

ABSTRACT
The enzyme glutaminyl-peptide cyclotransferase-like protein (QPCTL) catalyzes the formation of pyroglutamate residues at the NH2-terminus of proteins, thereby influencing their biological properties. A number of studies have implicated QPCTL in the regulation of chemokine stability. Furthermore, QPCTL activity has recently been shown to be critical for the formation of the high-affinity SIRPα binding site of the CD47 “don’t-eat-me” protein. Based on the latter data, interference with QPCTL activity —and hence CD47 maturation—may be proposed as a means to promote anti-tumor immunity. However, the pleiotropic activity of QPCTL makes it difficult to predict the effects of QPCTL inhibition on the tumor microenvironment (TME). Using a syngeneic mouse melanoma model, we demonstrate that QPCTL deficiency alters the intra-tumoral monocyte-to-macrophage ratio, results in a profound increase in the presence of pro-inflammatory cancer-associated fibroblasts (CAFs) relative to immunosuppressive TGF-β1-driven CAFs, and leads to an increased IFN and decreased TGF-β transcriptional response signature in tumor cells. Importantly, the functional relevance of the observed TME remodeling is demonstrated by the synergy between QPCTL deletion and anti PD-L1 therapy, sensitizing an otherwise refractory melanoma model to anti-checkpoint therapy. Collectively, these data provide support for the development of strategies to interfere with QPCTL activity as a means to promote tumor-specific immunity.
KEYWORDS: QPCTL, tumor micro-environment, cancer-associated fibroblasts, genetically modified mouse model
Introduction
Regulation of immune cell activity at sites of infection or cancer growth frequently occurs through a balance of signals that are received by immune activating and immune inhibitory receptors.1 For example, while activation of myeloid cells, including neutrophils, macrophages and monocytes, often occurs through Fc receptor signaling, such activation can be prevented through the simultaneous engagement of ITIM/ITSM-containing inhibitory receptors such as SIRPα. Specifically, binding of the “don’t-eat-me” signal CD47, which is widely expressed on hematopoietic and non-hematopoietic cells, to the SIRPα receptor on myeloid cells has been shown to result in decreased myeloid effector function, including suppression of target cell phagocytosis by macrophages and tumor cell killing by neutrophils.2–4
The inhibitory capacity of CD47 is dependent on the maturation of its SIRPα binding site by the ER-resident enzyme QPCTL.5,6 Similar to its secreted family member QPCT, QPCTL catalyzes the cyclization of N-terminal glutamine and glutamic acid residues on target proteins into a pyroglutamate residue (pGlu).7,8 As shown by structural analysis, the pGlu residue at the N-terminus of CD47 contributes to the interaction surface with SIRPα9and, through genetic screening, it was shown that the activity of QPCTL is critical for the formation of this residue, making this enzyme a key regulator of the high-affinity CD47-SIRPα-binding site.6 In line with this, prevention of pGlu formation on CD47, either by genetic knock-out or small-molecule inhibition, leads to reduced SIRPα binding and increased macrophage- and neutrophil-dependent killing of antibody-opsonized target cells. Based on its role in regulating CD47-SIRPα signaling, and the possibility to develop small-molecule inhibitors of enzymatic activity, QPCTL forms a potentially interesting target in cancer immunotherapy.
In addition to CD47, chemokines, such as CCL2 and CX3CL1 have been identified as QPCTL and/or QPCT substrates.10–12 The formation of the N-terminal pGlu on CCL2 was shown to increase its in vivo activity, both by conferring resistance to aminopeptidases and by increasing its capacity to induce CCR2 signaling.10 Likewise, pGlu-modified CX3CL1 appears to show an increased capacity to promote CX3CR1 signaling in vitro.12 Finally, around 600 human proteins harbor a N-terminal glutamine or glutamic acid residue after the predicted signal peptide cleavage site, and it is plausible that additional QPCTL/QPCT substrates exist amongst this group of proteins.13
Because of its known or potential role in the post-translational modification of different immune- or tumor cell-related molecules, it is difficult to predict the overall effects of QPCTL inhibition on the tumor microenvironment (TME), and the poor pharmacokinetics of available QPCT/QPCTL inhibitors such SEN1776 has precluded evaluation of such effects by small-molecule inhibition. To address this question, we have generated a QPCTL-deficient mouse model and combined it with QPCTL-deficient tumor cells, to map the effects of QPCTL deficiency of either cellular compartment on the composition of the TME. The obtained data reveal that QPCTL deficiency results in a skewing of the macrophage–monocyte ratio, causes an approximately 20-fold change in the balance between TGF-β-producing myofibroblastic cancer-associated fibroblasts (myCAFs) and cytokine-secreting inflammatory CAFs (iCAFs), and shifts tumor cells from a TGF-β-responding to an IFN-responding state. Collectively, these changes convert the TME to pro-inflammatory environment that sensitizes tumors to anti-PD-L1 therapy. Together, these results suggest that therapeutic manipulation of QPCTL activity may synergize with current cancer immunotherapies.
Genetic deletion of QPCTL does not significantly alter the immune compartment
Previous work has indicated that genetic deletion of QPCTL can be used to study its function in vivo;10,14 however, the possible effects of QPCTL inactivation on the TME has not been studied. To determine how blockade of QPCTL activity alters the TME, we applied CRISPR-Cas9 mediated gene-editing to generate a QPCTL-deficient (QPCTL−/−) C57BL/6 mouse strain that is compatible with commonly used tumor models. To first determine whether QPCTL deficiency results in abrogation of pGlu formation on CD47, peripheral blood cells from QPCTL−/− mice and WT littermates were stained with mouse SIRPα and an anti-mouse CD47 antibody that recognizes CD47 independent of pyroglutamate formation.6 As compared to WT littermates, blood cells of QPCTL−/− mice displayed significantly decreased SIRPα binding, thereby providing the first evidence that QPCTL is also a critical CD47 modifier in vivo (Figure 1a-b).
Figure 1.
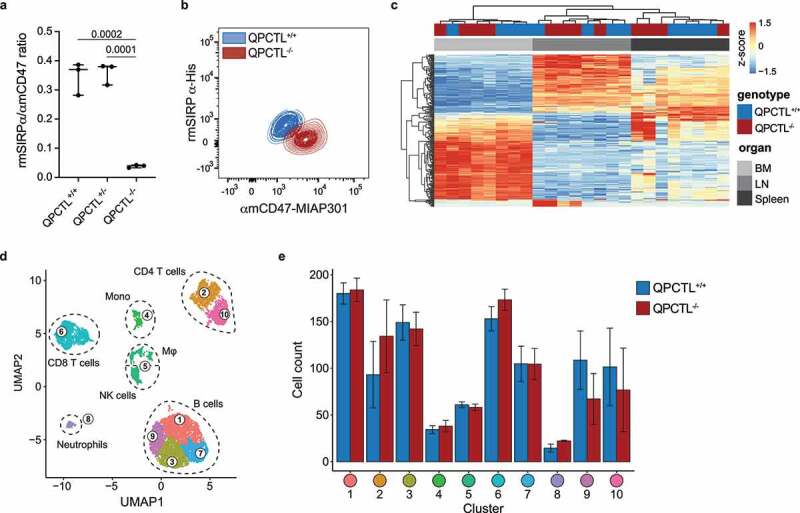
Generation and characterization of QPCTL-deficient mice. (a) Ratio of recombinant mouse (rm)SIRPα-His and anti-mouse (αm)CD47 antibody (clone MIAP301) binding to blood cells from QPCTL+/+, QPCTL+/-, and QPCTL−/− mice, as measured by flow cytometry. Dots depict the ratio of rmSIRPα-His/αmCD47-MIAP301 mean fluorescence intensity (MFI) on blood cells from individual mice, group medians are indicated and whiskers represent min/max, n = 3 mice per group. (b) Flow cytometry plot depicting data described in panel a for blood cells from a representative QPCTL+/+ and QPCTL−/− mouse. (c) Heatmap depicting hierarchical clustering performed on the 1,000 most differentially expressed genes in bone marrow (BM), lymph node (LN) and spleen samples from QPCTL+/+ and QPCTL−/− mice. (d-e) Unbiased Euclidean distance-based clustering of immune cells obtained from spleens of QPCTL+/+ and QPCTL−/− mice. UMAP 2-dimensional projection (d) depicts the obtained clusters. Cell counts of both genotypes within each cluster are depicted (e). Bars indicate group means, error bars represent standard error of the mean. P values were determined by one-way ANOVA followed by Tukey’s HSD test (a) or by two-sided Student’s T test with Bonferroni correction for multiple testing (e). Significant P values (< 0.05) are indicated in the plots. Data are representative of 3 independent experiments (a-b), or were obtained in a single experiment (c-d). UMAP, Uniform Manifold Approximation and Projection.
To understand how QPCTL deficiency influences steady-state immune cell frequencies and gene expression, QPCTL−/− and QPCTL+/+ littermates were subjected to histopathological, transcriptomic and flow cytometric analysis. Histopathological assessment of QPCTL−/− mice revealed no significant morphological aberrations relative to littermate controls (Supplementary Data 1), and gene expression analysis of spleen, lymph nodes, and bone marrow revealed no genotype-specific transcriptional changes (Figure 1c, Supplementary Fig 1a), indicating that QPCTL deficiency does not result in major alterations in steady-state immune activity. Likewise, no substantial differences in cell counts or immune cell population frequencies were observed in blood (Supplementary Fig. 1b-c). In spleen, a modest increase in the frequency of NK cells of total non-myeloid cells and a decrease in the fraction of activated cells of total CD4+ T cells was observed in QPCTL−/− mice, but no significant changes in other immune cell-type frequencies were identified (Supplementary Fig. 1d). The absence of substantial differences in immune cell frequencies was corroborated by unbiased hierarchical clustering of cells obtained from QPCTL+/+ and QPCTL−/− spleen samples (Figure 1d and Supplementary Fig. 1e).
QPCTL-deficiency alters macrophage-monocyte-ratios in the TME
To test whether systemic QPCTL deficiency influences immune cell infiltration in the TME, QPCTL+/+ and QPCTL−/− mice were inoculated with wild-type (QPCTL-WT) and QPCTL knock-out (QPCTL-KO) B16F10 melanoma cells (Supplementary Fig. 2a), respectively, and TMEs were analyzed by flow cytometry 14–16 days post inoculation. QPCTL-proficient and -deficient tumors grew with similar kinetics and were similarly infiltrated by large numbers of myeloid cells (Supplementary Fig. 2b-c). Importantly, within the myeloid subset, QPCTL-deficient TMEs exhibited a significant higher frequency of macrophages and a substantially increased macrophage-monocyte (Mφ-Mo) ratio (Figure 2a). Although no other significant changes within the immune infiltrate could be detected (Supplementary Fig. 2d), an increase in the frequency of B cells, a trend toward a decrease in monocytes, and a decrease in the frequency of CD4+ T cells within the non-myeloid immune cell subset was observed in peripheral blood samples from QPCTL-deficient tumor-bearing mice (Supplementary Fig. 2e). Also, when immune infiltrates of QPCTL-deficient and -proficient TMEs were analyzed through unbiased hierarchical clustering, an increase in F4/80+ cells (macrophages) and a decrease in Ly6Chigh cells (monocytes) in QPCTL-deficient TMEs was observed (Figure 2b-c, Supplementary Fig. 2f).
Figure 2.
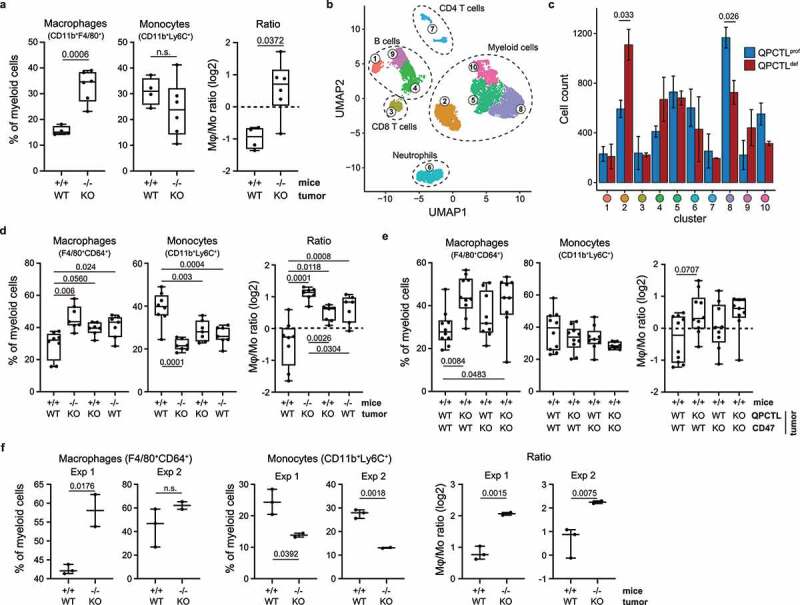
Tumor and host QPCTL deficiency alters the Mφ-Mo ratio in the TME. (a) Frequency of macrophages and monocytes of myeloid cells (CD11b+), and Mφ-Mo ratio, in the TME of QPCTL+/+ (n = 4) and QPCTL−/− (n = 6) mice inoculated with QPCTL-WT or QPCTL-KO B16F10 melanoma cells, respectively. Tumors were analyzed between 14–16 days post inoculation. (b) UMAP visualizing 30,000 cells sampled from the data shown in a. 5,000 cells were randomly drawn from each sample (n = 3 mice per group) prior to analysis. Colors indicate clusters obtained by Euclidean distance-based hierarchical clustering, cluster phenotype is shown in Supplementary Fig 2 f. (c) Contribution of cells from QPCTL-proficient (n = 3) and QPCTL-deficient (n = 3) TMEs to each cluster shown in panel b. Bars indicate group means, error bars represent standard error of the mean. (d) Frequency of macrophages and monocytes of myeloid cells (CD11b+), and Mφ-Mo ratio, in the TME of QPCTL+/+ and QPCTL−/− mice inoculated with either QPCTL-WT or QPCTL-KO B16F10 melanoma cells (n = 7–8 per group). Tumors were analyzed between 12–14 days post inoculation. (e) Frequency of macrophages and monocytes of myeloid cells (CD11b+), and Mφ-Mo ratio, in the TME of QPCTL+/+ mice inoculated with QPCTL-WT, QPCTL-KO, CD47-KO, or CD47/QPCTL double-KO (dKO) B16F10 cells. Tumors were analyzed between 14–16 days post inoculation. (f) Frequency of macrophages and monocytes of myeloid cells (CD11b+), and Mφ-Mo ratio, in the TME of QPCTL+/+ and QPCTL−/− mice inoculated with QPCTL-WT and QPCTL-KO MC38 cells, respectively. Data from 2 independent experiments are shown (n = 5 per experiment). Tumors were analyzed at 22 (experiment 1) or 29 (experiment 2) days post inoculation. Dots indicate measurements from individual mice, group medians are indicated and whiskers represent min/max. P values were determined by two-sided Student’s T test without (a, f) or with Bonferroni correction for multiple testing (c), or by one-way ANOVA followed by Tukey’s HSD test (d, e). Significant P values (< 0.05) are indicated in the plots. For all boxplots, dots represent individual mice, group median and 25th/ 75th percentiles are indicated by the box, whiskers indicate min/max. Data are representative of at least 2 independent experiments (a, d, f), or were obtained in a single experiment (b, c, e). UMAP, uniform manifold approximation and projection.
To determine whether the increased Mφ-Mo-ratio could be attributed to a lack of QPCTL activity in either host or tumor cells, QPCTL+/+ and QPCTL−/− mice were inoculated with either QPCTL-WT or QPCTL-KO melanoma tumor cells (Supplementary Fig. 3a). Both tumor and host QPCTL deficiency led to an increased Mφ-Mo-ratio, but the most profound increase in Mφ-Mo-ratios was observed when QPCTL activity was lacking in both cell compartments (Figure 2d, Supplementary Fig. 3b-c). In blood, the most pronounced differences in immune cell frequencies were found when comparing tumor-bearing versus non-tumor-bearing animals—independent of QPCTL activity—emphasizing that QPCTL deficiency does not impact the systemic immune compartment in a major way (Supplementary Fig. 3d). To explore to what extent loss of pGlu-modified CD47 contributed to the altered intra-tumoral Mφ-Mo balance, wild-type mice were inoculated with QPCTL-, CD47- or double-KO tumor cells. Absence of CD47 resulted in Mφ frequencies that were numerically higher than observed in recipients of WT B16 tumor cells, but to a lower extent than observed in recipients of QPCTL-KO cells, and did not significantly alter Mφ-Mo-ratios (Figure 2e). This suggests that, if the CD47-modifying activity of QPCTL contributes to the altered myeloid cell composition in these tumors, it likely plays a minor role.
To test whether the role of QPCTL as a modifier of the TME extended to other tumor models, QPCTL+/+ and QPCTL−/− mice were inoculated with QPCTL-WT and QPCTL-KO MC38 colon carcinoma cells, respectively. Following tumor outgrowth, a profound increase in Mφ-Mo-ratio was observed in QPCTL-deficient TMEs (Figure 2f, Supplementary Fig. 4a-b), while no significant changes were detected in other immune cell subsets (data not shown). Together, these data indicate that QPCTL affects the TME composition in at least two different tumor models, and that combined tumor and host QPCTL deficiency leads to a significant increase in Mφ-Mo-ratio.
QPCTL deficiency is associated with suppressed intra-tumoral melanogenesis and cell metabolism
Having established that QPCTL deficiency is associated with an alteration in intra-tumoral immune cell composition, we set out to investigate the effect of QPCTL deficiency on the tumor cell and stromal cell compartment of the TME. RNA sequencing of CD45-negative cell fractions from QPCTL-deficient and -proficient TMEs showed that QPCTL deficiency resulted in differential expression of a substantial set of genes (Figure 3a, Supplementary Fig. 5a), and network analysis revealed that expression of multiple genes involved in melanogenesis (Dct, Tyrp1, and Gpnmb) was reduced in QPCTL-deficient melanomas (Figure 3b). Assessment of the expression level of a broader set of genes involved in melanogenesis likewise showed dampening of this pathway (Figure 3c). Interestingly, these transcriptional changes coincided with the functional abrogation of melanin production, as evidenced by a loss of pigmentation of QPCTL-deficient tumors (Supplementary Fig. 5b). Expression of a network of genes involved in cell cycle (Mapk1, Akt1) and cell metabolism (Pgk1, Atp5a1, and Oxct1) was additionally found to be decreased in CD45-negative cells in the QPCTL-deficient setting (Figure 3b, d, Supplementary Fig. 5c). While dampened expression of these gene sets was consistently observed, effect sizes were small and had no discernible effect on tumor outgrowth (Supplementary Fig. 2b-c).
Figure 3.
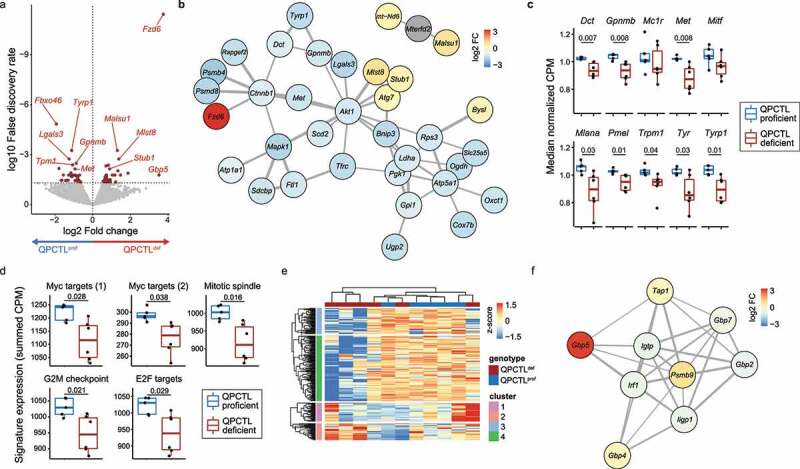
QPCTL deficiency results in suppression of melanogenesis and cell metabolism. mRNA sequencing was performed on sorted CD45-negative cells from QPCTL-proficient (n = 5) and QPCTL-deficient (n = 6) B16F10 TMEs. Tumors were harvested at day 14 post inoculation. (a) Differential gene expression analysis comparing CD45-negative cells obtained from QPCTL deficient versus QPCTL-proficient TMEs. Genes with a false discovery rate (FDR) < 0.05 are indicated in red. Selected genes are indicated in the plot. (b) network analysis (StringDB) performed on all significantly (FDR < 0.05) differentially expressed genes. Genes with a medium interaction strength (> 0.4) are included. Line thickness indicates interaction strength. Nodes are colored based on log2 fold differences obtained in a. (c) Transcript abundance of selected genes in the melanogenesis pathway. Boxplots indicate group median and 25th /75th percentiles, whiskers indicate the interquartile range multiplied by 1.5, dots signify individual samples. (d) Signature expression of cell cycle-associated hallmark signatures from MSigDB, calculated as the summed CPM of all genes within each signature. Boxplots indicate group median and 25th /75th percentiles, whiskers indicate the interquartile range multiplied by 1.5, dots signify individual samples. (e) Hierarchical clustering of the 1,000 most differentially expressed genes across all samples, depicted as a row-normalized heatmap. (f) Network analysis (StringDB) performed on genes from cluster 2 (e). Genes with a medium interaction strength (> 0.4) are included. Line thickness indicates interaction strength. Nodes are colored based on log2 fold differences obtained in panel a. P values were determined by one-way ANOVA followed by Tukey’s HSD test (c, d). Significant P values (< 0.05) are indicated in the plots. Data are representative of 2 independent experiments. CPM, counts per million; MSigDB, Molecular Signatures Database.
To further explore putative transcriptional alterations in the CD45-negative compartment as a result of QPCTL deletion, obtained transcriptomes were clustered based on the top 1,000 most variable genes, revealing a cluster of genes that was enriched in 3 out of 6 QPCTL-deficient samples (gene cluster 2, Figure 3e). Network analysis performed on this cluster showed that this gene set contained a small network comprised of IFN-induced genes (Gbp family members, Tap1, Irf1; Figure 3f). Interestingly, previous studies have shown that IFNγ can act as a suppressor of melanogenesis in B16F10 melanoma cells,15–17 a notion that potentially links this IFN signature to the observed decrease in tumor pigmentation.
Single cell transcriptomic profiling reveals remodeling of the tumor micro-environment by QPCTL deficiency
The above data indicate that QPCTL inactivation affects both immune cell composition of the TME and the transcriptome of non-immune cells at the tumor site. With the aim to potentially link these two observations, we inoculated QPCTL+/+ and QPCTL−/− mice with QPCTL-WT and QPCTL-KO B16F10 cells, respectively, and performed scRNAseq of both immune cells and non-immune cells from the resulting QPCTL-deficient and -proficient TMEs. Applying the MetaCell algorithm18 on 13,093 transcriptomes derived from 6 TMEs (3 QPCTL-proficient, 3 QPCTL-deficient), showed 3 transcriptionally divergent cell supertypes, reflecting immune cells (Ptprc, Itgam), fibroblasts (Col1a1, Acta2), and tumor cells (Mlana, Pmel) (Supplementary Fig. 6). To identify cell type-specific changes that accompany QPCTL deficiency, each of these supertypes was subsequently re-clustered and analyzed separately.
To investigate which of the transcriptional changes observed in the CD45-negative compartment could be mapped to the tumor cell compartment, this supertype was re-grouped into transcriptionally disparate MCs (Figure 4a, Supplementary figure 7a), and the relative contribution of cells from either QPCTL-deficient mice or QPCTL-proficient samples to the different MCs was examined. Strikingly, 1 MC (MC12) was nearly exclusively observed in QPCTL-deficient samples (Figure 4b). Examination of the marker genes of MC12 showed a prominent presence of IFN-induced transcripts Ifitm3, B2m, Bst2, and H2-D1 (Figure 4c), and gene-set enrichment analysis performed on MC12 marker genes identified both IFNγ and IFNα response as the strongest enriched gene-sets (Figure 4d, Supplementary figure 7b-c). IFNγ and IFNα/β response signatures show a high level of overlap,19–21 making it difficult to assign the observed response to either cytokine. To potentially deconvolute these signatures, and diagnose other putative cytokine response signatures, the CytoSig model19 was applied to MC12. In line with the GSEA, this analysis detected transcription response signatures of all IFN types, with the highest score being observed for IFNγ (Supplementary figure 7d). CytoSig additionally identified a reduction in TGF-β signaling in MC12, and retrospective analysis of the bulk RNAseq data revealed reduced expression of TGF-β responsive genes in QPCTL-deficient TMEs (Supplementary figure 7e). Furthermore, differential gene expression analysis showed that QPCTL deficiency was associated with a reduced TGF-β and an increased IFN responsive signature (Figure 4e-g) across all MCs, potentially indicating either an altered abundance of—or sensitivity to—these cytokines. Notably, in vitro sensitivity of B16F10 cells to both TGF-β and IFNγ was unaltered by QPCTL deficiency (Supplementary figure 7f-i), arguing in favor of an altered abundance of these cytokines in the TME.
Figure 4.
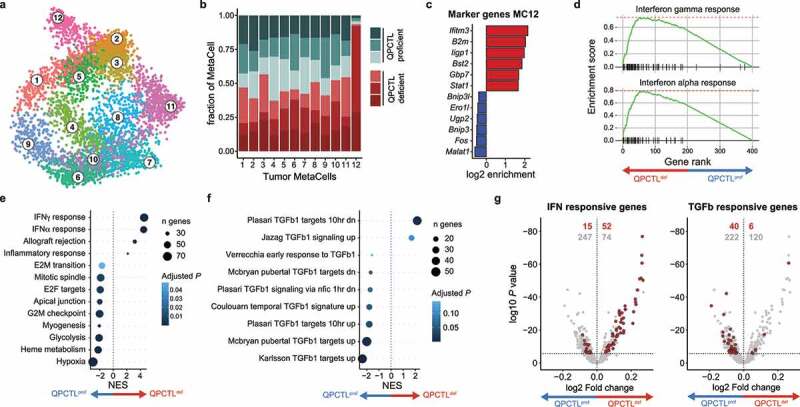
QPCTL deficiency leads to an increased IFN- and decreased TGF-β-response signature in tumor cells. scRNA sequencing was performed on sorted live cells from QPCTL-proficient (n = 3) and QPCTL-deficient (n = 3) B16F10 TMEs. Tumors were harvested at day 14 post inoculation. (a) 2-dimensional MetaCell projection of the tumor cell compartment. Single cells are colored by MetaCell. (b) Stacked bar chart depicting the sample composition of each tumor cell MetaCell. Cell counts from each sample were normalized to 1,000 cells. (c) Enrichment of marker genes (6 highest and lowest expressed) in tumor cell MetaCell 12. (d) Gene set enrichment analysis performed on the top and bottom 200 genes expressed by MC12 (see Supplementary Fig. 7b). Gene-enrichment plots for the IFNγ and IFNα response gene-sets are depicted. (e-f) Differential gene expression analysis comparing tumor cells derived from QPCTL-proficient and QPCTL-deficient TMEs, followed by gene set enrichment analysis using either hallmark (e) or immunologic signature (f) gene sets from MSigDB. Results obtained from the immunologic signature gene sets were filtered for those containing “TGFb”. Gene sets with a P < .05 are shown. (g) Volcano plots depicting differential gene expression analysis. Horizontal line indicates an adjusted P value cutoff of 0.05. IFN (left) or TGF-β (right) signature genes are highlighted in red (see Supplementary Table 1 for signature genes). Red numbers denote quantity of significant differentially expressed genes within the signature, gray numbers denote the quantity of remaining differentially expressed genes. Depicted data were obtained in a single experiment, consisting of 6 mice. NES, normalized enrichment score; MSigDB, Molecular Signatures Database.
Next, the immune cell compartment was grouped into 11 MetaCells (MCs), classified as either CD3+ lymphocytes (Cd8a, Cd3e), dendritic cells (Ccr7, H2-Aa), or macrophages/monocytes (Adgre1, Fcgr1; Figure 5a-b, Supplementary Fig. 8a). The macrophage/monocyte (Mφ/Mo) MCs could be further subdivided into two groups that were marked by high expression of either Ccr2 and Itga4 (Mφ/Mo-1 subgroup) or Ms4a7 and Pf4 (Mφ/Mo-2 subgroup; Figure 5b-c), suggestive of a blood- versus tissue-derived origin.22–25 Analysis of the contribution of cells from QPCTL-deficient and -proficient TMEs to individual MCs indicated that QPCTL deficiency changed the relative abundance of the different cell states that jointly comprised the Mφ/Mo-1 subgroup (Supplementary Fig. 8b). As intra-tumoral Mφ/Mo cells can exist within a continuum of transcriptional cell states,26,27 pseudotime analysis was performed on Mφ/Mo subgroup 1 to examine if such a continuum could be observed, and whether this was linked to QPCTL deficiency. This analysis demonstrated a strong continuous association between pseudotime (i.e. cell state) and sample-origin (Figure 5d). To investigate the transcriptional changes underlying this association, genes were clustered based on their expression kinetics across pseudotime (Supplementary Fig. 8c), revealing a gradual loss of expression of Mo-related genes Ly6c2 and Plac8 across pseudotime (Figure 5e), with the lowest expression levels found in the area that contained the highest fraction of cells from QPCTL TMEs. At the same time, an increase in transcripts linked to ‘inflammation-resolutory’ Mφ’s (Mrc1, Timp2), antigen-presentation (H2-Aa, Cd74) and Mφ effector function (Eps8, Ctsd, Ecm1, and Lipa)28–31 was observed. Consistent with these findings, Mφ/Mo cells that were more dominant in QPCTL-deficient samples had reduced expression of monocyte-associated transcripts, and displayed transcriptional similarity to previously identified tumor-associated macrophage subsets32 (Supplementary Fig. 8d). Together with the observed Mφ-Mo skewing (Figure 2), these data argue in favor of a model in which QPCTL deficiency in the TME leads to transcriptional changes that drive Mo-to-Mφ conversion. Moreover, the intra-tumoral macrophages identified by flow cytometry (Figure 2) exhibited high-level surface expression of MHC class II (H2-Aa), CD206 (Mrc1) and CCR2 (Ccr2), linking these cells to the Mφ/Mo population identified through scRNAseq (Supplementary Fig. 8e).
Figure 5.
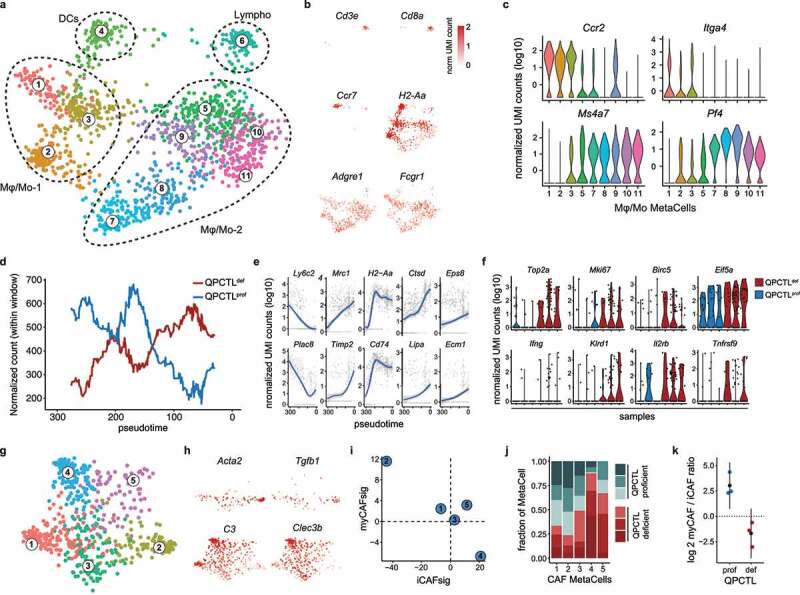
QPCTL deficiency alters the immune cell compartment and CAF polarization in the TME. scRNA sequencing was performed on sorted live cells from QPCTL-proficient (n = 3) and QPCTL-deficient (n = 3) B16F10 TMEs. Tumors were harvested at day 14 post inoculation. (a, b) 2-dimensional MetaCell projection of the immune cell compartment. Single cells are colored by metacell (a), or normalized UMI count (b) of selected genes. (c) Violin plots depicting normalized UMI counts of selected genes across Mφ/Mo MCs. (d,e) Slingshot trajectory analysis performed on Mφ/Mo subset 1 (MC1, 2 and 3). (d) QPCTL-deficient or QPCTL-proficient TMEs replicates were pooled, and normalized cell counts were tallied within windows of 60 cells wide, sliding 1 cell per frame. Lines indicate normalized cell counts within each window. (e) Normalized UMI counts of selected genes that are significantly associated with pseudotime. Blue lines indicate general additive linear models, grayed areas indicate confidence intervals, gray dots represent single cells. (f) Violin plots depicting normalized UMI counts of selected genes within the CD3+ lymphoid cell MetaCell (MC6). (g, h) 2-dimensional metacell projection of the fibroblast compartment. Single cells are colored by MetaCell (g), or normalized UMI count (h) of selected genes. (i) Enrichment of iCAF and myCAF signatures (Supplementary Table 2) in each CAF MetaCell. Signature values represent summed log2 transformed enrichment values, calculated using the MetaCell algorithm. (j) Stacked bar chart depicting sample composition of each CAF MetaCell. Cell counts from each sample were normalized to 1,000 cells. (k) myCAF/iCAF ratio detected in QPCTL-proficient and -deficient TMEs. Colored dots indicate individual mice, black dots indicate means, whiskers indicate the standard deviation. Depicted data were obtained in a single experiment, consisting of 6 mice. iCAF, inflammatory cancer-associated fibroblast; myCAF, myofibroblastic cancer-associated fibroblast; UMI, unique molecular identifier.
Congruent with the flow cytometric analysis, no differences in the frequencies of CD3+ lymphoid cells derived from QPCTL-deficient versus QPCTL-proficient TMEs could be observed in the scRNAseq dataset. However, as activated lymphoid cells are potent producers of IFN, we queried whether transcriptional features associated with lymphocyte activation were detected more frequently in lymphocytes from QPCTL-deficient TMEs. Interestingly, expression of genes associated with TCR-triggering (Ifng, Il2rb, and Tnfrsf9) and cell cycle activity (Top2a, Mki67, and Birc5) was detected more frequently in cells derived from the QPCTL-deficient tumors (Figure 5f). Likewise, lymphocytes derived from QPCTL-deficient TMEs showed increased expression of the chemokine genes Ccl3, Ccl4, and Ccl5, and of the T cell activation-related genes Ly6a, Nkg7, and Gzmb (Supplementary Fig. 8f). While the increase in Ifng gene expression in lymphocytes in QPCTL-deficient samples was only modest, the parallel observation of other aspects of lymphocyte activation in these samples suggests that these cells may, at least in part, be responsible for the IFN responsive signature that is observed in the tumor cell compartment.
Diverse subsets of cancer-associated fibroblasts (CAFs) that possess distinct immunomodulatory functions have been reported in the TME of different cancer types,33 and two highly distinct populations—termed TGF-β-producing myofibroblastic CAFs (myCAFs) and IL-1-driven inflammatory CAFs (iCAFs)—have been identified in a recent set of cross-species studies.34,35 As a reduced TGF-β response signature was identified as one of the characteristics of tumor cells in QPCTL-deficient tumors, we next asked whether QPCTL deficiency affected CAF polarization. MetaCell-based clustering within the fibroblast cell supertype resulted in five transcriptionally distinct MCs (Figure 5g, Supplementary Fig. 8g). Assessment of transcripts known to be involved in CAF function showed that MC2 was enriched for Tgfb1 transcripts and several myCAF markers (e.g. Acta2 and Itgb1), whereas MC4 exhibited more pronounced expression of genes involved in functional inhibition of TGF-β (Ltbp1, Dcn) and multiple iCAF markers (C3, Clec3b; Figure 5g, Supplementary Fig. 8h). In line with this, analysis of signature enrichment-scores pertaining to these two subsets showed that MC2 and MC4 scored the highest for either the myCAF or iCAF signature, respectively (Figure 5h). Moreover, increased surface expression of the myCAF and iCAF markers CD29/ITGB1 and Ly6C was detected on MC2 and MC4, respectively (Supplementary Fig. 8i). Strikingly, MC2 and MC4 displayed the highest depletion and enrichment in QPCTL-deficient and -proficient TMEs (Figure 5i), resulting in a 20-fold increase in the iCAF/myCAF ratio in QPCTL-deficient TMEs (Figure 5j). Thus, in the absence of QPCTL activity, polarization of fibroblasts toward TGF-β-producing myCAFs is reduced in favor of the more pro-inflammatory iCAFs.
QPCTL deficiency enhances susceptibility of B16F10 melanoma to anti-PD-L1 treatment
The B16F10 melanoma model commonly shows a poor response to single-agent PD1/PD-L1 checkpoint blockade.36,37 Having observed that QPCTL deficiency alters the TME to a more proinflammatory state, we hypothesized that QPCTL deletion may modulate the sensitivity of B16F10 tumors to such PD-1/PD-L1 blocking therapies. To test this, QPCTL+/+ and QPCTL−/− mice were inoculated with QPCTL-WT and QPCTL-KO B16F10 cells, respectively. Upon tumor formation (6 days post-inoculation) mice were treated with either an anti-PD-L1 or isotype control antibody. In QPCTL-proficient animals, tumor growth progressed rapidly and was not influenced by anti-PD-L1 therapy (Figure 6a). In contrast, anti-PD-L1 therapy did result in improved tumor control in QPCTL-deficient mice (Figure 6a), and led to a prolonged survival in approximately 50% of animals (Figure 6b), providing direct evidence that the TME modulatory effect of QPCTL removal has functional consequences.
Figure 6.
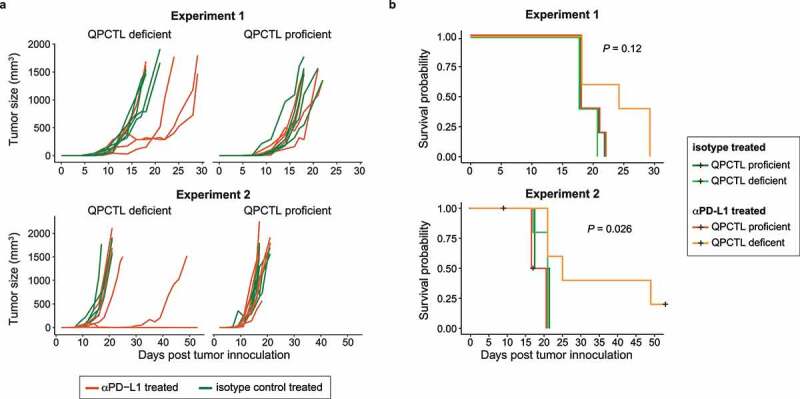
QPCTL deficiency sensitizes the tumor microenvironment to anti-PD-L1 treatment. QPCTL+/+ and QPCTL−/− mice were inoculated with QPCTL-WT and QPCTL-KO B16F10 melanoma cells, respectively. Each group subsequently received either anti-PD-L1 or isotype control antibody treatment at day 7, 9 and 11 post tumor inoculation. (a) Tumor growth curves, assessed until day 50 post tumor inoculation. Lines represent individual mice. Data from two experiments are depicted (n = 5 per group). (a) Survival probabilities of mice treated with anti-PD-L1 or isotype control antibody in a QPCTL-proficient and -deficient setting. Black plus-signs indicate censored events. Data from two experiments are depicted (n = 5 per group). Global P values were determined by log-rank test (b). Data from 2 independent experiments are depicted.
Discussion
QPCTL activity is known to influence the properties of a number of molecules that are active in the TME and may potentially influence additional—as of yet unidentified —substrates. To obtain a global view of the cumulative effects of QPCTL activity on the host’s immune response to tumor growth, we made use of a QPCTL−/− mouse model in combination with syngeneic QPCTL-KO tumor cell lines. We conclude that inactivation of QPCTL alters Mφ-Mo abundance, increases IFN pathway activity relative to TGF-β pathway activity, and leads to a profound increase in iCAFs relative to myCAF in the TME (Supplementary Fig. 9a). In line with the observed skewing of the TME to a pro-inflammatory state that is induced by QPCTL deficiency, we demonstrate that such deficiency leads to the sensitization of B16F10 melanomas to anti-PD-L1 treatment.
The current study has the following limitations: 1) Germline deletion of QPCTL may potentially lead to developmental alterations that influence the host’s response to tumor challenge, for example, affecting the capacity of certain CAF or immune subsets to differentiate, independent of QPCTL activity during tumor outgrowth. However, the absence of clear phenotypic alterations at baseline, and the fact that an increased Mφ-Mo-ratio was observed in wild-type mice challenged with QPCTL-deficient tumor cells, argue against this possibility. 2) In the present study, we have aimed to model the effects of depletion of QPCTL activity on the tumor micro-environment, whereas glutaminyl cyclase inhibitors will, based on the similarity of their active sites, likely inhibit both QPCTL and QPCT activity.38,39 Such inhibition of QPCT may be relevant as siRNA-mediated suppression of QPCT has been shown to reduce expression of CCL2, CX3CL1 and CD54/ICAM.12 In future work, dual inactivation of QPCTL and QPCT may form a means to test this.
The depletion of monocytes that we observe in QPCTL-deficient TMEs may potentially be explained by a decreased functionality of the CCL2-CCR2 signaling axis. In pre-clinical models of breast cancer, the CCL2-CCR2 axis has been shown to influence the abundance of monocytes in primary tumors40 and metastatic lesions.41 Furthermore, monocyte recruitment was found to be reduced after thioglycolate challenge of mice that were either QPCTL-deficient or treated with QPCT/QPCTL inhibitors.10 However, it is important to note that at high concentrations, pyroGlu-CCL2 and unmodified CCL2 demonstrate similar chemotactic activity,10 and the effect of impaired pyro-glutamylation of CCL2 will therefore depend on local concentrations.
Contrary to expectations, we observed a relative increase in macrophage frequencies in QPCTL-deficient TMEs. Transcriptomic profiling of intra-tumoral Mφ/Mo cells revealed that these macrophages expressed monocyte-associated molecules (e.g. Ccr2 and Itga4) and pseudotime analysis suggests the existence of intermediate Mφ/Mo cell states, together arguing in favor of their monocytic origin. In prior work, abrogation of the CCR2-CCL2 signaling axis in monocytes has been shown to strongly reduce the accumulation of intra-tumoral macrophages.42 Based on these data, we propose that the boosting of intra-tumoral macrophages by QPCTL inactivation occurs through a mechanism that is independent of CCL2, and is potentially driven by an accelerated monocyte-to-macrophage differentiation program.
Having observed a number of independent alterations in the TME that are induced by QPCTL deficiency, it is of interest to speculate on the possible causal relationship between these individual changes. One possible scenario (Supplementary Fig. 9b) is that suppression of the CCR2-CCL2 axis due to the lack of QPCTL activity causes an early reduction in the influx of monocytes, which have been shown to form a major source of TGF-β.43 Such an initial deficit in TGF-β abundance could potentially limit myCAF polarization and favor differentiation toward iCAFs, relieving TGF-β-driven suppressive effects on myeloid and lymphoid effector cells. Notably, TGF-β can act as a suppressor of IFNγ production by NK cells44,45 and CD8+ T cells,46,47 and increased abundance of activation-associated transcripts was observed in lymphocytes from QPCTL-deficient TMEs, together suggesting that this cell pool may be the source of the observed IFN-response signature. Genetic ablation experiments (Figure 2) indicated that CD47 does not play a significant mechanistic role in the TME-modulatory effects of QPCTL in the B16 melanoma model. However, as the CD47/SIRPα axis acts primarily through inhibition of activating signals of ITAM-containing receptors, such as activating Fc receptors on myeloid cells, it is plausible that the observed synergy between QPCTL deficiency and treatment with an opsonizing anti-PD-L1 antibody is at least partially mediated through its effect on the CD47 pathway.
In summary, our data provide evidence that removal of QPCTL activity can shift the TME from an immunosuppressive (monocyte skewed, myCAF, TGF-β) toward a pro-inflammatory (macrophage skewed, iCAF, IFN) milieu, and acts synergistically with anti-PD-L1 therapy to enhance tumor control and survival. If this TME-remodeling effect can also be achieved through pharmacological inhibition of QPCTL activity in human cancers, such inhibitors may offer potential in combination treatment strategies that include checkpoint blocking antibodies and/or tumor-opsonizing antibodies.
Material and methods
Mice. C57/Bl6JR mice were obtained from Janvier. QPCTL−/− mice carrying an 811 bp deletion in exon 2 of the Qpctl gene were generated on the C57BL/6JRj background using pronuclear microinjection in mice zygotes with a CRISPR/Cas9 mixture (50 ng/μl Cas9 RNA and 25 ng/μl sgRNA, in water). The sgRNA was targeted to the second exon of the Qpctl gene (5’-GCACAATCAATAAGGGACGC-3’). QPCTL−/− mice and QPCTL+/+ mice were identified by PCR using the following primers: Fwd_KO (5’-GTTTTAGGGATGGATGCCGC-3’), located before the 811 bp deletion, Fwd_WT (5’-GGACTCCTAGTAGGCAACGG-3’), located in the 811 bp deletion, and Rev (5’-GGCTGTTTTGGGATCTTCGG-3’), located after the 811 bp deletion.
Evaluation of mouse blood cell counts. Whole blood of mice was collected by heart puncture and total cell counts were determined using a DxH500 Hematology Analyzer (Beckman Coulter).
Peripheral blood collection and preparation. Whole blood of mice was collected into heparin-coated tubes by heart puncture or tail vein puncture at indicated time points. Samples were incubated twice for 5 min in erythrocyte-lysis buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.4), and washed once in staining buffer (0.5% BSA in PBS). Cells were then used for antibody staining, as described below.
Cell lines. B16F10 cells and MC38-AMS cells were kindly provided by D. Peeper (Division of Molecular Oncology & Immunology, Oncode Institute, The Netherlands Cancer Institute, Amsterdam, The Netherlands). The MC38-AMS cell line is a variant of the MC38 cell line available from Kerafast. Whole-exome sequencing was performed to compare the MC38-AMS and MC38-Kerafast line, and data have been uploaded to the Sequence Read Archive. B16F10 and MC38-AMS cells were cultured in DMEM (Gibco) supplemented with 8% FCS and penicillin-streptomycin. Cells were cultured at 37°C and 5% CO2.
CRISPR/Cas9-mediated generation of CD47 and QPCTL knockout cells. To generate QPCTL- knockout (KO), CD47-KO, and WT control B16F10 cell lines, cells were transfected with pLentiCRISPR v.2 vector encoding sgRNA targeting the murine QPCTL (5’- TATTGATTGTGCGACCCCCG-3’) or CD47 (5’- AGCAACAGCGCCGCCGCCAA-3’) gene, or left untransfected. Culture medium of transfected cells was supplemented the next day with 2 μg ml–1 puromycin for at least 2 days. Selected cells were expanded, and subsequently sorted on the basis of αmCD47-MIAP301lo mSIRPα-Fclo phenotype (in case of CD47 knockout), or αmCD47-MIAP301hi mSIRPα-Fclo phenotype (in case of QPCTL knockout), in order to obtain bulk knockout populations. WT control B16F10 cells were sorted based on morphology gating only. Next, single cells were isolated and expanded, and approximately 50 knockout or wild-type clones were pooled to obtain pure knockout or wild-type populations. To generate CD47/QPCTL double KO (dKO) cell lines, B16F10 QPCTL KO cell lines were transfected with pLentiCRISPR v.2 vector encoding sgRNA targeting the murine CD47 gene. One day after transfection, culture medium was supplemented with 2 μg ml–1 puromycin for at least 2 days. Single cells were isolated and expanded, and 12 clones were pooled to obtain knockout populations. To generate QPCTL-KO and control MC38-AMS cell lines, cells were transduced with pLentiCRISPR v.2 vector encoding sgRNA targeting the murine QPCTL gene or a non-targeting control gRNA. 2 days after transduction, culture medium was supplemented with 2 ug ml−1 puromycin for at least 4 days. Next, single cells were isolated and expanded, and 12 knock-out or control clones were pooled to obtain cell populations for further use. Gene disruption was validated by sequence analysis of the relevant gene locus by TIDE48 analysis and, in case of CD47, by flow cytometry.
Tumor challenge. To analyze the effect of QPCTL deficiency in both host and tumor cells or in host cells only, 8- to 25-week-old male and female QPCTL−/− or wild-type QPCTL+/+ littermate controls were injected with 2 × 105 of the indicated B16F10 tumor cell line in a 100 μL solution of PBS (Lonza) and Matrigel (Corning) (1:1) in the right flank on day 0. To analyze the effect of tumor cell CD47 deficiency or CD47 and QPCTL-double deficiency, 9–12-week-old C57/Bl6JR (female; Janvier) were injected with 2 × 105 of the indicated B16F10 tumor cell line. To analyze the effect of QPCTL deficiency in both host and tumor cells in MC38-AMS tumors, 8–25-week-old QPCTL−/− or wild-type QPCTL+/+ littermate controls were injected with 5 × 105 of the indicated MC38-AMS tumor cell line. Tumors were measured 3 times a week, and mice were sacrificed 13–17 days (B16F10 tumors) or 21–29 days (MC38 tumors) after tumor challenge. Mice with a tumor volume equal or below 40 mm3 were excluded and tumors used for subsequent flow cytometry analyses ranged from 75 to 1436 mm3 (B16F10 tumors) or 112.5–786.5 mm3 (MC38 tumors).
TME single-cell preparation. Tumors were fragmented on ice and were subsequently digested in DMEM (10 ml per tumor) supplemented with collagenase IV (2 mg ml−1, Sigma Aldrich) and DNase I (50 μg ml−1, Sigma Aldrich) for 30 min at 37°C. Subsequently, 40 ml DMEM supplemented with 8% FCS was added per tumor, and cell suspensions were passed through 100 μm strainers. Next, samples were incubated for 5 minutes in erythrocyte-lysis buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM EDTA, pH 7.4), and washed once in staining buffer (0.5% BSA in PBS). Tumor single-cell suspensions were then counted and used for antibody staining.
Flow cytometry. Cell surface CD47 was assessed by staining of blood immune cells with the anti-mouse CD47 antibody MIAP301 at a dilution of 1:100 or 1:200 plus His-tagged recombinant mouse SIRPα (rmSIRPα-His) (4, 12, or 36 μg ml–1), in PBS containing 0.5% (w/v) BSA (Sigma) and 0.2% (w/v) sodium azide (Sigma) (FACS buffer) for 30 min at room temperature, protected from light. After two washes with FACS buffer, cells were stained with a fluorochrome-labeled anti-His antibody at a dilution of 1:100 or 1:200 for 30 min at 4°C in FACS buffer, while protected from light. Cells were then washed with FACS buffer, and DAPI, propidium iodide, or 7-AAD Viability Staining Solution (eBioscience) was added to allow dead cell exclusion. Antibodies used to analyze immune cells in tumor single-cell suspensions are listed in Supplementary Table 3. Measurements were performed on an LSRII, LSRFortessa, or FACSCanto II (BD Biosciences). Data were analyzed using FACS Diva software (BD Biosciences) and FlowJo software.
Unbiased flow cytometry data analysis. Samples were preprocessed using FlowJo software, compensating for spectral overlap, selecting IR-Dye−CD45+ single cells and removing outlier cells. Further analysis was performed in R, implementing the FlowCore package.49 Samples were subsampled to obtain 10,000 or 30,000 total cells for spleen or tumor analysis, respectively. Next, a logicle (biexponential) transformation was applied to the measured fluorescence intensities. Uniform Manifold Approximation and Projection (UMAP) was used for dimension reduction, and subsequently used for hierarchical clustering by Euclidean distance (Ward’s method). Relative contributions of cells derived from QPCTL−/− and QPCTL+/+ samples to each of the clusters was then assessed.
IFNγ and TGF-β sensitivity of B16F10 cell lines. For IFNγ sensitivity testing, QPCTL-WT or QPCTL-KO cell lines were seeded on 6-well plates at 50,000 cells per well, incubated at 37°C for 3 hours to allow cells to adhere, and subsequently treated with indicated amounts of IFNγ (Mouse IFN-gamma Recombinant Protein, ThermoFisher Scientific) for 21 hours. Cells were then harvested with trypsin-EDTA (Gibco), washed twice with PBS, and examined either through flow-cytometry or western blotting. For TGF-β sensitivity testing, QPCTL-WT or QPCTL-KO cell lines were seeded on 6-well plates at 50,000 cells per well and incubated at 37°C for 16 hours. Next, cells were pre-incubated at 37°C for 4 hours in culture medium containing 0.2% FCS, and subsequently incubated at 37°C with indicated quantities of recombinant mouse TGF-β1 protein (R&D systems) for 1 hour at in culture medium containing 0.2% FCS. Cells were then harvested with trypsin-EDTA (Gibco), washed twice with PBS, and examined through western blotting. For flow-cytometry cells were stained with anti-PD-L1-BV421 and anti-H2-Kb-PE (both 1:100 dilution, see Supplementary Table 3) for 15 minutes at room temperature, washed twice with FACS buffer, and analyzed on an LSR Fortessa (BD Biosciences). For western blot analyses, cells were incubated on ice for 30 minutes in 200 μl RIPA buffer (1% Triton X100, 0.1% Sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 10 mM Tris pH 8, 140 mM NaCl) supplemented with Halt™ Protease and Phosphatase Inhibitor Cocktail (ThermoFisher Scientific), followed by pulse mixing on a Vortex Genie (Scientific Industries). Lysates were then centrifuged at 20,000x g for 20 minutes at 4°C and protein concentrations in the resulting supernatants were determined using Pierce™ BCA Protein Assay Kit (ThermoFisher Scientific) according to manufacturer’s protocol. Next, equal amounts of protein were processed using the Novex NuPAGE Electrophoresis system (Thermo Fisher Scientific) and Trans-Blot Turbo Transfer system (Bio-Rad) according to the manufacturers’ instructions. Membranes were blocked using Western Blocking Reagent (Roche) for 1 hours, and subsequently stained overnight at 4°C with indicated primary antibodies diluted in Western Blocking Reagent (see Supplementary Table 3 for antibody information) followed by 2 hours at 4°C with either anti-rabbit or anti-mouse secondary antibodies conjugated to HRP (see Supplementary Table 3 for antibody information). Western blots were imaged using the ChemiDoc MP Imaging System (Bio-Rad).
RNA sequencing. RNA was extracted from the indicated frozen tissues using the RNeasy Mini Kit (Qiagen). Cell populations isolated by FACS were washed once in PBS, and subsequently lysed in RLT buffer (Qiagen). Whole-transcriptome sequencing samples were prepared with the TruSeq Stranded mRNA Kit (Illumina). Paired-end 50 bp sequencing was performed on a NovaSeq 6000 system (S1 flowcell, Illumina), obtaining an average of 18 × 106 reads per sample. Reads were aligned to the pre-built GRCm38 genome_snp_tran reference using HISAT2,50 and transcript counts were obtained using an in-house generated pipeline (GenSum, https://github.com/NKI-GCF/gensum). Differential gene expression analysis was performed using the edgeR package.51 Network analysis was performed using the stringDB database, applying the igraph package for visualization.
Single-cell RNA sequencing analyses. Single-cell digests of QPCTL−/− and QPCTL+/+ TMEs were generated as outlined above. Cells were stained with IR-Dye for dead cell exclusion and with anti-mouse TotalSeq™ Hashtag antibodies (TotalSeq-A0301-06, Biolegend), pooled in equal numbers, and were single-cell sorted on a BD Fusion cell sorter. Single-cell RNA isolation and library preparation was performed according to the manufacturer’s protocol of the 10X Genomics Chromium™ Single Cell 3’ kit, and the cDNA library was sequenced on the NextSeqTM550 Sequencing System (Illumina). A total of ~3.7x108 reads resulted in the detection of 14,888 cells with a median of 3,344 detected genes per cell. Feature-barcode matrices were generated using the Cell Ranger software of the 10X Genomics Chromium™ pipeline. Further processing was subsequently performed using the MetaCell18 and Seurat R packages.52 Cells that contained less than 500 UMIs or had a mitochondrial transcript fraction of >0.2 were removed. Next, variable genes across the dataset were identified with a normalized variance/mean threshold at 0.1 and a down-sampled coverage threshold at 80, yielding 1,021 genes. These genes were subsequently used as anchors to search for gene–gene correlations across the dataset, and genes with correlations of >0.1 were included. The obtained genes were then clustered into 50 separate gene-modules, and each was annotated manually (Supplementary Table 4).
To identify the major cell types, a feature-gene list was compiled of gene-modules that contained marker genes for various cell types (modules 10, 17, 19, 21, 24, 26, 29, 30, 33, 39, 40, 44, 47, 48, 49; Supplementary Table 3), and these feature-genes were used to generate MetaCells. The obtained MetaCells were then classified as either immune cells, fibroblasts, or tumor cells, as shown in Supplementary figure 6. MetaCells that contained significant expression of marker genes from multiple cell types were identified as ‘doublet MetaCells’, and excluded from further analysis.
Subsequent analysis was performed within each individual cell type. In brief, doublet detection was performed using the HTOdemux function of Seurat, setting the positive quantile at 0.99. Cells containing either a high amount of UMIs (UMI-thresholds: Immune [10,000], fibroblast [11,000], tumor cell [30,000]) or gene-counts (gene-count-thresholds: Immune [3,000], fibroblast [4,000], tumor cell [5,700]) were considered doublets and excluded. Feature genes used for cell type-specific MetaCell generation were obtained using the mcell_gset_filter_varmean and mcell_gset_filter_cov functions implemented in the Metacell package. These features genes were filtered for genes involved in cell cycle (gene-module 7 and 20) and ribosomal proteins (gene-module 2).
For all plots showing normalized UMI counts, a center log ratio normalization was applied, as implemented in the Seurat package.52 To calculate sample fractions within MetaCells, cell counts were first normalized to 10,000 cells within each sample-hashtag to allow comparisons.
Pseudotime analysis was performed using the Slingshot algorithm.53 Gene-level general additive models were fitted to feature-genes used for MetaCell generation applying the fitGAM function from the TradeSeq R package,54 setting knots at 5. Only genes that associated significantly (adjusted P value < .05) with pseudotime were used in subsequent analysis. Genes were then clustered based on expression kinetics across pseudotime based on Euclidean distance (Supplementary Table 5). To assess sample composition across pseudotime, the three replicates from QPCTL−/− or QPCTL+/+ TMEs were analyzed together. To allow pooling of replicates, cell counts were normalized to the total number of cell counts within each sample. Normalized cell counts were then tallied within windows of 60 cell-codes wide, sliding 1 cell-code per frame. Differential gene expression analysis was performed using the FindMarkers function implemented in Seurat. Wilcoxon Rank Sum test was used to obtain log2 fold changes.
For comparison of Immune MetaCell 1–3 to the external monocyte/macrophage cell clusters, the scRNAseq dataset from Gubin et al.32 was retrieved from the Gene Expression Omnibus (GSE119352). The external data was subsequently filtered for monocyte/macrophage cell clusters (Mac_s1–5), and normalized through centered log-ratio transformation. Differentially expressed genes within each cluster was then determined by comparing each cluster to all others applying Wilcoxon Rank Sum test (FindAllMarks function, Seurat R package). Twenty-five marker genes were then selected for each MetaCell, defined as the 25 most enriched genes within that MetaCell. Each of these gene-sets was then used to compute a similarity score with each of the Mac_s clusters. Each similarity score was calculated by filtering the Wilcoxon Rank Sum test results of a given Mac_s cluster for a marker gene-set, followed by a weighted sampling of the log2 transformed fold change values (sampling 10,000 times, with replacement, weighted by the MetaCell gene-enrichment value), and finally averaging (median) the obtained values.
Anti-PD-L1 checkpoint blockade. 8- to 25-week-old male and female QPCTL−/− or wild-type QPCTL+/+ littermate controls were injected with 2x105 QPCTL-KO or QPCTL-WT B16F10 cells, respectively, in a 100 μL solution of PBS (Lonza) and Matrigel (Corning) (1:1) in the right flank on day 0. At day 7 post-inoculation mice were randomized into treatment and control groups, stratified by QPCTL status and sex. Mice in the treatment group received intra-peritoneal injections of 200 μg InVivoPlus anti-mouse PD-L1 (clone 10F.9G2, BioXcell) at day 7, 9 and 11 post tumor inoculation. Mice in the control group received intra-peritoneal injections of 200 μg InVivoPlus rat anti-keyhole limpet hemocyanin (IgG2b isotype control, clone LTF-2, BioXcell) at day 7, 9 and 11 post tumor inoculation. Tumor sizes were measured every 2-3 days. Tumor growth and survival was monitored until humane end-point (1500 m3 tumor size) was reached.
Pathology. For histopathological analyses, 2 μm-thick hematoxylin-eosin-stained sections were prepared from formalin-fixed, paraffin-embedded murine tissues, including skin, spleen, thymus, lymph nodes, liver, pancreas, gastrointestinal tract, heart, lung, kidneys, testes, ovaries, accessory sex glands, bone marrow (sternum and extremity), and muscles. Sections were evaluated and scored by an animal pathologist blinded to animal genotype.
Statistical analysis. All statistical analyses were performed either with R (V4.0.5, ‘Shake and Throw’) or Graphpad (V8.4.1, Prism software). All statistical test were two-sided, unless otherwise indicated. Differences were considered statistically significant if P < .05. The n values used to calculate statistics, the type of replicates and the relevant significant P values are noted in the figure legends.
Ethical compliance. All animal experiments were approved by the Animal Welfare Committee of the Netherlands Cancer Institute (NKI), in accordance with national guidelines. Animals were maintained in the animal department of the NKI, housed in individually ventilated cage systems under specific-pathogen-free conditions, and received food and water freely. Mice were used at 8–25 weeks of age.
Supplementary Material
Acknowledgments
We thank members of the Schumacher lab, Ferenc Scheeren (Leiden University Medical Centre) and Leila Akkari (Netherlands Cancer Institute) for helpful discussions, and the Netherlands Cancer Institute – Antoni van Leeuwenhoek (NKI/AVL) Transgenic unit, Preclinical Intervention Unit and flow facility for technical support and input. We thank the NKI animal pathology facility for technical support, and Sjoerd Klarenbeek for histopathological assessment of QPCTL−/− mice and discussions. We thank the NKI genomics core facility for processing, and providing input on, sequencing data presented in this work, and for analysis of the MC38-AMS exome data. This work was supported by ERC AdG SENSIT (grant agreement ID 742259) to T.N.S.
Funding Statement
This work was supported by the European Research Council [742259].
Author contributions
M.E.W.L. and K.B. conceived the project, designed and performed experiments, interpreted data, curated data and co-wrote the manuscript. M.T. designed and performed experiments. N.P, J.S, B.S and M.B. performed experiments. L.K established the QPCTL−/− strain. T.N.S. conceived the project, designed experiments, interpreted data and co-wrote the manuscript.
Disclosure statement
M.E.W.L. and T.N.S. are inventors on a patent application that covers manipulation of the CD47-SIRPα axis via QPCTL. M.E.W.L. is a consultant for Third Rock Ventures, outside of this work. T.N.S. is advisor to and holds equity in Scenic Biotech that develops QPCTL inhibitors. T.N.S. is consultant for Third Rock Ventures and advisor to and stockholder in Allogene Therapeutics, Asher Bio, Merus, and Neogene Therapeutics, all outside of this work.
Data availability
Transcriptomic data presented in the manuscript have been deposited to the Gene Expression Omnibus, and can be accessed as series GSE180201. Exome data for MC38-Kerafast and MC38-AMS have been deposited to the Sequence Read Archive, and can be accessed as project PRJNA753254. R scripts used to produce key figures in the manuscript have been submitted to GitHub (https://github.com/kasbress/QPCTL_Project). Statistical source data has been deposited to Zenodo (https://doi.org/10.5281/zenodo.6335363).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Rumpret M, Drylewicz J, Ackermans L.J, et al. Functional categories of immune inhibitory receptors. Nat Rev Immunol. 2020;20(12):771–14. doi: 10.1038/s41577-020-0352-z. [DOI] [PubMed] [Google Scholar]
- 2.Jaiswal S, Jamieson CHM, Pang WW, Park CY, Chao MP, Majeti R, Traver D, van Rooijen N, Weissman IL.. CD47 Is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271–285. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD, van Rooijen N, Weissman IL. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138(2):286–299. doi: 10.1016/j.cell.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao XW, van Beek E.M, Schornagel K, et al. CD47–signal regulatory protein-α (SIRPα) interactions form a barrier for antibody-mediated tumor cell destruction. Proc National Acad Sci. 2011;108(45):18342–18347. doi: 10.1073/pnas.1106550108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cynis H, Rahfeld J-U, Stephan A, Kehlen A, Koch B, Wermann M, Demuth H-U, Schilling S. Isolation of an Isoenzyme of human glutaminyl cyclase: retention in the golgi complex suggests involvement in the protein maturation machinery. J Mol Biol. 2008;379(5):966–980. doi: 10.1016/j.jmb.2008.03.078. [DOI] [PubMed] [Google Scholar]
- 6.Logtenberg MEW, Jansen JHM, Raaben M, Toebes M, Franke K, Brandsma AM, Matlung HL, Fauster A, Gomez-Eerland R, Bakker NAM, et al. Glutaminyl cyclase is an enzymatic modifier of the CD47- SIRPα axis and a target for cancer immunotherapy. Nat Med. 2019;25(4):612–619. doi: 10.1038/s41591-019-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schilling S, Niestroj AJ, Rahfeld J-U, Hoffmann T, Wermann M, Zunkel K, Wasternack C, Demuth H-U. Identification of human Glutaminyl Cyclase as a Metalloenzyme POTENT INHIBITION BY IMIDAZOLE DERIVATIVES AND HETEROCYCLIC CHELATORS*. J Biol Chem. 2003;278(50):49773–49779. doi: 10.1074/jbc.M309077200. [DOI] [PubMed] [Google Scholar]
- 8.Stephan A, Wermann M, von Bohlen A, Koch B, Cynis H, Demuth H-U, Schilling S. Mammalian glutaminyl cyclases and their isoenzymes have identical enzymatic characteristics. Febs J. 2009;276(22):6522–6536. doi: 10.1111/j.1742-4658.2009.07337.x. [DOI] [PubMed] [Google Scholar]
- 9.Hatherley D, Graham SC, Turner J, Harlos K, Stuart DI, Barclay AN. Paired receptor specificity explained by structures of signal regulatory proteins alone and complexed with CD47. Mol Cell. 2008;31(2):266–277. doi: 10.1016/j.molcel.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 10.Cynis H, Hoffmann T, Friedrich D, et al. The isoenzyme of glutaminyl cyclase is an important regulator of monocyte infiltration under inflammatory conditions. Embo Mol Med. 2011;3(9):545–558. doi: 10.1002/emmm.201100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Y-L, Huang K.F, Kuo W.C, et al. Inhibition of glutaminyl cyclase attenuates cell migration modulated by monocyte chemoattractant proteins. Biochem J. 2012;442(2):403–412. doi: 10.1042/BJ20110535. [DOI] [PubMed] [Google Scholar]
- 12.Kehlen A, Haegele M, Böhme L, et al. N-terminal pyroglutamate formation in CX3CL1 is essential for its full biologic activity. Bioscience Rep. 2017;37(4). doi: 10.1042/BSR20170712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mair B, Aldridge PM, Atwal RS, Philpott D, Zhang M, Masud SN, Labib M, Tong AHY, Sargent EH, Angers S, et al. High-throughput genome-wide phenotypic screening via immunomagnetic cell sorting. Nat Biomed Eng. 2019;3(10):796–805. doi: 10.1038/s41551-019-0454-8. [DOI] [PubMed] [Google Scholar]
- 14.Becker A, Eichentopf R, Sedlmeier R, Waniek A, Cynis H, Koch B, Stephan A, Bäuscher C, Kohlmann S, Hoffmann T, et al. IsoQC (QPCTL) knock-out mice suggest differential substrate conversion by glutaminyl cyclase isoenzymes. Biol Chem. 2016;397(1):45–55. doi: 10.1515/hsz-2015-0192. [DOI] [PubMed] [Google Scholar]
- 15.Natarajan VT, Ganju P, Singh A, Vijayan V, Kirty K, Yadav S, Puntambekar S, Bajaj S, Dani PP, Kar HK, et al. IFN-γ signaling maintains skin pigmentation homeostasis through regulation of melanosome maturation. P Natl Acad Sci Usa. 2014;111(6):2301–2306. doi: 10.1073/pnas.1304988111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou J, Ling J, Wang Y, Shang J, Ping F. Cross-talk between interferon-gamma and interleukin-18 in melanogenesis. J Photochem Photobiology B Biology. 2016;163:133–143. doi: 10.1016/j.jphotobiol.2016.08.024. [DOI] [PubMed] [Google Scholar]
- 17.Son J, Kim M, Jou I, Park KC, Kang HY. IFN- γ inhibits basal and α -MSH-induced melanogenesis. Pigm Cell Melanoma R. 2013;27(2):201–208. doi: 10.1111/pcmr.12190. [DOI] [PubMed] [Google Scholar]
- 18.Baran Y, Bercovich A, Sebe-Pedros A, Lubling Y, Giladi A, Chomsky E, Meir Z, Hoichman M, Lifshitz A, Tanay A, et al. MetaCell: analysis of single-cell RNA-seq data using K-nn graph partitions. Genome Biol. 2019;20(1):206. doi: 10.1186/s13059-019-1812-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang P, Zhang Y, Ru B, Yang Y, Vu T, Paul R, Mirza A, Altan-Bonnet G, Liu L, Ruppin E, et al. Systematic investigation of cytokine signaling activity at the tissue and single-cell levels. Nat Methods. 2021;18(10):1181–1191. doi: 10.1038/s41592-021-01274-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu S-Y, Sanchez DJ, Aliyari R, Lu S, Cheng G. Systematic identification of type I and type II interferon-induced antiviral factors. Proc National Acad Sci. 2012;109(11):4239–4244. doi: 10.1073/pnas.1114981109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gough DJ, Messina NL, Hii L, Gould JA, Sabapathy K, Robertson APS, Trapani JA, Levy DE, Hertzog PJ, Clarke CJP, et al. Functional crosstalk Between Type I And Ii interferon Through The regulated expression of STAT1. Plos Biol. 2010;8(4):e1000361. doi: 10.1371/journal.pbio.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zimmerman KA, Bentley MR, Lever JM, Li Z, Crossman DK, Song CJ, Liu S, Crowley MR, George JF, Mrug M, et al. Single-Cell RNA sequencing identifies candidate renal resident macrophage gene expression signatures across species. J Am Soc Nephrol Jasn. 2019;30(5):767–781. doi: 10.1681/ASN.2018090931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qian J, Olbrecht S, Boeckx B, Vos H, Laoui D, Etlioglu E, Wauters E, Pomella V, Verbandt S, Busschaert P, et al. A pan-cancer blueprint of the heterogeneous tumor microenvironment revealed by single-cell profiling. Cell Res. 2020;30(9):745–762. doi: 10.1038/s41422-020-0355-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mattiola I, Tomay F, De Pizzol M, Silva-Gomes R, Savino B, Gulic T, Doni A, Lonardi S, Astrid Boutet M, Nerviani A, et al. The macrophage tetraspan MS4A4A enhances dectin-1-dependent NK cell-mediated resistance to metastasis. Nat Immunol. 2019;20(8):1012–1022. doi: 10.1038/s41590-019-0417-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, Dhara S, Simpson K, Gardner EE, Iacobuzio-Donahue CA, et al. Macrophage ontogeny underlies differences in tumor-specific education in brain malignancies. Cell Reports. 2016;17(9):2445–2459. doi: 10.1016/j.celrep.2016.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arlauckas S, Oh N, Li R, Weissleder R, Miller MA. Macrophage imaging and subset analysis using single-cell RNA sequencing. Nanotheranostics. 2021;5(1):36–56. doi: 10.7150/ntno.50185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, Qin S, Zhang L, Ouyang H, Du P, et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021;184(3):792–809.e23. doi: 10.1016/j.cell.2021.01.010. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y-J, Hsieh M-Y, Chang MY, Chen H-C, Jan M-S, Maa M-C, Leu T-H. Eps8 protein facilitates phagocytosis by increasing TLR4-MyD88 protein interaction in lipopolysaccharide-stimulated macrophages. J Biol Chem. 2012;287(22):18806–18819. doi: 10.1074/jbc.M112.340935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fan W, Liu T, Chen W, Hammad S, Longerich T, Hausser I, Fu Y, Li N, He Y, Liu C, et al. ECM1 prevents activation of transforming growth factor β, hepatic stellate cells, and fibrogenesis in mice. Gastroenterology. 2019;157(5):1352–1367.e13. doi: 10.1053/j.gastro.2019.07.036. [DOI] [PubMed] [Google Scholar]
- 30.Huang S-C-C, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15(9):846–855. doi: 10.1038/ni.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu T, Gan S, Zhu Q, et al. Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nat Commun. 2019;10(1):4353. doi: 10.1038/s41467-019-12384-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, Noguchi T, Arthur CD, Meng W, Alspach E, et al. High-Dimensional analysis delineates myeloid and lymphoid compartment remodeling during successful immune-checkpoint cancer therapy. Cell. 2018;175(4):1014–1030.e19. doi: 10.1016/j.cell.2018.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mhaidly R, Mechta-Grigoriou F. Fibroblast heterogeneity in tumor micro-environment: role in immunosuppression and new therapies. Semin Immunol. 2020;48:101417. doi: 10.1016/j.smim.2020.101417. [DOI] [PubMed] [Google Scholar]
- 34.Öhlund D, Handly-Santana A, Biffi, G, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214(3):579–596. doi: 10.1084/jem.20162024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, Teinor JA, Belleau P, Biffi G, Lucito MS, et al. Cross-Species single-Cell analysis of pancreatic ductal adenocarcinoma reveals antigen-Presenting cancer-Associated fibroblasts. Cancer Discov. 2019;9(8):1102–1123. doi: 10.1158/2159-8290.CD-19-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wculek SK, Amores-Iniesta J, Conde-Garrosa R, Khouili SC, Melero I, Sancho D. Effective cancer immunotherapy by natural mouse conventional type-1 dendritic cells bearing dead tumor antigen. J Immunother Cancer. 2019;7(1):100. doi: 10.1186/s40425-019-0565-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sánchez-Paulete AR, Cueto FJ, Martínez-López M, Labiano S, Morales-Kastresana A, Rodríguez-Ruiz ME, Jure-Kunkel M, Azpilikueta A, Aznar MA, Quetglas JI, et al. Cancer immunotherapy with immunomodulatory Anti-CD137 and Anti–PD-1 monoclonal antibodies requires BATF3-Dependent dendritic cells. Cancer Discov. 2015;6(1):71–79. doi: 10.1158/2159-8290.CD-15-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jimenez-Sanchez M, Lam W, Hannus M, Sönnichsen B, Imarisio S, Fleming A, Tarditi A, Menzies F, Ed Dami T, Xu C, et al. siRNA screen identifies QPCT as a druggable target for huntington’s disease. Nat Chem Biol. 2015;11(5):347–354. doi: 10.1038/nchembio.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lues I, Weber F, Meyer A, et al. A phase 1 study to evaluate the safety and pharmacokinetics of PQ912, a glutaminyl cyclase inhibitor, in healthy subjects. Alzheimer’s Dementia Transl Res Clin Interventions. 2015;1(3):182–195. doi: 10.1016/j.trci.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arwert EN, Harney AS, Entenberg D, Wang Y, Sahai E, Pollard JW, Condeelis JS. A unidirectional transition from migratory to perivascular macrophage is required for tumor cell intravasation. Cell Reports. 2018;23(5):1239–1248. doi: 10.1016/j.celrep.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qian B-Z, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tymoszuk P, Evens H, Marzola V, Wachowicz K, Wasmer M-H, Datta S, Müller-Holzner E, Fiegl H, Böck G, van Rooijen N, et al. In situ proliferation contributes to accumulation of tumor-associated macrophages in spontaneous mammary tumors. Eur J Immunol. 2014;44(8):2247–2262. doi: 10.1002/eji.201344304. [DOI] [PubMed] [Google Scholar]
- 43.Gupta A, Budhu S, Fitzgerald K, Giese R, Michel AO, Holland A, Campesato LF, van Snick J, Uyttenhove C, Ritter G, et al. Isoform specific anti-TGFβ therapy enhances antitumor efficacy in mouse models of cancer. Commun Biology. 2021;4(1):1296. doi: 10.1038/s42003-021-02773-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-β controls T helper type 1 cell development through regulation of natural killer cell interferon-γ. Nat Immunol. 2005;6(6):600–607. doi: 10.1038/ni1197. [DOI] [PubMed] [Google Scholar]
- 45.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Cañellas A, Hernando-Momblona X, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–543. doi: 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- 46.Park BV, Freeman ZT, Ghasemzadeh A, Chattergoon MA, Rutebemberwa A, Steigner J, Winter ME, Huynh TV, Sebald SM, Lee SJ, Pan F, Pardoll DM, Cox AL, et al. TGF-β1-mediated smad3 enhances PD-1 expression on antigen-specific T cells in cancer. Cancer Discov. 2016;6(12):1366–1381. doi: 10.1158/2159-8290.CD-15-1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas DA, Massagué J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8(5):369–380. doi: 10.1016/j.ccr.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 48.Brinkman EK, Chen T, Amendola M, Steensel BV. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42(22):e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hahne F, LeMeur N, Brinkman RR, Ellis B, Haaland P, Sarkar D, Spidlen J, Strain E, Gentleman R. flowCore: a Bioconductor package for high throughput flow cytometry. BMC Bioinformatics. 2009;10(1). doi: 10.1186/1471-2105-10-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37(8):907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robinson MD, McCarthy DJ, Smyth G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinform Oxf Engl. 2009;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184(13):3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, Dudoit S. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics. 2018;19(1):477. doi: 10.1186/s12864-018-4772-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berge KVD, Roux de Bézieux H, Street K, Saelens W, Cannoodt R, Saeys Y, Dudoit S, Clement L. Trajectory-based differential expression analysis for single-cell sequencing data. Nat Commun. 2020;11(1):1201. doi: 10.1038/s41467-020-14766-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Transcriptomic data presented in the manuscript have been deposited to the Gene Expression Omnibus, and can be accessed as series GSE180201. Exome data for MC38-Kerafast and MC38-AMS have been deposited to the Sequence Read Archive, and can be accessed as project PRJNA753254. R scripts used to produce key figures in the manuscript have been submitted to GitHub (https://github.com/kasbress/QPCTL_Project). Statistical source data has been deposited to Zenodo (https://doi.org/10.5281/zenodo.6335363).