

Abstract
Retinal gene therapy using RNA interference (RNAi) to silence targeted genes requires both efficacy and safety. Short hairpin RNAs (shRNAs) are useful for RNAi, but high expression levels and activity from the co-delivered passenger strand may cause undesirable cellular responses. Ago2-dependent shRNAs (agshRNAs) produce no passenger strand activity. To enhance efficacy and to investigate improvements in safety, we have generated VEGFA-targeting agshRNAs and microRNA (miRNA)-embedded agshRNAs (miR-agshRNAs) and inserted these RNAi effectors in Pol II/III-driven expression cassettes and lentiviral vectors (LVs). Compared with corresponding shRNAs, agshRNAs and miR-agshRNAs increased specificity and safety, while retaining a high knockdown efficacy and abolishing passenger strand activity. The agshRNAs also caused significantly smaller reductions in cell viability and reduced competition with the processing of endogenous miR21 compared with their shRNA counterparts. RNA sequencing (RNA-seq) analysis of LV-transduced ARPE19 cells revealed that expression of shRNAs in general leads to more changes in gene expression levels compared with their agshRNA counterparts and activation of immune-related pathways. In mice, subretinal delivery of LVs encoding tissue-specific miR-agshRNAs resulted in retinal pigment epithelium (RPE)-restricted expression and significant knockdown of Vegfa in transduced RPE cells. Collectively, our data suggest that agshRNAs and miR-agshRNA possess important advantages over shRNAs, thereby posing a clinically relevant approach with respect to efficacy, specificity, and safety.
Keywords: Non-coding RNAs, Dicer-independent shRNAs, Pol II-driven miRNA scaffold, RNA interference, agshRNA, agoshRNA, off-target effects, VEGF, retinal gene therapy, in vivo efficacy
Graphical abstract
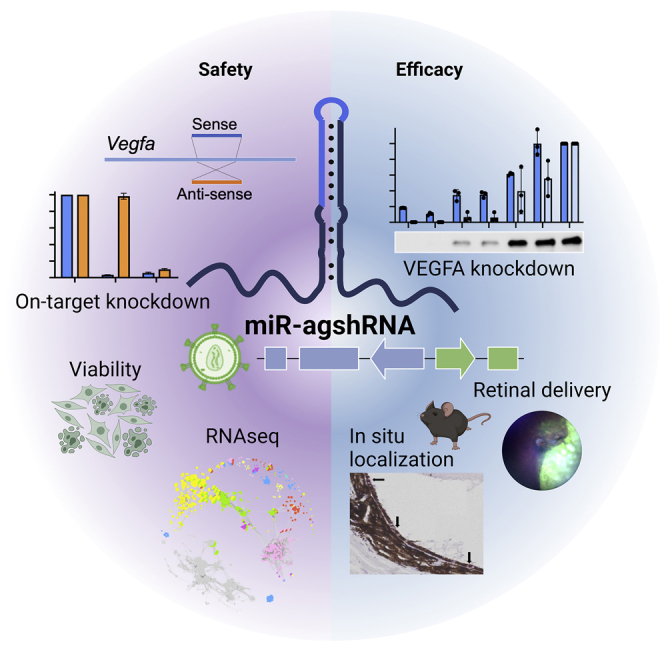
RNAi-based gene therapy requires both efficacy and safety. For the first time, Alsing et al. demonstrate that targeted knockdown of Vegfa using Pol III-driven Dicer-independent shRNAs (agshRNAs) or Pol II-driven pri-miRNA-embedded agshRNAs, poses a clinically relevant approach with respect to efficacy, specificity, and safety.
Introduction
RNA interference (RNAi) is a powerful mechanism that allows post-transcriptional regulation of a selected target gene through complementary base pairing between a small regulatory RNA and the target mRNA.1,2 The use of RNAi-based therapeutics including promoter-driven short hairpin RNAs (shRNAs) holds a great potential for use in gene therapy applications.3 Several concerns regarding the use of RNAi must, however, be considered carefully. These include (1) the risk of affecting the expression levels of non-target genes through off-target effects caused by the guide strand or the co-delivered passenger strand of the hairpin, (2) the risk of competing with the processing of endogenous microRNAs (miRNAs) through saturation of the miRNA processing pathways, and (3) the risk of initiating a cellular response to the overexpression of an exogenous small regulatory RNA.
The use of Dicer-independent shRNAs (Ago2-dependent shRNAs [agshRNAs]), which produce only a single effector strand,4 may offer compelling advantages compared with classical shRNAs. Notably, the absence of a co-delivered passenger strand is expected to reduce the risk of off-target effects caused by loading of this strand into the RNA-induced silencing complex (RISC).5,6
High-level expression of exogenous shRNAs in vivo has previously been described to cause severe toxicity in several organ systems and, in some cases, even lethality,7, 8, 9, 10 the cause of which has been speculated to be overloading of the endogenous miRNA processing machinery. The agshRNA biogenesis utilizes parts of the miRNA processing machinery, but Dicer is not needed and there is evidence that agshRNA-like species are preferentially or exclusively loaded into the catalytically competent Ago2.11, 12, 13 Hence, Ago1, Ago3, and Ago4 loading of endogenous miRNAs may be unaffected. Furthermore, several studies suggest that endogenous miRNAs are affected to a lesser extent by the expression of an agshRNA-like RNAi effector than by a Dicer-dependent shRNA.6,14 Nonetheless, the expression level of the RNAi effector will likely be the most important factor determining the impact on the endogenous miRNAs.
Another potential concern that must be addressed regarding the therapeutic use of RNAi effectors is the risk of eliciting an immune response. Activation of the interferon pathway has previously been detected in cells transduced with lentiviral vectors (LVs) encoding classical shRNAs.15,16 This activation was, however, found to be dependent on retinoic-acid-inducible gene I (RIG-I) recognition of the 5′ triphosphate, which is present in the RNA polymerase III (Pol III) transcripts15 but not in Drosha-processed precursor miRNA (pre-miR)-like species.
The agshRNA design that is used in this study builds upon previously published design guidelines17 based on the endogenous Dicer-independent miR451 (hsa-mir-451a) featuring a stem of 17 base pairs, which allows bypass of Dicer processing.4,18,19 agshRNAs are instead cleaved by Ago2 at their 3′ arm to produce a single guide strand,4,5,20 while classical shRNAs are processed by Dicer into a guide strand and a passenger strand.18,21,22 In our recent study,23 we described the embedding of agshRNAs into primary miRNA (pri-miRNA) scaffolds of miR451 and miR324, which accommodate the incorporation of the agshRNAs, allowing tunable RNA polymerase II (Pol II)-driven expression of the agshRNAs, while preserving knockdown efficacy. We have named these chimeric pri-miRNA-embedded agshRNAs miR-agshRNAs and use the term RNAi effector to describe classical shRNA, agshRNAs, or miR-agshRNAs.
In this study, we explored the efficacy, specificity, and safety of two vascular endothelial growth factor A (VEGFA)-targeting agshRNA-shRNA pairs and a non-targeting pair using reporter-based in vitro assays and a VEGFA-expressing cell line. Additionally, we investigated the Pol II-driven miR-agshRNA versions of the most potent VEGFA-targeting agshRNA. We incorporated the RNAi effector expression cassettes into LVs and show that they can be efficiently packaged and delivered, and that knockdown efficacy can be retained. Furthermore, we successfully used an LV encoding a tissue-specific promoter for the expression of a Vegfa-targeting miR-agshRNA and confirmed the in vivo expression in retinal pigment epithelium (RPE) cells and significant knockdown of Vegfa after a single subretinal injection. Lastly, we investigated the global effects of overexpression of the agshRNAs and classical shRNAs in an RPE cell line by means of RNA sequencing (RNA-seq) and identified affected pathways that include immune-related pathways that were activated in shRNA-treated cells.
Results
Characterization of two VEGFA-targeting agshRNA-shRNAs pairs, Pol II-driven miR-agshRNAs, and their competition with an endogenous miRNA
We designed two VEGFA-targeting agshRNAs (agsh12 and agsh13) and corresponding shRNAs (Figures 1A and S1). Both agshRNA-shRNA pairs target human VEGFA and murine Vegfa. The classical (Dicer-dependent) shRNAs were designed so that the predicted guide strand sequence based on the 5′ and the loop counting rules for Dicer processing24,25 are identical in the corresponding agshRNA and shRNA (Figures 1A and S1). The agshS1 and shS1, which target the HIV-1 tat-rev transcript site 1 (S1),26 serve as non-targeting controls.
Figure 1.

agshRNAs provide efficacy, lack of passenger strand activity, and reduced competition with an endogenous miRNA compared with classical shRNAs
(A) The U6-driven RNAi effector expression cassette. Sequence and predicted secondary structure of the agsh12 and the classical sh12 with expected Ago2 and Dicer cleavage sites. The predicted guide strand seed sequence (blue) and the predicted passenger strand seed sequence (orange) are shown in bold. The 13 and S1 agshRNA-shRNA pairs are shown in Figure S1. (B) Knockdown efficacy of the U6-driven RNAi effectors in HEK293 cells measured using co-transfected dedicated dual luciferase reporters with the Renilla luciferase (Rluc) fused to a Vegfa or S1 target sequence in the sense or the AS direction (see also Figure 1D) and the firefly luciferase (Fluc) serving as an internal control for normalization. Data for each individual experiment are presented in Figures S2A–S2C. (C) The competition of the U6-driven RNAi effectors with the endogenous miR21 was investigated in HEK293 cells using a co-transfected dual luciferase reporter with a perfect miR21 target sequence. An increase in the expression of the miR21-sensitive reporter may be interpreted as reduced functional miR21 levels caused by RNAi effector-mediated competition. The H1-driven miR21-specific tough decoy (TuD21), which has previously been shown to efficiently reduce miR21-mediated silencing,27 was included as a positive control, along with an empty control vector for normalization of this control. (D) Left: schematics of the Vegfa mRNA and the position of target region 12 and 13, which were fused to the Rluc of the psiCHECK dual luciferase reporters in the sense or the AS direction to create the dedicated luciferase reporters psiCHECK2-mVEGF-T12-Sense/AS or psiCHECK2-mVEGF-T13-Sense/AS. Right: schematics of the HIV-1 tat transcript with the S1 target sequence that was fused to the psiCHECK reporter in the sense or the AS direction to create psiCHECK2-S1-Sense/AS. The psiCHECK reporters also encoded Fluc. Rluc/Fluc ratio is the mean of triplicates ± SD normalized to a control. (C) A one-way ANOVA was performed followed by Šídák's multiple comparisons test between each of the agshRNAs and their corresponding shRNAs. The multiplicity-adjusted p values are reported. ∗∗∗∗p <0.0001.
The agshRNAs and shRNAs were expressed using a modified human Pol III U6 snRNA (U6) promoter and the knockdown efficacy and passenger strand activity of each pair was evaluated using dedicated Vegfa or HIV-1 tat-rev sense and antisense (AS) luciferase reporters (Figures 1B, 1D, and S2A–S2C). A sense reporter knockdown efficacy of up to 95% was observed, with the agshRNAs producing similar or improved knockdown compared with their corresponding shRNA (Figure 1B). The passenger strand activity, as measured by the knockdown of the AS reporter, was abolished in all agshRNAs, while the shRNAs produced 45%–90% AS reporter knockdown (Figure 1B).
To investigate how the expression of agshRNAs or classical shRNAs competes with the processing of endogenous miRNAs, we employed a reporter containing a perfect miR21 (hsa-mir-21) target (Figure 1C). Endogenous miR21 has previously been shown to silence the reporter in HEK293 cells.27 By co-transfecting the miR21-sensitive reporter with the U6-driven RNAi effectors, we observed increases in reporter expression ranging from 60% to 220% (Figure 1C). We attribute this increased reporter expression to a decrease in endogenous miR21-mediated silencing caused by competition. For each of the three corresponding agshRNA-shRNA pairs, the competition was more pronounced for the shRNA than for the corresponding agshRNA. For example, the sh13 produced a ∼160% increase in the expression of the miR21-sensitive reporter, whereas the agsh13 only caused a ∼60% increase (p < 0.0001) (Figure 1C). Nevertheless, there were also large differences between the pairs. The H1-driven miR21-specific tough decoy (TuD21), which has been shown to efficiently reduce miR21-mediated knockdown,27 was included as a positive control.
Having established the knockdown efficacy of the U6-driven agshRNAs, we went on to explore the Pol II-driven miR-agshRNAs. We investigated the hsa-mir-451a (miR451)-embedded and hsa-mir-324 (miR324)-embedded agsh12 (miR451-12 and miR324-12, respectively; Figures 2A and S1). The miR451-S1 (miR451-embedded agshS1) and miR451 (endogenous miR451, which does not target VEGFA) were included as controls (Figure S1). miR451-12 and miR324-12 both produced a sense reporter knockdown of ∼75%, while there was no knockdown of the AS reporter (Figure 2B), in line with the results in our previous publication.23
Figure 2.

Pol II-driven miR-agshRNAs produce potent knockdown and cause no competition with endogenous miR21
(A) The CMV-driven miR-agshRNA expression cassette. Sequence and predicted secondary structure of the miR451-12 with expected Ago2 and Drosha cleavage sites. The pri-miR451 scaffold includes ∼100 nt on both sides of the hairpin. The miR451-S1, miR324-12, and the endogenous miR451 are shown in Figure S1. (B) The knockdown efficacy of the CMV-driven RNAi effectors was investigated using a co-transfected dedicated dual luciferase Vegfa reporter in HEK293 cells. (C) The effect of the expression of the CMV-driven RNAi effectors on the knockdown efficacy of the endogenous miR21 was investigated in HEK293 cells as in Figure 1C. A tough decoy with an irrelevant target miRNA (TuDIrr) was included as a control for normalization. Rluc/Fluc ratio is the mean of triplicates ± SD normalized to a control.
While we have shown that the agshRNAs and miR-agshRNAs are processed Dicer independently,23 the miR-agshRNAs were expected to be processed by Drosha, yielding the pre-miRNA451-like agshRNA. This was confirmed using a Drosha-deficient HCT116 cell line28 in which the cytomegalovirus (CMV)-driven and U1 snRNA Pol II promoter (U1)-driven miR451-12 and miR324-12 produced no knockdown (Figures S3A and S3B), while the knockdown efficacy of the U6-driven agsh12 was unaffected (Figure S3C).
We assessed the competition of the CMV-driven RNAi effectors, endogenous miR30a (hsa-mir-30a), endogenous miR451, miR451-12, and miR324-12 with the endogenous miR21 using the experimental design described above. Despite the use of a strong viral promoter, the CMV-driven RNAi effectors produced only negligible changes in the miR21-sensitive reporter levels, translating into largely unaffected functional miR21 levels and no competition detectable with this experimental design (Figure 2C).
Lentiviral delivery of U6-driven agshRNAs produces potent knockdown of VEGFA in vitro
To allow lentiviral delivery, we introduced the U6-driven RNAi effectors into a pCCL-based LV.29 The resulting LVs are designated LV/U6-[RNAi effector] in the following. The U6-driven cassette was inserted in a unidirectional orientation with the PGK-EGFP cassette in the vector (Figure 3A). This orientation has previously been shown to be preferable in terms of obtaining high LV titers.30 Each of the LV preparations produced functional titers within the range of 4–70 × 106 infectious units (IU)/mL in the viral supernatant as well as clearly detectable levels of EGFP upon transduction (data not shown).
Figure 3.

LVs encoding the Pol III-driven RNAi effectors produce potent VEGFA knockdown but also affect viability
(A) A schematic vector map of the pCCL-based lentiviral transfer vector encoding the U6-driven RNAi effectors. CMV, cytomegalovirus promoter; cPPT, central polypurine tract; EGFP, enhanced green fluorescent protein; ΔU3, LTR unique 3′ region (U3) with self-inactivating deletion; PGK, phosphoglycerate kinase 1 promoter; Ψ, packaging signal; R, LTR repeat region; RRE, Rev-responsive element; T6, T-rich Pol III termination signal; U5, LTR unique 5′ region; U6, U6 snRNA promoter; WPRE, woodchuck hepatitis virus post-transcriptional regulatory element. (B and C) Knockdown efficacy of LVs encoding the U6-driven 12 and 13 agshRNA-shRNA pairs. HEK293 cells were transduced with the LVs encoding the RNAi effectors and then transfected with dedicated dual luciferase reporters. Rluc/Fluc ratio is the mean of triplicates ± SD normalized to agshS1. (D and E) Western blot-assessed VEGFA levels in 293-hVEGFA cells transduced with the LVs encoding the U6-driven RNAi effectors, quantification, and representative blot. The intracellular VEGFA levels are presented relative to the total protein content, while the extracellular samples are volume normalized. Mean of triplicates ± SD, normalized to NT control. (F) RT-qPCR quantification of the mRNA levels of VEGFA in 293-hVEGFA cells transduced with the LVs encoding the U6-driven RNAi effectors. Geometric mean of the PPIA-normalized VEGFA fold change (FC) with geometric SD relative to NT. (G) MTT-based cell number assessment of ARPE19 cells 5 days post transduction with the LVs encoding the U6-driven RNAi effectors. The background-subtracted absorbance at 570 nm relative to the untreated negative control (NC) is presented. Means of quintuplicates ± SD. The NT and NC are in triplicates. (G) A one-way ANOVA was performed followed by Šídák's multiple comparisons test between each of the agshRNAs and their corresponding shRNAs. The multiplicity-adjusted p values are reported. ∗∗∗∗p <0.0001.
The knockdown efficacy was evaluated using the dedicated Vegfa luciferase reporters. For the LV/U6-agsh12 and LV/U6-sh12, a sense reporter knockdown efficacy of ∼90%–95% was retained, comparable with the transfection-based assays (compare Figures 3B and 1B). The sh12 produced ∼35% AS reporter knockdown, while the agsh12, as expected, produced no AS reporter knockdown (Figure 3B). The LV/U6-agsh13 and LV/U6-sh13 produced ∼65% sense reporter knockdown (Figure 3C). The sh13 produced ∼60% knockdown of the AS reporter, whereas the agsh13 produced a small reduction in the expression of the AS reporter (∼15%) (Figure 3C). The LV/U6-agshS1 and LV/U6-shS1 produced ∼75%–80% S1-sense reporter knockdown, and the shS1 produced ∼30% S1-AS reporter knockdown (Figure S4A).
The efficacy of the LVs encoding the VEGFA-targeting RNAi effectors was further investigated using a Flp-In-293-based cell line with stable high-level expression of human VEGFA16531 (293-hVEGFA). In these cells, the LV/U6-agsh12 or LV/U6-sh12 reduced the intracellular levels of VEGFA by ∼80% or ∼90%, respectively (Figures 3D and 3E), and the LV/U6-agsh13 and LV/U6-sh13 produced a decrease of ∼65% (Figures 3D and 3E). For both VEGFA-targeting agshRNA-shRNA pairs, the reduction of extracellular VEGFA was more pronounced, reaching ∼95% or more (Figures 3D and 3E). The observed reduction in the extracellular levels of VEGFA compared with the non-transduced (NT) control may in part be ascribed to the reduced number of cells at the time of harvest caused by the LV transduction. The knockdown efficacy was also detectable in the VEGFA mRNA levels of the 293-hVEGFA cells where the LV/U6-agsh12 and LV/U6-sh12 reduced the levels by ∼80% and ∼70% respectively and the LV/U6-agsh13 and LV/U6-sh13 by ∼65% and 50%, respectively (Figure 3F).
In our studies, we observed a decrease in viability in the cells transduced with the LVs encoding some of the U6-driven RNAi effectors, which became evident several days post transduction, most pronouncedly for the sh12. To investigate how the sustained expression of the RNAi effectors affected proliferation and viability, an RPE cell line (ARPE19) was transduced with the LVs encoding the U6-driven RNAi effectors and the viability was assessed 5 days post transduction using the MTT cell proliferation assay. Compared with the untreated control (NC), the sh12 and sh13 both caused the largest reduction in cell number (∼75%). The agsh12 and agsh13 caused smaller reductions in viability compared with their corresponding shRNA (∼50% and ∼25%, respectively, p < 0.0001 for both). The agshS1 and shS1 caused ∼65% or ∼35% reduction in viability, respectively (p < 0.0001). No reduction in viability was detected in an NT control treated with the transduction-enhancing reagent Polybrene (Figure 3G).
LVs encoding Pol II-driven miR-agshRNAs produce VEGFA knockdown in 293-hVEGFA cells
A cassette encoding the U1-driven endogenous miR451, miR451-12, or miR324-12 was inserted into the pCCL-based lentiviral transfer vector in a manner similar to the U6-driven RNAi effectors (Figure 4A). This orientation has also previously been shown to be preferable in terms of obtaining high LV titers.32 We have previously shown that the knockdown efficacy of U1-driven miR451-12 and miR324-12 was superior to or comparable with the CMV-driven miR-agshRNAs.23 These LVs will be designated LV/U1-[miR-agshRNA]. Each LV yielded a titer within the range of 1.8–5.4 × 106 IU/mL and clearly detectable levels of EGFP upon transduction (Figure S5B).
Figure 4.

LVs encoding the U1-driven miR-agshRNAs produce knockdown of VEGFA and no reduction in viability
(A) A schematic vector map of the pCCL-based lentiviral transfer vector encoding the U1-driven RNAi effectors. U1, U1 snRNA promoter; U1t, U1 terminator box. (B) HEK293 cells were transduced with LVs encoding minimal Vegfa target 12 reporters (see Figure S5A). Then they were transduced with the LVs encoding the U1-driven RNAi effectors, and the Fluc activity was measured after 3 days. Mean Fluc levels (relative light units [RLU]) of triplicates ± SD, normalized to HEK293 cells transduced with the reporter only. See Figure S5B for flow cytometry evaluation. (C) RT-qPCR quantification of the mRNA levels of VEGFA in the 293-hVEGFA cells transduced with the LV encoding the indicated U1-driven RNAi effector. Geometric mean of the PPIA-normalized VEGFA FC relative to the NT control with geometric SD. (D and E) Western blot-assessed VEGFA levels in 293-hVEGFA cells transduced with the LV encoding the U1-driven RNAi effectors, quantification, and representative blot. The intracellular VEGFA levels are presented relative to the total protein content, while the extracellular samples are volume normalized. Mean of triplicates ± SD, normalized to the NT samples. (F) MTT-based cell number assessment of ARPE19 cells 5 days post transduction with the LVs encoding the indicated U1-driven miR451 or miR-agshRNAs. The background-subtracted absorbance at 570 nm relative to the untreated NC is presented. Mean of quintuplicates ± SD. The NT and NC are in triplicates.
To evaluate the knockdown efficacy of the LVs encoding the U1-driven miR-agshRNAs in a stable reporter cell line, we transduced HEK293 cells with an LV encoding a fluorescent reporter (E2Crimson) and firefly luciferase (Fluc) followed by a minimal agsh12-target in the sense or AS direction allowing evaluation of knockdown through the Fluc activity or E2Crimson expression (Figure S5A). Based on the reduction in Fluc activity, the U1-driven miR451-12 and miR324-12 both produced ∼60% knockdown of the sense reporter, and 10%–15% reduction in the expression of the AS reporter, compared with the reporter cells without RNAi effector (Figure 4B). The E2Crimson fluorescent reporter followed the same pattern, with 25%–30% reduction in the sense reporter median fluorescence intensity (MFI) for both scaffolds (Figure S5B) and no reduction for the AS reporter (Figure S5B).
The knockdown of VEGFA by the LV/U1-miR451-12 and LV/U1-miR324-12 was further investigated using the 293-hVEGFA cell line where the miR451-12 and miR324-12 reduced VEGFA mRNA levels by ∼20% or ∼25%, respectively (Figure 4C). The effect on VEGFA protein levels was more pronounced; miR451-12 and miR324-12 decreased the intracellular VEGFA levels by ∼30%–35% (Figures 4D and 4E) and the extracellular VEGFA levels were reduced by ∼70% or ∼60%, respectively, compared with non-transduced 293-hVEGFA cells (NT). However, it is possible that part of the observed reduction in extracellular VEGFA might be ascribed to the reduced number of cells at the time of harvest caused by the transduction.
The effects of sustained expression of the U1-driven miR-agshRNAs on the viability of ARPE19 cells was investigated using the MTT assay. The viability was affected by the transduction in all cases, but no large differences were observed between the endogenous miR451, miR451-12, and miR324-12 where the reduction in cell number compared with the untreated control (NC) was in the range of ∼25%–35% for all (Figure 4F). An NT control treated with Polybrene reagent was included, which in this experiment produced a reduction in viability of 25% (Figure 4F).
LVs encoding VMD2-driven anti-VEGFA miR-agshRNAs produce knockdown of Vegfa and allow RPE-specific expression in vivo
To demonstrate that our miR-agshRNA system can be employed with a tissue-specific Pol II promoter, and to prepare for the in vivo application of the anti-VEGFA miR-agshRNA, we implemented a design that we have previously used for LV delivery of RNAi effectors,33 placing the RPE-specific vitelliform macular dystrophy 2 (VMD2) promoter driving the miR451-S1 or the miR451-12 in a back-to-back configuration with the PGK promoter, allowing simultaneous expression of EGFP (Figure 5A). These LVs will be designated LV/VMD2-[RNAi effector]. Titers of 2.0–2.3 × 108 IU/mL were obtained after ultracentrifugation, and EGFP expression was clearly detectable upon transduction (Figure S5C).
Figure 5.

In Vivo knockdown of Vegfa by LVs encoding the VMD2-driven miR451-12
(A) A schematic vector map of the pCCL-based lentiviral transfer vector encoding the VMD2-driven RNAi effectors inserted back to back with the PGK-EGFP expression cassette. pA, bovine growth hormone polyadenylation signal; VMD2, vitelliform macular dystrophy-2 promoter. (B) Melanoma cells were transduced with LVs encoding minimal Vegfa target 12 reporters (see Figure S5A). Then they were transduced with the ultracentrifuged LVs encoding the VMD2-driven RNAi effectors, and the Fluc activity was measured after 3 days. Mean Fluc levels (RLU) of triplicates ± SD, normalized to melanoma cells transduced with the reporter only. See Figure S5C for flow cytometry evaluation. (C) The RPE cells of mice injected with the LV/VMD2-miR451-12 or LV/VMD2-miR451-S1 were harvested at day 14 p.i. Prior to FACS, RPE cells from three injected or two contralateral uninjected eyes were pooled. The two upper panels show pools of RPE cells from three eyes injected with the LV/VMD2-miR451-S1 or LV/VMD2-miR451-12, respectively. The lower panel shows a pool of RPE cells from two uninjected eyes. The EGFP+ cells were identified based on fluorescence measured in the EGFP detector (530/30 nm, x axis) and proportional fluorescence measured in the neighboring PE detector (585/42 nm, y axis). An equal level of fluorescence in the EGFP and PE detectors placing the cells in the diagonal of the plots was interpreted as autofluorescence, and these cells were excluded. The percentages of EGFP+ RPE cells in each pool are indicated. The full gating strategy is shown in Figure S7. (D) Twelve mice were injected in each group, producing four pools, but only three pools in each group yielded sufficient EGFP+ cells for purification of RNA. RNA was purified from the sorted pools, and Vegfa mRNA was quantified using RT-qPCR. Actb-normalized Vegfa FC relative to uninjected control eyes with the geometric mean indicated. Each data point represents a pool of FACS-sorted RPE cells. (D) The data was log transformaed and a one-way ANOVA was performed followed by the uncorrected Fisher's least significant difference (LSD) test between the LV/VMD2-miR451-S1 EGFP+ group and the LV/VMD2-miR451-12 EGFP+ group.
The efficacy of the VMD2-driven miR451-12 was evaluated in a melanoma-based reporter cell line, as the VMD2 promoter is active in these cells.34 We applied the LV-based minimal Vegfa target 12 reporter introduced above (Figure S5A) to create a melanoma-based reporter cell line. Using this cell line, the LV/VMD2-miR451-12 produced ∼40% knockdown of the sense reporter, and no knockdown of the AS reporter (Figure 5B) based on the Fluc activity. The sense reporter MFI was reduced ∼34% (Figure S5C), while no reduction was observed for the AS reporter (Figure S5C).
To evaluate the expression of the VMD2-driven miR451-12 and the knockdown of Vegfa in vivo, we performed subretinal injections with the LV/VMD2-miR451-12 and LV/VMD2-miR451-S1 in C57BL/6J mice. Using this route of delivery with VSV-G pseudotyped LV, the transduction is limited almost exclusively to the RPE cells.35 First, we compared the Vegfa mRNA expression levels in RPE cells from the whole eye cup day 56 post injection (p.i.). We compared eyes from mice that had received the LV/VMD2-miR451-12 or LV/VMD2-miR451-S1, and uninjected eyes. The Vegfa mRNA levels were reduced in the RPE cells in both groups compared with uninjected eyes, but we detected no reduction in the LV/VMD2-miR451-12 group compared with the LV/VMD2-miR451-S1 group (Figure S6A). To investigate the expression of the vector, we quantified the level of pri-miR451-agshRNA transcript and EGFP mRNA (Figure S6B). The pri-miR451-agshRNA was detected in three out 10 mice injected with the LV/VMD2-miR451-12 and in four out of 10 mice injected with the LV/VMD2-miR451-S1 (Figure S6B), and the expression generally correlated with the EGFP levels as expected (Figures S6B–S6D).
To account for the variability of the transduction profile across the retina after subretinal injections as shown by the different sizes of EGFP-positive area in fundoscopy (Figures S6C and S6D), we refined our experimental approach; 14 days after subretinal injections of the LV/VMD2-miR451-12 and LV/VMD2-miR451-S1, we isolated the transduced EGFP+ RPE cells using fluorescence-activated cell sorting (FACS), allowing us to detect changes in the Vegfa mRNA levels specifically in the transduced cells (Figures 5C and S7). The Vegfa mRNA level was reduced by ∼55% in the EGFP+ RPE cells from mice injected with the LV/VMD2-miR451-12 compared with EGFP+ RPE cells from mice injected with the LV/VMD2-miR451-S1 (p = 0.0288) (Figure 5D). Similarly, a ∼55% reduction was observed in the EGFP+ RPE cells compared with the EGFP− RPE cells from mice injected with the LV/VMD2-miR451-12 (Figure 5D). Unexpectedly, the Vegfa levels were reduced by 45%–55% both in the EGFP+ and EGFP− RPE cells from LV/VMD2-miR451-S1-injected eyes, and in the EGFP− RPE cells from LV/VMD2-miR451-12-injected eyes, compared with uninjected eyes (Figure 5D, see also the section “discussion”).
The expression of the VMD2-driven miR451-12 in vivo was further investigated using miRNAscope, a novel advanced method for detection of small RNAs. We examined cross sections of one of the eyes injected with the LV/VMD2-miR451-12, which had a high level of EGFP expression in the RPE cells at day 57 p.i. when the mice were sacrificed for this analysis (Figures 6A–6C and 6E). The miR451-12 signal was detected in several RPE cells, although the detection of the red chromogenic signal was impeded slightly by the presence of naturally occurring pigment in the RPE cells (Figures 6D, 6F, and 6G). The miR451-12 signal was also detectable as a more easily distinguishable red fluorescent signal (Figure 6H). Importantly, no signal was detected in other cell types in the retina or choroid (Figures 6D and 6F–6H). We examined the EGFP expression in the sections adjacent to the ones that were used for detection of miR451-12 and confirmed that the miR451-12 signals co-localized with EGFP-positive RPE cells (Figures 6E and 6F). The analysis of retinal cross sections of an LV/VMD2-miR451-S1-injected eye showed no miR451-12 signal inside or outside the EGFP-positive area (Figures 6I and S8). Thus, our data demonstrate that we can use a tissue-specific promoter for expression of the miR451-12 and achieve expression in vivo.
Figure 6.

RPE-specific expression of the VMD2-driven miR451-12 in vivo
(A and B) Brightfield fundoscopy and EGFP detection of the LV/VMD2-miR451-12-injected eye, which was used for the in situ detection of miR451-12 57 days p.i. The expected position of the cross section used in the analysis is indicated with the dashed line. Fifty-seven days p.i. (C) Formalin-fixed and paraffin-embedded cross section of the LV/VMD2-miR451-12-injected eye, harvested at day 57 p.i. EGFP signal and DAPI staining. (D) Cross section adjacent to the section in (C), which was used for chromogenic in situ detection of the miR451-12 with the miRNAscope probe detecting the agsh12 guide strand. Brightfield image with hematoxylin counterstain. (E) Magnification of the EGFP-positive section indicated in (C). (F) Magnification of the section indicated in (D). The arrows show the location of the red chromogenic signal indicating the detection of miR451-12. (G and H) Magnification of the section indicated in (F) with detection of the chromogenic or fluorescent signal of the miR451-12 probe. (I) Formalin-fixed and paraffin-embedded cross section of the LV/VMD2-miR451-S1-injected eye (NC), harvested at 57 days p.i., treated with the miRNAscope probe detecting the agsh12 guide strand. Brightfield image with hematoxylin counterstain. (I′) EGFP signal in the LV/VMD2-miR451-S1-injected eye, in the section adjacent to the one shown in (I). See also Figure S8.
Sequence-dependent and Sequence-independent Effects of overexpression of RNAi effectors in ARPE19 cells
Having confirmed the efficacy of the LVs encoding the agshRNAs or miR-agshRNAs, we sought to further evaluate the specificity of the agshRNAs compared with the corresponding classical shRNAs, as well as the more general effects of overexpression of the agshRNAs and shRNAs. To this end, we employed RNA-seq of ARPE19 cells transduced with the LVs encoding the U6-driven VEGFA-targeting agshRNA-shRNA pairs (12 and 13), and the non-targeting S1 pair.
First, we investigated the number of differentially expressed genes compared with the LV/U6-agshS1-transduced ARPE19 cells, which served as a reference throughout these analyses. For both VEGFA-targeting agshRNA-shRNA pairs, the number of downregulated genes was larger in the cells transduced with the LVs encoding the shRNA compared with the corresponding agshRNA (Figures 7A and S9A). The number of upregulated genes was higher in the cells transduced with the LV/U6-agsh12 compared with sh12, although the highest number of upregulated genes was observed in the cells transduced with the LV/U6-sh13 (Figures 7B and S9B). The sh13 also produced the largest changes in expression levels, with ∼23% of the downregulated genes and ∼34% of the upregulated genes reaching a log2 fold change (log2FC) >1 (Figures 7A and 7B), whereas with the agsh13 only ∼9% of the downregulated genes and ∼17% of the upregulated genes reached a log2FC >1 (Figures 7A and 7B). The magnitude of the changes of the expression levels for the agsh12 and sh12 was comparable, with ∼15% of downregulated genes and ∼22% of the upregulated genes having a log2FC >1 (Figures 7A and 7B). The shS1 caused the fewest downregulated and upregulated genes and produced the smallest changes in expression levels (Figures 7A and 7B), which is not surprising considering the shared guide strand with the agshS1 reference sample. Taken together, these data show that the expression of a classical shRNA in general leads to more changes in gene expression levels compared with its agshRNA counterpart, although these effects unsurprisingly are also highly sequence dependent.
Figure 7.

Changes in gene expression caused by LVs encoding the U6-driven RNAi effectors
(A) Log2 fold change (log2FC) distribution for the genes that were downregulated in the ARPE19 cells transduced with the LVs encoding the U6-driven RNAi effectors, compared with the cells transduced with the LV/U6-agshS1. The inset shows the number of downregulated genes compared with the cells transduced with the LV/U6-agshS1. (B) As in (A), but with upregulated genes. EDCF, empirical cumulative distribution function.
To investigate the effects of overexpression of RNAi effectors, both in terms of sequence-dependent effects and pathways that were affected by sequence-independent or sequence-dependent effects, we employed a self-organizing map (SOM) analysis of the 3,000 most variably expressed genes among differentially expressed genes in pairwise comparisons (Figure 8A). In this analysis, the genes were divided into nodes with genes that had a similar expression profile across the samples. For example, the turquoise node in the upper left corner contains genes with a low level of expression in the LV/U6-sh13-transduced cells (represented by the red wedge), and a high level of expression in the LV/U6-shS1-transduced cells (pale orange wedge) (Figure 8A). The nodes were then grouped into clusters containing nodes with similar gene expression profiles, such as the three turquoise nodes in the upper left corner, which constitute cluster 1 (Figure 8A).
Figure 8.

LVs encoding the U6-driven RNAi effectors produce a wide range of sequence-dependent and sequence-independent effects
(A) Self-organizing map (SOM) analysis of the 3,000 most differentially expressed genes among differentially expressed genes in pairwise comparisons in the ARPE19 cells transduced with the LVs encoding the U6-driven RNAi effectors. Genes with similar expression profiles are grouped in the circular nodes. Nodes with similar profiles are organized in clusters. The polar area charts inside the nodes represent the expression profile of the genes in the node across the different samples. (B) Enrichment plot showing the enriched gene ontology biological processes (GO-BP) terms in the clusters from the SOM analysis. Captions for enriched pathways in selected clusters are shown (see Table S1). (C) Genes that contain heptamer seed site matches (HSMs, matching nt 2–8 of the RNAi effector strand) in their 3′UTR were identified for each predicted guide and passenger strand. The fraction of genes containing HSMs relative to both the number of genes in the SOM cluster and the total number of HSMs in the expressed genes was calculated for each SOM cluster and strand (normalized fraction). Arrows designate bars mentioned in the text.
To discover the pathways to which the genes in a particular cluster belong, we performed Gene Ontology (GO) enrichment analysis for GO biological process (GO-BP) terms. Figure 8B shows the enriched pathways that were identified in the different clusters in an enrichment plot with colors corresponding to the SOM clusters and descriptions of key pathways (see also Table S1). Furthermore, to address the question of sequence-specific off-target effects of the RNAi effectors, we explored the number of guide and passenger strand heptamer seed matches (HSMs, matching nt 2–8 of the RNAi effector strand) in the 3′UTR region of the genes in the different clusters. For each cluster, we determined the number of genes that contained a particular HSM and calculated the fraction of HSM-containing genes relative to both the total number of HSMs for this guide or passenger strand in all expressed genes and to the number of genes in the cluster (normalized fraction, Figure 8C). Notably, the number of HSMs that were identified in the genes expressed in the ARPE19 cells differed remarkably from a few hundred for the 13 and S1 guide strands to several thousand genes (Figure S9C).
Some of the clusters that were identified in the SOM analysis appeared to be composed largely of genes that were downregulated in the cells transduced with the LV encoding the agshRNA or the corresponding shRNA. An example of this is cluster 3, which contains genes with a low level of expression in the agsh12 and sh12 samples (Figure 8A, light purple nodes). Indeed, the largest normalized fraction of genes containing 12 guide HSMs that may be directly targeted and hence downregulated by off-target effects of the shared 12 guide strand was found in cluster 3 (Figure 8C). Accordingly, as sequence-specific off-target effects are expected to affect genes independently of their function and of the pathways in which they are involved, no GO-BP terms were enriched in this cluster.
Similarly, cluster 11 included genes with a low expression level in the agshS1- and shS1-treated samples (Figure 8A, mint green nodes), which may represent genes that are downregulated by off-target effects of the shared S1 guide strand. Accordingly, the highest normalized fraction of genes containing S1 guide HSMs was found in cluster 11 (Figure 8C), and no GO-BP terms were enriched in this cluster. Clusters 1 and 6 (Figure 8A, turquoise and orange nodes) included genes with a low level of expression in the sh13 or sh12 samples, respectively, which might represent genes that were downregulated by the sh13 or sh12 passenger strand. In agreement with this interpretation, the highest normalized fraction of genes containing sh13 passenger strand HSMs was found in cluster 1 (Figure 8C), indicating sh13 passenger strand off-target effects.
Interestingly, the genes in cluster 12 with a high level of expression in the sh12- and sh13-treated cells (Figure 8A, mustard yellow nodes) were involved mainly in immune-related pathways and particularly antiviral mechanisms (Figure 8B). Similarly, several immune system process-related GO-BP terms were enriched among the genes in cluster 7 (Figure 8B), which was composed mainly of genes that were highly expressed in the sh13 samples (Figure 8A, green nodes). Thus, the high expression level of immune system-related genes in cluster 12 and 7 in the sh12- and/or sh13-treated cells may be linked to a class-specific effect shared by the classical targeting shRNAs.
Overall, the transduction with an LV encoding a Pol III-driven RNAi effector, whether an agshRNA or a classical shRNA, produced a wide range of effects that were reflected in the changes in the gene expression levels. Some of the changes may be attributed to sequence-specific off-target effects or downstream effects of these, while others are more likely a result of a more general cellular response to the overexpression of an RNAi effector. Furthermore, the on-target efficacy does not seem to be correlated with the off-target effects, as exemplified by the sh13, which produces a wide range of off-target effects but only moderate on-target knockdown efficacy.
Discussion
The present study investigates the use of Dicer-independent RNAi effectors, and in particular tissue-specific Pol II-driven miR-agshRNAs for future therapeutic applications. We have shown that the agshRNAs and miR-agshRNAs produce knockdown of VEGFA in vitro, and that they may provide important advantages in terms of specificity and safety, including reduced competition and improved viability compared with classical shRNAs. Additionally, we have shown that the miR-agshRNAs are expressed in RPE cells in vivo following subretinal delivery of an LV encoding a Vegfa-targeting miR-agshRNA driven by the VMD2 promoter and produce significant knockdown of Vegfa in vivo.
Numerous investigations have explored the features that determine the efficacy of Dicer-independent RNAi effectors.5,13,17,20,36, 37, 38, 39, 40 While these have provided highly useful design guidelines, empirical evaluation of the efficacy is still needed. In comparison with classical shRNAs, we observe an overall increase in the efficacy of the agshRNAs, both in reporter-based assays and on the level of VEGFA, in agreement with other studies comparing classical and Dicer-independent RNAi effectors.4,14 The efficacy is nevertheless highly sequence dependent. Importantly, we detected significant knockdown of Vegfa in vivo specifically in the EGFP+ RPE cells from mice injected with the LV/VMD2-miR451-12. The observed ∼55% reduction in Vegfa levels compared with EGFP+ RPE cells from mice injected with the LV/VMD2-miR451-S1 was superior even to the knockdown produced in vitro in the melanoma reporter cell line. The reduction in Vegfa levels in the LV/VMD2-miR451-S1-treated group and the EGFP− RPE cells in the LV/VMD2-miR451-12 group compared with uninjected control eyes might be ascribed to effects of the injection or transduction process, which may affect the overall health of the RPE cells and reduce the expression of Vegfa. In contrast, we did not detect knockdown by the LV/VMD2-miR451-12 in the RNA isolated from RPE cells from the whole eye cup, likely because the Vegfa expression level in the untransduced RPE cells occluded the detection of knockdown in the transduced cells, which comprised only a fraction of the RPE cells as estimated by the EGFP-positive area detected by fundoscopy. This finding underscores the importance of isolating the transduced cells to obtain an accurate measure of the in vivo knockdown efficacy. While, surprisingly, no reduction of VEGFA mRNA was detectable in the ARPE19 cells transduced with the LVs encoding the U6-driven RNAi effectors in vitro (Figure S9D), we ascribe this finding to the modest expression level of VEGFA in the ARPE19 cells.
The utility of an RNAi effector tool is, however, determined not only by the efficacy but also by the level of undesired effects of the RNAi effector, including off-target effects, competition with endogenous miRNAs, and unanticipated cellular responses such as activation of immune system processes. The data presented in this study indicate that agshRNAs and miR-agshRNAs offer compelling advantages in these regards.
A well-known potential pitfall for RNAi-based therapeutics is the risk of off-target effects caused by guide or passenger strand seed sequence complementarity to the 3′UTR of genes other than the intended target gene, thereby affecting their expression level through miRNA-like off-target effects.41 agshRNAs may intrinsically produce fewer sequence-specific off-target effects, since passenger strand off-target effects are avoided. Additionally, the preferential loading of the agshRNAs into Ago211, 12, 13 may contribute to the increased specificity of the agshRNAs, since off-target effects caused by partial complementarity to the 3′UTR have been coupled to the association of the RNAi effector with the catalytically incompetent Ago1, 3, and 4.12,42 Indeed, a recent study by Shang et al. reported a high level of specificity of an agshRNA-like RNAi effector.6 Conversely, a study by Sun et al. suggested that there may be an increased risk of guide strand off-target effects by agshRNA-like RNAi effectors, due to a higher mismatch tolerance.37 Generally, our data indicate that the agshRNAs cause fewer off-target effects than a corresponding shRNA, comparing the number of downregulated genes between the agshRNA and corresponding shRNA. The striking difference between the number of identified HSMs, however, suggests that the sequence of the RNAi effector also plays a major role in determining the off-targeting potential.
To predict the off-targeting potential of an RNAi effector, several tools exist (e.g., the siSPOTR tool).43 Although prediction of off-targets can provide valuable information, off-targeting should also be investigated in the specific cell type or tissue in which the RNAi effector will be expressed, and at the dose where the RNAi effector has the desired efficacy, to detect the true effects of RNAi effector expression, both sequence dependent and sequence independent. This is evident when considering the sh13, which, although it has the fewest guide and passenger HSMs compared with the sh12 and shS1, produces a wide range of cellular responses.
In the present study, we explored the off-target effects using RNA-seq-based transcriptome analysis of cells treated with LVs encoding the U6-driven RNAi effectors. We investigated the number of downregulated genes compared with the cells transduced with the LV encoding the agshS1. Naturally, this means that any gene that is designated as downregulated in a particular sample might as well be upregulated in the cells transduced with the LV/U6-agshS1 and vice versa, but agshS1, being a non-targeting control with few HSMs, nevertheless provides a common reference point that allows internal comparisons between the RNAi effectors. To explore the sequence-specific off-target effects of the RNAi effectors, we utilized the information from the SOM analysis of the 3,000 most variably expressed genes among differentially expressed genes in pairwise comparisons and compared the normalized fraction of HSMs for each of the guide and passenger strands of the RNAi effectors in all the clusters. While this approach excludes both less differentially expressed genes and shorter hexamer seed matches, our data indicated that, in some cases, sequence-specific off-target effects were indeed at play, and, importantly, we were able to distinguish these effects from the multitude of other effects that we observed in this analysis.
Although the concerns for off-target effects are certainly valid, issues regarding the RNAi effector competition with the processing of endogenous miRNAs may be equally important to address, and high-level expression of exogenous RNAi effectors has been described to cause severe toxicity in vivo.7, 8, 9, 10 The nuclear karyopherin XPO5, which exports pre-miRNAs as well as shRNAs, has been identified as a common saturable factor,7,44 but other studies suggest that the competition takes place at several levels of processing from XPO5 to RISC incorporation.45 Our data indicate that the agshRNAs overall compete less with the expression of endogenous miRNAs than the corresponding shRNAs, as exemplified by the competition with miR21 in HEK293 cells. This observation is corroborated by several studies,6,14 and may be attributed to the bypass of Dicer and the preferential loading into Ago211, 12, 13 leaving Ago1, Ago3, and Ago4 loading largely unaffected. The pattern was, however, less clear for the upregulated genes in the RNA-seq analysis of the ARPE19 cells transduced with the LVs encoding the U6-driven RNAi effectors, which might include genes that are targets of out-competed endogenous miRNAs. The level of competition is evidently influenced not only by the structural features of the RNAi effector but also by the sequence, as reported previously.45 Nevertheless, Pol III-driven RNAi effectors are likely to be unsuitable for some applications. With our experimental design, we observe no competition caused by the Pol II-driven miR-agshRNAs, echoing the effects of shRNA incorporation into an miRNA backbone.45
In addition to the sequence-specific off-target effects and the competition with the processing of endogenous miRNAs, we observed an effect on immune system-related pathways in the ARPE19 cells that were transduced with the LV/U6-sh12 and LV/U6-sh13. Induction of interferon-based immune responses elicited by shRNAs delivered through transient plasmid transfections16 or through LVs15,16 has been described previously. While the induction of an interferon response appears to be linked to the recognition of the 5′ triphosphate of the U6-driven transcript by RIG-I,15 sequence-specific effects also seem to be at play,15 which might explain why no similar response was observed in the LV/U6-shS1-transduced cells. Noticeably, we did not observe any such induction of interferon-based immune responses in the cells transduced with the LVs encoding the agshRNAs. Although we cannot rule out that this was owing to the sequence composition of the agshRNAs, it is possible that the structural features of the agshRNAs preclude the activation of an immune response. Additionally, the 5′ triphosphate is not present in the Drosha-processed miR-agshRNAs, and there is no indication that the miR-agshRNAs will elicit a similar response.
The sustained expression of the U6-driven RNAi effectors caused an unexpected reduction in proliferation or viability as shown by the reduced number of ARPE19 cells 5 days post transduction. While this effect appeared to be dependent both on the sequence and on the type of RNAi effector, the sh12 and sh13 caused the largest reduction in viability. We speculate that this may be linked to the activation of immune system-related and antiviral pathways described above. Importantly, the sustained expression of miR451-12 did not appear to cause a similar reduction in proliferation.
To be useful for therapeutic applications, RNAi effectors must have a high intrinsic efficacy that allows the use of controlled tissue-specific Pol II expression and a minimal dose of delivery vector while retaining therapeutic effect. Additionally, the safety and specificity, including the consideration of competition with endogenous miRNAs and of the risk of eliciting an immune response, is of the utmost importance. We have previously shown that VMD2-driven intron-embedded Vegfa-targeting miRNA-embedded shRNAs reduce the Vegfa levels and Vegfa-driven formation of choroidal neovascularization in a mouse model,33,46 but additional studies are required to further investigate the efficacy of the miR-agshRNAs in an in vivo model. Furthermore, application of atypical RNAi effectors in the form of mirtrons has recently been pursued for RNA knockdown/replacement therapy.47 Our present findings, combined with the fact that bicistronic adeno-associated viruses (AAVs) encoding RNAi effectors and proteins have been engineered,46 suggest that application of our miR-agshRNAs in RNA replacement gene therapy might add additional advantages over current technologies, facilitating broad treatment options of dominantly inherited diseases.
In the present study, we deliver an anti-VEGFA miR-agshRNA with an LV, supporting the notion that the miR-agshRNA system is easily adaptable for numerous targets,23 and, owing to the small size of the expression cassette, other delivery systems will likely be equally useful, thus expanding the therapeutic applicability. Collectively, our data suggest that the agshRNAs, and particularly the Pol II-driven miR-agshRNAs, possess important advantages in terms of efficacy, high intrinsic specificity, safety, and versatility over classical shRNAs, making them highly suitable for future gene therapy-based applications both inside and outside of the eye.
Materials and methods
Naming and design of the agshRNA-shRNA pairs
The VEGFA-targeting agsh12 was described in our previous publication with the name agsh12.3.23 We have in this study renamed the agsh12.3 as agsh12, and redesigned the corresponding classical shRNA, sh12, so that the predicted guide strand sequence based on the 5′ and the loop counting rules for Dicer24,25 is the same for agsh12 and sh12 (Figures 1A and S1). Similarly, the miR451- and miR324-embedded agsh12.3 have been renamed miR451-12 and miR324-12, respectively. The VEGFA-targeting agsh13 has also been introduced in our previous publication23 and is here investigated along with the corresponding sh13. Both agshRNA-shRNA pairs were designed to target human VEGFA and murine Vegfa.
For the sh13, an A-U base pair was swapped at position 20 from the 5′ end of the hairpin to avoid a premature U6 termination signal. The predicted guide strand sequence was not affected by this. The agshS1 and shS1 target the HIV-1 tat-rev transcript (site 1).26 Please note that the shS1 refers to an shRNA with a 21-bp stem, different from the shS1 presented in our previous publication.23 The guide strand of agshS1 contains a target mismatch at position 1 to allow an A at position +1. The shS1 guide strand contains target mismatches at positions 1, 19, and 20 to allow a G at position +1 for initiation of U6 transcription. The predicted shS1 passenger strand contains target mismatches at positions 1, 19, and 20. These mismatches were deemed unlikely to influence efficacy. See also Figure S1.
Plasmid construction
PsiCHECK reporter plasmids were constructed as previously described,23,26,48 and the psiCHECK-S1-AS was generated by inserting the S1 target sequence of the psiCHECK-S1 reporter in the AS direction, 3′ of the Rluc in the psiCHECK2 vector using XhoI and NotI sites, using double-stranded oligos with compatible overhangs.
For generation of Pol III expression vectors, agshRNA or shRNA constructs were cloned directionally into the pFRT-U6 expression plasmid49 as double-stranded oligos, using BglII- and XhoI-compatible overhangs as described previously.23 The pFRT-U6 expression plasmid encodes a modified human U6 snRNA promoter, containing modifications at position −4 and −2, creating a BglII site allowing direct cloning of RNAi effector oligonucleotides.49 The generation of the U1 and CMV-driven miR-agshRNA pcDNA3.1-based expression vectors has been described previously,23 as well as the H1-driven tough decoys (TuDs).27
For generation of the lentiviral transfer plasmids, a third-generation pCCL-based lentiviral transfer plasmid encoding PGK-driven EGFP, named pCCL-PGK-EGFP-MCS, was employed. pCCL-PGK-EGFP-MCS was constructed in two steps. First an “empty” vector, named pCCL-PGK-MCS, with two unique restriction sites upstream of the PGK promoter (ClaI and BsiWI) and seven unique restriction sites downstream of the PGK promoter (BamHI, SalI, XbaI, ApaI, XmaI, MluI, and XhoI) was created by excising the ClaI/XhoI PGK-EGFP fragment from pCCL-PGK-EGFP50 and inserting a restriction-site-flanked PGK promoter PCR fragment made using restriction-site-tagged primers (see Table S2). Next, EGFP was reintroduced into pCCL-PGK-MCS at the BamHI/XbaI sites as a PCR fragment made using restriction-site-tagged primers (see Table S2). Of note, the MCS did not affect transfer as pCCL-PGK-EGFP-MCS titers were equal to the original pCCL-PGK-EGFP (data not shown).
The lentiviral transfer plasmids encoding U6-driven RNAi effectors were constructed by PCR amplifying a region spanning the promoter as well as the RNAi effectors and T5 termination signal using primers incorporating BsiWI and ClaI sites. The pCCL-PGK-EGFP-MCS was cleaved with BsiWI and ClaI, and the PCR-amplified expression cassette was inserted, orienting the promoters in a unidirectional fashion. The lentiviral transfer plasmids encoding U1-driven RNAi effectors were constructed in a similar manner, inserting a PCR-amplified region spanning the promoter as well as the RNAi effectors and U1 terminator box from the appropriate pcDNA3.1-U1 expression plasmid with primer-incorporated BsiWI and ClaI sites into the BsiWI and ClaI cleaved pCCL-PGK-EGFP-MCS.
The lentiviral transfer plasmids encoding the VMD2-driven miR451-S1 and miR451-12 were constructed using a previously cloned pCCL-based lentiviral transfer plasmid encoding the CMV-driven miR451-12 with the CMV promoter in a back-to-back configuration with the PGK promoter in the pCCL-PGK-EGFP-MCS. This was produced by inserting a PCR-amplified region spanning the CMV promoter as well as the RNAi effectors and bovine growth hormone polyadenylation signal from the pcDNA3.1-CMV-miR451-12 into the pCCL-PGK-EGFP-MCS using BsiWI and ClaI restriction sites. The lentiviral transfer plasmids encoding the CMV-driven miR451-12 was in turn cleaved with Kpn2I and SalI, allowing replacement of the CMV-PGK fragment with a PCR amplified VMD2-PGK fragment with compatible overhangs, effectively replacing the CMV promoter with the VMD2 promoter. The pCCL-PGK-EGFP-VMD2-miR451-S1 was generated by amplifying the miR451-S1 from pcDNA3.1-CMV-miR451-S1 with SalI and BclI tagged primers and inserting the fragment into the SalI and BclI cleaved pCCL-PGK-EGFP-VMD2-miR451-12.
The lentiviral transfer plasmids for production of the LV-based minimal Vegfa target 12 reporters, named pCCL-PGK-E2Crimson-P2A-Fluc-min12-sense and pCCL-PGK-E2Crimson-P2A-Fluc-min12-AS, were constructed by inserting a minimal Vegfa target 12 sequence in the sense or AS direction into the pCCL-PGK-E2Crimson-P2A-Fluc plasmid (Aagaard, unpublished) with MluI and XhoI. The minimal target sequence consisted of a short part of the Vegfa T12 sequence containing the predicted sh12 and agsh12 guide strand target in the sense or the AS direction with five to eight additional nucleotides 5′ to 3′. All plasmids were verified using restriction analysis and sequencing of inserts. Cloning oligo and primer sequences may be found in Table S2.
Cell culturing
All cell lines were kept at 37°C and 5% v/v CO2. HEK293 (catalog no. CRL-1573; American Type Culture Collection [ATCC], Manassas, VA, USA), HEK293T (catalog no. CRL-11268; ATCC), 293-Flp-In-hVEGFA,31 and the CD86 wild-type (WT) melanoma cell line51 were kept in Dulbecco’s modified Eagle’s medium (DMEM). ARPE19 cells (catalog no. 2302; ATCC) were kept in 50/50% DMEM/F12 medium. HCT116 cells and HCT116 Drosha knockout cells were purchased from Korean Collection for Type Cultures (KCTC, Jeongeup, Korea, reference numbers BP1230983 and BP1230984, respectively) and kept in McCoy's 5a medium (Sigma, St. Louis, MO, USA). All media were supplemented with 10% fetal calf serum (FCS), 2 mM glutamine, 100 U/mL penicillin, and 0.1 mg/mL streptomycin. F12 media and supplements were purchased from Sigma-Aldrich [Sigma]). DMEM medium was purchased from Sigma, Lonza Bioscience (Basel, Switzerland), or Gibco (Thermo Fisher Scientific, Waltham, MA, USA).
Dual luciferase reporter assay
All dual luciferase reporter assays were performed using the Dual-Glo Luciferase Assay System (Promega, Madison, WI, USA) according to the manufacturer's protocol. Luminescence was detected in an opaque white 96-well plate using a multi-sample plate reading luminometer (MicroLuminat plus LB 96V, Berthold Technologies, Bad Wildbad, Germany). Data are presented as averages of Rluc/Fluc ratios or Fluc values of triplicates normalized to an empty or non-targeting control.
For the transfection-based Vegfa reporter knockdown assays, HEK293, HCT116, or HCT116 Drosha KO cells were seeded in a white 96-well plate at a density of 3,000, 3,000, or 4,000 cells/well, respectively. The next day, the cells were co-transfected with 6 ng of psiCHECK2 reporter plasmid (psiCHECK2-mVEGF-T12 or psiCHECK2-mVEGF-T1323), and 34 ng of plasmid encoding an RNAi effector, using XtremeGENE 9 (Roche, Basel, Switzerland) according to the manufacturer's protocol. Luminescence was evaluated 2 days post transfection.
The transfection-based miR21 competition assays were inspired by an experimental design described by Ma et al.14 HEK293 cells were seeded at a density of 10,000 cells/well and co-transfected the following day with 100 ng of psiCHECK2 reporter plasmid (psiCHECK2-miR21perfect, encoding the Rluc fused to a perfect miR21 target48) and 100 ng of plasmid encoding the RNAi effector. Luminescence was evaluated 2 days post transfection.
PsiCHECK reporter-based assays evaluating the efficacy of the LVs encoding the U6-driven RNAi effectors were performed as follows: HEK293 cells were seeded in a white 96-well plate at a density of 2,000 cells/well. The next day, the cells were transduced with the LVs at a multiplicity of infection (MOI) of 25. The following day, the cells were co-transfected with 6 ng of psiCHECK reporter plasmid (psiCHECK2-mVEGF-T12 Sense/AS or psiCHECK2-mVEGF-T13 Sense/AS23) and 34 ng of pcDNA3.1(+) stuffer plasmid, and the luminescence was evaluated 2 days post transfection.
LV reporter-based assays evaluating the efficacy of the LVs encoding the Pol II-driven RNAi effectors were performed as follows: HEK293 or melanoma cells were transduced with the LV reporter using an MOI of 20 or 10, respectively, more than 3 weeks before analysis, to generate the reporter cells. The HEK293 or melanoma reporter cells were seeded at a density of 2,000 cells/well or 3,000 cells/well respectively, in a white 96-well plate for evaluation of luminescence. The next day, 3 days before luminescence evaluation, the HEK293 reporter cells were transduced with the LVs encoding the U1-driven RNAi effectors using an MOI of 25 and the melanoma reporter cells were transduced with the LVs encoding the VMD2-driven RNAi effectors at an MOI of 35. For both experiments, the Fluc was evaluated using the Dual-Glo reagent of the Dual-Glo Luciferase Assay System according to the manufacturer's protocol and a flow cytometry analysis was performed in parallel (see below).
Lentivirus production and titration
For each LV preparation 1 × 107 HEK293T cells were seeded in a 15-cm dish on day 1. On day 2, the cells were co-transfected with 6.75 μg of pRSV-REV, 8.43 μg of pMD.2G (all LVs were VSV-G pseudotyped), 29.25 μg of pMDIg/p-RRE, and 29.25 μg of lentiviral transfer plasmid using CaPO4 transfection. Medium was replaced with medium containing 1 mM sodium butyrate (Sigma) and 2% FCS on day 3. Supernatants containing the viral particles were harvested on day 4 by filtering the medium of the producer cells through a 0.45-μm filter (Sarstedt, Nürnbrecht, Germany). For production of ultracentrifuged LV preparations for subretinal injections, approximately 300 mL of viral supernatant was prepared as described above and centrifuged through a 5% sucrose cushion at 25,000 rpm in an SW28 or SW27 rotor for 2 h at 4°C. Pellets were resuspended in Hanks Balanced Salt Solution (HBSS, Gibco, Thermo Fisher), and the preparations were purified and concentrated by centrifugation through an Amicon Ultra-15 Centrifugal Filter Unit (100-kDa MWCO) (Millipore, Sigma) according to the manufacturer's protocol.
A functional LV titer was determined using a qPCR-based approach. One-hundred thousand HEK293 cells were seeded. The next day, the cells were transduced with three serial dilutions of each filtered viral supernatant or ultracentrifuged viral preparation supplemented with 8 μg/mL Polybrene (hexamethrine bromide, Sigma) and cells from one of the wells were counted to estimate the number of cells at the time of transduction. Medium was replaced the following day and genomic DNA (gDNA) was harvested 4 days post transduction using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). qPCR samples were prepared using Maxima Probe qPCR Master Mix (Thermo Fisher Scientific) according to the manufacturer's protocol, and appropriate primers and probes (see Table S2) and 50–100 ng of the harvested gDNA per reaction. The qPCR and data analysis were performed using the LightCycler 480 instrument and software (Roche). The WPRE copy number per cell was determined by relating the sample values of WPRE and albumin to a plasmid-based standard curve and assuming two copies of albumin per cell. The viral titers were determined based on the three replicates and the number of cells.
For all transduction experiments (except transduction of the melanoma cells with the LV/VMD2-miR451-S1 and LV/VMD2-miR451-12), transductions were performed using 8 μg/mL Polybrene, and the medium was replaced the day after transduction. NT controls were also treated with Polybrene. As all titers were estimated using HEK293 cells, the actual MOI of ARPE19 and melanoma cells may deviate from the calculated MOI.
VEGFA detection by western blot analysis
One-hundred thousand 293-hVEGFA cells were seeded in six-well plates. The next day, they were transduced with the LVs at an MOI of 25. Lysates were harvested 3 days post transduction using RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific) supplemented with cOmplete Mini Protease Inhibitor Cocktail (Roche) according to the manufacturer's protocol, except incubation time on ice was 15 min. Medium was filtered through 0.20-μm filters (Sarstedt). Forty micrograms of total protein of the lysates or 27 μL of medium were loaded on a 4%–15% Mini-PROTEAN TGX gel with one-quarter v/v 4× Laemmli sample buffer (Sigma) with 50 mM DTT after denaturation for 5 min at 95°C and electrophoresed. For the lysates, a stain-free gel was used, which, after electrophoresis, was activated for 45 s using the ChemiDoc MP imaging system (Bio-Rad, Hercules, CA) before transfer to a polyvinylidene fluoride (PVDF) membrane (Bio-Rad) and visualization of total protein using the ChemiDoc MP imaging system (Bio-Rad). The membrane was blocked for 1 h at room temperature (RT) in 5% w/v skim milk powder (VWR, Radnor, PA) in Tris-buffered saline (TBS) buffer (Thermo Fisher Scientific) with 0.1% Tween 20 (Sigma) (TBS-T). The membrane was incubated overnight at 4°C with a Rb-anti-VEGF antibody (ab46154, Abcam, Cambridge, UK) at a concentration of 1:2,000 with constant rocking before washing three times for 5 min in TBS-T and incubation with a horseradish peroxidase (HRP)-conjugated goat-anti-rabbit antibody (Dako, Agilent, Agilent Technologies, Santa Clara, CA) in a concentration of 1:10,000 for 1 h at RT. Detection was done using Clarity Western enhanced chemiluminescence (ECL) substrate (Bio-Rad) and imaging was done using the ChemiDoc MP imaging system. Data analysis was performed using Image Lab software (Bio-Rad). Samples were analyzed in triplicates.
Purification of RNA from cell lines
For the purification of RNA from 293-hVEGFA cells, 50,000 cells were seeded in 12-well plates. The next day, they were transduced in triplicates with the LVs at an MOI of 25. Three days post transduction, RNA was harvested using the RNeasy mini plus kit (Qiagen) according to the manufacturer's protocol and frozen at −80°C. The RNA concentration was determined using the NanoDrop One and cDNA was synthesized from 0.161 μg of RNA in a 10 μL reaction volume using the iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer's protocol.
RT-qPCR analyses
For the detection of knockdown of VEGFA mRNA in 293-hVEGFA cells, the qPCR was prepared using RealQ Plus Master Mix Green (Ampliqon, Odense, Denmark) according to the manufacturer's protocol, using 3 μL of the diluted (1:60) cDNA synthesis reaction as template in a total reaction volume of 10 μL and a primer concentration of 1 μM. Cycling conditions were set according to the manufacturer's protocol, with 10 s of denaturation, 20 s of annealing, and 30 s of elongation for 40 cycles, and an annealing temperature of 57°C. For all samples, a DNA contamination control without reverse transcriptase was included. All reactions, except negative controls, were analyzed in technical triplicates, and a standard curve made using a mix of cDNA samples in 5-fold serial dilutions was included to determine the PCR efficiency, which was 90% for VEGFA and 92% for PPIA, which was used as a reference gene for normalization. The qPCR and data analysis were performed using the LightCycler 480 instrument and software (Roche, release 1.5.1.62) and Excel 365 for Windows. Cq values were determined with the second derivative max method. The specificity was confirmed using melt curve analysis and/or gel electrophoresis. For both genes, the range of Cq values was between 20 and 27.5, while the no-template control was above the detection limit of Cq 35. Replicates that had a difference >0.5 in Cq values compared with the other replicates were excluded from the analysis. In addition to PPIA, HPRT1 and GAPDH were evaluated as reference genes, and PPIA was selected as the most stable reference gene using NormFinder for Excel.52 For each of the genes, concentrations were calculated from the Cq values using the efficiency calculated from the standard curve. Then a relative quantity (RQ) was calculated by relating the concentrations for each sample to the concentration of the control samples. The RQ for VEGFA was then normalized using the RQ for PPIA. The qPCR data analysis was based on the method described by Taylor et al.53 Primers for VEGFA were designed using NCBI's Primer-BLAST with the forward primer spanning an exon-exon junction, producing an amplicon of 165 bp. The PPIA, HPRT1, and GAPDH primers have been previously validated.54
The quantification of the Vegfa and EGFP mRNA and pri-miR451-agsh transcripts in the RPE cells from the whole eye cups was performed similarly. The qPCR was prepared using 3 μL of the diluted (1:30) cDNA synthesis reaction. The annealing temperature was 58°C for all primer sets, except the EGFP primers, where it was 60°C. The PCR efficiency was 106% for Vegfa, 99% for EGFP, 96% for the pri-miR451-agsh transcript, and 88% for Actb, which was used as a reference gene for normalization. The qPCR was performed using the LightCycler 480 II instrument (Roche). The range of Cq values was between 25.5 and 31.5 for Atcb and Vegfa. In addition to Actb, Hprt and Rn18s were evaluated as reference genes, and Actb was selected as the most stable reference gene using NormFinder for Excel.52
The quantification of the Vegfa mRNA in the FACS-sorted murine RPE cells was performed similarly, using the same primers for Vegfa and using Atcb as a reference gene for normalization, but with another primer set (Actb 2, see Table S3). Fifty amplification cycles were used, but Cq values above 35 were not considered. If the difference in the technical triplicates values exceeded 0.5 Cq, the most extreme value was excluded. The annealing temperature for both primer sets was 58°C, and the primer concentration was 0.5 μM. The standard curve from the previous experiment with RPE cells was used, and the efficiency was 98% for Vegfa and 98% for Actb. The qPCR was prepared using 3 μL of the diluted (1:6) cDNA synthesis reaction. The range of Cq values was between 22 and 34.5 for Atcb and Vegfa.
All primer sequences are available in Table S3.
Flow cytometry analysis
LV reporter-based assays evaluating the efficacy of the LVs encoding the Pol II-driven RNAi effectors based on the reduction of E2Crimson expression (Figure S5) were performed as follows: HEK293 cells or melanoma cells were transduced with the LV reporter using an MOI of 20 or 10, respectively, to generate reporter cell lines. The reporter cells were seeded at a density of 100,000 cells/well in a six-well plate or 50,000 cells/well in a 12-well plate for flow cytometry analysis. The next day, 3 days before flow cytometry analysis, the HEK293 or melanoma reporter cells were transduced with the LVs encoding the U1-driven or VMD2-driven RNAi effectors using an MOI of 25 or 35, respectively. On the day of analysis, adherent cells were harvested by trypsinization and centrifugation. The cells were washed twice in phosphate buffered saline (PBS) (BioWest, Nuaillé, France), centrifuged, resuspended in PBS, and kept at 4°C until analysis shortly after harvest. The samples were analyzed using a Novocyte 2100YB flow cytometer and the NovoExpress Software (both ACEA Biosciences, Agilent Technologies).
MTT assay
ARPE19 cells were seeded at a density of 1,000 cells/well in a transparent 96-well plate. The next day, the cells were transduced at an MOI of 30 with the LVs encoding the U6-driven or U1-driven RNAi effectors. Untreated controls (NC) and controls treated with Polybrene only (NT) were included. Transductions were performed in quintuplicates, while controls were in triplicates. Four days post transduction, a standard curve of ARPE19 cells was seeded at a density ranging from 3,125 to 100,000 cells/well and medium was replaced for all wells. Five days post transduction, the MTT assay was performed using the MTT cell proliferation assay kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer's protocol, measuring absorbance at 570 nm using the ELx808 Absorbance Reader (BioTek, Agilent Technologies). The background absorbance measured in wells containing only medium was subtracted from all measurements, and the absorbance relative to the untreated controls was reported. The standard curve was used to ensure that the cell number was within or near the linear range of measurement.
Animals
Animals were handled as previously described.46 Briefly, 8-week-old male C57BL/6J mice were purchased from Janvier Labs (Le Genest-Saint-Isle, France) and kept on a 12 h/12 h light/dark cycle at the Animal Facilities at the Department of Biomedicine, Aarhus University, Denmark with ad libitum access to water and Altromin maintenance feed (Altromin, Brogaarden, Denmark). Prior to subretinal injection or in vivo fundoscopy, mice were anesthetized with an intraperitoneal injection of ketamine and medetomidine hydrochloride (Ketador 60–100 mg/kg [Richter Pharma AG, Wels, Austria] and Cepetor/Domitor 0.8–1.2 mg/kg [ScanVet Animal Health A/S, Fredensborg, Denmark]), and the pupils were dilated with a drop of 1% tropicamide solution (Mydriacyl, Alcon Nordic A/S, Copenhagen, Denmark). Eyes were kept lubricated with a carbomer eye gel (Viscotears Alcon Nordic). After the procedures, mice were brought out of anesthesia with atipamezole 0.5–1 mg/kg (Antisedan, Orion Pharma, Copenhagen, Denmark). Mice were treated with a subcutaneous injection of carprofen (Rimadyl Bovis Vet, Zoetis Finland Oy, Helsinki, Finland) (5 mg/kg) just before or immediately after the subretinal injection and 3 days after via their drinking water (3.33 mg/100 mL) for pain relief. Animals were handled in accordance with the Statement for the Use of Animals in Ophthalmic and Vision Research from the Association for Research in Vision and Ophthalmology (ARVO). All animal experiments were performed under the approval of The Danish Animal Inspectorate (case# 2020-15-0201–00745).
Subretinal injections
Mice were injected unilaterally with 4.13 × 105 IU (or 4.54 × 105 IU of the LV/VMD2-miR451-S1 for the short-term experiment with FACS) LV in a total volume of 2 μL as described previously.46 For the study of miR451-12 expression in vivo with miRNAScope, eight mice received the LV/VMD2-miR451-S1 or LV/VMD2-miR451-12 (four mice in each group). For the study of Vegfa expression in RPE cells from the entire eye cup, pri-miR451-agshRNA transcript expression, and EGFP expression, 20 mice received the LV/VMD2-miR451-S1 or LV/VMD2-miR451-12 (10 mice in each group). For the study of Vegfa knockdown in FACS-sorted RPE cells, 24 mice received the LV/VMD2-miR451-S1 or LV/VMD2-miR451-12 (12 mice in each group). Fundoscopic examinations were carried out with the Micron IV imaging system (Phoenix Research Laboratories, Pleasanton, CA) to detect the EGFP expression. In each experiment, a fundoscopic examination was performed immediately before the sacrifice of the animals (day 57 for miRNAScope, day 56 for RNA purification from RPE cells in the whole eye cup, or day 14 for RNA purification from FACS-sorted RPE cells).
Isolation of primary mouse RPE cells for FACS
On day 14 p.i., mice were sacrificed by cervical dislocation, the eyes were enucleated, and the RPE cells were purified as follows: the eyes were placed in ice-cold HBSS, periocular tissue was removed, and subsequently the cornea and lens were removed by a circumferential incision. The eyecups were placed in hyaluronidase solution (1 mg/mL hyaluronidase [Sigma] in HBSS) at 37°C for 45 min. Then the eyecups were incubated in HBSS for 30 min on ice. The neural retina was removed, and the remaining eyecup was placed in a 0.25% trypsin/EDTA in HBSS solution (Sigma) at 37°C for 45 min. The eyecups were transferred to FCS (Sigma) and the RPE cells were detached by shaking the eyecup in a 20% FCS in HBSS solution. The solution with the RPE cells, the trypsin/EDTA solution, and the FCS were transferred to a 15-mL Falcon tube. RPE cells from three injected eyes or two uninjected eyes were pooled in the same tube. The pools were centrifuged at 2,300 rpm for 2 min at RT and the supernatant was discarded. The pellet was resuspended in a 0.05% trypsin/EDTA solution and incubated in a water bath at 37°C for 1 min. Four milliliters of FACS buffer (1% BSA [VWR], 2.5 mM EDTA [Sigma], 25 mM HEPES [Alfa Aesar, Haverhill, MA] in HBSS) was added to the RPE cell solution, which was transferred to a 100-μm cell strainer (Falcon, Thermo Fisher Scientific). The strainer was washed three times with 1 mL of FACS buffer, and the RPE cell solution was centrifuged for 2 min at 2,300 rpm. The RPE cell pellet was resuspended in FACS buffer, and the solution was kept at 4°C until analysis. The contralateral eyes from six mice (four from the LV/VMD2-miR451-S1 group and two from the LV/VMD2-miR451-12 group) were used as uninjected controls.
FACS of primary murine RPE cells
The purified RPE cells were sorted on a three-laser (375 nm, 488 nm, 633 nm) BD FACSAria III (BD Biosciences, Franklin Lakes, NJ). The cell population was identified based on forward scatter (FSC) and side scatter (SSC), and FSC and SSC doublets were excluded. The EGFP+ cells were identified based on fluorescence measured in the EGFP detector (530/30 nm) and in the neighboring PE detector (585/42 nm). An equal level of fluorescence in both the EGFP and PE detectors was interpreted as autofluorescence, and these cells were excluded. The gating strategy is shown in Figure S7.
Purification of RNA from murine RPE cells
For the purification of RNA from RPE cells from the whole eye cup: on day 56 p.i., the mice were sacrificed by cervical dislocation, the eyes were enucleated, and the RNA was extracted from the RPE cells as previously described.46 The contralateral eyes from five mice from each group were used as uninjected controls. The RNA was eluted in a volume of 10 μL and kept at −20°C for no more than 1 day before cDNA synthesis. For the cDNA synthesis, 2.5 μL of the eluate was used in a 10-μL reaction using the iScript cDNA synthesis kit (Bio-Rad) according to the manufacturer's protocol.
For the purification of RNA from FACS sorted RPE cell pools were centrifuged for 3 × 5 min at 300, 500, and 1,000 × g, the supernatant was removed, and the cell pellet was frozen at −80°C until RNA purification. RNA was purified using the RNeasy Micro Kit (Qiagen) according to the manufacturer's protocol, except the gDNA eliminator column was not used, and DNAse treatment was not performed on the column. The RNA was eluted in a volume of 14 μL and treated with DNAfree (Thermo Fisher Scientific) according to the manufacturer's protocol, before cDNA was synthesized with 5 μL of the DNAse-treated RNA using the iScript cDNA synthesis kit (Bio-Rad) as described above. The RT-qPCR analyses are described above.
Detection of miR451-12 in RPE cells with chromogenic in situ hybridization
The cellular localization of miR451-12 was investigated by chromogenic in situ hybridization (CISH) using miRNAscope high-definition (HD) RED procedure (Advanced Cell Diagnostics [ACD], Hayward, CA). The protocol provided by the manufacturer and the recommended conditions were followed. Mice were sacrificed 57 days p.i. and the eyes were enucleated and immediately transferred to 4% formalin solution for fixation. Paraffin embedding and sectioning was performed at the Department of Pathology, Aarhus University Hospital. In brief, for the miRNAscope analysis, 5-μm thick paraffin sections from formalin-fixed and paraffin-embedded blocks of LV/VMD2-miR451-12 and LV/VMD2-miR451-S1-transduced mouse retinas were pretreated, post-fixated, and hybridized with a custom-designed miRNAscope probe targeting the predicted guide strand of the miR451-12 (5′-AGUAGGAAGCUCAUCUCUCC-3′). The miR451-12-binding probe was detected using six amplification steps combined with red-signal detection and counterstaining with Gill's Hematoxylin I solution. Data were collected using an Olympus VS120 slide scanner and analyzed using cellSens Entry 2.3, QuPath software version 0.2.3, and ImageJ software version 1.53c for Windows.
Statistical analysis
GraphPad Prism version 9.2.0 was used for statistical analysis of the competition, MTT, and RT-qPCR assays. For the competition and MTT assays, a one-way ANOVA was performed followed by Šídák's multiple comparisons test between each of the agshRNAs and their corresponding shRNAs. The multiplicity-adjusted p values were reported. For RT-qPCR assay, the data were log transformed before a one-way ANOVA was performed followed by the uncorrected Fisher's least significant difference (LSD) test between the LV/VMD2-miR451-S1 EGFP+ group and the LV/VMD2-miR451-12 EGFP+ group.
RNA-seq and analyses
One-hundred and twenty-five thousand ARPE19 cells were seeded in six-well plates 1 day before transduction with the LVs at an MOI of 20. The cells were harvested 3 days post transduction. The cells were lysed using the QIAzol lysis reagent (Qiagen) and RNA was extracted using phenol-chloroform extraction and propan-2-ol/ethanol precipitation. Four replicates were included for each LV. The RNA purity, integrity, and concentration were analyzed on the Agilent 2100 Bioanalyzer instrument (Agilent Technologies), and only RNA with an RNA integrity number (RIN) value of 10 was used for sample preparation.
Libraries were prepared using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina with the NEBNext Poly(A) mRNA Magnetic Isolation Module (New England Biolabs, Ipswich, MA) according to the manufacturer's protocol, substituting SPRIselect beads for AMPure XP beads (Beckman Coulter, Brea, CA). The libraries were then analyzed on the Agilent 2100 Bioanalyzer instrument, and with the KAPA Library Quantification Kit for Illumina Platforms (Roche).
Following sequencing, reads were trimmed using bbduk.sh55 and mapped to the human hg38 genome using STAR56 version 2.6.1a and to the human transcriptome using salmon57 version 0.14.1, in both cases using Ensembl version 93 gene annotations.58 Gene counts were obtained from transcript counts using tximport59 and differential expression determined using DESeq260 and apeglm.61 To identify gene clusters, we employed SOMs using the kohonen package62,63 and analyzed biological pathways using clusterProfiler64 followed by enrichplot65 for plotting. Briefly, we first identified significantly differentially expressed genes with respect to treatment with the two RNAi effector sequences (12 or 13), mode of delivery (agsh or sh), and the interaction between these. We then identified the 3,000 most variably expressed genes across all samples and used these in our clustering analysis.
Acknowledgments
The authors would like to thank Tina Hindkjaer and Kamilla Zahll Hornbek for their excellent technical support. Flow cytometry analysis and FACS were performed at the FACS Core Facility, Aarhus University, Denmark, and the authors would like to thank staff at the core facility for their support. Paraffin embedding and sectioning were performed at the Department of Pathology, Aarhus University Hospital. This work was supported by the Faculty of Health Sciences, Aarhus University (PhD scholarship to S.A.), the Danish Eye Research Foundation (T.J.C.), Aase and Ejnar Danielsen’s Foundation (T.J.C.), Bagenkop-Nielsens Myopifond, Svend Helge Schrøder og hustru Ketty Lydia Larsen Schrøders fond (T.J.C.), Maskinfabrikant Jochum Jensen og hustru Mette Marie Jensen F. Poulsens Mindelegat (S.A.), and The Velux Foundation (T.J.C.).
Author contributions
Writing – original draft, S.A., A.L.A., L.A., and T.J.C.; writing – reviewing & editing, S.A., A.L.A., E.G.J., T.K.D., U.A., L.S., B.S.A, L.A., and T.J.C.; supervision, T.J.C., L.A., L.S., B.S.A., and A.L.A.; investigation, S.A., T.K.D., E.G.J., U.A., and A.L.A.; methodology, S.A., T.K.D., L.A., T.J.C., A.L.A., L.S., and B.S.A.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.02.019.
Contributor Information
Lars Aagaard, Email: aagaard@biomed.au.dk.
Thomas J. Corydon, Email: corydon@biomed.au.dk.
Supplemental information
References
- 1.Fire A., Xu S., Montgomery M.K., Kostas S.A., Driver S.E., Mello C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 2.Elbashir S.M., Harborth J., Lendeckel W., Yalcin A., Weber K., Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 3.Bajan S., Hutvagner G. RNA-based therapeutics: from antisense oligonucleotides to miRNAs. Cells. 2020;9 doi: 10.3390/cells9010137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Y.P., Schopman N.C., Berkhout B. Dicer-independent processing of short hairpin RNAs. Nucleic Acids Res. 2013;41:3723–3733. doi: 10.1093/nar/gkt036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herrera-Carrillo E., Berkhout B. Dicer-independent processing of small RNA duplexes: mechanistic insights and applications. Nucleic Acids Res. 2017;45:10369–10379. doi: 10.1093/nar/gkx779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shang R., Zhang F., Xu B., Xi H., Zhang X., Wang W., Wu L. Ribozyme-enhanced single-stranded Ago2-processed interfering RNA triggers efficient gene silencing with fewer off-target effects. Nat. Commun. 2015;6:8430. doi: 10.1038/ncomms9430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grimm D., Streetz K.L., Jopling C.L., Storm T.A., Pandey K., Davis C.R., Marion P., Salazar F., Kay M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 8.Beer S., Bellovin D.I., Lee J.S., Komatsubara K., Wang L.S., Koh H., Borner K., Storm T.A., Davis C.R., Kay M.A., et al. Low-level shRNA cytotoxicity can contribute to MYC-induced hepatocellular carcinoma in adult mice. Mol. Ther. 2010;18:161–170. doi: 10.1038/mt.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McBride J.L., Boudreau R.L., Harper S.Q., Staber P.D., Monteys A.M., Martins I., Gilmore B.L., Burstein H., Peluso R.W., Polisky B., et al. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. U S A. 2008;105:5868–5873. doi: 10.1073/pnas.0801775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martin J.N., Wolken N., Brown T., Dauer W.T., Ehrlich M.E., Gonzalez-Alegre P. Lethal toxicity caused by expression of shRNA in the mouse striatum: implications for therapeutic design. Gene Ther. 2011;18:666–673. doi: 10.1038/gt.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harwig A., Kruize Z., Yang Z., Restle T., Berkhout B. Analysis of AgoshRNA maturation and loading into Ago2. PLoS One. 2017;12:e0183269. doi: 10.1371/journal.pone.0183269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dueck A., Ziegler C., Eichner A., Berezikov E., Meister G. microRNAs associated with the different human Argonaute proteins. Nucleic Acids Res. 2012;40:9850–9862. doi: 10.1093/nar/gks705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang J.S., Maurin T., Lai E.C. Functional parameters of Dicer-independent microRNA biogenesis. RNA. 2012;18:945–957. doi: 10.1261/rna.032938.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ma H., Zhang J., Wu H. Designing Ago2-specific siRNA/shRNA to avoid competition with endogenous miRNAs. Mol. Ther. Nucleic Acids. 2014;3:e176. doi: 10.1038/mtna.2014.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kenworthy R., Lambert D., Yang F., Wang N., Chen Z., Zhu H., Zhu F., Liu C., Li K., Tang H. Short-hairpin RNAs delivered by lentiviral vector transduction trigger RIG-I-mediated IFN activation. Nucleic Acids Res. 2009;37:6587–6599. doi: 10.1093/nar/gkp714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bridge A.J., Pebernard S., Ducraux A., Nicoulaz A.L., Iggo R. Induction of an interferon response by RNAi vectors in mammalian cells. Nat. Genet. 2003;34:263–264. doi: 10.1038/ng1173. [DOI] [PubMed] [Google Scholar]
- 17.Sun G., Yeh S.Y., Yuan C.W., Chiu M.J., Yung B.S., Yen Y. Molecular properties, functional mechanisms, and applications of sliced siRNA. Mol. Ther. Nucleic Acids. 2015;4:e221. doi: 10.1038/mtna.2014.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siolas D., Lerner C., Burchard J., Ge W., Linsley P.S., Paddison P.J., Hannon G.J., Cleary M.A. Synthetic shRNAs as potent RNAi triggers. Nat. Biotechnol. 2005;23:227–231. doi: 10.1038/nbt1052. [DOI] [PubMed] [Google Scholar]
- 19.Ge Q., Ilves H., Dallas A., Kumar P., Shorenstein J., Kazakov S.A., Johnston B.H. Minimal-length short hairpin RNAs: the relationship of structure and RNAi activity. RNA. 2010;16:106–117. doi: 10.1261/rna.1894510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y.P., Karg M., Harwig A., Herrera-Carrillo E., Jongejan A., van Kampen A., Berkhout B. Mechanistic insights on the dicer-independent AGO2-mediated processing of AgoshRNAs. RNA Biol. 2015;12:92–100. doi: 10.1080/15476286.2015.1017204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brummelkamp T.R., Bernards R., Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 22.Paddison P.J., Caudy A.A., Bernstein E., Hannon G.J., Conklin D.S. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002;16:948–958. doi: 10.1101/gad.981002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaadt E., Alsing S., Cecchi C.R., Damgaard C.K., Corydon T.J., Aagaard L. Efficient knockdown and Lack of passenger strand activity by dicer-independent shRNAs expressed from Pol II-driven MicroRNA scaffolds. Mol. Ther. Nucleic Acids. 2019;14:318–328. doi: 10.1016/j.omtn.2018.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park J.E., Heo I., Tian Y., Simanshu D.K., Chang H., Jee D., Patel D.J., Kim V.N. Dicer recognizes the 5' end of RNA for efficient and accurate processing. Nature. 2011;475:201–205. doi: 10.1038/nature10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu S., Jin L., Zhang Y., Huang Y., Zhang F., Valdmanis P.N., Kay M.A. The loop position of shRNAs and pre-miRNAs is critical for the accuracy of dicer processing in vivo. Cell. 2012;151:900–911. doi: 10.1016/j.cell.2012.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aagaard L.A., Zhang J., von Eije K.J., Li H., Saetrom P., Amarzguioui M., Rossi J.J. Engineering and optimization of the miR-106b cluster for ectopic expression of multiplexed anti-HIV RNAs. Gene Ther. 2008;15:1536–1549. doi: 10.1038/gt.2008.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hollensen A.K., Bak R.O., Haslund D., Mikkelsen J.G. Suppression of microRNAs by dual-targeting and clustered Tough Decoy inhibitors. RNA Biol. 2013;10:406–414. doi: 10.4161/rna.23543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim Y.-K., Kim B., Kim V.N. Re-evaluation of the roles of DROSHA, Exportin 5, and DICER in microRNA biogenesis. Proc. Natl. Acad. Sci. U S A. 2016;113:E1881–E1889. doi: 10.1073/pnas.1602532113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dull T., Zufferey R., Kelly M., Mandel R.J., Nguyen M., Trono D., Naldini L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li M.J., Bauer G., Michienzi A., Yee J.K., Lee N.S., Kim J., Li S., Castanotto D., Zaia J., Rossi J.J. Inhibition of HIV-1 infection by lentiviral vectors expressing Pol III-promoted anti-HIV RNAs. Mol. Ther. 2003;8:196–206. doi: 10.1016/s1525-0016(03)00165-5. [DOI] [PubMed] [Google Scholar]
- 31.Holmgaard A.B., Askou A.L., Jensen E.G., Alsing S., Bak R.O., Mikkelsen J.G., Corydon T.J. Targeted knockout of the Vegfa gene in the retina by subretinal injection of RNP complexes containing Cas9 protein and modified sgRNAs. Mol. Ther. 2021;29:191–207. doi: 10.1016/j.ymthe.2020.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chung J., Zhang J., Li H., Ouellet D.L., DiGiusto D.L., Rossi J.J. Endogenous MCM7 microRNA cluster as a novel platform to multiplex small interfering and nucleolar RNAs for combinational HIV-1 gene therapy. Hum. Gene Ther. 2012;23:1200–1208. doi: 10.1089/hum.2012.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Askou A.L., Benckendorff J.N.E., Holmgaard A., Storm T., Aagaard L., Bek T., Mikkelsen J.G., Corydon T.J. Suppression of choroidal neovascularization in mice by subretinal delivery of multigenic lentiviral vectors encoding anti-angiogenic MicroRNAs. Hum. Gene Ther. Methods. 2017;28:222–233. doi: 10.1089/hgtb.2017.079. [DOI] [PubMed] [Google Scholar]
- 34.Askou A.L., Aagaard L., Kostic C., Arsenijevic Y., Hollensen A.K., Bek T., Jensen T.G., Mikkelsen J.G., Corydon T.J. Multigenic lentiviral vectors for combined and tissue-specific expression of miRNA- and protein-based antiangiogenic factors. Mol. Ther. Methods Clin. Dev. 2015;2:14064. doi: 10.1038/mtm.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Balaggan K.S., Ali R.R. Ocular gene delivery using lentiviral vectors. Gene Ther. 2012;19:145–153. doi: 10.1038/gt.2011.153. [DOI] [PubMed] [Google Scholar]
- 36.Herrera-Carrillo E., Harwig A., Liu Y.P., Berkhout B. Probing the shRNA characteristics that hinder Dicer recognition and consequently allow Ago-mediated processing and AgoshRNA activity. RNA. 2014;20:1410–1418. doi: 10.1261/rna.043950.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun G., Wang J., Huang Y., Yuan C.W., Zhang K., Hu S., Chen L., Lin R.J., Yen Y., Riggs A.D. Differences in silencing of mismatched targets by sliced versus diced siRNAs. Nucleic Acids Res. 2018;46:6806–6822. doi: 10.1093/nar/gky287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herrera-Carrillo E., Gao Z.L., Harwig A., Heemskerk M.T., Berkhout B. The influence of the 5΄-terminal nucleotide on AgoshRNA activity and biogenesis: importance of the polymerase III transcription initiation site. Nucleic Acids Res. 2017;45:4036–4050. doi: 10.1093/nar/gkw1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Herrera-Carrillo E., Harwig A., Berkhout B. Influence of the loop size and nucleotide composition on AgoshRNA biogenesis and activity. RNA Biol. 2017;14:1559–1569. doi: 10.1080/15476286.2017.1328349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herrera-Carrillo E., Harwig A., Berkhout B. Toward optimization of AgoshRNA molecules that use a non-canonical RNAi pathway: variations in the top and bottom base pairs. RNA Biol. 2015;12:447–456. doi: 10.1080/15476286.2015.1022024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Birmingham A., Anderson E.M., Reynolds A., Ilsley-Tyree D., Leake D., Fedorov Y., Baskerville S., Maksimova E., Robinson K., Karpilow J., et al. 3' UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat. Methods. 2006;3:199–204. doi: 10.1038/nmeth854. [DOI] [PubMed] [Google Scholar]
- 42.Wu L., Fan J., Belasco J.G. Importance of translation and nonnucleolytic ago proteins for on-target RNA interference. Curr. Biol. 2008;18:1327–1332. doi: 10.1016/j.cub.2008.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boudreau R.L., Spengler R.M., Hylock R.H., Kusenda B.J., Davis H.A., Eichmann D.A., Davidson B.L. siSPOTR: a tool for designing highly specific and potent siRNAs for human and mouse. Nucleic Acids Res. 2013;41:e9. doi: 10.1093/nar/gks797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yi R., Doehle B.P., Qin Y., Macara I.G., Cullen B.R. Overexpression of exportin 5 enhances RNA interference mediated by short hairpin RNAs and microRNAs. RNA. 2005;11:220–226. doi: 10.1261/rna.7233305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Castanotto D., Sakurai K., Lingeman R., Li H., Shively L., Aagaard L., Soifer H., Gatignol A., Riggs A., Rossi J.J. Combinatorial delivery of small interfering RNAs reduces RNAi efficacy by selective incorporation into RISC. Nucleic Acids Res. 2007;35:5154–5164. doi: 10.1093/nar/gkm543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Askou A.L., Alsing S., Benckendorff J.N.E., Holmgaard A., Mikkelsen J.G., Aagaard L., Bek T., Corydon T.J. Suppression of choroidal neovascularization by AAV-based dual-acting antiangiogenic gene therapy. Mol. Ther. Nucleic Acids. 2019;16:38–50. doi: 10.1016/j.omtn.2019.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Orlans H.O., McClements M.E., Barnard A.R., Martinez-Fernandez de la Camara C., MacLaren R.E. Mirtron-mediated RNA knockdown/replacement therapy for the treatment of dominant retinitis pigmentosa. Nat. Commun. 2021;12:4934. doi: 10.1038/s41467-021-25204-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aagaard L., Amarzguioui M., Sun G., Santos L.C., Ehsani A., Prydz H., Rossi J.J. A facile lentiviral vector system for expression of doxycycline-inducible shRNAs: knockdown of the pre-miRNA processing enzyme Drosha. Mol. Ther. 2007;15:938–945. doi: 10.1038/sj.mt.6300118. [DOI] [PubMed] [Google Scholar]
- 49.Aagaard L., Bjerregaard B., Kjeldbjerg A.L., Pedersen F.S., Larsson L.I., Rossi J.J. Silencing of endogenous envelope genes in human choriocarcinoma cells shows that envPb1 is involved in heterotypic cell fusions. J. Gen. Virol. 2012;93:1696–1699. doi: 10.1099/vir.0.041764-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jakobsen M., Stenderup K., Rosada C., Moldt B., Kamp S., Dam T.N., Jensen T.G., Mikkelsen J.G. Amelioration of psoriasis by anti-TNF-alpha RNAi in the xenograft transplantation model. Mol. Ther. 2009;17:1743–1753. doi: 10.1038/mt.2009.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Corydon T.J., Haagerup A., Jensen T.G., Binderup H.G., Petersen M.S., Kaltoft K., Vestbo J., Kruse T.A., Borglum A.D. A functional CD86 polymorphism associated with asthma and related allergic disorders. J. Med. Genet. 2007;44:509–515. doi: 10.1136/jmg.2007.049536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Andersen C.L., Jensen J.L., Ørntoft T.F. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64:5245–5250. doi: 10.1158/0008-5472.Can-04-0496. [DOI] [PubMed] [Google Scholar]
- 53.Taylor S.C., Nadeau K., Abbasi M., Lachance C., Nguyen M., Fenrich J. The ultimate qPCR experiment: producing publication quality, reproducible data the first time. Trends Biotechnol. 2019;37:761–774. doi: 10.1016/j.tibtech.2018.12.002. [DOI] [PubMed] [Google Scholar]
- 54.Liu X., Xie J., Liu Z., Gong Q., Tian R., Su G. Identification and validation of reference genes for quantitative RT-PCR analysis of retinal pigment epithelium cells under hypoxia and/or hyperglycemia. Gene. 2016;580:41–46. doi: 10.1016/j.gene.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 55.Bushnell, B. BBMap. 2014 Sourceforge.net/projects/bbmap.
- 56.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Giron C.G., et al. Ensembl 2018. Nucleic Acids Res. 2018;46:D754–D761. doi: 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Soneson C., Love M.I., Robinson M.D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 2015;4:1521. doi: 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhu A., Ibrahim J.G., Love M.I. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics. 2019;35:2084–2092. doi: 10.1093/bioinformatics/bty895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wehrens R., Buydens L.M.C. Self- and super-organizing maps in R: the kohonen package. J. Stat. Stofw. 2007;21:1–19. doi: 10.18637/jss.v021.i05. [DOI] [Google Scholar]
- 63.Wehrens R., Kruisselbrink J. Flexible self-organizing maps in kohonen 3.0. J. Stat. Stofw. 2018;87:1–18. doi: 10.18637/jss.v087.i07. [DOI] [Google Scholar]
- 64.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu G. 2021. Enrichplot: Visualization of Functional Enrichment Result. R package version 1.12.1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.