

Abstract
Introduction
Composite phaeochromocytoma is a tumour containing a separate tumour of neuronal origin in addition to a chromaffin cell tumour. This study reports on two cases from a single centre’s records and presents a systematic literature review of composite phaeochromocytomas.
Methods
In addition to describing 2 case reports, a systematic search of the Medline database from inception up to April 2020 was done for human case reports on composite phaeochromocytomas. Relevant titles and/or abstracts were screened, and full texts were reviewed to identify appropriate studies. Data was extracted and a descriptive analysis of presentation, clinical features, management strategies and outcomes was performed. The quality of included studies was assessed using a critical appraisal checklist.
Results
There were 62 studies included, with a total of 94 patients. Of 91 patients where data was available, the median (range) age of patients was 48 (4–86) years. Of 90 patients where information was provided, 57% were female. In at least 28% of patients, a genetic cause was identified. Common presenting features include abdominal pain, palpable mass, cardiovascular and gastrointestinal symptoms. The most common tumour component with phaeochromocytoma is ganglioneuroma; other components include ganglioneuroblastoma, neuroblastoma and malignant peripheral nerve sheath tumours. In patients with follow-up data (n=48), 85% of patients were alive and well at a median (range) follow-up time of 18 (0.5–168) months.
Conclusion
Composite phaeochromocytoma is a rare tumour, with a significant genetic predisposition. This review summarises available epidemiological data, which will be useful for clinicians managing this rare condition.
Keywords: Phaeochromocytoma, Ganglioneuroma, Ganglioneuroblastoma, Neuroblastoma, Schwannoma, Adrenal, Incidentaloma, Composite tumours
Introduction
Phaeochromocytomas are chromaffin cell tumours characterised by the excessive production and secretion of catecholamines. These tumours usually arise in the adrenal medulla but occasionally from chromaffin cells of the sympathetic ganglia; here they are called paragangliomas. These tumours occur in approximately 0.5–2 patients per 1000 with hypertension [1]. Diagnosis is typically made between the third and fifth decades; up to 30% of phaeochromocytomas have a genetic predisposition including syndromes such as von Hippel-Lindau syndrome, neurofibromatosis type 1 (NF-1) and multiple endocrine neoplasia (MEN) syndrome type II [2, 3].
Approximately 15% of phaeochromocytomas are malignant [4]. Patients may present with sustained or paroxysmal hypertension, with associated symptoms such as headaches, sweating, palpitations and tremor [4], caused by the excessive release of catecholamines. Initial biochemical investigations include plasma and/or urine catecholamine and metanephrine levels. If these are elevated, imaging to locate a potential tumour includes CT and/or MRI scans. If these scans are inconclusive, nuclear medicine imaging using radiotracers 123I-metaiodobenzylguanidine (MIBG scan) [1] and more recently gallium-68 dotatate PET (68GA-PET) scans may also be useful [5, 6].
Surgery (usually by laparoscopy) after preoperative alpha blockade is the recommended intervention. Alpha blocker therapy is traditionally used reduce the risk of perioperative cardiovascular complications [1, 7]. The prognosis after surgery is very good for benign tumours; the 5-year survival rate is 95%; however, this drops to 50% in malignant tumours [1]. Treatment of metastatic disease is not well understood due to tumour rarity. Chemotherapy, radionuclide agents such as iobenguane 131I, tyrosine kinase inhibitors such as sunitinib and immunotherapeutic agents such as pembrolizumab have been reported; however, clinical trials for these treatments are ongoing [8]. Long-term follow-up is important in both benign and malignant pathology to ensure that recurrences are detected promptly [1].
Ganglioneuromas are rare tumours of autonomic ganglion cells of the nervous system. They are usually benign and often arise in the posterior mediastinum and retroperitoneum. They rarely occur in the adrenal gland, accounting for 0.3–2% of all adrenal incidentalomas [9]. Most adrenal ganglioneuromas are discovered incidentally on CT scans as they are largely asymptomatic and hormonally inactive. However, 30% of patients with ganglioneuromas are found to have raised plasma and urinary metanephrines [10]. Diagnosis can only be confirmed on histology after resection and prognosis is extremely good [9, 10].
Rarely, phaeochromocytomas may be part of a composite tumour [11], where there is another type of tumour (usually of the same embryological origin, i.e. neural crest) present. Tumour types that co-exist with phaeochromocytomas are reported to include ganglioneuromas, schwannomas and ganglioneuroblastomas [12, 13]. There are only a few documented cases in the literature, leading to uncertainty in the understanding of the pathogenesis and natural history of these conditions.
The aim of this study was to report cases of composite phaeochromocytomas seen in one centre and to undertake a systematic review of the published literature to increase the understanding of the epidemiology and clinical outcomes of these rare tumours.
Methods
The histology reports of all patients who underwent resection of phaeochromocytoma over a 19-year period were reviewed to identify patients with composite tumours (defined as a tumour including phaeochromocytoma as at least one of several components). Two patients were identified in a review of 115 reports.
A systematic review of literature was performed to identify all reports of patients with composite tumours including phaeochromocytoma. The online database Medline was searched (on April 16, 2020), via search engine PubMed using the following combination of keywords:
Phaeochromocytoma
Composite OR combined OR incidental OR complex OR co-existing OR coincidental OR associated
Paraganglioma OR ganglioneuroma OR neurofibroma OR schwannoma OR ganglioneuroblastoma OR neurilemmoma OR neuroendocrine carcinoma
The titles and/or abstracts of all articles retrieved by the search were reviewed independently by two authors to include original human studies on patients with composite tumours with phaeochromocytoma as one component. Studies on non-composite tumours, animal studies and those not written in the English language were excluded.
Full texts of articles considered suitable for inclusion were reviewed against the same criteria. The bibliography of included papers was also screened. Data from all included studies on demographics, clinic-pathological features and outcomes were collected in an excel spreadsheet and analysed. Included studies were also critically appraised using the Joanne Briggs Institute (JBI) critical appraisal checklist for case reports [14]. One point was awarded for each question if appropriate. Scores were reported as a percentage of the total applicable questions.
Descriptive analyses included reporting of frequencies (or percentages) for categorical data; median (range) for nonparametric, continuous data and mean (SD) for parametric, continuous data.
As this was a systematic review of literature and presentation of case studies where all identifiable details have been removed, no formal permission has been obtained from the research department and patient consent was not deemed necessary.
Results
Case presentations
Case 1
A 69-year-old gentleman with learning difficulty, hypertension and neurofibromatosis type I presented in 2019 with haematuria. Abdominal examination only showed some neurofibromas and a café au lait lesion. Investigations revealed no definite cause, but CT scan of the urinary tract demonstrated left (4.5 cm) and right (2 cm) adrenal nodules. Biochemical testing demonstrated raised plasma and urine metanephrines, confirming a diagnosis of phaeochromocytoma. I-123 MIBG scans demonstrated uptake in both glands, but more on the left side. Following discussions in a multi-disciplinary team meeting, the patient and his family, left adrenalectomy alone was considered to avoid life-long steroid treatment. Regular surveillance of metanephrine levels was considered preferable to steroid treatment, as compliance with medications was an issue. After preoperative alpha blockade and with perioperative steroids, a left laparoscopic adrenalectomy was performed uneventfully. He was discharged on a low dose of phenoxybenzamine and bisoprolol. Histology showed features of a composite phaeochromocytoma-ganglioneuroma (Fig. 1) with a PASS score of 5/20. The patient and his metanephrine levels 19 months after surgery were satisfactory.
Fig. 1.

Low power view (×4) showing pheochromocytoma (solid black arrow) and ganglioneuroma (solid white arrow) as part of the composite phaeochromocytoma. The inset shows chromogranin A staining at ×10 magnification
Case 2
A 75-year-old gentleman was incidentally found to have a 6-cm left adrenal lesion on CT scan for a new diagnosis of prostate cancer. He had hypertension and gout. A further fluorodeoxyglucose (FDG) PET scan demonstrated raised uptake and biochemistry confirmed raised metanephrine levels, confirming the diagnosis of phaeochromocytoma. After alpha blockade, the patient underwent an uneventful laparoscopic left adrenalectomy. Histology showed a composite tumour with two components—phaeochromocytoma and ganglioneuroma—with a PASS score of 4/20 (Figs. 2 and 3). He was well at 25 months following surgery.
Fig. 2.

Low power view (×4) with background adrenal (solid black arrow) in the left upper part of the picture and composite pheochromocytoma on the right. The inset is a high-power view (×10) showing pheochromocytoma on the left (solid black arrow) and ganglioneuroma on the right (solid white arrow)
Fig. 3.

Low power view (4x) showing S100 staining highlighting the Schwann cells and the sustentacular cells of the composite pheochromocytoma (solid black arrows) but sparing the ganglion cells (solid white arrow) of the ganglioneuroma
Data from these two cases are summarised in Table 1.
Table 1.
Summary of two cases from host institution
| Case 1 | Case 2 | |
|---|---|---|
| Gender | Male | Male |
| Age at presentation (years) | 69 | 75 |
| Composite tumour component | Ganglioneuroma | Ganglioneuroma |
| Underlying genetic syndrome | NF-1 | None |
| Presentation details | Haematuria | Incidental finding |
| Imaging modality | CT, MIBG | CT, FDG-PET |
| Primary management | Laparascopic adrenalectomy | Laparascopic adrenalectomy |
| Tumour diameter (cm) | 4.5 | 6 |
| Tumour laterality | Left | Left |
| Tumour PASS score | 5/20 | 4/20 |
| Outcome | Alive without disease | Alive without disease |
| Follow-up (months) | 19 | 25 |
The process of inclusion and exclusion of articles for this review is shown in a modified PRISMA flow diagram (Fig. 4). In total, 62 studies published between the years 1943 and 2017 involving 94 patients were included [15–73]. Of these studies, 51 were single-case reports. Of the 90 patients where gender information was available; there were 39 males (42%) and 51 females (54%). The median (range) age of patients where this information was available (n=91) was 48 (4–86) years. Data on patient demographics, tumour size and histology, underlying genetic syndrome and presentation details is presented in Table 2. Data on genetic syndromes and on composite tumour components are displayed in Fig. 5 and Fig. 6, respectively. Data on imaging modalities used, primary management, tumour laterality and removal method, patient outcomes and follow-up times is displayed in Table 3.
Fig. 4.

Modified PRISMA flow diagram showing the process of inclusion and exclusion of articles included in this review
Table 2.
Summary of demographic, clinical presentation, histology and underlying genetic syndrome in patients with composite phaeochromocytoma
| Category (n=number of patients where data is available) | Classification | Number of patients |
|---|---|---|
| Gender (n=90) | Male | 39 (42%) |
| Female | 51 (54%) | |
| Age (n=91) | Median (range): 48 (4–86) | |
| Composite tumour component histology (n=94) | Ganglioneuroma | 61 (65%) |
| Ganglioneuroblastoma | 15 (16%) | |
| Neuroblastoma | 10 (11%) | |
| Schwannoma | 1 (1%) | |
| Other* | 7 (7%) | |
| Underlying genetic syndrome (n=94) | None | 68 (73%) |
| NF-1 | 18 (19%) | |
| MEN 2A | 4 (4%) | |
| von Hippel-Lindau syndrome | 2 (2%) | |
| WDHA syndrome2 | 2 (2%) | |
| Presentation details (n=74) | Incidental finding | 35 (47%) |
| Abdominal pain/palpable mass | 24 (32%) | |
| Hypertension | 18 (24%) | |
| Headaches | 14 (19%) | |
| Weight loss | 14 (19%) | |
| Diarrhoea | 12 (16%) | |
| Palpitations and/or tachycardia | 10 (14%) | |
| Nausea and/or vomiting | 5 (7%) | |
| Sweating | 4 (5%) | |
| Anxiety | 3 (4%) | |
| Dysuria/haematuria | 3 (4%) | |
*Other includes: “neuroendocrine carcinoma” (n=1), “ganglion cells in clusters” (n=1), “malignant peripheral nerve sheath tumour” (MPNST—n=3), “MPNST sustentaculoma” (n=1), “MPNST-rhabdomyosarcoma—Triton tumour” (n=1)
Fig. 5.
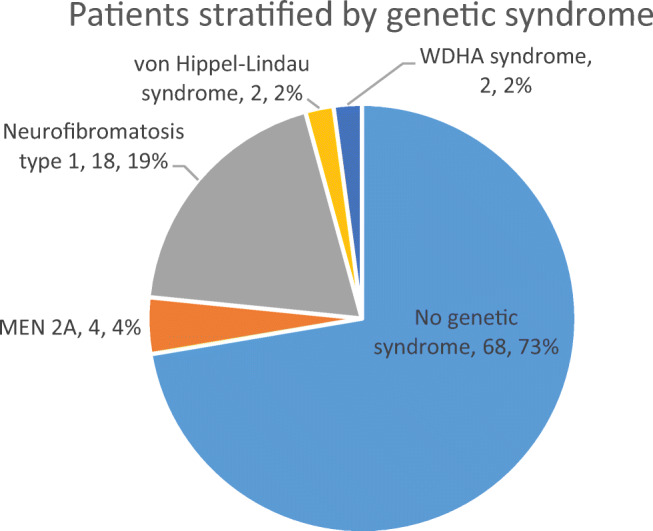
Pie chart showing patients with composite phaeochromocytoma in the review stratified by underlying genetic syndrome
Fig. 6.

Pie chart showing patients with composite phaeochromocytoma included in the review, stratified by composite tumour components
Table 3.
Imaging modalities used, primary management and clinical outcomes of patients with composite phaeochromocytoma
| Category (n=number of patients where data is available) | Classification | Number of patients | |
|---|---|---|---|
| Imaging modality (n=57) | CT | 45 (78%)—solitary modality in 24 (42%) | |
| MRI | 15 (26%) | ||
| MIBG | 21 (37%)—with CT in 11 (19%), with MRI in 5 (9%) | ||
| Ultrasound | 4 (7%) | ||
| Primary management (n=74) | Adrenalectomy | 66 (90) | |
| None—autopsy after death | 6 (8%) | ||
| Radiotherapy | 1 (1%) | ||
| Chemotherapy (for neuroblastoma metastases) + adrenalectomy | 1 (1%) | ||
| Tumour diameter (n=81) | Median (range): 4.98cm (1.5–26) | ||
| Tumour laterality (n=59) | Left (n=23, 39%) | Laparoscopic | 4 (7%) |
| Unspecified | 19 (32%) | ||
| Right (n=33, 56%) | Laparoscopic | 5 (8%) | |
| Unspecified | 28 (48%) | ||
| Bilateral (n=3, 5%) | Laparoscopic | 3 (5%) | |
| Unspecified | 8 (14%) | ||
| Outcomes (n=70) | Alive without disease | 55 (79%) | |
| Alive with metastases | 1 (1%) | ||
| Death from composite tumour | 10 (14%) | ||
| Death from other disease* | 4 (6%) | ||
| Follow up time (n=48) | Median (range): 18 months (2 weeks–14 years) | ||
| Time between primary management and death (n=7) | Median (range): 8 months (3–168 months) | ||
*Other causes of death include colorectal cancer (n=1), non-small cell lung cancer (n=1), bladder cancer (n=1) and myocardial infarction (n=2)
Left-sided operations were performed in 23 instances with 4 specifically described as laparoscopic procedures; the remaining 19 were unspecified. Right-sided operations were performed in 33 patients, with 5 specifically described as laparoscopic procedures; the remaining 28 were unspecified. In 8 procedures, the laterality was not mentioned. Three of these procedures were explicitly described as laparoscopic; the remaining 5 were unspecified. In 3 patients, bilateral resections were performed; one was explicitly described as an open method, while the remaining two were unspecified.
In 48 patients where follow-up information was available, most (85%) patients remained alive without disease at follow-up. Follow-up times ranged from 2 weeks to 14 years, with a median value of 18 months. The median (range) time of death for the 7 of 9 patients where information was available was 8 with a range of (3–168) months.
Of 62 studies, the median (range) score was 100% (range 0–100%); 41 scored 100% in the JBI classification (Table 4). There was a study that scored zero, which was a larger series of adrenal lesions that included 3 patients with composite tumours, but provided no relevant information [16].
Table 4.
JBI critical appraisal of case reports
| JBI Question No. | Score |
|---|---|
| 1. Were the patients’ demographic characteristics clearly described? | 61/62 (98%)* |
| 2. Was the patient’s history clearly described and presented as a timeline? | 53/61 (85%) |
| 3. Was the current clinical condition of the patient on presentation clearly described? | 55/61 (91%) |
| 4. Were diagnostic tests or assessment methods and the results clearly described? | 47/61 (77%) |
| 5. Was the intervention(s) or treatment procedure(s) clearly described? | 52/59 (88%) |
| 6. Was the post-intervention clinical condition clearly described? | 50/58 (86%) |
| 7. Were adverse events (harms) or unanticipated events identified and described? | 27/30 (90%) |
| 8. Does the case report provide takeaway lessons? | 58/62 (94%) |
E.g. of the 62 studies for which question 1 was applicable, 61 studies satisfied the question (98%)
Discussion
Composite phaeochromocytoma tumours are extremely rare [9]. Two patients were identified in this unit over a 19-year period, in addition to the 94 patients identified in this review.
Genetic syndromes including neurofibromatosis 1, MEN 2A, von Hippel Lindau syndrome and watery-diarrhoea hypokalaemia-achlorhydria (WDHA) syndrome were only identified in 28% of patients, similar to patients with phaeochromocytoma alone—a review of 314 patients with phaeochromocytoma 27.4% with an underlying genetic cause [74].
The pathogenesis of composite tumours is unclear. Apart from coincidental occurrence, alteration in the microenvironment of one tumour may favour the formation of a second tumour arising in the same area [75].
Although not currently included in treatment guidelines [7], recent small studies have shown 68Ga-dotatate PET scans to be more specific than MIBG in the diagnosis of phaeochromocytoma and paraganglioma (PPGL) tumours [5, 6]. However, this imaging modality is not widely available.
Of 94 case reports, only seven reported use of alpha blocker therapy before surgery. Alpha blocker therapy pre-surgery is currently recommended in all patients with functional phaeochromocytoma-paragangliomas (PGGLs) to reduce the risk of hypertensive crisis [7]. However recent studies have shown that alpha blockade may not have any effect on intraoperative blood pressure or mortality [76–79].
This review is fairly comprehensive, but it is not possible to make clear recommendations on management based on a review of case studies. Another limitation of our review is the discrepancy in reporting quality amongst the case reports.
Conclusion
This review provides comprehensive demographic and clinical information on composite phaeochromocytomas published in the literature. These tumours affect men and women equally, with the majority of diagnoses occurring between the third and fifth decades. Clinical presentation can be classified into two main categories: cardiovascular and gastrointestinal. Adrenalectomy is the gold standard treatment and prognosis is good; however, these tumours remain extremely rare and their occurrence should prompt consideration for genetic testing.
Author’s contributions
Study conception and design: Keerthan Dhanasekar and Saba P Balasubramanian; acquisition of data: Keerthan Dhanasekar, Saba P Balasubramanian, Vivek Visakan and Fawzia Tahir; analysis and interpretation of data: Keerthan Dhanasekar, Saba P Balasubramanian and Vivek Visakan; drafting of manuscript: Keerthan Dhanasekar, Saba P Balasubramanian and Vivek Visakan; critical revision of manuscript: Keerthan Dhanasekar, Saba P Balasubramanian, Vivek Visakan and Fawzia Tahi.
Declarations
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Manger WM, Eisenhofer G. Pheochromocytoma: diagnosis and management update. Curr Hypertens Rep. 2004;6:477–484. doi: 10.1007/s11906-004-0044-2. [DOI] [PubMed] [Google Scholar]
- 2.Manea M, Marcu D, Bratu O, et al. Pheochromocytoma – clinical manifestations, diagnosis and current perioperative management. J Mind Med Sci. 2019;6:243–247. doi: 10.22543/7674.62.P243247. [DOI] [Google Scholar]
- 3.Widimský J. Recent advances in the diagnosis and treatment of pheochromocytoma. Kidney Blood Press Res. 2006;29:321–326. doi: 10.1159/000097262. [DOI] [PubMed] [Google Scholar]
- 4.Manger WM, Gifford RW. Pheochromocytoma. J Clin Hypertens Greenwich Conn. 2002;4:62–72. doi: 10.1111/j.1524-6175.2002.01452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jing H, Li F, Wang L, et al. Comparison of the 68Ga-DOTATATA PET/CT, FDG PET/CT, and MIBG SPECT/CT in the evaluation of suspected primary Pheochromocytomas and Paragangliomas. Clin Nucl Med. 2017;42:525–529. doi: 10.1097/RLU.0000000000001674. [DOI] [PubMed] [Google Scholar]
- 6.Chang CA, Pattison DA, Tothill RW, et al. (68)Ga-DOTATATE and (18)F-FDG PET/CT in Paraganglioma and Pheochromocytoma: utility, patterns and heterogeneity. Cancer Imaging Off Publ Int Cancer Imaging Soc. 2016;16:22. doi: 10.1186/s40644-016-0084-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lenders JWM, Duh Q-Y, Eisenhofer G, et al. Pheochromocytoma and Paraganglioma: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2014;99:1915–1942. doi: 10.1210/jc.2014-1498. [DOI] [PubMed] [Google Scholar]
- 8.Jimenez C (2018) Treatment for patients with malignant Pheochromocytomas and Paragangliomas: a perspective from the hallmarks of Cancer. Front Endocrinol 9. 10.3389/fendo.2018.00277 [DOI] [PMC free article] [PubMed]
- 9.Mylonas KS, Schizas D, Economopoulos KP. Adrenal ganglioneuroma: what you need to know. World J Clin Cases. 2017;5:373–377. doi: 10.12998/wjcc.v5.i10.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erem C, Fidan M, Civan N, et al. Hormone-secreting large adrenal ganglioneuroma in an adult patient: a case report and review of literature. Blood Press. 2014;23:64–69. doi: 10.3109/08037051.2013.796103. [DOI] [PubMed] [Google Scholar]
- 11.Hu J, Wu J, Cai L, et al. Retroperitoneal composite pheochromocytoma-ganglioneuroma : a case report and review of literature. Diagn Pathol. 2013;8:63. doi: 10.1186/1746-1596-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rao RN, Singla N, Yadav K. Composite pheochromocytoma-ganglioneuroma of the adrenal gland: a case report with immunohistochemical study. Urol Ann. 2013;5:115–118. doi: 10.4103/0974-7796.110011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shida Y, Igawa T, Abe K, et al. Composite pheochromocytoma of the adrenal gland: a case series. BMC Res Notes. 2015;8:257. doi: 10.1186/s13104-015-1233-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moola S, Munn Z, Tufanaru C, et al (2020) Chapter 7: Systematic reviews of etiology and risk. In: JBI Manual for Evidence Synthesis. https://wiki.jbi.global/display/MANUAL/Appendix+7.4+Critical+appraisal+checklist+for+case+reports
- 15.Gupta S, Zhang J, Erickson LA. Composite Pheochromocytoma/Paraganglioma-Ganglioneuroma: a Clinicopathologic study of eight cases with analysis of succinate dehydrogenase. Endocr Pathol. 2017;28:269–275. doi: 10.1007/s12022-017-9494-3. [DOI] [PubMed] [Google Scholar]
- 16.Perrino CM, Ho A, Dall CP, Zynger DL. Utility of GATA3 in the differential diagnosis of pheochromocytoma. Histopathology. 2017;71:475–479. doi: 10.1111/his.13229. [DOI] [PubMed] [Google Scholar]
- 17.Efared B, Atsame-Ebang G, Tahirou S, et al. Bilateral pheochromocytoma with ganglioneuroma component associated with multiple neuroendocrine neoplasia type 2A: a case report. J Med Case Rep. 2017;11:208. doi: 10.1186/s13256-017-1364-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sousa NV, Marques de Oliveira LC, Cortez PJO, et al. A rare case of Ganglioneuroblastoma encapsulated in Pheochromocytoma. Acta Med (Hradec Kralove) 2016;59:67–69. doi: 10.14712/18059694.2016.92. [DOI] [PubMed] [Google Scholar]
- 19.Namekawa T, Utsumi T, Imamoto T, et al. Composite pheochromocytoma with a malignant peripheral nerve sheath tumor: case report and review of the literature. Asian J Surg. 2016;39:187–190. doi: 10.1016/j.asjsur.2012.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Miettinen M, Saari A. Pheochromocytoma combined with malignant schwannoma: unusual neoplasm of the adrenal medulla. Ultrastruct Pathol. 1988;12:513–527. doi: 10.3109/01913128809032236. [DOI] [PubMed] [Google Scholar]
- 21.Suenaga S, Ichiyanagi O, Ito H, et al. Expression of extracellular signal-regulated kinase 5 and Ankyrin repeat domain 1 in composite Pheochromocytoma and Ganglioneuroblastoma detected incidentally in the adult adrenal gland. Intern Med Tokyo Jpn. 2016;55:3611–3621. doi: 10.2169/internalmedicine.55.7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kimura N, Watanabe T, Fukase M, et al. Neurofibromin and NF1 gene analysis in composite pheochromocytoma and tumors associated with von Recklinghausen’s disease. Mod Pathol Off J U S Can Acad Pathol Inc. 2002;15:183–188. doi: 10.1038/modpathol.3880513. [DOI] [PubMed] [Google Scholar]
- 23.Juarez D, Brown RW, Ostrowski M, et al (1999) Pheochromocytoma associated with neuroendocrine carcinoma. A new type of composite pheochromocytoma. Arch Pathol Lab Med 123:1274–1279. 10.1043/0003-9985(1999)123<1274:PAWNC>2.0.CO;2 [DOI] [PubMed]
- 24.Franquemont DW, Mills SE, Lack EE. Immunohistochemical detection of neuroblastomatous foci in composite adrenal pheochromocytoma-neuroblastoma. Am J Clin Pathol. 1994;102:163–170. doi: 10.1093/ajcp/102.2.163. [DOI] [PubMed] [Google Scholar]
- 25.Zhang B-Y, Zhao M, Li B, Zhang J-M. Diverse proportion in composite pheochromocytoma-ganglioneuroma may induce varied clinical symptom: comparison of two cases. Int J Clin Exp Pathol. 2015;8:15369–15374. [PMC free article] [PubMed] [Google Scholar]
- 26.Comstock JM, Willmore-Payne C, Holden JA, Coffin CM. Composite pheochromocytoma: a clinicopathologic and molecular comparison with ordinary pheochromocytoma and neuroblastoma. Am J Clin Pathol. 2009;132:69–73. doi: 10.1309/AJCPN76VTIGWPOAG. [DOI] [PubMed] [Google Scholar]
- 27.Brady S, Lechan RM, Schwaitzberg SD, et al. Composite pheochromocytoma/ganglioneuroma of the adrenal gland associated with multiple endocrine neoplasia 2A: case report with immunohistochemical analysis. Am J Surg Pathol. 1997;21:102–108. doi: 10.1097/00000478-199701000-00011. [DOI] [PubMed] [Google Scholar]
- 28.Tran L, Fitzpatrick C, Cohn SL, Pytel P. Composite tumor with pheochromocytoma and immature neuroblastoma: report of two cases with cytogenetic analysis and discussion of current terminology. Virchows Arch Int J Pathol. 2017;471:553–557. doi: 10.1007/s00428-017-2225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kikuchi Y, Wada R, Sakihara S, et al. Pheochromocytoma with histologic transformation to composite type, complicated by watery diarrhea, hypokalemia, and achlorhydria syndrome. Endocr Pract Off J Am Coll Endocrinol Am Assoc Clin Endocrinol. 2012;18:e91–e96. doi: 10.4158/EP11370.CR. [DOI] [PubMed] [Google Scholar]
- 30.Hu J, Wu J, Cai L, et al. Retroperitoneal composite pheochromocytoma-ganglioneuroma : a case report and review of literature. Diagn Pathol. 2013;8:63. doi: 10.1186/1746-1596-8-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tohme CA, Mattar WE, Ghorra CS. Extra-adrenal composite pheochromocytoma-ganglioneuroma. Saudi Med J. 2006;27:1594–1597. [PubMed] [Google Scholar]
- 32.Gullu S, Gursoy A, Erdogan MF, et al. Multiple endocrine neoplasia type 2A/localized cutaneous lichen amyloidosis associated with malignant pheochromocytoma and ganglioneuroma. J Endocrinol Investig. 2005;28:734–737. doi: 10.1007/BF03347557. [DOI] [PubMed] [Google Scholar]
- 33.Khan AN, Solomon SS, Childress RD. Composite pheochromocytoma-ganglioneuroma: a rare experiment of nature. Endocr Pract Off J Am Coll Endocrinol Am Assoc Clin Endocrinol. 2010;16:291–299. doi: 10.4158/EP09205.RA. [DOI] [PubMed] [Google Scholar]
- 34.Mezitis SGE, Geller M, Bocchieri E, et al. Association of pheochromocytoma and ganglioneuroma: unusual finding in neurofibromatosis type 1. Endocr Pract Off J Am Coll Endocrinol Am Assoc Clin Endocrinol. 2007;13:647–651. doi: 10.4158/EP.13.6.647. [DOI] [PubMed] [Google Scholar]
- 35.Gupta R, Sharma A, Arora R, Vijayaraghavan M. Composite phaeochromocytoma with malignant peripheral nerve sheath tumour and rhabdomyosarcomatous differentiation in a patient without von Recklinghausen disease. J Clin Pathol. 2009;62:659–661. doi: 10.1136/jcp.2009.064790. [DOI] [PubMed] [Google Scholar]
- 36.Majumder S, Grabska J, Trikudanathan G, et al. Functional “composite” pheochromocytoma-ganglioneuroma presenting as a pancreatic mass. Pancreatol Off J Int Assoc Pancreatol IAP Al. 2012;12:211–214. doi: 10.1016/j.pan.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 37.Bernini GP, Moretti A, Mannelli M, et al. Unique association of non-functioning pheochromocytoma, ganglioneuroma, adrenal cortical adenoma, hepatic and vertebral hemangiomas in a patient with a new intronic variant in the VHL gene. J Endocrinol Investig. 2005;28:1032–1037. doi: 10.1007/BF03345345. [DOI] [PubMed] [Google Scholar]
- 38.Gong J, Wang X, Chen X, et al. Adrenal and extra-adrenal nonfunctioning composite pheochromocytoma/paraganglioma with immunohistochemical ectopic hormone expression: comparison of two cases. Urol Int. 2010;85:368–372. doi: 10.1159/000317312. [DOI] [PubMed] [Google Scholar]
- 39.Sakaguchi N, Sano K, Ito M, et al. A case of von Recklinghausen’s disease with bilateral pheochromocytoma-malignant peripheral nerve sheath tumors of the adrenal and gastrointestinal autonomic nerve tumors. Am J Surg Pathol. 1996;20:889–897. doi: 10.1097/00000478-199607000-00013. [DOI] [PubMed] [Google Scholar]
- 40.Tatekawa Y, Muraji T, Nishijima E, et al. Composite pheochromocytoma associated with adrenal neuroblastoma in an infant: a case report. J Pediatr Surg. 2006;41:443–445. doi: 10.1016/j.jpedsurg.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 41.Candanedo-González FA, Alvarado-Cabrero I, Gamboa-Domínguez A, et al. Sporadic type composite pheochromocytoma with neuroblastoma: clinicomorphologic, DNA content and ret gene analysis. Endocr Pathol. 2001;12:343–350. doi: 10.1385/ep:12:3:343. [DOI] [PubMed] [Google Scholar]
- 42.Choi E-K, Kim W-H, Park K-Y. A case of a composite adrenal medullary tumor of pheochromocytoma and ganglioneuroma masquerading as acute pancreatitis. Korean J Intern Med. 2006;21:141–145. doi: 10.3904/kjim.2006.21.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vlenterie M, Flucke U, Hofbauer LC, et al. Pheochromocytoma and gastrointestinal stromal tumors in patients with neurofibromatosis type I. Am J Med. 2013;126:174–180. doi: 10.1016/j.amjmed.2012.07.022. [DOI] [PubMed] [Google Scholar]
- 44.Min KW, Clemens A, Bell J, Dick H. Malignant peripheral nerve sheath tumor and pheochromocytoma. A composite tumor of the adrenal. Arch Pathol Lab Med. 1988;112:266–270. [PubMed] [Google Scholar]
- 45.Hyatt E, Andreotti R. Society of radiologists in ultrasound 2013 Toshiba residents program. Ultrasound Q. 2014;30:205–207. doi: 10.1097/RUQ.0000000000000090. [DOI] [PubMed] [Google Scholar]
- 46.Thiel EL, Trost BA, Tower RL. A composite pheochromocytoma/ganglioneuroblastoma of the adrenal gland. Pediatr Blood Cancer. 2010;54:1032–1034. doi: 10.1002/pbc.22436. [DOI] [PubMed] [Google Scholar]
- 47.Ch’ng ES, Hoshida Y, Iizuka N, et al. Composite malignant pheochromocytoma with malignant peripheral nerve sheath tumour: a case with 28 years of tumour-bearing history. Histopathology. 2007;51:420–422. doi: 10.1111/j.1365-2559.2007.02781.x. [DOI] [PubMed] [Google Scholar]
- 48.Watanabe T, Noshiro T, Kusakari T, et al. Two cases of pheochromocytoma diagnosed histopathologically as mixed neuroendocrine-neural tumor. Intern Med Tokyo Jpn. 1995;34:683–687. doi: 10.2169/internalmedicine.34.683. [DOI] [PubMed] [Google Scholar]
- 49.Onozawa M, Fukuhara T, Minoguchi M, et al. Hypokalemic rhabdomyolysis due to WDHA syndrome caused by VIP-producing composite pheochromocytoma: a case in neurofibromatosis type 1. Jpn J Clin Oncol. 2005;35:559–563. doi: 10.1093/jjco/hyi139. [DOI] [PubMed] [Google Scholar]
- 50.Chetty R, Duhig JD. Bilateral pheochromocytoma-ganglioneuroma of the adrenal in type 1 neurofibromatosis. Am J Surg Pathol. 1993;17:837–841. doi: 10.1097/00000478-199308000-00009. [DOI] [PubMed] [Google Scholar]
- 51.Matias-Guiu X, Garrastazu MT. Composite phaeochromocytoma-ganglioneuroblastoma in a patient with multiple endocrine neoplasia type IIA. Histopathology. 1998;32:281–282. doi: 10.1046/j.1365-2559.1998.0372g.x. [DOI] [PubMed] [Google Scholar]
- 52.Lau SK, Chu PG, Weiss LM. Mixed cortical adenoma and composite pheochromocytoma-ganglioneuroma: an unusual corticomedullary tumor of the adrenal gland. Ann Diagn Pathol. 2011;15:185–189. doi: 10.1016/j.anndiagpath.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 53.Ercolino T, Becherini L, Valeri A, et al. Uncommon clinical presentations of pheochromocytoma and paraganglioma in two different patients affected by two distinct novel VHL germline mutations. Clin Endocrinol. 2008;68:762–768. doi: 10.1111/j.1365-2265.2007.03131.x. [DOI] [PubMed] [Google Scholar]
- 54.Lee Y, Tan LYR, Ho YH, Leow MKS (2017) Giant phaeochromocytoma presenting with an acute stroke: reappraising phaeochromocytoma surveillance for the neurofibromatosis type 1 phakomatosis. BMJ Case Rep. 10.1136/bcr-2017-222553 [DOI] [PMC free article] [PubMed]
- 55.Salmi J, Pelto-Huikko M, Auvinen O, et al. Adrenal pheochromocytoma-ganglioneuroma producing catecholamines and various neuropeptides. Acta Med Scand. 1988;224:403–408. doi: 10.1111/j.0954-6820.1988.tb19603.x. [DOI] [PubMed] [Google Scholar]
- 56.Moore PJ, Biggs PJ. Compound adrenal medullary tumor. South Med J. 1995;88:475–478. doi: 10.1097/00007611-199504000-00020. [DOI] [PubMed] [Google Scholar]
- 57.George DJ, Watermeyer GA, Levin D, et al. Composite adrenal phaeochromocytoma-ganglioneuroma causing watery diarrhoea, hypokalaemia and achlorhydria syndrome. Eur J Gastroenterol Hepatol. 2010;22:632–634. doi: 10.1097/MEG.0b013e328311a697. [DOI] [PubMed] [Google Scholar]
- 58.Mahajan H, Lee D, Sharma R, et al. Composite phaeochromocytoma-ganglioneuroma, an uncommon entity: report of two cases. Pathology (Phila) 2010;42:295–298. doi: 10.3109/00313021003636451. [DOI] [PubMed] [Google Scholar]
- 59.Nakagawara A, Ikeda K, Tsuneyoshi M, et al. Malignant pheochromocytoma with ganglioneuroblastoma elements in a patient with von Recklinghausen’s disease. Cancer. 1985;55:2794–2798. doi: 10.1002/1097-0142(19850615)55:12<2794::aid-cncr2820551213>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 60.Nagashima F, Hayashi J, Araki Y, et al. Silent mixed ganglioneuroma/pheochromocytoma which produces a vasoactive intestinal polypeptide. Intern Med Tokyo Jpn. 1993;32:63–66. doi: 10.2169/internalmedicine.32.63. [DOI] [PubMed] [Google Scholar]
- 61.Tanaka T, Yoshimi N, Iwata H, et al. Fine-needle aspiration cytology of pheochromocytoma-ganglioneuroma of the organ of Zuckerkandl. Diagn Cytopathol. 1989;5:64–68. doi: 10.1002/dc.2840050112. [DOI] [PubMed] [Google Scholar]
- 62.Balázs M. Mixed pheochromocytoma and ganglioneuroma of the adrenal medulla: a case report with electron microscopic examination. Hum Pathol. 1988;19:1352–1355. doi: 10.1016/s0046-8177(88)80292-2. [DOI] [PubMed] [Google Scholar]
- 63.Wilsher MJ. Metachronous malignant composite phaeochromocytoma and pancreatic mucinous cystadenoma in a patient with neurofibromatosis type 1. Pathology (Phila) 2011;43:170–174. doi: 10.1097/PAT.0b013e32834274a3. [DOI] [PubMed] [Google Scholar]
- 64.Satake H, Inoue K, Kamada M, et al. Malignant composite pheochromocytoma of the adrenal gland in a patient with von Recklinghausen’s disease. J Urol. 2001;165:1199–1200. doi: 10.1016/S0022-5347(05)66471-3. [DOI] [PubMed] [Google Scholar]
- 65.Nigawara K, Suzuki T, Tazawa H, et al. A case of recurrent malignant pheochromocytoma complicated by watery diarrhea, hypokalemia, achlorhydria syndrome. J Clin Endocrinol Metab. 1987;65:1053–1056. doi: 10.1210/jcem-65-5-1053. [DOI] [PubMed] [Google Scholar]
- 66.Steen O, Fernando J, Ramsay J, Prebtani APH. An unusual case of a composite Pheochromocytoma with Neuroblastoma. J Endocrinol Metab. 2014;4:39–46. doi: 10.14740/jem.v4i1-2.211. [DOI] [Google Scholar]
- 67.Contreras LN, Budd D, Yen TS, et al. Adrenal ganglioneuroma-pheochromocytoma secreting vasoactive intestinal polypeptide. West J Med. 1991;154:334–337. [PMC free article] [PubMed] [Google Scholar]
- 68.Hirasaki S, Kanzaki H, Okuda M, et al. Composite paraganglioma-ganglioneuroma in the retroperitoneum. World J Surg Oncol. 2009;7:81. doi: 10.1186/1477-7819-7-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yoshimi N, Tanaka T, Hara A, et al. Extra-adrenal pheochromocytoma-ganglioneuroma. A case report. Pathol Res Pract. 1992;188:1098–1100. doi: 10.1016/S0344-0338(11)81261-6. [DOI] [PubMed] [Google Scholar]
- 70.Kimura N, Miura Y, Miura K, et al. Adrenal and retroperitoneal mixed neuroendocrine-neural tumors. Endocr Pathol. 1991;2:139–147. doi: 10.1007/BF02915454. [DOI] [PubMed] [Google Scholar]
- 71.Pathmanathan N, Murali R. Composite phaeochromocytoma with intratumoural metastatic squamous cell carcinoma. Pathology (Phila) 2003;35:263–265. doi: 10.1080/0031302031000151028. [DOI] [PubMed] [Google Scholar]
- 72.Lisewski D, Ryan S, Lim EM, et al. Concomitant compostite adrenal phoechromocytoma, multipte gastric stromal tumours and pseudohermaphrodism in a patient with von Recklinghausen’s disease. Int Semin Surg Oncol ISSO. 2006;3:11. doi: 10.1186/1477-7800-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dawson DW, Tapp E. Aompound tumour of the adrenal medulla. J Pathol. 1969;97:231–233. doi: 10.1002/path.1710970207. [DOI] [PubMed] [Google Scholar]
- 74.Amar L, Bertherat J, Baudin E, et al. Genetic testing in Pheochromocytoma or functional Paraganglioma. J Clin Oncol. 2005;23:8812–8818. doi: 10.1200/JCO.2005.03.1484. [DOI] [PubMed] [Google Scholar]
- 75.Sung CT, Shetty A, Menias CO, et al. Collision and composite tumors; radiologic and pathologic correlation. Abdom Radiol N Y. 2017;42:2909–2926. doi: 10.1007/s00261-017-1200-x. [DOI] [PubMed] [Google Scholar]
- 76.Schimmack S, Kaiser J, Probst P, et al. Meta-analysis of α-blockade versus no blockade before adrenalectomy for phaeochromocytoma. Br J Surg. 2020;107:e102–e108. doi: 10.1002/bjs.11348. [DOI] [PubMed] [Google Scholar]
- 77.Brunaud L, Boutami M, Nguyen-Thi P-L, et al. Both preoperative alpha and calcium channel blockade impact intraoperative hemodynamic stability similarly in the management of pheochromocytoma. Surgery. 2014;156:1410–1417. doi: 10.1016/j.surg.2014.08.022. [DOI] [PubMed] [Google Scholar]
- 78.Groeben H, Nottebaum BJ, Alesina PF, et al. Perioperative α-receptor blockade in phaeochromocytoma surgery: an observational case series. Br J Anaesth. 2017;118:182–189. doi: 10.1093/bja/aew392. [DOI] [PubMed] [Google Scholar]
- 79.Shao Y, Chen R, Shen Z, et al. Preoperative alpha blockade for normotensive pheochromocytoma: is it necessary? J Hypertens. 2011;29:2429–2432. doi: 10.1097/HJH.0b013e32834d24d9. [DOI] [PubMed] [Google Scholar]