

Abstract
The clinical course of trilateral retinoblastoma can be unpredictable, and expressivity of germline RB1 variants may vary during development. We describe an unexpected fatal case of trilateral retinoblastoma with an intracranial tumor in an unusual location and discuss genetic copy number analyses as a useful diagnostic tool with therapeutic potential.
Keywords: DNA copy number analysis, intracranial tumor, RB1, retinoblastoma, trilateral
The clinical course of trilateral retinoblastoma can be unpredictable, and expressivity of germline RB1 variants may vary during development.

1. INTRODUCTION
Retinoblastoma is a rare childhood cancer of the retina (incidence 1 in 16,000 live births). In high income countries, survival rate is >95%. 1
Retinoblastoma arises from a cell that nearly always harbors pathogenic variants in both copies of the RB1 gene, a first and second “hit” as described by Knudson in 1971 (Knudson's two‐hit hypothesis). 2 In non‐heritable retinoblastoma, both RB1 variants arise in the developing retina, whereas in heritable retinoblastoma, a germline RB1 variant is present. The majority of heritable retinoblastoma patients have a de novo RB1 variant, as only 10% inherit the variant from a parent (autosomal dominant transmission). 2 Patients with heritable retinoblastoma have a significantly increased risk for other primary cancers later in life, including osteosarcoma, melanoma, and pinealoblastoma. 1 , 3
Retinoblastoma can affect one eye (unilateral) or both eyes (bilateral). Children with bilateral disease have a pathogenic germline RB1 variant (hence heritable retinoblastoma), but some children with heritable retinoblastoma only develop unilateral disease, due to variation in penetrance and expressivity. Trilateral retinoblastoma refers to an intracranial tumor associated with heritable retinoblastoma and occurs in 3,5% of patients with heritable retinoblastoma. 4
In this case study of trilateral retinoblastoma, we describe a patient with an intracranial tumor in an unusual location and a challenging clinical investigation process involving extensive genetic testing.
2. CASE HISTORY
A 15‐month‐old boy presented with deviation of the right eyeball that had been slowly progressing since onset one month earlier. There was no family history of retinoblastoma, and the patient had an unremarkable prior history with normal growth and developmental milestones. The diagnosis of retinoblastoma was made on the basis of ophthalmoscopy and ultrasound of the right eye that revealed large, calcified tumor masses. The anterior chamber was normal. A baseline brain MRI was performed and revealed no extraocular extensions or trilateral disease (Figure 1).
FIGURE 1.
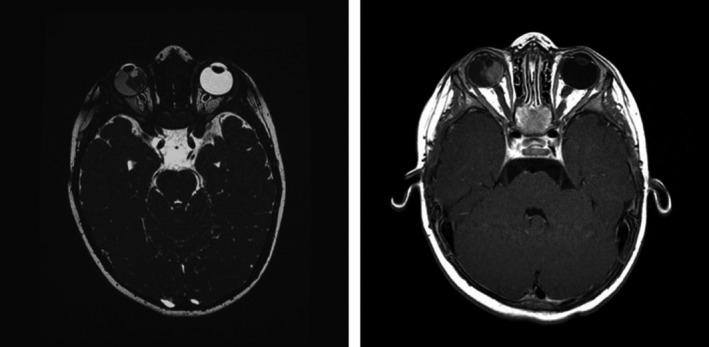
Baseline brain MRI at age 15 months
The eye was staged Group E according to International Intraocular Retinoblastoma Classification, 5 cT3 TNMH. 6 Pathology showed retinoblastoma with minimal invasion of the choroid, and no extrascleral tumor cells, invasion of the optic nerve, or tumor cells in the anterior chamber, pT2a (Figure 2). 6 The patient was treated according to guidelines with surgical enucleation of the right eye, without adjuvant chemo‐ or radiation therapy. 7 Because of the risk of contralateral retinoblastoma, the patient was monitored with retinal examination under anesthesia every 4 weeks.
FIGURE 2.
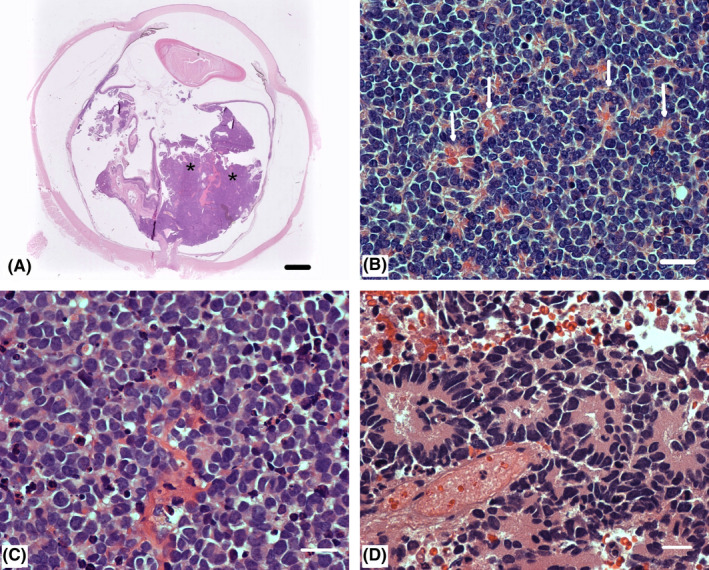
Histopathology images of the retinal (A and B) and cerebral (c and d) tumors. A. The overview of the right eye shows an intraocular tumor of the retina (asterisks) with both endo‐ and exophytic growth and with minimal invasion of the choroid and no extrascleral extension (bar = 800 μm). B. The retinal tumor cells are seen with round or oval hyperchromatic nuclei and surrounded by a scant amount of cytoplasm. Homer‐Right rosettes are seen (white arrows): The tumor is consistent with a retinoblastoma (bar = 40 μm). C. The cerebral tumor shows poorly differentiated round tumor cells with hyperchromatic nuclei. In general, the tumor shows a diffuse growth pattern (bar = 40 μm). D. In small areas of the cerebral tumor, rosettes are also seen (bar = 40 μm). The cerebral tumor is consistent with a retinoblastoma
A germline deletion encompassing exon 2 and at least part of exon 1 of RB1 was detected on DNA extracted from blood. The deletion was not detected in DNA from blood from either of the parents. However, mosaicism in either parent could not be ruled out, and the couple was offered prenatal testing in the future pregnancies.
Almost 2 years after the primary diagnosis, at age 37 months, the patient was admitted to a local pediatric center with a left side peripheral facial nerve palsy and vomiting. In the 8 weeks prior to admission, the patient had experienced hoarseness, fatigue, weight loss, and a dry cough that did not respond to antibiotic treatment. The parents had noticed a lump on the left side of the neck 3 weeks prior to admission, which had been growing.
The patient was transferred to a specialized children's oncology unit. MRI of the neuroaxis revealed a large tumor (3.5 × 5.5 × 5.5 cm) in the cerebellopontine angle, subependymal tumor elements in the rim of the lateral ventricle anterior horns, several drop metastases in the thoracolumbar spinal canal, and enlarged lymph nodes on the left side of the neck (Figure 3). There were no signs of tumor masses in the left eye or the corpus pineale.
FIGURE 3.
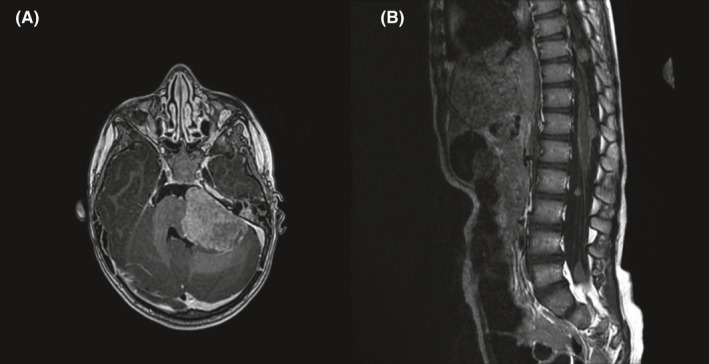
Trilateral retinoblastoma diagnosed at age 37 months. MRI shows (A) a large tumor (3.5 × 5.5 × 5.5 cm) in the cerebellopontine angle and subependymal tumor elements in the rim of the lateral ventricle anterior horns and (B) several drop metastases in the thoracolumbar spinal canal
A surgical decompression with partial resection of the cerebral tumor was performed, and pathology review confirmed the diagnosis of a malignant neuroblastic tumor that could represent either a medulloblastoma or a retinoblastoma, the latter being the most likely diagnosis due to the patient history. The patient was treated with eight cycles of intravenous and intrathecal chemotherapy, followed by high‐dose chemotherapy and stem cell reinfusion. Brain MRIs showed regression throughout the treatment, and a supplemental positron emission tomography (PET) scan performed at one‐month follow‐up showed no pathological activity in residual lesions in the left cerebellopontine angle and the spinal canal. The three‐month follow‐up brain and spine MRI showed no changes.
Five months after treatment was completed, the patient was admitted with sudden‐onset vomiting, headache, fatigue, and loss of balance. MRI showed multiple infra‐ and supratentorial processes bilaterally, and several small nodules in the ventricles. Aspergillosis was suspected, but biopsy showed malignant cells that were confirmed to be retinoblastoma (Figure 2). The patient received palliative care and died 2 weeks later, at 50 months of age.
3. MOLECULAR GENETIC INVESTIGATIONS
A heterozygous deletion of exon 2, and at least part of exon 1, of RB1 was detected in DNA from the patient's blood. Quantitative multiplex PCR (QM‐PCR) confirmed that only one copy of exon 2 was present. For exon 1, three sets of non‐overlapping primers showed only one copy, the first primer being located at the position NM_000321.2(RB1): c.101. Analysis of the RB1 promoter and exon 3 revealed 2 copies.
It was suggested that the deletion might be a reduced‐expressivity variant, as the patient was only unilaterally affected at the time, and it is known that some exon 1 variants show reduced penetrance due to utilization of alternative transcription start sites. 8 , 9 The germline deletion was also detected at a heterozygous allele frequency in the patient's primary, intraocular tumor; a second RB1 pathogenic variant was not identified after sequencing all RB1 coding sequence and flanking noncoding regions, methylation‐sensitive PCR analysis of the RB1 promoter, 10 and copy number analysis by multiplex ligation‐depended probe amplification (MLPA, SALSA P047‐D1 RB1 MRC‐Holland, Amsterdam, The Netherlands). To confirm presence of tumor DNA in the intraocular tumor sample, analysis for well‐recognized retinoblastoma somatic copy number changes in the genes KIF14, DEK, E2F3, CDH11, and MYCN was performed using QM‐PCR. 11 , 12 The analysis showed three copies of DEK and four copies of MYCN, confirming the presence of tumor DNA.
When the patient was diagnosed with an intracranial tumor, further genetic analyses were performed to help determine whether the tumor was a primary tumor (trilateral retinoblastoma) or a metastasis. MLPA analysis of the intracranial tumor identified homozygous loss of RB1 exons 1–2 (Table 1) as well as a gain of RB1 exons 3–27. This result potentially suggests that the intracranial tumor DNA may have developed by loss of the normal RB1 allele and two rounds of endoreduplication of the abnormal copy (loss of heterozygosity, LOH). LOH is frequently the second mutational event leading to the development of a tumor in both retinoblastomas as well as in other cancers. 13 Analysis for somatic copy number changes in the KIF14, DEK, E2F3, CDH11, and MYCN genes showed differences in comparison with the intraocular tumor (Table 2), suggesting that the intraocular and the intracranial tumors were independent in origin, predisposed by the presence of the germline RB1 variant.
TABLE 1.
Comparison of genetic test results
| Sample | RB1 allele 1 | RB1 allele 2 |
|---|---|---|
| Intracranial tumor | del1‐>2 | del1‐>2 |
| Eye tumor | del1‐>2 | Normal |
| Blood | del1‐>2 | Normal |
TABLE 2.
Genetic copy number analysis of the patient's tumors
| Tumor sample | Copy numbers | ||||
|---|---|---|---|---|---|
| KIF14 | DEK | E2F3 | CDH11 | MYCN | |
| Intracranial | 5 | 6 | 5 | 2 | 5 |
| Eye | 2 | 3 | 2 | 2 | 4 |
4. DISCUSSION
We describe a clinical case of trilateral retinoblastoma that is atypical in several aspects. Non‐pineal tumors in trilateral retinoblastoma usually occur synchronously, and almost exclusively in the supra‐ or parasellar regions. 14 , 15 , 16 Accordingly, we have identified only two previous cases with a non‐pineal tumor not located to the supra‐ or parasellar regions. 17 , 18 As described in our presented case, the tumors were instead located in relation to the fourth ventricle and cerebellum. In both previous cases, however, the patients had been treated for bilateral retinoblastoma prior to the diagnosis of metachronous, trilateral retinoblastoma. The first case, presented by Finelli et al. in 1995, describes a girl with a family history of retinoblastoma, who was diagnosed with bilateral retinoblastoma at age 7 weeks, at which point a brain CT was normal. At age five months, she had developed bilateral recurrent ocular disease and an MRI of the brain showed a mass in the fourth ventricle, extending from the inferior vermian region along the right cerebellar hemisphere. Histopathology showed a primitive neuroectodermal tumor with neuronal differentiation. 17 The second case, presented by Elias et al. in 2001, describes a girl who was diagnosed with retinoblastoma of the right eye at age 9 months, and a second primary neoplasm of the left eye one month later. MRI of the brain was normal. Cytogenetic investigations revealed a germline deletion on chromosome 13 with breakpoints at q12.3 and q21.3, thus encompassing the RB1 gene that originated from a balanced chromosomal rearrangement in the mother. At age 4 years, the patient had no sign of recurrent eye disease, but had developed a large, symptomatic tumor located to the midline cerebellum. She died months later from disseminated disease with spinal involvement. Autopsy and thorough histopathological assessment were performed, and the cerebellar tumor was found to be a medulloblastoma that originated separately from the ocular tumors, based on histomorphological and immunocytochemical features. 18 In our presented case, the same conclusion was reached by supplement of genetic copy number analysis, to confirm the diagnosis of trilateral retinoblastoma with a primary tumor located to the cerebellopontine angle.
In addition to being a diagnostic tool, characterization of somatic alterations is the basis of personalized oncology therapies, developed to improve treatment outcomes and minimize long‐term sequelae by selectively targeting cancer cells. For some cancer types, targeted treatments are well established, such as the use of tyrosine kinase inhibitors in Philadelphia chromosome‐positive leukemia 19 and the use of PARP inhibitors in ovarian‐ and breast cancers harboring somatic or germline pathogenic variants in certain genes, including BRCA1 and BRCA2. 20 Pediatric cancer tumors in general—and retinoblastomas in particular—harbor less somatic genomic alterations than cancerous tumors in adults, but the potential for targeted treatments is promising. 19 , 21 , 22 Among possible targets being studied in retinoblastoma are E2F3 and MYCN, 23 which were amplified in the tumors of the patient described in this case report.
More than 50% of trilateral retinoblastomas can be diagnosed by a baseline brain MRI in relation to the primary diagnosis of retinoblastoma, 4 and this is now generally accepted to be the standard approach. 7 , 24 Whether further screening for trilateral retinoblastoma should be implemented remains unsolved. In a recent meta‐analysis by De Jong et al., it is estimated that if a brain MRI screening was implemented, with scans every six months from diagnosis to age 36 months, it would take at least 311 scans to detect one asymptomatic pineal trilateral retinoblastoma and 776 scans to save a single life. 24 The authors also found that there is no association between age at diagnosis of intraocular‐ and pineal retinoblastoma, or between the laterality of intraocular retinoblastoma and the age at diagnosis of pineal trilateral retinoblastoma. They suggested that their findings could be due to an independent development of intraocular and pineal retinoblastoma, and that penetrance and expressivity of the germline RB1 variant may vary during development.
Similarly, in a review by Yamanaka et al., no difference in latency period between intraocular and tertiary retinoblastoma of any location was found, when comparing patients according to laterality of their intraocular retinoblastoma. 15 In line with these findings, our report of a patient with non‐pineal trilateral retinoblastoma demonstrates an apparent variation in expressivity. The patient presented with unilateral and localized intraocular disease, was found to carry a seemingly reduced‐expressivity RB1 germline variant, and was expected to have a good prognosis. However, the patient developed aggressive tertiary disease that initially responded to treatment but recurred shortly after.
In conclusion, the clinical course of trilateral retinoblastoma can be unpredictable. Whether screening for trilateral retinoblastoma should be implemented is still a subject of debate, and no international consensus exists. Genetic analysis can be a useful diagnostic tool in challenging clinical investigations of trilateral retinoblastoma and has therapeutic potential.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
Saga Elise Eiset and Pernille Axél Gregersen drafted the manuscript. Mikkel Funding, Hilary Racher, Steffen Heegaard, Brenda Gallie, and Steen F. Urbak contributed to relevant parts of the manuscript. All authors reviewed and approved the final manuscript.
ETHICAL APPROVAL
None.
CONSENT
Informed consent was obtained from both of the patient's parents for publication of this case report and clinical images. Written documentation of consent in accordance with the institution's patient consent policy is available upon request.
ACKNOWLEDGMENTS
We are very grateful to the family for providing their consent for publication.
Eiset SE, Funding M, Racher H, et al. Metachronous, non‐pineal, trilateral retinoblastoma in a patient with a seemingly reduced‐expressivity RB1 germline deletion. Clin Case Rep. 2022;10:e05498. doi: 10.1002/ccr3.5498
Funding information
None
DATA AVAILABILITY STATEMENT
None.
REFERENCES
- 1. Dimaras H, Corson Timothy W, Cobrinik David, et al. Retinoblastoma. Nat Rev Dis Primers. 2015;1:15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Knudson AG Jr. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68(4):820‐823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gregersen PA, Olsen MH, Urbak SF, et al. Incidence and mortality of second primary cancers in danish patients with retinoblastoma, 1943–2013. JAMA Netw Open. 2020;3(10):e2022126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Jong MC, Kors WA, de Graaf P, Castelijns JA, Moll AC, Kivelä T. The incidence of trilateral retinoblastoma: a systematic review and meta‐analysis. Am J Ophthalmol. 2015;160(6):1116‐1126. [DOI] [PubMed] [Google Scholar]
- 5. Linn Murphree A. Intraocular retinoblastoma: the case for a new group classification. Ophthalmol Clin North Am. 2005;18(1):41‐53. [DOI] [PubMed] [Google Scholar]
- 6. Mallipatna AGB Chévez‐Barrios P, et al. Retinoblastoma. In Amin MB, Edge S, Greene F, Byrd DR, Brookland RK, Washington MK, Gershenwald JE, Compton CC, Hess KR, Sullivan DC, Jessup JM, Brierley JD, Gaspar LE, Schilsky RL, Balch CM, Winchester DP, Asare EA, Madera M, Gress DM, Meyer LR, (Eds.). AJCC Cancer Staging Manual. Springer; 2017:819‐831. https://link.springer.com/book/9783319406176 [Google Scholar]
- 7. NOPHO . Nordiske retningslinjer for utredning og behandling av Retinoblastomer (Nordic Guidelines for Retinoblastoma). 2019; NOPHO Retinoblastoma Meeting September 2019]. Available from: https://www.nopho.org/member_pages/member_area/workinggroups/retinoblastom/Nordiske%20retningslinjer%20for%20utreding%20og%20behandling%20av%20Retinoblastom_v2_2016‐05.pdf
- 8. Lohmann DR, Brandt B, Höpping W, Passarge E, Horsthemke B. Distinct RB1 gene mutations with low penetrance in hereditary retinoblastoma. Hum Genet. 1994;94(4):349‐354. [DOI] [PubMed] [Google Scholar]
- 9. Sanchez‐Sanchez F, Ramírez‐Castillejo C, Weekes DB, et al. Attenuation of disease phenotype through alternative translation initiation in low‐penetrance retinoblastoma. Hum Mutat. 2007;28(2):159‐167. [DOI] [PubMed] [Google Scholar]
- 10. Zeschnigk M, Lohmann D, Horsthemke B. A PCR test for the detection of hypermethylated alleles at the retinoblastoma locus. J Med Genet. 1999;36(10):793‐794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gallie BL, Campbell C, Devlin H, Duckett A, Squire JA Developmental basis of retinal‐specific induction of cancer by RB mutation. Cancer Res. 1999;59(7 Suppl):1731s‐1735s. [PubMed] [Google Scholar]
- 12. Bowles E, Corson TW, Bayani J, et al. Profiling genomic copy number changes in retinoblastoma beyond loss of RB1. Genes Chromosomes Cancer. 2007;46(2):118‐129. [DOI] [PubMed] [Google Scholar]
- 13. Zhu X, Dunn JM, Goddard AD, et al. Mechanisms of loss of heterozygosity in retinoblastoma. Cytogenet Cell Genet. 1992;59(4):248‐252. [DOI] [PubMed] [Google Scholar]
- 14. de Jong MC, Kors WA, de Graaf P, Castelijns JA, Kivelä T, Moll AC. Trilateral retinoblastoma: a systematic review and meta‐analysis. Lancet Oncol. 2014;15(10):1157‐1167. [DOI] [PubMed] [Google Scholar]
- 15. Yamanaka R, Hayano A, Takashima Y. Trilateral retinoblastoma: a systematic review of 211 cases. Neurosurg Rev. 2019;42(1):39‐48. [DOI] [PubMed] [Google Scholar]
- 16. Kivela T. Trilateral retinoblastoma: a meta‐analysis of hereditary retinoblastoma associated with primary ectopic intracranial retinoblastoma. J Clin Oncol. 1999;17(6):1829‐1837. [DOI] [PubMed] [Google Scholar]
- 17. Finelli DA, Shurin SB, Bardenstein DS. Trilateral retinoblastoma: two variations. AJNR Am J Neuroradiol. 1995;16(1):166‐170. [PMC free article] [PubMed] [Google Scholar]
- 18. Elias WJ, Lopes MBS, Golden WL, Jane JA, Gonzalez‐Fernandez F. Trilateral retinoblastoma variant indicative of the relevance of the retinoblastoma tumor‐suppressor pathway to medulloblastomas in humans. J Neurosurg. 2001;95(5):871‐878. [DOI] [PubMed] [Google Scholar]
- 19. Burdach SEG, Westhoff MA, Steinhauser MF, Debatin KM. Precision medicine in pediatric oncology. Mol Cell Pediatr. 2018;5(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mateo J, Lord Cj, Serra V, et al. A decade of clinical development of PARP inhibitors in perspective. Ann Oncol. 2019;30(9):1437‐1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kooi IE, Mol BM, Massink MPG, et al. Somatic genomic alterations in retinoblastoma beyond RB1 are rare and limited to copy number changes. Sci Rep. 2016;6:25264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grobner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555(7696):321‐327. [DOI] [PubMed] [Google Scholar]
- 23. Kaewkhaw R, Rojanaporn D. Retinoblastoma: Etiology, Modeling, and Treatment. Cancers (Basel). 2020;12(8):2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. de Jong MC, Kors WA, Moll AC, et al. Screening for pineal trilateral retinoblastoma revisited: a meta‐analysis. Ophthalmology. 2020;127(5):601‐607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
None.