liver and therefore, facilitates the absorption and the excretion of the drug. It is thus not surprising that, in our analysis, patients co‐medicated with an inhibitor of OATP2B1 had lower CL/F than the rest of the population, as atorvastatin has been identified as an OATP2B1 substrate. 16 However, it has been also shown that OATP1B1 and OATP1B3 are the major atorvastatin uptake transporters of atorvastatin in the liver and that OATP2B1 probably plays a more important role in other tissues where other OATP transporters are underrepresented. 16 Interestingly, Knauer et al. have demonstrated that OATP2B1 is expressed on the sarcolemmal membrane of human skeletal muscle fiber and that its expression increases intracellular accumulation and toxicity of statins. 13 In that way, co‐administration of inhibitors might potentially counteract this influx activity and protect against drug myocyte accumulation. It is thus not clear whether co‐administration of OATP2B1 inhibitors would be harmful (decreased CL/F) or beneficial (decreased muscle accumulation) for the patient but this finding supports recommendations that co‐administration of commonly prescribed OATP2B1 inhibitors, such as L‐thyroxin, should be considered in clinical decisions and warrants further studies.
Our study has several obvious limitations. Because the available concentration‐time data in the investigation cohort were sparse and collected in an ambulatory environment, the development of the structural model had to be supported by previously collected rich datasets. This approach might have biased the results as the cohorts are not homogenous and the analytical techniques used were not the same. Furthermore, because of the sparse nature of sampling and the limited amount of data, the population values of the intercompartmental clearance, the central volume of distribution, and the peripheral volume of distribution were characterized by high shrinkage values that, because of the potential distribution bias, limits the assessment of the impact of covariates on these parameters. Because the precision of the estimated CL/F parameter in the investigation cohort was quite good (RSE = 6.2%) and the estimation was comparable to previous PopPK studies, we decided thus to focus our covariates analysis on CL/F even if because of the high shrinkage value on this PK parameter, the analysis of the effect of selected covariates on CL/F must be interpreted with caution. Another limitation of our study is that time‐varying covariates were summarized over the entire observation period as it was not possible to characterize interoccasion PK variability in any of the datasets. In addition, even if different atorvastatin dosages were considered in the present study, due to the limited amount of collected data per dosage scheme, it was not possible to evaluate the linearity of atorvastatin PKs. Finally, we were not able to model metabolite PK data and to link it with atorvastatin acid PopPK model. Our approach has thus the limitation to not consider other active metabolites.
In conclusion, our study demonstrates that PopPK modeling can be useful in daily clinics to estimates a patient’s atorvastatin clearance. Even if not directly implementable in clinical routine, our results suggest that notifying the clinician with this information might help in identifying patients at risk of myalgia and gives indication about the potential responsiveness to atorvastatin therapy in complement to cholesterol measurements.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
G.S., A.A., and L.E. wrote the manuscript. G.S., A.A., J.L.B., V.H., and L.E. designed the research. G.S., A.P., G.G.M., E.H., J.‐L.B., V.H., and L.E. performed the research. G.S., A.P., G.G.M., and L.E. analyzed the data. G.S., A.P., G.G.M., and N.P. contributed new reagents/analytical tools.
Supporting information
Table S1
Fig S1
Stillemans G, Paquot A, Muccioli GG, et al. Atorvastatin population pharmacokinetics in a real‐life setting: Influence of genetic polymorphisms and association with clinical response. Clin Transl Sci. 2022;15:667–679. doi: 10.1111/cts.13185
Funding information
This work was supported by the Fonds pour la Formation à la Recherche dans l’Industrie et dans l’Agriculture (FRIA) under Grant Number FC16749 to G.S. and FC33437 to E.H., and by grants from the Fonds national de la Recherche scientifique [FRS‐FNRS] (Grants J018317 and J006920).
REFERENCES
- 1. Ference BA, Ginsberg HN, Graham I, et al. Low‐density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38:2459‐2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Piepoli MF, Hoes AW, Agewall S, et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: the Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts)Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR). Eur Heart J. 2016;37:2315‐2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Adams SP, Tsang M, Wright JM. Atorvastatin for lowering lipids. Cochrane Database Syst Rev. 2015. 10.1002/14651858.CD008226.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Abd TT, Jacobson TA. Statin‐induced myopathy: a review and update. Expert Opin Drug Saf. 2011;10:373‐387. [DOI] [PubMed] [Google Scholar]
- 5. Lennernas H. Clinical pharmacokinetics of atorvastatin. Clin Pharmacokinet. 2003;42:1141‐1160. [DOI] [PubMed] [Google Scholar]
- 6. Stern RH, Yang B‐B, Hounslow NJ, MacMahon M, Abel RB, Olson SC. Pharmacodynamics and pharmacokinetic‐pharmacodynamic relationships of atorvastatin, an HMG‐CoA reductase inhibitor. J Clin Pharmacol. 2000;40:616‐623. [PubMed] [Google Scholar]
- 7. Thompson PD, Clarkson PM, Rosenson RS. & National Lipid Association Statin Safety Task Force Muscle Safety Expert, P. An assessment of statin safety by muscle experts. Am J Cardiol. 2006;97:69C‐76C. [DOI] [PubMed] [Google Scholar]
- 8. Cilla DD Jr, Whitfield LR, Gibson DM, Sedman AJ, Posvar EL. Multiple‐dose pharmacokinetics, pharmacodynamics, and safety of atorvastatin, an inhibitor of HMG‐CoA reductase, in healthy subjects. Clin Pharmacol Ther. 1996;60:687‐695. [DOI] [PubMed] [Google Scholar]
- 9. Jacobsen W, Kuhn B, Soldner A, et al. Lactonization is the critical first step in the disposition of the 3‐hydroxy‐3‐methylglutaryl‐CoA reductase inhibitor atorvastatin. Drug Metab Dispos. 2000;28:1369‐1378. [PubMed] [Google Scholar]
- 10. Skottheim IB, Gedde‐Dahl A, Hejazifar S, Hoel K, Asberg A. Statin induced myotoxicity: the lactone forms are more potent than the acid forms in human skeletal muscle cells in vitro. Eur J Pharm Sci. 2008;33:317‐325. [DOI] [PubMed] [Google Scholar]
- 11. Kearney AS, Crawford LF, Mehta SC, Radebaugh GW. The interconversion kinetics, equilibrium, and solubilities of the lactone and hydroxyacid forms of the HMG‐CoA reductase inhibitor, CI‐981. Pharm Res. 1993;10:1461‐1465. [DOI] [PubMed] [Google Scholar]
- 12. Chen C, Mireles RJ, Campbell SD, et al. Differential interaction of 3‐hydroxy‐3‐methylglutaryl‐coa reductase inhibitors with ABCB1, ABCC2, and OATP1B1. Drug Metab Dispos. 2005;33:537‐546. [DOI] [PubMed] [Google Scholar]
- 13. Knauer MJ, Urquhart BL, Meyer zu Schwabedissen HE, et al. Human skeletal muscle drug transporters determine local exposure and toxicity of statins. Circ Res. 2010;106:297‐306. [DOI] [PubMed] [Google Scholar]
- 14. Ellis LC, Hawksworth GM, Weaver RJ. ATP‐dependent transport of statins by human and rat MRP2/Mrp2. Toxicol Appl Pharmacol. 2013;269:187‐194. [DOI] [PubMed] [Google Scholar]
- 15. Shitara Y, Sugiyama Y. Pharmacokinetic and pharmacodynamic alterations of 3‐hydroxy‐3‐methylglutaryl coenzyme A (HMG‐CoA) reductase inhibitors: drug‐drug interactions and interindividual differences in transporter and metabolic enzyme functions. Pharmacol Ther. 2006;112:71‐105. [DOI] [PubMed] [Google Scholar]
- 16. Vildhede A, Karlgren M, Svedberg EK, et al. Hepatic uptake of atorvastatin: influence of variability in transporter expression on uptake clearance and drug‐drug interactions. Drug Metab Dispos. 2014;42:1210‐1218. [DOI] [PubMed] [Google Scholar]
- 17. Hirota T, Fujita Y, Ieiri I. An updated review of pharmacokinetic drug interactions and pharmacogenetics of statins. Expert Opin Drug Metab Toxicol. 2020;16:809‐822. [DOI] [PubMed] [Google Scholar]
- 18. Du Y, Wang S, Chen Z, Sun S, Zhao Z, Li X. Association of SLCO1B1 polymorphisms and atorvastatin safety and efficacy: a meta‐analysis. Curr Pharm Des. 2018;24:4044‐4050. [DOI] [PubMed] [Google Scholar]
- 19. Mladenovska K, Daka Grapci A, Vavlukis M, et al. Influence of SLCO1B1 polymorphisms on atorvastatin efficacy and safety in Macedonian subjects. Pharmazie. 2017;72:288‐295. [DOI] [PubMed] [Google Scholar]
- 20. Zhang L, Lv H, Zhang Q, et al. Association of SLCO1B1 and ABCB1 genetic variants with atorvastatin‐induced myopathy in patients with acute ischemic stroke. Curr Pharm Des. 2019;25:1663‐1670. [DOI] [PubMed] [Google Scholar]
- 21. Woo HI, Kim SR, Huh W, Ko JW, Lee SY. Association of genetic variations with pharmacokinetics and lipid‐lowering response to atorvastatin in healthy Korean subjects. Drug Des Devel Ther. 2017;11:1135‐1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kadam P, Ashavaid TF, Ponde CK, Rajani RM. Genetic determinants of lipid‐lowering response to atorvastatin therapy in an Indian population. J Clin Pharm Ther. 2016;41:329‐333. [DOI] [PubMed] [Google Scholar]
- 23. Qu KK, Zhang C‐N, Dong L‐X, Wang S‐S, Zhang Z‐D, Zhang L. Association of ABCB1 polymorphisms with lipid homeostasis and liver injury response to atorvastatin in the Chinese population. Can J Physiol Pharmacol. 2020;98:15‐22. [DOI] [PubMed] [Google Scholar]
- 24. Mirosevic Skvrce N, Šarinić VM, Šimić I, Ganoci L, Katanec DM, Božina N. ABCG2 gene polymorphisms as risk factors for atorvastatin adverse reactions: a case‐control study. Pharmacogenomics. 2015;16:803‐815. [DOI] [PubMed] [Google Scholar]
- 25. Yue YH, Bai X‐D, Zhang H‐J, et al. Gene polymorphisms affect the effectiveness of atorvastatin in treating ischemic stroke patients. Cell Physiol Biochem. 2016;39:630‐638. [DOI] [PubMed] [Google Scholar]
- 26. Wei KK, Zhang LR. Interactions between CYP3A5*3 and POR*28 polymorphisms and lipid lowering response with atorvastatin. Clin Drug Investig. 2015;35:583‐591. [DOI] [PubMed] [Google Scholar]
- 27. Prado Y, Zambrano T, Salazar LA. Transporter genes ABCG2 rs2231142 and ABCB1 rs1128503 polymorphisms and atorvastatin response in Chilean subjects. J Clin Pharm Ther. 2018;43:87‐91. [DOI] [PubMed] [Google Scholar]
- 28. Leon‐Cachon RBR, Ascacio‐Martínez JA, Gamino‐Peña ME, et al. A pharmacogenetic pilot study reveals MTHFR, DRD3, and MDR1 polymorphisms as biomarker candidates for slow atorvastatin metabolizers. BMC Cancer. 2016;16:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsamandouras N, Guo Y, Wendling T, Hall S, Galetin A, Aarons L. Modelling of atorvastatin pharmacokinetics and the identification of the effect of a BCRP polymorphism in the Japanese population. Pharmacogenet Genomics. 2017;27:27‐38. [DOI] [PubMed] [Google Scholar]
- 30. Ramakumari N, Indumathi B, Katkam SK, Kutala VK. Impact of pharmacogenetics on statin‐induced myopathy in South‐Indian subjects. Indian Heart J. 2018;70(Suppl 3):S120‐S125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Behdad N, Kojuri J, Azarpira N, Masoomi A, Namazi S. Association of ABCB1 (C3435T) and ABCC1 (G2012T) polymorphisms with clinical response to atorvastatin in Iranian patients with primary hyperlipidemia. Iran Biomed J. 2017;21:120‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rajput TA, Naveed AK, Farooqi ZR, Khan S. Effects of two functionally important SLCO1B1 gene polymorphisms on pharmacokinetics of atorvastatin. Pak J Pharm Sci. 2017;30:1363‐1370. [PubMed] [Google Scholar]
- 33. Drogari E, Ragia G, Mollaki V, et al. POR*28 SNP is associated with lipid response to atorvastatin in children and adolescents with familial hypercholesterolemia. Pharmacogenomics. 2014;15:1963‐1972. [DOI] [PubMed] [Google Scholar]
- 34. Becker ML, Elens LLFS, Visser LE, et al. Genetic variation in the ABCC2 gene is associated with dose decreases or switches to other cholesterol‐lowering drugs during simvastatin and atorvastatin therapy. Pharmacogenomics J. 2013;13:251‐256. [DOI] [PubMed] [Google Scholar]
- 35. Wang Y, Tian Y, Lv P, et al. The effect of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and 2‐hydroxyatorvastatin in healthy Chinese people. Pharmazie. 2017;72:365‐368. [DOI] [PubMed] [Google Scholar]
- 36. Pasanen MK, Fredrikson H, Neuvonen PJ, Niemi M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin Pharmacol Ther. 2007;82:726‐733. [DOI] [PubMed] [Google Scholar]
- 37. Birmingham BK, Bujac SR, Elsby R, et al. Impact of ABCG2 and SLCO1B1 polymorphisms on pharmacokinetics of rosuvastatin, atorvastatin and simvastatin acid in Caucasian and Asian subjects: a class effect? Eur J Clin Pharmacol. 2015;71:341‐355. [DOI] [PubMed] [Google Scholar]
- 38. Lee YJ, Lee MG, Lim LA, Jang SB, Chung JY. Effects of SLCO1B1 and ABCB1 genotypes on the pharmacokinetics of atorvastatin and 2‐hydroxyatorvastatin in healthy Korean subjects. Int J Clin Pharmacol Ther. 2010;48:36‐45. [DOI] [PubMed] [Google Scholar]
- 39. Daka A, Dimovski A, Kapedanovska A, et al. Effects of single nucleotide polymorphisms and haplotypes of the SLCO1B1 gene on the pharmacokinetic profile of atorvastatin in healthy Macedonian volunteers. Pharmazie. 2015;70:480‐488. [PubMed] [Google Scholar]
- 40. Duan P, Zhao P, Zhang L. Physiologically Based Pharmacokinetic (PBPK) modeling of pitavastatin and atorvastatin to predict Drug‐Drug Interactions (DDIs). Eur J Drug Metab Pharmacokinet. 2017;42:689‐705. [DOI] [PubMed] [Google Scholar]
- 41. Li S, Yiqun YU, Jin Z, et al. Prediction of pharmacokinetic drug‐drug interactions causing atorvastatin‐induced rhabdomyolysis using physiologically based pharmacokinetic modelling. Biomed Pharmacother. 2019;119:109416. [DOI] [PubMed] [Google Scholar]
- 42. Morse BL, Alberts JJ, Posada MM, et al. Physiologically‐based pharmacokinetic modeling of atorvastatin incorporating delayed gastric emptying and acid‐to‐lactone conversion. CPT Pharmacometrics Syst Pharmacol. 2019;8:664‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Reig‐López J, García‐Arieta A, Mangas‐Sanjuán V, Merino‐Sanjuán M. Current evidence, challenges, and opportunities of physiologically based pharmacokinetic models of atorvastatin for decision making. Pharmaceutics. 2021;13:709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang T. Physiologically based pharmacokinetic modeling of disposition and drug‐drug interactions for atorvastatin and its metabolites. Eur J Pharm Sci. 2015;77:216‐229. [DOI] [PubMed] [Google Scholar]
- 45. Knebel W, Gastonguay MR, Malhotra B, et al. Population pharmacokinetics of atorvastatin and its active metabolites in children and adolescents with heterozygous familial hypercholesterolemia: selective use of informative prior distributions from adults. J Clin Pharmacol. 2013;53:505‐516. [DOI] [PubMed] [Google Scholar]
- 46. Narwal R, Akhlaghi F, Asberg A, Hermann M, Rosenbaum SE. Development of a population pharmacokinetic model for atorvastatin acid and its lactone metabolite. Clin Pharmacokinet. 2010;49:693‐702. [DOI] [PubMed] [Google Scholar]
- 47. Schwartz JB, Verotta D. Population analyses of atorvastatin clearance in patients living in the community and in nursing homes. Clin Pharmacol Ther. 2009;86:497‐502. [DOI] [PubMed] [Google Scholar]
- 48. Dostalek M, Sam W‐J, Paryani KR, et al. Diabetes mellitus reduces the clearance of atorvastatin lactone: results of a population pharmacokinetic analysis in renal transplant recipients and in vitro studies using human liver microsomes. Clin Pharmacokinet. 2012;51:591‐606. [DOI] [PubMed] [Google Scholar]
- 49. Hermann M, Bogsrud M, Molden E, et al. Exposure of atorvastatin is unchanged but lactone and acid metabolites are increased several‐fold in patients with atorvastatin‐induced myopathy. Clin Pharmacol Ther. 2006;79:532‐539. [DOI] [PubMed] [Google Scholar]
- 50. Lemahieu WP, Hermann M, Asberg A, et al. Combined therapy with atorvastatin and calcineurin inhibitors: no interactions with tacrolimus. Am J Transplant. 2005;5:2236‐2243. [DOI] [PubMed] [Google Scholar]
- 51. FDA . Drug development and drug interactions: table of substrates, inhibitors and inducers. 2021.
- 52. pharmacothérapeutique, C.b.d.i . Répertoire Commenté Des Médicaments (2020).
- 53. Karlgren M, Vildhede A, Norinder U, et al. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): influence of protein expression on drug‐drug interactions. J Med Chem. 2012;55:4740‐4763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. van de Steeg E, Kleemann R, Jansen HT, et al. Combined analysis of pharmacokinetic and efficacy data of preclinical studies with statins markedly improves translation of drug efficacy to human trials. J Pharmacol Exp Ther. 2013;347:635‐644. [DOI] [PubMed] [Google Scholar]
- 55. FDA . FDA label ‐ Atorvastatin.
- 56. Kameyama Y, Yamashita K, Kobayashi K, Hosokawa M, Chiba K. Functional characterization of SLCO1B1 (OATP‐C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15+C1007G, by using transient expression systems of HeLa and HEK293 cells. Pharmacogenet Genomics. 2005;15:513‐522. [DOI] [PubMed] [Google Scholar]
- 57. Turner RM, Fontana V, Zhang JE, et al. A Genome‐wide association study of circulating levels of atorvastatin and its major metabolites. Clin Pharmacol Ther. 2020;108:287‐297. [DOI] [PubMed] [Google Scholar]
- 58. Skottheim IB, Bogsrud MP, Hermann M, Retterstol K, Asberg A. Atorvastatin metabolite measurements as a diagnostic tool for statin‐induced myopathy. Mol Diagn Ther. 2011;15:221‐227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Fig S1