

Abstract
Phenylketonuria (PKU) is an inborn error caused by deficiencies in phenylalanine (Phe) metabolism. Mutations in the phenylalanine hydroxylase (PAH) gene are the main cause of the disease whose signature hallmarks of toxically elevated levels of Phe accumulation in plasma and organs such as the brain, result in irreversible intellectual disability. Here, we present a unique approach to treating PKU deficiency by using an mRNA replacement therapy. A full-length mRNA encoding human PAH (hPAH) is encapsulated in our proprietary lipid nanoparticle LUNAR and delivered to a Pahenu2 mouse model that carries a missense mutation in the mouse PAH gene. Animals carrying this missense mutation develop hyperphenylalanemia and hypotyrosinemia in plasma, two clinical features commonly observed in the clinical presentation of PKU. We show that intravenous infusion of LUNAR-hPAH mRNA can generate high levels of hPAH protein in hepatocytes and restore the Phe metabolism in the Pahenu2 mouse model. Together, these data establish a proof of principle of a novel mRNA replacement therapy to treat PKU.
Keywords: MT: Delivery Strategies, mRNA, replacement, lipid nanoparticle, PKU, phenylketonuria, liver, LNP, therapy, hepatocytes, PAH
Graphical abstract
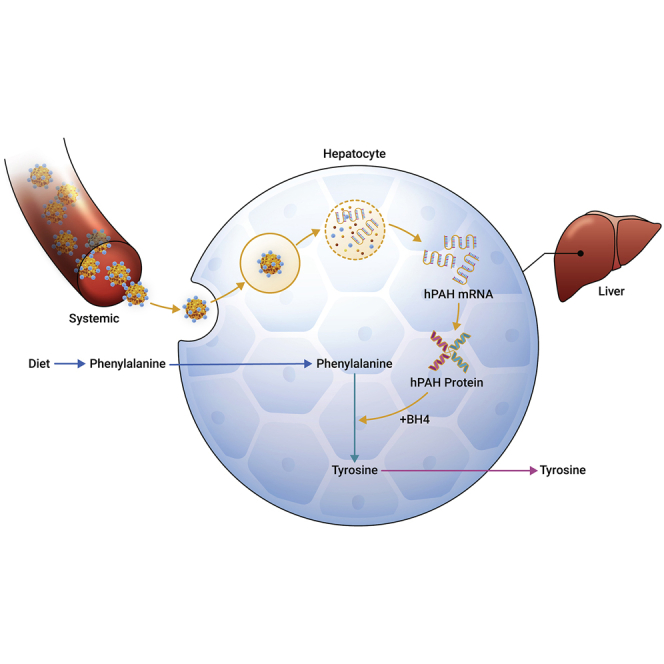
Perez-Garcia-Trelles and colleagues have developed an mRNA replacement therapeutic approach to treat the underlying cause of phenylketonuria in patients carrying a phenylalanine hydroxylase mutation.
Introduction
Phenylketonuria (PKU) is an autosomal recessive disease caused by abnormalities in phenylalanine (Phe) metabolism.1, 2, 3, 4 PKU results from mutations in the phenylalanine hydroxylase (PAH) gene that encodes for a hepatic enzyme responsible for breaking down the dietary Phe into Tyrosine (Tyr), a critical precursor in the synthesis of neurotransmitters such as dopamine, epinephrine, or norepinephrine. PAH is a homotetrameric protein that needs the catalytic activity of the cofactor tetrahydrobiopterin (BH4) to be functional. Defects in BH4 also develop into mild forms of PKU. In these mild forms of PKU, oral administration of sapropterin dihydrochloride (Kuvan), a synthetic form of BH4, is able to normalize circulating Phe levels with the support of a Phe-restricted diet.5, 6, 7 For classic PKU, the only approved drug is pegvaliase (Palynziq)2; however, it is associated with inconvenient dosing and side effects.6,7
Normal blood levels of Phe are <120 μmol/L, with higher Phe levels leading to a phenotypic mosaic ranging from mild hyperphenylalanemia to moderate PKU, where the most severe type, classic PKU, is defined by levels of Phe in blood >1,200 μmol/L.2,4 If left untreated, PKU leads to severe and irreversible intellectual disabilities. Therefore, early detection and preventive standard of care is fundamental for these patients to maintain Phe levels in the blood within a normal therapeutic range (120–360 μmol/L). The implementation of neonatal screenings for PKU has allowed an early detection and initiation of Phe-restricted diets to prevent the development of irreversible neurological defects. However, for older children and adults it is difficult to adhere to a strict diet, which creates severe problems for patients who eat a high-protein meal and are at risk of Phe plasma peaks.2,4 Despite living with specific dietary restrictions, there are still high unmet needs for PKU patients to normalize Phe levels, reduce the number of hyperphenylalanemic peaks, and prevent any irreversible impact on their bodies.
We discuss below the proof-of-concept of a novel mRNA therapeutic approach for PKU. We use the Pahenu2 mice8 as a model and subjected to the treatment, intravenously (i.v.), of a codon-optimized human PAH (hPAH) mRNA encapsulated in our lipid nanoparticle (LNP) LUNAR (LUNAR-hPAH mRNA).9 We have previously shown that LUNAR is 5× more efficient in delivering mRNA than formulations carrying MC3, which is the ionizable lipid of the Food and Drug Administration (FDA)-approved drug Onpattro.10 Delivering a full-length copy of the PAH mRNA to the liver will replenish the functional PAH protein pool, reestablishing the Phe metabolism and reducing the toxic levels of Phe in blood, thereby ameliorating the progression of the disease. An mRNA therapeutic approach could potentially provide a longer and effective solution to PKU patients compared with the current approved standard of care. We show how LUNAR-hPAH mRNA primarily targets hepatocytes, producing high levels of hPAH protein that efficiently restores Phe plasma levels in the PAHenu2 mouse model. These data strongly support LUNAR-hPAH mRNA as an efficient mRNA replacement therapy for PKU.
Results
Pahenu2 mice: a PKU model
The Pahenu2 mouse model is characterized by severe reduction in the activity of the enzyme PAH, responsible for the conversion of plasma Phe into Tyr.1,8 This model recapitulates the hyperphenyalanemia observed in classic PKU patients that courses with high levels of Phe and decreased levels of Tyr in plasma,1,8 making it optimal as a preclinical animal model for PKU studies.1, 2, 3
Plasma levels of Phe and Tyr were tested in wild-type (WT) and mutant BTBR-Pahenu2 mice, with either a heterozygous or homozygous mutation in the Pahenu2 allele (Figure S1). Since Phe levels are strongly influenced by diet, we compared both groups under a normal (Teklad Global 18% protein, Envigo RMX, Inc) versus a special Phe-free diet (Phe-free diet, Teklad Custom Diet TD.170012, Envigo RMX, Inc). At the beginning of the study (day 1), mice were maintained in a normal diet and water ad libitum; on day 2, a cohort of mice were shifted to the special Phe-free diet with water supplemented with limited amount of Phe (30 mg/L Phe; Sigma-Aldrich). Plasma levels of Phe and Tyr were monitored daily for a week (Figure S1).
WT and Pahenu2 heterozygous mice fed on normal diet, maintained plasma Phe levels to approximately 100 to 120 μmol/L throughout the study (Figure S1A). These Phe levels were similar to those seen for Tyr in these animals (e.g., Phe:Tyr ratios approximately equal 1, Figure S1B). In contrast, PAH homozygous mice provided with the normal diet exhibited plasma Phe levels of approximately 2,000 to 2,500 μmol/L throughout the study, a 20-fold increase compared with WT and Pahenu2 heterozygous Phe levels (Figure S1A). Similarly, plasma levels of Tyr in the Pahenu2 homozygous mice were approximately 40 μmol/L, while Phe:Tyr ratios were exceeding 50 (Figures S1A and S1B).
For mice maintained on the special diet (Phe-free diet, and L-Phe-water), WT and Pahenu2 heterozygous mice exhibited plasma Phe levels of approximately 40 to 80 μmol/L throughout the study (Figure S1A). These Phe levels were similar to those seen for Tyr in these animals (e.g., Phe:Tyr ratios approximately equal 1, Figure S1A), consistent with what has been observed in animals under the normal diet. In contrast, plasma Phe levels in Pahenu2 homozygous mice dropped over 6-fold, from approximately 2,000 μmol/L at the beginning of the diet to ∼350 μmol/L within 1 day after shifting to the special diet (Figure S1A). These animals maintained decreased plasma Phe levels throughout the rest of the study. Tyr levels in the homozygous mice resembled those in WT and Pahenu2 heterozygous mice, with Phe:Tyr ratios of approximately 2 to 8 (Figures S1A and S1B). These effects demonstrated that maintenance of Pahenu2 homozygous mice on a low-Phe diet can counteract PKU symptoms by providing a reduction in both plasma Phe concentrations and Phe:Tyr ratios (Figures S1A and S1B).
To determine the dynamics of Phe inducing a hyperphenylalanemic status, animals were subjected to a Phe challenge on the last day of the study (Figure S1C). On day 7, animals were subcutaneously dosed with vehicle (PBS) or 100 mg/kg of L-Phe. One hour post-dose, animals were subjected to terminal bleeding. When assessed 1 hour after the Phe challenge, Pahenu2 homozygous mice that were maintained on the normal diet demonstrated a mild increase in Phe plasma levels (<50%) when compared with pre-dosing (Figure S1C). Among mice maintained on the special diet, plasma Phe levels at 1 hour post-Phe challenge were approximately 5-fold higher than those observed pre-dosing (Figure S1C). In comparison, WT and Pahenu2 heterozygous mice maintained on the special diet exhibited <2-fold elevation in plasma Phe levels after the Phe challenge when compared with pre-dosing (Figure S1C). Notably, Tyr levels were largely unchanged in any of the animals at 1 hour post-Phe challenge; therefore, changes in Phe:Tyr ratios largely tracked with changes in Phe levels (Figure S1C). These data suggested that a single subcutaneous administration of Phe to Pahenu2 homozygous mice fed a limited-Phe diet provided elevated circulating Phe levels, consistent with a hyperphenylalanemic stage, that was not observed in either WT or Pahenu2 heterozygous mice.
These data confirmed that while WT and Pahenu2 heterozygous mice can regulate and metabolize exogenously administered Phe, PAH deficiency in Pahenu2 homozygous mice is responsible for the hyperphenylalanemic stage under normal diet.
Optimizing hPAH mRNAs to generate higher protein levels: a codon and UTR screening
Sequence optimization offers the advantage to generate mRNAs that express higher and long-lasting protein levels.10, 11, 12, 13, 14, 15
We designed two main optimization strategies for in vitro screening and selection of hPAH mRNAs that include coding (Figure 1) and UTR (Figure 2) optimization approaches. The selection of the mRNAs was based on mRNAs that produce higher hPAH protein levels over the human reference sequence.
Figure 1.

Coding region screening unveils optimized mRNA expressing high levels of PAH
(A) Diagram summarizing the screening approaches used to generate improved coding regions for hPAH. (B) Screening of codon-optimized compounds (comp1 to comp7) in mouse and human hepatocyte cell lines at 24, 48, and 72 h. Reference (Ref) sequence was used as a baseline control. Plots were normalized to untransfected wells. (C) Comp8 and comp9 that lack any PAH regulatory domains were screened in human primary hepatocytes at 24 and 72 h. PAH levels were quantified as mean fluorescence intensity (MFI) and plotted. (D) Screening of novel coding region approaches based on comp7 (comp10 to comp17) on human primary hepatocytes at 24 and 72 h. PAH levels were quantified as MFI and plotted. (E) Screening of selected protein stability (comp18 to comp20) and codon optimization sequences (comp2 and comp7) in human primary hepatocytes at 24 and 72 h. N1-methyl-pseudouridine (N1) and 5-methoxyuridine (5M) versions of the sequences were also screened. PAH levels were quantified as MFI and plotted. In (C–E), mock refers to endogenous levels. (F) Final screening on top expressers from the different coding region optimization approaches. A time course up to 9 h was done on human primary hepatocytes, where PAH protein levels were quantified by western blot and plotted. Reference sequence was used as a comparator. Plot was normalized to untransfected (Unt). N1 chemistry was used in all the compounds screened. All the samples were done in triplicate for all the experiments.
Figure 2.

Selection of PAH mRNA candidates based on 5′ and 3′ UTRs library screening
(A) Diagram summarizing the additional UTR arm used in the selection of the PAH compounds. (B) Representative plot of a selection of UTR-based compounds screened in a human hepatocyte cell line at 24 and 48 h. PAH protein levels were quantified by in-cell western, normalized to the reference sequence (dashed line, Ref Seq) and plotted. (C and D) (C) Table summarizing the 5′ and 3′ UTRs selected and screened in (D). (D) Transfection of the selected UTR combos in human primary hepatocytes, normalized to total protein content and plotted at both 24 and 48 h post-transfection. All the samples were done in triplicate for all the experiments.
For the coding region, we focused on strategies increasing translational efficiency, as well as mRNA and protein stability (Figure 1A). Codon optimization was used to screen compounds in mouse Hepa1-6 and human Hep3B cell lines for improved protein expression levels and duration across a time course (Figure 1B). Human reference sequence (NCBI Reference Sequence: NM_000277.3) was an unoptimized hPAH mRNA used as internal control for expression levels comparison. The trend observed in both mouse and human cell lines was similar, with higher protein levels over reference sequence in human cell lines at 24 h. By 72 h, most of the expression levels were baselined to un-transfected, suggesting a short protein half-life (Figure 1B). Additional optimization strategies, based on mRNA and protein stability approaches, were implemented on the selected optimized hPAH mRNAs (Figures 1C–1F). Recent literature has shown that certain hPAH domains might not be required for a functional protein, and their deletion might improve protein stability.11,12 We tested this approach by designing hPAH mRNAs without the regulatory domains. These compounds (e.g., compounds 8–9) were screened in human primary hepatocytes but they were poorly expressed in vitro when compared with the mock (endogenous, Figure 1C) and, therefore, were not used for further optimization studies. Additional protein stabilization designs (e.g., phosphomimetics, ubiquitination, etc., Figures 1D and 1E) were incorporated on selected codon-optimized hPAH mRNAs (e.g., compound 7 in Figures 1A and 1B), which were tested in vitro using human primary hepatocytes at different timepoints (Figures 1E and 1F). The protein levels observed indicate that codon optimization, when combined with proper protein stabilization approaches, may have a beneficial effect in generating compounds with improved PAH expression profiles and longer duration in vitro (e.g., compounds 19–20 over compound 7 in Figure 1F).
For our second strategy in the optimization process, we designed a library composed of more than 300 UTRs encompassing combinations of both 5′ and 3′ UTRs on the same hPAH coding region (Figure 2A). UTR compounds were screened for protein expression at 24 and 48 h in a human Hep3B cell line (representative plot in Figure 2B). The selection of the most optimal UTR combinations was made based on 1) peak height of protein expression at 24 h, and 2) percentage of retained protein expression at 48 h (Figures 2B and 2C). A pool of 15 compounds was selected and tested for a final screening in human primary hepatocytes at both 24 and 48 h (Figure 2D). The same selection criteria were applied to select the top UTR combo (compound 1 in Figure 2D).
The selection of the final hPAH mRNA leads was done by combining the top UTR combo (Figure 2) with the selected coding region sequences (Figure 1).
To confirm that the selected compounds derived from the coding and UTR screening approaches were functional in the disease model, we tested them for efficacy in the Pahenu2 homozygous mouse model (Figure S2). Animals were injected i.v. with a single dose of LUNAR formulations encapsulating the hPAH mRNAs. The same LUNAR profiling was used and the only differential factor was the mRNA. Animals were dosed at 10 mg/kg and plasma Phe levels were measured at different time points, from 6 to 96 h. The results showed a similar reduction in Phe levels in all the compounds analyzed, reaching baseline levels at 96 h (Figure S2A), except for mRNA4. The protein stability/codon-optimized hPAH mRNA (mRNA1) showed better and lasting reduction in Phe levels. No body weight changes were observed during the study (Figure S2B).
LUNAR-hPAH mRNAs express improved PAH protein levels in hepatocytes
The selected codon-optimized hPAH mRNAs were tested in WT mice to determine the levels of PAH protein in vivo (Figure 3). WT mice were dosed i.v. with a single dose of 3 mg/kg of LUNAR-hPAH mRNAs. Six and 24 hours post-dose, animals were dissected, and livers were extracted, and PAH levels were quantitated with western blot (qWB) analysis using a PAH antibody that cross-reacts with human and mouse. Protein analysis on the livers showed that 1) LUNAR formulations are being delivered to the liver, and 2) hPAH mRNAs are producing detectable and quantifiable levels of PAH protein (Figure 3E) when compared with controls. The 6-h time point shows a 2-fold increase in PAH protein levels compared with controls, whereas at 24 h is similar to endogenous.
Figure 3.

Delivery of LUNAR-mRNA (LUNAR1) carrying codon-optimized hPAH or EGFP mRNAs to hepatocytes in WT mice
(A) PAH protein detection by immunofluorescence (green) in vehicle or LUNAR-EGFP-treated mice.
(B) Images were quantified (n = 3–6, ∗∗∗p = 0.0001) and normalized by the number of nuclei (DAPI, blue stain on B). (C) Hepatocyte cells isolated from untreated and LUNAR-EGFP-treated mouse livers by cell sorting were imaged to detect EGFP fluorescence. (D) FACS analysis of mouse livers to identify the percentage of hepatocytes (top, CD95/EGFP) or non-hepatocytes (bottom, CD11b/EGFP) that have been targeted by LUNAR-EGFP (n = 5 mice per group in D and E). (E) Protein expression analysis by western blot (left) and quantification (right graph) of PAH levels in the liver of WT mice after 6 and 24 h post-dose with either PBS or 3 mg/kg of LUNAR (n = 4 mice per group). (F) Clinical chemistry to evaluate effect of LUNAR-hPAH mRNA (LUNAR1) delivery in liver and kidney markers: ALT, ALP, potassium, albumin, globulin, cholesterol, bilirubin, sodium (n = 15 mice per group). Scale bars, (A) 50 μm, (C) 30 μm.
Characterization of LUNAR formulations specificity targeting the liver was conducted in WT mice. Animals were i.v. injected with a 2.5 or 5 mg/kg dose of LUNAR-EGFP mRNA, and the assessment of EGFP distribution in hepatocytes was determined 24 h post-dose by immunohistochemistry (IHC) (Figures 3A and 3B). IHC analysis shows a robust expression of the EGFP protein across the liver (Figure 3A). Quantitative analysis indicates a significant (p = 0.0001) dose-dependent increase in EGFP counts in treated mice compared with vehicle (Figure 3B).
A parallel study was conducted in WT mice to determine the percentage of hepatocytes that express EGFP (Figures 3C and 3D). Animals were i.v. dosed with LUNAR-EGFP mRNA and, 24 h post-dose, animals were dissected, and perfused livers were recovered for evaluation by IHC and fluorescence-activated cell sorting (FACS) analysis. IHC analysis showed a robust increase in EGFP signal not observed in the mock (Figure 3C), whereas FACS analysis demonstrated that the fraction of EGFP+ cells increased from 1.34% of vehicle-dosed animals to approximately 47% in LUNAR-EGFP-dosed animals (Figure 3D). Similarly, the fraction of hepatocytes that express EGFP (EGFP+/CD95+) increased from approximately 1.2% in vehicle-dosed animals to approximately 66% in LUNAR-EGFP-dosed animals (Figure 3D), with minimal-to-no delivery to Kupffer cells. Overall, the data indicate an elevated distribution of EGFP in hepatocytes (EGFP+/CD95+) as observed by FACS and liver immunostaining for EGFP compared with vehicle controls (Figures 3A–3D).
Efficacy of a single dose of LUNAR-hPAH mRNAs in Pahenu2mice
Mixed-gender Pahenu2 homozygous mice were tested for efficacy of LUNAR-hPAH mRNA on reducing circulating Phe concentrations. Pahenu2 homozygous mice maintained on standard food and water ad libitum were i.v. dosed with either control article (LUNAR buffer) or 10 mg/kg of LUNAR-hPAH mRNA, alone, and in combination with 2 mg/mL of PAH's cofactor BH4 each day of dosing (Figures 4A and 4B). Treatment with LUNAR-hPAH mRNA was well tolerated with no apparent clinical observations, and nothing significant was observed in the clinical chemistry analysis of the serum samples (Figure S4). By 24 h, treatment with LUNAR-hPAH mRNA decreased plasma Phe levels in Pahenu2 homozygotes by approximately 20-fold post-dose, similar to the reduction of Phe levels obtained with the low-Phe diet regimen (Figure 4A), while gradually returning to pre-dose levels by 72 h post-dose. Dosing with BH4 did not appear to alter plasma Phe concentrations, either alone or in combination with LUNAR-hPAH mRNA, indicating that either BH4 is not a limiting factor for PAH enzyme activity introduced by exogenous hPAH mRNA, or that BH4 supplementation was provided at too low a concentration to saturate de novo PAH protein produced by the hPAH mRNA. Concomitantly, treatment with LUNAR-hPAH mRNA did not impact the Tyr levels, demonstrating that the effects associated with PAH mRNA were specific for Phe metabolism only (Figure 4B).
Figure 4.

LUNAR-hPAH mRNA pharmacodynamics in vivo following a single dose
(A and B) Phe plasma levels (A) and Phe:Tyr ratios (B) in Pahenu2 homozygous mice at different time points after a single dose of control (LUNAR buffer) or 10 mg/kg of LUNAR-hPAH mRNA (LUNAR 1) with or without the PAH cofactor BH4. Dashed lines represent values in untreated animals maintained on a special Phe-free diet (n = 3–4 mice in control groups and n = 5–6 in treated groups). (C) Phe plasma levels dose response in Pahenu homozygous mice after a single dose of LUNAR-hPAH mRNA (LUNAR 1) at different concentrations (3 and 10 mg/kg). Gray dashed line indicates average Phe levels in WT mice (n = 8 mice per group). (D) PAH protein quantification by MRM in WT and Pahenu2 homozygous mice from different strains after a single injection of LUNAR-hPAH mRNA (LUNAR 1) (n = 4 mice per group). (E) Plasma Phe levels in Pahenu2 homozygous mice at different time points after a single dose of control (LUNAR buffer) and 3 mg/kg of two different LUNAR formulations (LUNAR1, LUNAR2) encapsulating the same hPAH mRNA (n = 6–8 mice per group). (F and G) Liver and plasma LUNAR lipid concentration quantification at different time points (2 min, 15 min, 30 min, 1 h, 2 h, 6 h, 24 h, 48 h, 7 days, and 21 days post-dose) in WT mice after a single injection of 0.5 mg/kg of LUNAR1 and LUNAR2 mRNAs (n = 4 mice per time point). (G) Detail of the LUNAR lipid concentration during the first 24 h (dashed box in F).
A single-dose study was conducted to determine the percentage and duration of the Phe reduction in the Pahenu2 mice (Figure 4C). A 3- and a 10-mg/kg dose of LUNAR-hPAH mRNA were i.v. injected in a mixed-gender colony of homozygous Pahenu2 mice (Figure 4C). Plasma samples were collected at 6, 24, 48, and 60 h post-dose, and Phe levels were measured and compared with vehicle (LUNAR buffer). A dose-dependent reduction of >80% in Phe plasma levels compared with vehicle was observed at 6 h, with a persistent 30% reduction at approximately 40 h post-dose (Figure 4C).
A parallel study was run to establish physiological differences in PAH levels across different mouse strains when dosed with LUNAR-hPAH mRNA (Figure 4D). The different strains of mice included homozygous Pahenu2 and WT BTBR, WT C57Bl/6, and WT Balb/c and all animals were i.v. dosed with LUNAR-hPAH mRNA at a 3 mg/kg dose; note that homozygous Pahenu2 BTBR mice were also dosed at 10 mg/kg. Six hours post-dose, animals were euthanized, and livers were extracted for quantitative PAH protein analysis using multiple reaction monitoring (MRM) mass spectrometry. For the MRM, we developed heavy peptides specific for the human PAH protein that do not cross-react with the mice counterpart. A dose-dependent increase in hPAH protein levels was observed in the homozygous Pahenu2 BTBR mice (Figure 4D) that correlates with the dose-dependent reduction observed in Phe plasma levels at 6 h (Figure 4C) using a similar dosing regimen. The comparison at 3 mg/kg between the different strains of WT mice did not show any strain-specific differences in the PAH levels generated (Figure 4D).
Additional formulations were tested to improve efficacy in the Pahenu2 mice model (Figure 4E). In the example, we tested two LUNAR formulations carrying the same hPAH mRNA (LUNAR1 and 2). Homozygous Pahenu2 mice were i.v. dosed with either vehicle (LUNAR buffer) or 3 mg/kg of LUNAR-hPAH mRNA, and plasma Phe levels analyzed at 6, 24, 48, 72, and 96 h post-dose. LUNAR1 reduced plasma Phe levels approximately to 50% at 6 h and less than 30% at 24 h when compared with vehicle. In contrast, LUNAR2 reduced the plasma Phe levels to WT range by 24 h post-dose compared with vehicle; with >80% reduction in Phe levels persisting at 48 h and a 30% reduction at 96 h (Figure 4E). The differences observed between LUNAR1 and LUNAR2 carrying the same hPAH mRNA indicated that LUNAR2 is a >3-fold more potent lipid in reducing and sustaining low-Phe levels in liver than LUNAR1 (Figure 4E).
LUNAR is an LNP, and their components are biodegradable to minimize the risk of toxicity from frequent/chronic dosing. To determine the biodegradability profile of LUNAR, we dosed mice with a single dose of LUNAR-hPAH mRNA via i.v. administration. LUNAR1 and LUNAR2 were used. For both formulations, no lipid was detected in plasma 6 h post-dose. In liver, lipid was not detected 6 h post LUNAR1 injection, whereas LUNAR2-injected mice had very low lipid levels at 7 days, and lipid was not detectable at 21 days (Figures 4F and 4G). Less than 1% of lipids were detected at day 7 with LUNAR2, and none with LUNAR1, which is consistent with internal data. Due to the high biodegradability nature of both lipids, no accumulation risk is expected upon repeat dosing.
Efficacy of repeated dosing of LUNAR-hPAH mRNAs in Pahenu2 mice
Mixed-gender mice homozygous for Pahenu2 were i.v. dosed at 3-day intervals with five doses of either control article (LUNAR buffer) or 3 mg/kg of LUNAR-hPAH mRNA. Animals were fed on standard diet and water ad libitum throughout the entire study (Figure 5A). LUNAR-hPAH-treated animals showed a decrease in mean plasma Phe concentrations by at least 50% (relative to pre-treatment) at 6 h post-dose following repeat dosing (doses 1, 4, and 5). Plasma samples were not collected at 6 h post-dose on doses 2 and 3. Large variabilities in plasma Phe concentration were measured at multiple timepoints in both treated and control groups. However, despite the variabilities, the strongest effects were seen in males at 6 h post doses 4 and 5, when plasma fell 10- and 7-fold, respectively, when compared with baseline (Figure 5A). In females, the major reduction in plasma Phe was observed at 6 h post dose 5, when plasma Phe felt 4-fold compared with baseline (Figure 5A). The reductions in plasma observed in males exceeded those in females. Values returned to within 20% of baseline by 24 h after each administration. The gender differences are consistent with what has been reported for Phe-lowering therapies using the PKU mice model,13, 14, 15, 16, 17 which seems to be specific for the rodent model since it has not been reported in either non-human primates or in humans.17, 18, 19
Figure 5.

Plasma Phe pharmacodynamics following a repeating dosing of LUNAR-hPAH mRNA (LUNAR 1)
(A–C) Pahenu2 homozygous mice maintained on a standard diet and water ad libitum were dosed five times with either vehicle (LUNAR buffer) or 3 mg/kg of LUNAR-hPAH mRNA at 3-day intervals. (A) Plasma Phe levels were quantified every 24 h in males (left graph, lines) and females (right graph, dotted line) in vehicle (blue lane) and treated (orange) groups. Blue arrows in X axis indicate dosing times. (B and C) Plasma levels of Phe and Tyr (B) and Phe:Tyr ratios (C) after first, fourth, and fifth doses compared with pre-dose at 6, 24, 48, and 72 h post-dose. (D) Quantification of hPAH mRNA present in mouse liver at 6, 24, and 48 h after the first (red) or fifth (blue) dose by Quantigene. ∗∗∗p < 0.001, ∗∗p < 0.02, ∗p < 0.5 (unpaired t test). Levels of hPAH mRNA were normalized to endogenous PPIB. Arrows indicate each dose (n = 15 mice per group for graphs A, B, and C; on D n = 4–10 mice per group).
Plasma levels of Phe and Tyr (Figure 5B) and Phe:Tyr ratio (Figure 5C) were plotted at 6, 24, 48, and 72 h post doses 1, 4, and 5, and compared with pre-dose (Figures 5B and 5C). The results suggest a cumulative Phe-lowering effect on repeat dosing, reaching its highest at 6 h in each tested dose, with levels similar to pre-dosing at 48 h (Figure 5B). These Phe data indicate that LUNAR-hPAH mRNA induced a robust pharmacodynamic response in vivo (Figure S3). The Tyr levels inversely correlate to those of Phe, with highest levels of Tyr at 6 h, which contrasts with Phe levels being at its lowest (Figure 5B). This suggests an effective conversion of Phe into Tyr and confirms the metabolic restoration of the Phe pathway, which is also reciprocated by the Phe:Tyr ratio at each time point (Figure 5C). Tyr levels reached pre-dosing levels at 48 h.
Clinical chemistry revealed significant changes in a small number of markers, including decreased alkaline phosphatase (ALP) (females at 48 h), alanine aminotransferase (ALT) and potassium (males at 48 h), and albumin:globulin ratio (females at 48 h) (Figure S4), whereas an increase in cholesterol was observed at 6 h in both males and females, as well as unconjugated bilirubin (males at 24 h) and sodium:potassium ratio (males at 48 h) (Figure S4). Together, these changes did not suggest consistent effects on liver or kidney function (Figure S4).
To evaluate the stability of the hPAH mRNA in vivo, we quantified hepatic hPAH mRNA levels using the Quantigene assay. Liver samples were obtained from vehicle control and treated mice at 6, 24, and 48 h post doses 1 and 5 (Figure 5D). No hPAH mRNA levels were detected in the vehicle controls at any dose. Significant levels of hPAH mRNA were detected at 6 h post each dose in treated animals, with the highest levels of mRNA observed after the fifth dose (∗∗∗p < 0.001, Figure 5D). At 48 h, PAH mRNA levels were comparable with vehicle controls.
Discussion
PKU is a metabolic rare disease caused by mutations in the PAH gene.1, 2, 3, 4 Approximately 1,281 genetic variants have been described in the PKU disease database (http://www.biopku.org/home/pah.asp), where 950 are missense mutations.20 Based on clinical studies, approximately 20% of the total PKU patient population respond to oral administration of sapropterin.21 Overall, current standard of care is poor, with a strong emphasis on managing Phe-restricted diets2,4,22 to control plasma Phe levels. Clinically, a Phe-restricted diet has been effective in maintaining plasma Phe levels within a normal-like range to prevent the development of irreversible neurological disabilities.2 Caregiver feedback has suggested it is very difficult to adhere to a Phe-restricted diet, particularly during adolescence and adulthood,1, 2, 3, 4,23 increasing risk of unwanted spikes in Phe plasma levels, and subsequently recurrent hyperphenyalanemic peaks that can have consequences to the body.2
Current FDA treatments for PKU include two approved drugs: sapropterin21 and pegvaliase.2 Sapropterin is a synthetic form of the cofactor BH4, which is highly effective in mild PKU patients but also requires in most patients a Phe-restricted diet to fully normalize Phe levels. Pegvaliase is an oral form of pegylated-derivative of the bacterial enzyme phenylalanine ammonia-lyase (PEG-PAL) that metabolizes Phe to ammonia and trans-cinnamic acid, thus reducing its plasma levels.2,24,25 In contrast with PAH, PAL has the advantage of not requiring any coenzyme to metabolize Phe.24, 25, 26 However, the PEG-PAL effect does not last long, and it can potentially induce serious anaphylactic reactions in some of the patients; therefore, it is currently limited to adults.25, 26, 27
Recent advances in gene therapy have shown promising results in PKU mouse models with long-lasting reductions in Phe levels and partial liver correction.14, 15, 16, 17 Some of these therapeutic approaches are currently being tested in ongoing clinical trials with more efficient adeno-associated virus (AAV)-based delivery approaches to minimize effects associated with commonly known immune responses to the viral capsid, existing resistance to AAV, low transduction, or potential non-specific insertion.28 The neonate's population is likely not suitable to PKU gene therapeutic approaches due to the wash-out of the transgene in hepatocytes in a still growing liver.28, 29, 30 With all this in mind, novel therapeutic approaches to treat and prevent the severity of the PKU disease are needed.
An mRNA replacement therapy to treat PKU patients has several potential advantages over some of the DNA-based or enzymatic approaches31 discussed above. Modulating lipid composition of LUNAR nanoparticles allowed an enrichment toward hepatocytes, as was shown by the IHC and FACS data. A direct mRNA delivery to the cytoplasm of hepatocytes leads to immediate production of hPAH protein, bypassing the risk of potential unintended genomic integration, and allowing rapid efficacious responses. In addition to this, the use of chemically modified mRNAs with uridine derivatives enhances translation, reduces immunogenicity driven by sensors such as Toll-Like Receptors and nuclease recognition,32 making the mRNA therapy more suitable for repeat dosing.
The studies described herein show proof-of-concept of an mRNA replacement therapy for PKU using the Pahenu2 mice as a model. Codon-optimized and chemically modified hPAH mRNAs with different UTR combinations were selected after showing higher levels of PAH protein expression, than natural PAH mRNA, in a human primary hepatocyte screening. The selected compounds were encapsulated in LUNAR, a biodegradable LNP, and delivered i.v. specifically into hepatocytes as a single or repeat dose in the Pahenu2 mice. We showed that LUNAR-hPAH mRNA produces a functional hPAH protein in the Pahenu2 mouse liver that effectively reduces Phe levels without any adverse clinical signs, either after single or repeat doses in both males and females. An mRNA replacement, as a therapy for PKU deficiency, has many advantages: 1) it is a non-integrative approach, mRNA is delivered to the cytoplasm, not to the nuclei, of the hepatocytes bypassing any undesired off-target integration; 2) half-life of the mRNA is short; 3) mRNA uses the cell's own translational machinery to produce a newly generated protein; and 4) it allows for repeat dosing.
Multiple therapeutic approaches are being investigated to treat PKU; however, there is still not a therapeutic modality that offers a balance between the pathophysiology of the disease and an improved quality of life for the patient. An optimal therapeutic approach will require increasing the half-life of the PAH protein to reduce the dose frequency and improve the standard of care. mRNA technology allows quick engineering of mRNAs to produce more stable PAH mRNAs and/or modified PAH proteins with increased half-life.
Here, we show data supporting a novel PAH mRNA replacement therapy as an advantageous modality for the treatment of PKU. This therapy has the potential to eliminate dependency on a restricted diet, which may have a beneficial impact in the life of PKU patients by restoring their Phe metabolism, reducing the toxic Phe levels in plasma, and, therefore, reducing any risk of adverse events.
Material and methods
Codon optimization and UTR design
hPAH codon usage was optimized using algorithms developed at Arcturus Therapeutics; 5′ and 3′UTR sequences were extracted from the NCBI database or selected from Arcturus' proprietary UTR library and synthesized in a library of plasmids with different 5′/3′ UTR combinations.
In vitro transcription for mRNA synthesis
For a 200-μL in vitro transcription (IVT) reaction, we used the NEB HiScribe T7 In Vitro Transcription Kit (New England Biolabs, Ipswich, MA), which should yield about 1 mg of RNA. Briefly, a 2.5× NTP mix was prepared as required by thawing individual l00 mM NTP stocks (ATP, GTP, CTP, and UTP nucleotides, or chemically modified counterparts) and pooling them together. The 200-μL IVT reaction included about 2 to 4 μg of the DNA template, 10× IVT reaction buffer, 2.5× NTP mix, and 14 μL of T7 RNA polymerase mix. After mixing by pipetting, the reaction mixture was incubated at 37°C for 4 h. To degrade the DNA template, the IVT reaction was diluted with 700 μL of nuclease-free water and then 10× DNase I buffer and 20 μL of the RNase-free DNase I were added to the IVT mix and incubated at 37°C for 15 min. The synthesized RNA was then purified by a RNeasy Maxi column (Qiagen, Hilden, Germany), eluted in RNase-free water, and quantified by NanoDrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA).
Enzymatic capping of IVT-synthesized mRNA
For enzymatic capping, a 50× scaled-up version of NEB's one-step capping and 2′-O-methylation reaction was used for treating up to l mg of IVT transcripts. Briefly, mRNA was denatured at 65°C for 5 min and then snap chilled to relieve any secondary conformations. For the total 1-mL capping reaction, 1 mg denatured mRNA, 100 μL of 10× capping buffer, 50 μL of 10 mM GTP, 50 μL of 4 mM SAM, 50 μL of 10 U/mL vaccinia capping enzyme, and 50 μL of mRNA cap 2′-O-methyltransferase (50 U/mL) were combined and adjusted the final volume to 1 mL with nuclease-free water and incubated at 37°C for 1 h. The resulting cap 1 mRNA (m7GpppGm-mRNA) was purified on an RNeasy column, eluted with nuclease-free water, and quantified by the NanoDrop spectrophotometer. Purity of the mRNA was confirmed by denaturing urea polyacrylamide gel electrophoresis and a Fragment Analyzer system (Agilent, Santa Clara, CA).
Transfections and protein expression in cell culture
All different cell lines were transfected with mRNA in 96-well plates using Lipofectamine MessengerMax as the transfection reagent (Thermo Fisher Scientific) and following the manufacturer’s instruction for all transfections. Primary hepatocytes were plated in 96-well collagen-coated plates at least 8 h before transfection. DMEM medium containing 10% fetal bovine serum (FBS) was replaced immediately before beginning the transfection experiment. Medium was collected after desired time points and 100 μL fresh medium was added into each well. Medium was kept at −80°C until an ELISA assay for PAH was performed using the standard manufacturer protocol. At the desired time points, cells were either fixed or collected for protein analysis by in-cell western or western blot.
In-cell western
At the desire timepoints, medium was removed, and cells were fixed in 4% fresh paraformaldehyde (PFA) for 20 min. After washing with TBST, cells were permeabilized with Triton X-100 for 5 min five times. When permeabilization washes were complete, cells were incubated with the blocking buffer for 45 min. Primary antibody was then added and incubated for 1 h at room temperature. Following that, cells were washed several times in TBST, and then incubated for 1 h with the secondary antibody diluted in blocking buffer and containing the CellTag 700 stain. To finalize, cells were washed several times in TBST followed by a last wash in TBS. Then, the plate was imaged using the OdysseyCLx Imaging System (Li-COR Biosciences, Lincoln, NE), and data were normalized to the total number of cells labeled by the CellTag 700 stain.
PKU animal model
Pahenu2/J homozygous mice were obtained from The Jackson Laboratory and studies were performed in the Buck Institute Animal Facility. Prior to dosing, animals were group-housed (up to five per cage); from the time of dosing, animals were single-housed. Mice were housed in microisolator caging in ventilated racks. Environmental controls for the animal room generally targeted a temperature range of 23 ± 3°C and a relative humidity range of 50% ± 20% with a 12-h/12-h light/dark cycle. Throughout the study, the mice were offered Teklad Global 18% protein rodent diet (Envigo RMX, Inc.) and water ad libitum, with the exception of the studies in which a Phe-free diet was used (Teklad Custom Diet TD.170012; Envigo RMX, Inc).
For each of the separate cohorts, animals were assigned to groups by sex and by randomization designed to achieve similar group mean body weights. Each cohort of the study consisted of 28 mice of mixed gender (14 males, 14 females). All animals were aged 2 to 4 months on the day of dosing. All in vivo procedures involving animals were performed in accordance with guidelines established by the Institutional Animal Care and Use Committee.
Blood collection and clinical chemistry
Prior to dosing (“0 h”; all mice), and at, for example, 6, 24, 48, and 72 h post-dose (one-half of the animals in each group per time point, alternating at each time point), blood was collected from each animal by the submandibular route (for in-life bleeds) or by terminal cardiocentesis (for terminal bleeds only). For each time point, blood was collected into K2EDTA-containing tubes and processed to plasma by centrifugation. The resulting plasma was stored at −80°C until processed for bioanalytical analysis. Following the terminal bleeds, all animals were euthanized, and carcasses were discarded without further examination.
Serum chemistry profiles were obtained using the AU680 Chemistry System (Beckman Coulter). Serum levels of ALT activity, albumin, ALP activity, aspartate aminotransferase activity, bicarbonate, total bilirubin, direct bilirubin, blood urea nitrogen (BUN), calcium, cholesterol, creatine kinase activity, globulin, glucose, phosphorous, and total protein were determined enzymatically using reagents designed for the AU680 Chemistry System and following the manufacturer's instructions. Sodium, potassium, and chloride levels were determined by use of an ion-specific electrode that was part of the chemistry analyzer. Indirect bilirubin was calculated from the total and direct bilirubin levels. The sodium-to-potassium ratio (Na:K) was determined from the measured sodium and potassium levels.
Plasma Phe and Tyr concentration measurements
All plasma samples were assessed for Phe and Tyr concentrations by liquid chromatography tandem mass spectrometry (LC-MS/MS). All procedures associated with this analysis were conducted following a non-GLP method with predetermined acceptance criteria. In brief, for each sample, plasma was diluted 1:10 and 1:100 in water. An aliquot (10 μL) of each dilution was transferred to a microtiter plate. Protein was precipitated by combining with 29 volumes of methanol containing 1 μM of each of the internal standards (13C-6-Phe and 13C-6-Tyr). The precipitate was pelleted, and the supernatants were transferred to a fresh microtiter plate and subjected to LC-MS/MS. Chromatographic separation was performed on a Hypercarb 100∗2.1 (5 μm; 5-μL sample volume) column using gradient chromatography with water +0.1% formic acid as Mobile Phase A and a 50:50 mix of acetonitrile:isopropyl alcohol (50:50) + 0.1% formic acid as Mobile Phase B. MS used a QTrap 4,500 linear ion trap quadrupole (Model 5035162-B; AB Sciex Instruments) with a TurboSpray source at 650°C. The lower limits of quantitation were 1.2 and 0.4 μM for Phe and Tyr, respectively.
hPAH protein quantification in mouse liver by western blot
Protein extraction from liver tissue was done using Precellys Lysing Kit tubes and RIPA buffer including a cocktail of protease inhibitors. After lysing the tissue using Precellys 24 (Bertin Instrument, Bretonneux, France), samples were briefly sonicated and centrifuged and the supernatant was kept for standard western blot analysis. Immunoblot was performed on polyvinyl difluoride membranes. PAH was detected using goat anti-PAH polyclonal antibody (AbCam Cat.106805) and donkey anti-goat immunoglobulin G (H + L)-horseradish peroxidase c\Conjugate (Santa Cruz Biotechnology Cat. sc-2020) as the primary and secondary antibodies. Glyceraldehyde phosphate dehydrogenase (GAPDH; a housekeeping protein used as a loading control) was detected using mouse anti-GAPDH antibody (Abcam Cat. 125247) as the primary antibody; the blots were not stripped before re-probing. To account for non-specific binding, duplicate membranes were processed without primary antibody. Samples from WT (i.e., PAH+) BTBR mice were included to provide a positive control.
FACS analysis
Hepatocytes isolated from the collagenase-perfused livers were pelleted at 50 G and resuspended gently in 50 μL staining buffer (PBS supplemented with 0.5% FBS, 1 mM EDTA, 20 mM HEPES, and 0.01% NaN3). Cells were blocked by incubating for 10 min on ice with mouse Fc block (TruStain FcX [anti-mouse CD16/32] antibody; BioLegend Cat. #101320). Surface staining was performed using 5 μL of phycoerythrin-conjugated anti-CD95 (Fas) antibody (phycoerythrin [PE] anti-mouse CD95; Biolegend Cat # 152608) or PE-conjugated anti-CD11b (Fas) antibody (PE anti-mouse/human CD11b, Clone M1/70; Biolegend Cat # 101208). Stained cells were washed twice in PBS and analyzed by flow cytometry using a Beckman Coulter Cytoflex. CD95/PE-positive or CD11b/PE-positive cells were gated and intrinsic GFP fluorescence was used as the analytical parameter to infer EGFP expression in either hepatocyte or Kupffer cells subpopulations, respectively, for each liver.
IHC analysis
PFA-fixed liver lobes were dehydrated, processed, sectioned, and stained with a primary antibody against GFP (chicken anti-GFP; Abcam ab13970, Lot GR236651-24; at 1:1,000) and a complementary secondary antibody (donkey anti-chicken conjugated to Cy3; Jackson ImmunoResearch #703-165-155, Lot 130328; at 1:500). The sections were mounted on glass slides using ProLong Gold with DAPI and evaluated by fluorescence microscopy (Zeiss Axio Imager M2 microscope with respective excitation and emission at wavelengths of 493 and 520 nm for endogenous GFP and of 549 and 562 nm for Cy3). Images were recorded at ×10 using ZEN 2.3 software.
Nanoparticle formulations
LUNAR is a proprietary LNP-based delivery system of Arcturus Therapeutics. LNPs are prepared by mixing lipids in ethanol with mRNA in an acidic aqueous buffer using NanoAssembler microfluidic device (Precision Nanosystems, Vancouver, Canada) followed by a downstream purification process. Briefly, mRNA is dissolved in 5 mM citric acid buffer (pH 3.5). Lipids are dissolved in ethanol at a desired molar ratio. The molar lipid composition of LUNAR1 is 50% ATX lipid (Arcturus' proprietary ionizable cationic lipid), 7% DSPC (l ,2-distearoyl-sn-glycero-3-phosphocholine; Avanti Polar Lipids), 40% cholesterol (Avanti Polar Lipids), and 3% DMG-PEG2000 (1,2-Dimyristoyl-sn-glycerol, methoxypoly ethylene glycol, PEG chain molecular weight: 2000; NOF America Corporation); while LUNAR2 is 58% ATX, 7% DSPC, 33.5% cholesterol, and 1.5% DMG-PEG2000. The lipid and mRNA solutions are combined using the microfluidic device (Precision NanoSystems) at a flow ratio of 1:3 (ethanol: aqueous phase). The total combined flow rate is 12 mL/min. LNPs thus formed were purified by dialysis against phosphate buffer overnight, by using in Spectra/Por Flot-a-lyzer ready-to-use dialysis device, followed by concentration using Amicon Ultra-15 centrifugal filters (Merck Millipore Ltd.) to the desired concentration. Particle size of the LNPs is determined by dynamic light scattering (ZEN3600, Malvern Instruments). Encapsulation efficiency was calculated by determining unencapsulated small interfering RNA (siRNA) content by measuring the fluorescence upon the addition of RiboGreen (Molecular Probes) to the LNPs (Fi) and comparing this value to the total siRNA content that is obtained upon lysis of the LNPs by 1% Triton X-100 (Ft), where percentage of encapsulation = (Ft − Fi)/Ft × 100. All formulations exhibited encapsulation efficiencies of >90%, particle sizes in the range of 60 to 100 nm, and polydispersity indices of ≤0.2.
Pharmacodynamics and biodegradability of LNPs
Prior to dosing (“0 h”), and at different time points post-dose, plasma samples were prepared, and liver tissues were immediately frozen in liquid nitrogen. For lipid analysis, plasma and homogenized liver samples were mixed with organic solvents with spiked internal standard to precipitate proteins. After centrifugation, supernatant was diluted further with organic solvent before LC-MS analysis. Positive electrospray ionization was used, and MRM parameters were set up to specifically target the lipid and internal standard. Calibration standards prepared in plasma were used for quantitation. Quality control samples with spiked known amount of lipids were prepared in plasma and liver to control the precision and accuracy of the bioanalysis.
Statistical analysis
Where appropriate, values are expressed as means ± SD. Groups were compared by nonpaired two-tailed heteroscedastic t tests using GraphPad Prism software. A p value < 0.05 was considered significant.
Acknowledgments
We thank former and current employees at Arcturus Therapeutics who have contributed to the development of this therapeutic approach. The work included in this manuscript was performed using funds provided by Arcturus Therapeutics, Inc
Author contributions
C.G.P.G. and R.D.T. contributed equally to this study. C.G.P.G. and R.D.T. designed and performed research; P.L. generated mRNAs; S.C. and H.Y. performed the biodegradability assays; J.B.V., Y.B., and P.K. formulated mRNAs into LUNAR LNPs; M.S. performed in vivo studies; W.T. coordinated program activities; K.T. provided oversight of genetic tools and sequence designs; P.C. developed LUNAR technology; C.G.P.G. and R.D.T. wrote the paper.
Declaration of interests
The work included in this manuscript was performed using funds provided by Arcturus Therapeutics, Inc. The authors are current or former employees of Arcturus Therapeutics Inc.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.02.020.
Supplemental information
References
- 1.Scriver C.R., Kaufman S. McGraw-Hill; New York: 2001. Hyperphenylalaninemia: Phenylalanine Hydroxylase Deficiency; pp. 1661–1724. [Google Scholar]
- 2.Lichter-Konecki U., Vockley J. Phenylketonuria: current treatments and future developments. Drugs. 2019;79:495–500. doi: 10.1007/s40265-019-01079-z. [DOI] [PubMed] [Google Scholar]
- 3.Blau N., van Spronsen F.J., Levy H.L. Phenylketonuria. Lancet. 2010;376:1417–1427. doi: 10.1016/S0140-6736(10)60961-0. [DOI] [PubMed] [Google Scholar]
- 4.van Wegberg A.M.J., MacDonald A., Ahring K., Belanger-Quintana A., Blau N., Bosch A.M., Burlone A., Campistol J., Feillet F., Gizewska M., et al. The complete European guidelines on phenylketonuria: diagnosis and treatment. Orphanet J. Rare Dis. 2017;12:162. doi: 10.1186/s13023-017-0685-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levy H.L., Milanowski A., Chakrapani A., Cleary M., Lee P., Trefz F.K., Whitley C.B., Feillet F., Feigenbaum A.S., Debchuk J.D., et al. Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo-controlled study. Lancet. 2007;370:504–510. doi: 10.1016/S0140-6736(07)61234-3. [DOI] [PubMed] [Google Scholar]
- 6.Blau N., Belanger-Quintana A., Demirkol M., Feillet F., Giovannini M., MacDonald A., Trefz F.K., van spronsen F.J. Optimizing the use of sapropterin (BH(4)) in the management of phenylketonuria. Mol. Genet. Metab. 2009;96:158–163. doi: 10.1016/j.ymgme.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Blau N. Defining tetrahydrobiopterin (BH4)-responsiveness in PKU. J. Inherit. Metab. Dis. 2008;31:2–3. doi: 10.1007/s10545-007-9979-1. [DOI] [PubMed] [Google Scholar]
- 8.Shedlovsky A., McDonald J.D., Symula D., Dove W.F. Mouse models of human phenylketonuria. Genetics. 1993;134:1205–1210. doi: 10.1093/genetics/134.4.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramaswamy S., Tonnu N., Tachikawa K., Limphong P., Vega J.B., Karmali P.P., Chivukula P., Verma I.M. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. U S A. 2017;114:E1941–E1950. doi: 10.1073/pnas.1619653114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X., Goel V., Robbie G.J. Pharmacokinetics of patisiran, the first approved RNA interference therapy in patients with hereditary transthyretin-mediated amyloidosis. J. Clin. Pharmacol. 2019;60:573–585. doi: 10.1002/jcph.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Erlandsen H., Patch M.G., Gamez A., Straub M., Stevens R.C. Structural studies on phenylalanine hydroxylase and implications toward understanding and treating phenylketonuria. Pediatrics. 2003;112:1557–1565. [PubMed] [Google Scholar]
- 12.Jaffe E.K. New protein structures provide an updated understanding of phenylketonuria. Mol. Genet. Metab. 2017;121:289–296. doi: 10.1016/j.ymgme.2017.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davidoff A.M., Ng C.Y., Zhou J., Spence Y., Nathwani A.C. Sex significantly influences transduction of murine liver by recombinant adeno-associated viral vectors through an androgen-dependent pathway. Blood. 2003;102:480–488. doi: 10.1182/blood-2002-09-2889. [DOI] [PubMed] [Google Scholar]
- 14.Oh H.J., Park E.S., Kang S., Jo I., Jung S.C. Long-term enzymatic and phenotypic correction in the phenylketonuria mouse model by adeno-associated virus vector-mediated gene transfer. Pediatr. Res. 2004;56:278–284. doi: 10.1203/01.PDR.0000132837.29067.0E. [DOI] [PubMed] [Google Scholar]
- 15.Mochizuki S., Mizukami H., Ogura T., Kure S., Ichinohe A., Kojima K., Matsubara E., Kobayahi E., Okada T., Hoshika A., et al. Long-term correction of hyperphenylalaninemia by AAV-mediated gene transfer leads to behavioral recovery in phenylketonuria mice. Gene Ther. 2004;11:1081–1086. doi: 10.1038/sj.gt.3302262. [DOI] [PubMed] [Google Scholar]
- 16.Sarkissian C.N., Gamez A., Wang L., Charbonneau M., Fitzpatrick P., Lemontt J.F., Zhao B., Vellard M., Bell S.M., Henschell C., et al. Preclinical evaluation of multiple species of PEGylated recombinant phenylalanine ammonia lyase for the treatment of phenylketonuria. Proc. Natl. Acad. Sci. U S A. 2008;105:20894–20899. doi: 10.1073/pnas.0808421105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ahmed S.S., Rubin H., Wang M., Faulkner D., Sengooba A., Dollive S.N., Avila N., Ellsworth J.L., Lamppu D., Lobikin M., et al. Sustained correction of a murine model of phenylketonuria following a single intravenous administration of AAVHSC15-PAH. Mol. Ther. Methods Clin. Dev. 2020;17:568–580. doi: 10.1016/j.omtm.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ellsworth J.L., O'Callaghan M., Rubin H., Seymour A. Low seroprevalence of neutralizing antibodies targeting two clade F AAV in humans. Hum. Gene Ther. Clin. Dev. 2018;29:60–67. doi: 10.1089/humc.2017.239. [DOI] [PubMed] [Google Scholar]
- 19.Szypowska A., Franek E., Grzeszczak W., Filipow W., Zieba M., Kabicz P., Wieckowska B., Sykuy-Cegielska J., Taybert J. Medical care of patients with disorders of aromatic amino acid metabolism: a report based on the Polish National Health Fund data records. Pediatr. Endocrinol. Diabetes Metab. 2018;2018:118–125. doi: 10.5114/pedm.2018.80993. [DOI] [PubMed] [Google Scholar]
- 20.Williams R.A., Mamotte C.D., Burnett J.R. Phenylketonuria: an inborn error of phenylalanine metabolism. Clin. Biochem. Rev. 2008;29:31–41. [PMC free article] [PubMed] [Google Scholar]
- 21.Lee P., Treacy E.P., Crombez E., Wasserstein M., Waber L., Wolff J., Wendel U., Dorenbaum A., Bebchuk J., Christ-Schmidt H., et al. Safety and efficacy of 22 weeks of treatment with sapropterin dihydrochloride in patients with phenylketonuria. Am. J. Med. Genet. A. 2008;146A:2851–2859. doi: 10.1002/ajmg.a.32562. [DOI] [PubMed] [Google Scholar]
- 22.MacDonald A., Gokmen-Ozel H., van Rijn M., Burgard P. The reality of dietary compliance in the management of phenylketonuria. J. Inherit. Metab. Dis. 2010;33:665–670. doi: 10.1007/s10545-010-9073-y. [DOI] [PubMed] [Google Scholar]
- 23.Demirkol M., Gizewska M., Giovannini M., Walter J. Follow up of phenylketonuria patients. Mol. Genet. Metab. 2011;104:S31–S39. doi: 10.1016/j.ymgme.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 24.Hoskins J.A., Jack G., Wade H.E., Peiris R.J., Wright E.C., Starr D.J., Stern J. Enzymatic control of phenylalanine intake in phenylketonuria. Lancet. 1980;1:392–394. doi: 10.1016/s0140-6736(80)90944-7. [DOI] [PubMed] [Google Scholar]
- 25.Sarkissian C.N., Shao Z., Blain F., Peevers R., Su H., Heft R., Chang T.M., Scriver C.R. A different approach to treatment of phenylketonuria: phenylalanine degradation with recombinant phenylalanine ammonia lyase. Proc. Natl. Acad. Sci. U S A. 1999;96:2339–2344. doi: 10.1073/pnas.96.5.2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blau N., Longo N. Alternative therapies to address the unmet medical needs of patients with phenylketonuria. Expert Opin. Pharmacother. 2015;16:791–800. doi: 10.1517/14656566.2015.1013030. [DOI] [PubMed] [Google Scholar]
- 27.Kang T.S., Wang L., Sarkissian C.N., Gamez A., Scriver C.R., Stevens R.C. Converting an injectable protein therapeutic into an oral form: phenylalanine ammonia lyase for phenylketonuria. Mol. Genet. Metab. 2010;99:4–9. doi: 10.1016/j.ymgme.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kattenhorn L.M., Tipper C.H., Stoica L., Geraghty D.S., Wright T.L., Clark K.R., Wadsworth S.C. Adeno-associated virus gene therapy for liver disease. Hum. Gene Ther. 2016;27:947–961. doi: 10.1089/hum.2016.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manno C.S., Pierce G.F., Arruda V.R., Glader B., Ragni M., Rasko J.J., Ozelo M.C., Hoots K., Blatt P., Konkle B., et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 30.Cunningham S.C., Dane A.P., Spinoulas A., Alexander I.E. Gene delivery to the juvenile mouse liver using AAV2/8 vectors. Mol. Ther. 2008;16:1081–1088. doi: 10.1038/mt.2008.72. [DOI] [PubMed] [Google Scholar]
- 31.Magadum A., Kaur K., Zangi L. mRNA-based protein replacement therapy for the heart. Mol. Ther. 2019;27:785–793. doi: 10.1016/j.ymthe.2018.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kormann M.S., Hasenpusch G., Aneja M.K., Nica G., Flemmer A.W., Herber-Jonat S., Huppmann M., Mays L.E., Illenyi M., Schams A., et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 2011;29:154–157. doi: 10.1038/nbt.1733. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.