

Graphical abstract
Keywords: Argonaute, microRNA, Molecular dynamics simulations, Gene regulation
Abstract
Argonaute (AGO) proteins, the core of RNA-induced silencing complex, are guided by microRNAs (miRNAs) to recognize target RNA for repression. The miRNA–target RNA recognition forms initially through pairing at the seed region while the additional supplementary pairing can enhance target recognition and compensate for seed mismatch. The extension of miRNA lengths can strengthen the target affinity when pairing both in the seed and supplementary regions. However, the mechanism underlying the effect of the supplementary pairing on the conformational dynamics and the assembly of AGO–RNA complex remains poorly understood. To address this, we performed large-scale molecular dynamics simulations of AGO–RNA complexes with different pairing patterns and miRNA lengths. The results reveal that the additional supplementary pairing can not only strengthen the interaction between miRNA and target RNA, but also induce the increased plasticity of the PAZ domain and enhance the domain connectivity among the PAZ, PIWI, N domains of the AGO protein. The strong community network between these domains tightens the mouth of the supplementary chamber of AGO protein, which prevents the escape of target RNA from the complex and shields it from solvent water attack. Importantly, the inner stronger matching pairs between the miRNA and target RNA can compensate for weaker mismatches at the edge of supplementary region. These findings provide guidance for the design of miRNA mimics and anti-miRNAs for both clinical and experimental use and open the way for further engineering of AGO proteins as a new tool in the field of gene regulation.
1. Introduction
Argonaute (AGO) proteins play a critical role in all small-RNA-guided RNA silencing, pivotal in the regulation of gene expression and essential for cell proliferation, differentiation, and homeostasis [1], [2], [3]. MicroRNAs (miRNA), which are short non-coding RNAs of ∼ 22 nucleotides, function by guiding AGO proteins to interact with complementary target RNAs, yielding the RNA-induced silencing complex (RISC) [4], [5]. The formation of RISC can promote the translation repression of messenger RNA [6], [7] or the degradation of exogenous pathogen RNA [8]. In addition to the key role in cellular process, microRNA has also been widely used as an experimental tool [9], [10] and recent advances have gradually made miRNA therapeutics for clinical utility [11], [12], [13], [14].
There are four kinds of AGO proteins in human cells: AGO1, AGO2, AGO3, and AGO4. Among them, AGO2 is known as the only slicer [15]. AGO2 is a bi-lobed protein with a central cleft for binding of guide and target RNAs and is composed of four globular domains (N, PAZ, MID and PIWI) and two connective linker domains (L1 and L2) [16] (Fig. 1A and B). The 5′ and 3′ ends of miRNA are anchored at the MID and PAZ domain, respectively [17]. In the crystal structure of miRNA and target RNA bound to the AGO2 protein, the guide miRNA can be divided into five functional domains, including anchor (g1, from the miRNA 5′ end), seed (g2–g7 or g2–g8), central (g8–g13 or g9–g13), 3′ supplementary (g13–g17), and tail (g18–the end) [18] (Fig. 1C–E).
Fig. 1.

Overview of AGO2–RNA complex. (A) Domain organization of AGO2. (B) Surface representation of AGO2-guide (red)-target (black) complex, with AGO2 color coded as in (A). Nucleotide sequences of guide RNA (red) and target RNA (black) in the T1 systems (C), T2 systems (D) and T3 systems (E). The additional 22th and 23th nucleotides (pink) are indicated with arrows. Anchor (green), seed (cyan), central (deep blue), supplementary (gold) and tail (red) regions of guide RNA are shown in (D).
In miRNA targeting, initial pairing is formed in the seed region [19]. Seed region pairing alone is sufficient for target recognition [20] and ∼ 80% of miRNA interactions rely on seed complementarity [21]. Seed region is also the most evolutionarily conserved domain in animals [22]. Owing to the evolutionary conservation of seed region, miRNA family members share identical seed sequences. However, miRNAs in the same family have different targets derived from the determinant role of 3′ end pairing in target specificity [23], [24].
The 3′ end of miRNA can be divided into 3′ supplementary and tail [18] (Fig. 1D), and the supplementary pairing beyond seed region could efficiently regulate target mRNA levels [25]. The additional supplementary interaction can enhance target recognition or compensate for seed mismatch [26], [27]. Previous studies showed that the additional supplementary pairing could enhance target RNA binding affinities with varied abilities dependent on different miRNAs: ∼2-fold for let-7 [18], ∼7-fold for miR-21 [28] and >20-fold for miR-122 [29]. Biochemical experiments and mathematical model further elucidated that AGO proteins were highly occupied by seed-plus-supplementary-pairing targets even with the existence of overnumbered seed-pairing-only targets, revealing the differential targeting driven by supplementary pairing [27]. For the biochemical properties of supplementary pairing, catalytic kinetic analysis showed that two simultaneous mismatches (g13 and g17) introduced into pairing slightly weaken the target cleavage efficiency [20]. Competition assays also confirmed that g15 and g16 mismatches did not impair binding affinity of target RNA in mouse AGO2–RISC [18]. Consistent with the experimental results, target predicting algorithms implied that 3′ pairing effect was relatively insensitive to edge mismatches while sensitive to continuous Watson-Crick pairing [23]. In addition to the supplementary region, the miRNA’s 3′ tail region can pair with the target RNAs. This pairing paradigm in the tail region based on the seed and supplementary pairings could lead to the 3′ end destabilization of miRNAs because of the release of 3′ end from the PAZ domain [30], [31]. When in vivo instability of miRNAs, the guide miRNA may decay instead of target repression; this biological phenomenon is referred to as target-directed miRNA degradation (TDMD) [32], [33], [34].
MiRNA can exhibit heterogeneous 3′ ends due to the post-transcriptional 3′ addition of nucleotides which may stabilize miRNA or strengthen guide-target interaction [35], [36]. Recent biochemical data showed that for the seed-plus-supplementary pairing, the increased lengths of miRNA from 21mer to 23mer could enhance target affinity [29]. In contrast to seed-plus-supplementary pairing, only-seed pairing had no appreciable difference on target RNA affinities with extended lengths of miRNA. Based on the crystal structure of seed-plus-supplementary pairing AGO2 complex, the structural model with the supplementary pairing shows that the PAZ domain and the L2 loop shift away from the central cleft, thereby providing sufficient space to form a supplementary chamber for guide-target pairing [29]. The movement of the PAZ domain may create tension in the miRNA 3′ tail, and the increased target RNA affinity may be ascribed to the relieved tension at the miRNA 3′ tail, which facilitates the opening of supplementary chamber.
However, the underlying mechanism how the structure of the supplementary chamber and the AGO–target interactions influenced by the miRNA extensions remains unclear. Furthermore, the structural model of the supplementary pairing is unable to elucidate the mechanism how the mismatches at the edge of supplementary pairing had a minor effect on target binding and the 3′ pairing was relatively insensitive to the predicted thermodynamic stability. Although the crystal structure of supplementary pairing in the AGO complex has been solved, it is challenging to provide enough information for elucidating the mechanism of supplementary pairing with only one averaged snapshot because of the limited conformational changes. Molecular dynamics (MD) simulations that explore protein and nucleic acid conformational dynamics at the atomic level are critical for understanding of target repression and TDMD process [37], [38], [39], [40], [41], [42], [43]. In the recent work, we have used MD simulations to directly uncover biomolecular mechanisms and protein–ligand/protein or protein–DNA/RNA recognitions [44], [45], [46], [47], [48], [49], [50], [51], [52], [53], [54], [55], [56], [57]. Previous investigations on AGO utilizing large-scale MD simulations have successfully illuminated detailed mechanisms behind AGO-RNA interaction [58], [59], [60] and have been testified by experiments [61]. Thus, this method can further help to improve miRNA target site prediction algorithm which can improve the identification of genes directly regulated by miRNAs [62], [63] and to guide rational engineering of AGO complex for therapeutical implications.
Here, we performed a multiple, microsecond-scale length explicit MD simulations to investigate the effect of different miRNA lengths and miRNA-target pairing patterns on the target RNA binding affinities. Nine systems were built with different miRNA lengths and miRNA-target pairing patterns. The simulations revealed that with the extension of the miRNA lengths, the supplementary chamber opened a smaller range and fastened the mouth of the central cleft to prevent the escape of target RNA from the complex and protect it from the attacking from solvent. Our study further showed that the inner stronger matching pairs between the miRNA and target RNA can compensate for weaker mismatches at the edge of supplementary region and result in almost the same pairing effect between the guide and target RNAs. This study aims to shed light on the mechanistic understanding of the supplementary pairing paradigms, inspiring further research on target prediction algorithm and miRNA design for experimental and therapeutic use.
2. Materials and methods
2.1. Structure preparation for simulations
A total of nine systems were performed: three for seed-pairing (g2–g8)-only (T1 systems), three for seed-plus-supplementary (g13–g16)-pairing (T2 systems), and three for seed-plus-supplementary (g15–g19)-pairing (T3 systems). For systems with the same pairing pattern, miRNA length was 21mer, 22mer, and 23mer, respectively. The RNA sequences of the nine systems were shown in Fig. 1C–E.
2.1.1. Preparation of initial structures for modelling
The crystal structure of seed (g2–g8)-plus-supplementary (g13–g16)-pairing AGO2 complex (PDB ID 6N4O) [29] was selected as the initial structure for further modelling. The missing residues and atoms of AGO protein were added using the MODELLER program except the first 21 residues at the N-terminus. The missing g10 nucleotide of the guide RNA was filled up using the counterpart of the guide RNA based on the seed-pairing-only structure of AGO2 complex (PDB ID 4Z4D) [64] because of the similar central region of guide RNA in the two structures. The missing g19 was added through growing a nucleotide from g18 and ligating it with g20 using the Discovery Studio 2019. The missing t8 nucleotide in the target RNA was added by the counterpart of the target RNA from the TDMD structure of AGO2 complex (PDB ID 6NIT) [30]. After that, a nucleotide was prepended from t8 to t7 and t6 was added by lengthening from t5 using the Discovery Studio 2019. To remove the incompatibility of newly added nucleotides, 10,000 steps of minimization of nucleotides were performed using the steepest descent algorithm with the protein fixed. Then, a second-round minimization was performed without any restraints.
2.1.2. Construction of the seed-pairing-only systems
Three seed-pairing-only systems, including 21mer-T1, 22mer-T1, and 23mer-T1, were constructed. Based on the initial structure, mutations of nucleotides to form seed-pairing-only nucleic sequences and extensions of guide RNA from 21mer to 23mer were done using the Discovery Studio 2019. 10,000 steps of minimization were then performed using the steepest descent algorithm to optimize complex structures.
2.1.3. Construction of the seed-plus-supplementary (g13–g17)-pairing systems
Three seed-plus-supplementary (g13–g17)-pairing systems, including the 21mer-T2, 22mer-T2, and 23mer-T2, were constructed. First, to model the 21mer-T2 system, nucleotide mutation, nucleotide extension, and energy minimization were performed based on the initial structure. For the additional pairing of g17–t6 compared, a 20 ns MD simulation was performed to drive the guide and target pairing at the anticipative position. The prepared complex was solvated to a water box and Na + and Cl− were added to simulate normal saline environment. The system was then minimized and heated. In the production simulation, a pulling force was added of 3 kcal/mol/ Å2 to promote the pairing of g17 to t6 by restraining the distance between the N1 atom of g17 and the N3 atom of t6 and the distance between the N6 atom of g17 and the O4 atom of t6 within 3.5 Å. Next, to choose the most optimal conformation, average-linkage algorithm was carried out to extract the most representative structure based on the snapshots from the last 5 ns of simulations. We selected the coordinates of Cα atoms in the AGO2 protein and P atoms in the nucleotides as input for average-linkage algorithm. With the obtained structure of the 21mer-T1, the guide extension from 21mer to 23mer and energy minimizations were accomplished using the Discovery Studio 2019.
2.1.4. Construction of the seed-plus-supplementary (g15–g19)-pairing systems
Three seed-plus-supplementary (g15–g19)-pairing systems, including the 21mer-T3, 22mer-T3, and 23mer-T3, were constructed. First, to model the 21mer-T3 system, we mutated and remove redundant nucleotides using Discovery Studio 2019 from the structure of 21mer-T2. To enable the additional pairing of g18 and g19 in the 21mer-T3 compared to the 21mer-T2, a 20 ns simulation was performed as mentioned above. Pulling force was added using 3 kcal/mol/Å2 to promote the pairing. After a 20 ns simulation, the 3′ end of guide RNA was released from the PAZ domain. This phenomenon might due to the inappropriate force added. Since the pairing in the tail region may cause the release 3′ end of guide RNA, the 21mer RNA was too short to bear the tension upon additional 3′ pairing. Therefore, we chose 23mer-T2 to construct 23mer-T3 using the above method. We used average-linkage algorithm to extract the representative structure as the initial structure of the 23mer-T3 system. With the obtained structure of the 21mer-T3, the guide truncation from 23mer to 21mer and energy minimization were accomplished using the Discovery Studio 2019.
2.2. MD simulations
The above-mentioned systems were prepared with the LEaP program using ff14SB and ff99OL3 to describe the RNA-protein complexes [65], [66], [67]. The prepared complex was solvated to a truncated octahedron transferable intermolecular potential three point (TIP3P) water box [68] and Na + and Cl− were added to neutralize and simulate normal saline environment. Two rounds of minimization were performed in which the first round contained 5000 steps maximum minimization cycles with the protein and nucleic fixed and the second round contained 10,000 steps maximum minimization cycles with no constraints. Subsequently, all systems were heated from 0 to 300 K within 300 ps, followed by 700 ps equilibration running in a canonical ensemble (NVT). After all these preparations, 10 replicas of independent 1 μs simulations were performed with random velocities under isothermal isobaric (NPT) conditions, producing a total of 90 μs simulation trajectories for analysis. For the long-range electrostatic interactions, the particle mesh Ewald method [69] was used, while a 10 Å non-bonded cut-off was introduced for the short-range electrostatics and van der Waals interactions. Covalent bonds involving hydrogens were restrained using the SHAKE method [70].
2.3. Principal component analysis (PCA)
Principal component analysis (PCA) widely used in describing the kinetic process during simulation is a technique that can transform a series of potentially coordinated observations into orthogonal vectors. Among these vectors, the first two principal component (PC1 and PC2) provide the dominant motions during simulation [71]. In this analysis, PCs were generated based on coordinate covariance matrix of Cα atoms in the AGO protein using every 10 frames in all nine systems and these collected frames were all projected on the first and second principal components.
2.4. Generalized correlation analysis
Generalized correlation (GCij) analysis was applied to calculate the correlated motion. Compared to traditional Pearson correlation analysis, GCij analysis is independent of mutual orientations of the atomic fluctuations and enable the separation between linear and nonlinear contributions [72]. To reflect how much information of one atom’s position is provided by another, Mutual Information (MI) was introduced and calculated by:.
| (1) |
The right side of equation can be related to the more widely known measure of entropy which calculated by:.
| (2) |
To calculate based on the correlation between pairs of atoms:.
| (3) |
M[xi,xj] was further related to a more intuitive Pearson-like correlation coefficient GCij which can be calculated by:.
| (4) |
where d represents the dimensionality of xi and xj, which is 3 in our study. GCij calculation was done by g_correlation tool in Gromacs 3.3 [73] with the coordinates of Cα in each residue as input. The inter-domain correlation was calculated by accumulation of GCij between each residue in respective domains with a minimum threshold value of 0.50. Then, the accumulated value was normalized according to the maximum and minimum value in the counterpart in all nine systems.
2.5. Dynamic network analysis
Dynamic network analysis was performed to reflect the motion connection using the Network View plugin in VMD [74]. In our analysis, the Cα atoms in the AGO2 protein, the N1 atoms in the uracil and cytosine, and the N9 atoms in the adenine and guanine were chosen as nodes to represent their corresponding residues. Edges were drawn between nodes whose distances are within a cutoff of 4.5 Å for at least 75% of simulation time. The edge between nodes was calculated by:.
| (5) |
where i and j represent the two nodes. In addition, community, which means a combination of residues whose connections are stronger was calculated using the Girvan-Newman algorithm [75] through the gcommunities program in VMD.
2.6. Markov state model (MSM)
Markov state model (MSM) is a mathematical framework to describe the dynamics of time-series data [76], [77], [78]. It has been used as an invaluable tool in understanding the dynamics during MD simulation [79], [80]. In this analysis, MSM was used to differentiate conformational states during simulation and help to extract representative structures from each state. All MSM calculations were performed using the PyEMMA software. MSM transition matrix was first calculated based on the probability of transition between different states.
Implied timescale (ITS) test was performed to check the Markovian property and choose the proper lag time(τ). ITS as a function of τ can be calculated by:.
| (6) |
where λi means the eigenvalue gotten from MSM transition matrix of the ith process. Based on this, implied timescale plot can be drawn in which each curve represents an average transition time in one process. When the curve became approximately constant, the corresponding lag time is appropriate for following analysis and the system is memoryless (Markovian property). In this study, the lag time was set as 10 ns.
Every frame of each system was projected on the free energy plot according to the characteristic vectors (CV). Then, K-centers algorithm was applied to cluster the two-dimensional conformations into 300 microstates in each system. Based on these 300 microstates, PCCA + algorithm was performed to divide the microstates into certain states (can be thought as metastates). With the divided states, the Chapman–Kolmogorov test was computed to further testify the property of MSM.
Every microstate was labeled as a certain metastate. The frames near the K-centers (microstates) belonging to the certain state in energy landscape were extracted to constitute a new trajectory to represent the state. The structure that has the smallest cas (RMSD) with other frames in the trajectory was chosen as the representative structure.
3. Results
3.1. Distinct miRNA extensions affect conformational dynamics of AGO2–RNA complex
A total of nine systems were performed: three for seed-pairing (g2–g8)-only (T1 systems), three for seed-plus-supplementary (g13–g16)-pairing (T2 systems), and three for seed-plus-supplementary (g15–g19)-pairing (T3 systems). For systems with the same pairing pattern, miRNA length was 21mer, 22mer, and 23mer, respectively. The RNA sequences of the nine systems were shown in Fig. 1C–E. Each system had 1 μs × 10 independent runs with random initial velocities, leading to a cumulative simulation timescale of 90 μs. Such an extensive timescale has been proven efficient for exploring the biomacromolecule conformational landscapes and recognition processes. To explore the effect of RNA lengths on overall conformational dynamics, the Cα atoms root-mean-square deviation (RMSD) of the AGO2 protein relative to the crystal structure was calculated. No significant differences were found except for the 21mer-T3 system (Supplementary Fig. 1A–C). Compared to the 22mer-T3 and 23mer-T3 systems, a ∼ 0.5 Å RMSD increase in the 21mer-T1 system was observed, indicating a conformational variation compared with the initial structure. To explore the conformational variation, we extracted the representative structure of the 21mer-T3 system from the 10 replicas of MD simulations and found the release of the 3′ end of the miRNA from the PAZ domain in each replica (Supplementary Fig. 1D). This observation indicated that the length of the 21mer miRNA was insufficient to tolerate the increased tension created by the 3′ tail pairing.
To further uncover the influence of miRNA lengths on local conformational dynamics, the root-mean square fluctuation (RMSF) of each residue (represented by the Cα atoms for AGO2 and P atoms for miRNA) was calculated (Supplementary Figs. 2 and 3). To better show the differences, we subtracted RMSF of the 22mer and 23mer systems from the counterpart of 21mer systems in the three systems and projected the difference values on the protein structure (Fig. 2A–I). Inspection of the RMSF plot showed that the PAZ domain displayed the highest fluctuation and showed an increased plasticity with the extension of miRNAs, indicating the less constrained conformation of the AGO2 protein in response to the extended miRNAs. However, it should be noted that with the extension of the miRNA lengths from 22mer to 23mer, the RMSF of the PAZ domain decreased significantly in the region proximal to the miRNA tail. This can be attributed to the enhanced stability created by the additional binding of 23th nucleotide. In general, the RMSF variations in the PAZ domain induced by different miRNA lengths are originated from the combined actions of the tension release and the additional binding.
Fig. 2.

Conformational dynamics of AGO2–RNA complex. The averaged RMSF values are plotted on the 3D structures of the AGO2–RNA complex in the 21mer-T1 (A), 21mer-T2 (B) and 21mer-T3 (C) systems. The substations of RMSF values of the 22mer and 23mer systems from the counterpart of 21mer systems are plotted on the 3D structures (D–I). Positive regions (red) stand for higher RMSF values, whereas negative regions (blue) represent lower RMSF values. The significant fluctuation differences are highlighted with a dashed rectangle in each panel.
3.1.1. Extended miRNA fastens the mouth of supplementary chamber
To characterize the global conformational transition and essential dynamics in the simulations, principal component analysis (PCA) was performed. We projected all MD snapshots from 10 independent runs of each system on the two-dimensional plot according to the first two principal components (PC1 and PC2) (Fig. 3A–I) and the domain motions along the PC1 (Fig. 3J) and PC2 (Fig. 3K) were shown on the AGO2 protein.
Fig. 3.

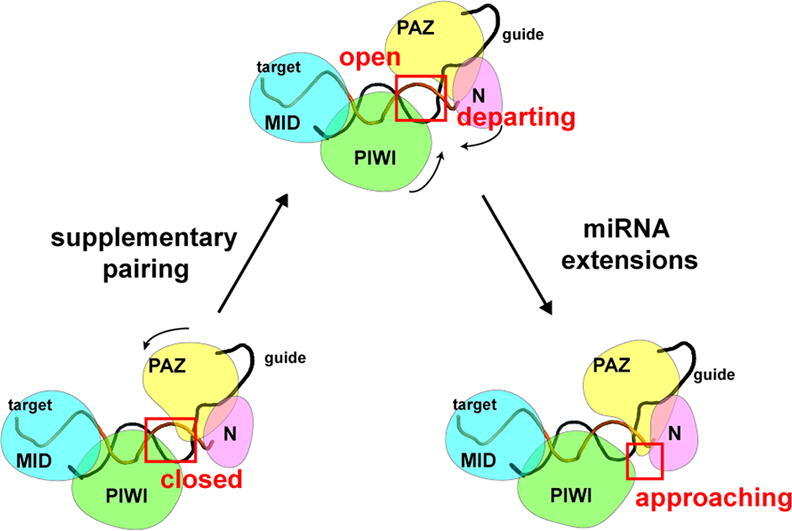
Global conformational transitions of AGO2–RNA complex. (A–I) Projections of the first and second principal components (PC1 vs PC2) from MD simulations of the AGO2 protein in nine systems. (J) The “departing” to “approaching” domain motion along the PC1. (K) The “open” to “closed” domain motion along the PC2. (L) Superposition of the AGO2 protein between the 23mer-T1 (gray) and the 23mer-T2 (gold) systems reveals the shift of the PAZ domain (indicated with arrow) to form supplementary chamber. (M) Distances between the E261 Cα atom from the PAZ domain and the D358 Cα atom from the L2 domain. The bars indicate the mean ± SEM values from 10 independent MD runs.
The pairing pattern can be distinguished based on the PC2 with the seed-pairing-only systems showing higher PC2 value. Along the PC2, the PAZ domain underwent large-amplitude motions towards the N domain and moved away from the L2 domain, which rendered the supplementary chamber changing from the “open” to “closed” conformations (Fig. 3K). This observation supported the notion that the PAZ domain shifted away from the N domain to provide sufficient space to form supplementary chamber. By superimposing the averaged structures of the 23mer-T1 system which represented the seed-pairing-only conformation and the 23mer-T2 system which represented the seed-plus-supplementary-pairing conformation, the shift of the PAZ domain concerted with the motion of the PC2 (Fig. 3L). However, PC2 values had no variation with the extended miRNA lengths, suggesting that miRNA lengths cannot affect the “open” to “closed” motions. To further elucidate this observation, we calculated the distance between the E261 Cα atom from the PAZ domain and the D358 Cα atom from the L2 domain, representing the shifted range of the PAZ domain from the L2 domain (Fig. 3L and M). The calculated distances in the seed-pairing-only systems were relatively larger than those in the seed-plus-supplementary pairing systems. However, no significant differences were found with the extension of the miRNA lengths in the additional supplementary pairing systems, indicating that the extended miRNA lengths did not affect the “open” to “closed” motions of the AGO2 protein in the seed-plus-supplementary pairing systems.
Compared to the PC2, the PC1 values changed with the extended miRNA lengths. In the seed-plus-supplementary-pairing systems, PC1 values increased with the extended miRNA lengths from 21mer to 23mer while this property was not observed in the seed-pairing-only systems (Fig. 3A–I). Along the PC1, both the PAZ and PIWI domains moved towards each other and yielded the supplementary chamber to open a smaller range, which can be called as the “departing” to “approaching” motions (Fig. 3J).
To further probe the influence of miRNA lengths on the “departing” to “approaching” motions, we projected all MD trajectories onto the two-dimensional surface according to the distances from the I61 Cα atom to the G836 Cα atom (d1) and the E333 Cα atom to the center of mass of Cα atoms in the A603, G604 and D605 (d2) (Fig. 4A–I and Supplementary Fig. 4). D1 represents the distance between the PAZ and PIWI domain which correlates to the “departing” to “approaching” motions while d2 represents the distance between the PAZ and N domain. Since the PAZ, PIWI, and N domains form the mouth of the supplementary chamber, d1 and d2 can reflect the supplementary chamber-opening degree. To differentiate the conformational states in the energy landscape, Markov state model (MSM) was applied (see Material and Methods) and only the states in the energy basin of free-energy landscape were analyzed. The implied timescale test and the Chapman–Kolmogorov test were calculated to testify the Markovian property (Supplementary Figs. 5 and 6). The proportion, the representative structure, and the representative trajectory of each state were calculated based on the MSM. In addition to the free-energy diagram, violin plots of d1 and d2 were also drawn to better compare between different systems (Supplementary Figures 7 and 8). Compared to the seed-plus-supplementary pairing systems, the range of both d1 and d2 parameters were generally larger in the seed-pairing-only systems, indicating the instability of the contacts between the PAZ, PIWI and N domains when pairing only in the seed region. The higher d2 parameter in the seed-pairing-only systems also reflected the larger distance between the PAZ and PIWI domains, which further suggested a contraction of the PAZ and PIWI domains with the additional supplementary pairing. According to the states extracted using MSM in the seed-pairing-only systems, with the increase of the d1 parameter, three states were observed: CX, CY, and CZ (Fig. 4A–C). The proportion and location of the three states did not change with the extended miRNA lengths and the projected area on the free-energy landscape was widely distributed and very similar in three systems, which reflected the conformational similarity of the AGO2 protein with different miRNA lengths in the seed-pairing-only systems. In the seed-plus-supplementary (g13-g17) pairing systems, two different states were observed: one with a smaller d1 parameter means a closer contact between the N and PIWI domains (CL), and the other was opposite (CR) (Fig. 4D–F). In the seed-plus-supplementary (g15-g19) pairing systems, three different states were observed (Fig. 4G–I). One had larger d1 and d2 parameters (CU), meaning a distant relationship between the PAZ, PIWI and N domains which only existed in the 21mer-T1 system. The other two states shared a similar d1 parameter and from the high to low d2 parameter was CM and CD, respectively, which merged into one state in the 22mer-T3 system. Except for the 21mer-T3 system whose 3′ end of miRNA was released from the PAZ domain, both d1 and d2 parameters broadly decreased with the extended miRNA lengths in the seed-plus-supplementary-pairing systems, indicating the shrinkage of the mouth of supplementary chamber encompassed by the PAZ, PIWI and N domains.
Fig. 4.

The closer contacts between the PAZ, PIWI and N domains induced by the miRNA extensions could shield the target RNA from solvent water attack. (A–I) Conformational landscapes generated using the d1 (distance from the I61 Cα atom to the G836 Cα atom) and d2 (distance from the E333 Cα atom to the center of mass of Cα atoms in the A603, G604 and D605) order parameters. Representative structures of (J) CL state and (K) CR state in the 22mer-T2 system, and (L) CM state and (M) CD state in the 23mer-T3 system. The nucleotides for calculating SASA was noted. SASAs of nucleotides noted above in different states of the T2 (N) and T3 (O) systems. * The CD* state in the 22mer-T3 system is merged by the CD state and the CM state.
By superimposing the representative structures of their respective dominant state in the T2 systems (CR for 21mer-T2 and 22mer-T2, and CL for 23mer-T2), we found that with the extended miRNA lengths, the PAZ, PIWI and N domains were getting closer to each other and wrapping around the target more tightly (Supplementary Figure 9). To show the conformational changes clearly, we superimposed the representative structures of the CL, CR, CM, and CU states. Compared to the CR conformation, the proximity of the PIWI and N domains in the CL conformation shielded the side of target RNA (Fig. 4J and K). In the CD conformation, the approaching of the PIWI and PAZ domains yielded the direct interactions between them, wrapping the target RNA more tightly and protecting it from solvent water (Fig. 4L and M). To quantify this change, we calculated the solvent accessible surface area (SASA) of the target nucleotide surrounded by the PAZ, PIWI and N domains which includes the A9–C12 of target RNA in the T2 systems and its counterpart U6–A9 of target RNA in the T3 systems. The SASA value decreased from the CR state to the CL state in the T2 systems and from the CM state to the CU state in the T3 systems (Fig. 4N and O and Supplementary Figure 10), indicating that the closer contacts between the PAZ, PIWI and N domains induced by the miRNA extensions could protect the target RNA from water solvent.
To discuss the linkage between these states to the target RNA regulation, we calculated the RMSDs between representative structures of each state and reported crystal structures (Supplementary Table 1). For RNA sequence and pairing pattern of target RNA and miRNA could affect the structure of AGO protein, we selected the crystal structure of seed-plus-supplementary-pairing AGO2 complex (PDB ID 6N4O) [29] as the representative structure of target inhibiting process and the crystal structure of only four central mismatches (bu4) pairing AGO2 complex (PDB ID 6NIT) [30] as the representative structure of target-directed microRNA degradation (TDMD) process to make RNA sequence and pairing pattern of the crystal structures as similar as possible to the simulation system. The RMSDs of whole protein and key domains (PAZ and PIWI domains) in AGO-RNA interaction between each representative structure and the crystal structures were calculated respectively. The result showed that RMSDs with target inhibiting conformation are smaller than that with TDMD conformation, correlated with the fact that systems we built were used to describe target inhibiting process rather than TDMD process. However, the phenomenon cannot be observed in the 23mer-T3 system. This can be ascribed to the huge difference in pairing pattern and RNA sequence between the 23mer-T3 system and the crystal structure 6N4O. The CR conformation has the lowest RMSDs with target inhibiting conformation among all states especially when calculating key domains, indicating that the crystal structure 6N4O adopted the CR conformation. This is consistent with the previous observation that CR state was the dominant conformation in the 21mer-T2 system for crystal structure 6N4O has 21mer miRNA and almost the same pairing pattern as the 21mer-T2 system.
Taken together, compared to the seed-pairing-only systems, the additional supplementary pairing could induce “closed” to “open” motions of the AGO2 protein and promote the proximity of the PAZ and PIWI domains. Further, the proximity of the PAZ, PIWI and N domains led to the shrinkage of the supplementary chamber and tightened the mouth of the chamber, which in turn prevented the escape of target RNA from the complex and shielded it from solvent water attack.
3.1.2. Pairing patterns and miRNA extensions influence the interaction of the target RNA with the AGO2 and miRNA
To examine how the pairing patterns and the miRNA extensions influenced the interaction of the target RNA with the AGO2 and miRNAs, we calculated the number of hydrogen bonds formed between AGO2–target RNA and miRNA–target RNA in each system. The cutoff distance and angle of hydrogen bonds are set as 3.5 Å and 135°, respectively. With the additional supplementary pairing, the number of hydrogen bonds between AGO2–target and miRNA–target increased compared to the seed-pairing-only systems (Fig. 5A). Additionally, the number of hydrogen bonds formed between target and protein in the T2 system was smaller than that in the T3 system while the number of hydrogen bonds formed between target and guide was just opposite. The deviation can be ascribed to the different pairing patterns and target RNA structures in the T2 and T3 systems. There are two more nucleotides pairing to miRNA at the 5′ end of target RNA in the T3 systems (pairing to g15-g19) compared to the T2 systems (pairing to g13-g17). The additional pairing pulled the 5′ end of target RNA from the PIWI domain of protein. Moreover, target2 has an additional 5′ tail compared to target3 (Fig. 1D and E), which can form additional hydrogen bonds with protein. To prove the explanation above, we calculated the hydrogen bonds formed between the PIWI domain and target RNA and between protein and the 5′ additional tail of target RNA (A1target, C2target and G3target) in the T2 and T3 systems (Supplementary Figure 11). We could observe that the 5′ additional tail of target RNA in the T2 systems provided ∼ 6 additional hydrogen bonds with AGO2 in contrast with the T3 systems and the difference of the protein- target RNA hydrogen bonds between the T2 and T3 systems is also about 6 (Fig. 5A). The T2 systems had about seven more hydrogen bonds between target RNA and the PIWI domain than the T3 system and the 5′ additional tail of the T2 systems mainly formed hydrogen bonds with the PIWI domain. Based on these, we speculated that hydrogen bonds formed between the 5′ additional tail in the T2 systems provided the most deviation in the number of target RNA-protein hydrogen bonds between the T2 and T3 systems. The number of hydrogen bonds between miRNA and target RNA in the 21mer-T3 system was slightly larger relative to the 22mer-T3 and 23mer-T3 systems. This can be explained that the release of the 3′ end of the guide RNA relieved the conformational limitation of the PAZ domain on its tail and it could adopt a more suitable conformation to pair with the target RNA.
Fig. 5.

The interaction of the target RNA with the AGO2 and miRNA. (A) The number of hydrogen bonds formed between AGO2–target RNA and miRNA–target RNA. Pairing occupancy of nucleotide pairs with more than 3% variation as the miRNA extensions in the T2 (B) and T3 (C) systems.
To further elucidate the pairing between the target and guide RNAs, the pairing occupancy of each nucleotide pair was calculated using the average of occupancy of each hydrogen bond formed between the nucleotide pair. Nucleotide pairs with more than 3% variation in the pairing occupancy as the miRNA extensions were shown in Fig. 5B and C. The pairing occupancy at the edge of supplementary pairing which contains U17miRNA–A6target in the T2 systems and U19miRNA–A1target and G18miRNA–C2target in the T3 systems increased slightly with the miRNA extensions except for the 21mer-T3 system in which the 3′ tail of the guide RNA released from the PAZ domain. The opening angles of the U17miRNA–A6target in the T2 systems and the U19miRNA–A1target in the T3 systems were further calculated to describe the pairing at the edge of supplementary region (Supplementary Figure 12). With the miRNA extensions, the opening angle became smaller, suggesting a better pairing at the edge of supplementary region. The pairing at the edge of supplementary region depends on the tail of miRNA’s motions towards the target RNA, which may increase the tension in the 3′ end of miRNA. Hence, with the extended miRNA lengths, the tension can be reduced and therefore the miRNA can move together with the PAZ domain towards the target RNA. In addition to the edge pairing, the pairing occupancy of other sites seemed to vary less regularly, but generally compensated for the variation in the edge pairing caused by the miRNA extensions. This was correlated with the findings that the extended miRNA lengths did not affect the sum of hydrogen bonds between the target RNA and miRNA.
3.1.3. The effect of pairing patterns and miRNA extensions on the correlated motions and community networks
Using Generalized correlation analysis, we provided an overview of the inter-residue and inter-domain correlated motions in the nine systems (see Material and Methods). The results showed that the additional supplementary pairing significantly weakened the overall correlated motions (Supplementary Figure 13A–J). Additionally, the overall correlated movements also enhanced with the miRNA extensions. Further investigation on the interdependent motions between different domains indicated that the increased correlated motions between the PAZ domain and other domains (especially the MID and PIWI domains) were more significant in the context of overall enhanced domain connectivity with the miRNA extensions (Fig. 6A–I). All these observations, except the 21mer-T3 system in which the 3′ release of miRNA from the PAZ domain, reshaped the overall correlation structure and strengthened the correlated motions compared to other seed-plus-supplementary-pairing systems. These results were consistent with the proximity of the PAZ, PIWI and N domains with the extended miRNA lengths, implying that miRNA extensions could enhance the domain connectivity between the PAZ, PIWI and N domains to promote the proximity of these domains to wrap around the target RNA more tightly.
Fig. 6.

Inter-domain correlation of AGO2. The inter-domain correlation was calculated by accumulation of GCij between each residue within respective domains in the (A) 21mer-T1, (B) 22mer-T1, (C) 23mer-T1, (D) 21mer-T2, (E) 22mer-T2, (F) 23mer-T2, (G) 21mer-T3, (H) 22mer-T3 and (I) 23mer-T3 systems.
Community network analysis was performed to uncover the correlation network in different systems (see Material and Methods). Each community was visualized as colored circles whose area is proportional to the number of residues it contains. The strength of inter-community connection was represented by the width of sticks connecting communities. In general, communities of different systems were similar (Fig. 7A–I) and the detailed configurations of the communities in each system were shown in Supplementary Figure 14 to reveal the relationship between domains and communities. The PIWI domain consisted of Communities C, D, and F while the community F only existed in the T2 systems. The Community F diminished from the 21mer-T2 system to the 22mer-T2 system and even vanished in the 23mer-T2 system, suggesting a better motion integrity of the PIWI domain with the miRNA extensions. As for the nucleic acids, they were segmented into communities in pairs, playing a role of connecting communities along the way. In terms of where each nucleotide belonging, the 5′ non-pairing nucleotides of target RNA in the T2 systems gradually joined Community I and moved together with miRNAs during simulations with the extension of miRNA lengths from 21mer to 23mer (Supplementary Figure 12D–F), indicating better interactions between target and guide RNAs induced by the miRNA extensions.
Fig. 7.

Community network of AGO2–RNA complex. The community network in the (A) 21mer-T1, (B) 22mer-T1, (C) 23mer-T1, (D) 21mer-T2, (E) 22mer-T2, (F) 23mer-T2, (G) 21mer-T3, (H) 22mer-T3 and (I) 23mer-T3 systems. Areas of the circles represent the numbers of residues in corresponding communities, and the widths of sticks connecting communities represent the intercommunity connections.
For the connectivity between communities, we focused on the connection between Community I (main part of the PAZ domain) and Community C (main part of the PIWI domain). In the seed-pairing-only systems, the connection between the Community I and Community C was made through two additional communities which were Community G/H and Community D (Fig. 7A–C). However, with the additional supplementary pairing, the connection was through one additional community which was Community G/H, except for the 21mer-T2 system whose connection network was influenced by the additional community F (Fig. 7D–I). This analysis suggested that the supplementary pairing can promote the information flow between the PAZ and PIWI domains. In the seed-plus-supplementary pairing systems, the connection between Community I and C was also strengthened with the miRNA extensions through the enhancement of connections along the way while this change cannot be observed in the seed-pairing-only systems. These results indicated that the miRNA extensions can produce stronger information transduction between the PAZ and PIWI domains, which was in consistence with the generalized correlation analysis revealing that the miRNA extensions can enhance the domain connectivity between the PAZ and PIWI domains.
4. Discussion
The human AGO2 protein, acting by guiding miRNA to recognize target RNA, plays a critical role in gene expression regulation. Here, using MD simulations, we investigated how the pairing pattern and miRNA lengths influence target affinity at the atomic level, thereby providing further guidance for the design of miRNA for experimental and therapeutic use.
We observed the release of the 3′ end of the miRNA from the PAZ domain in the 21mer-T3 system, indicating that the length of the 21mer miRNA was insufficient to tolerate the increased tension created by the 3′ tail pairing. However, when building the 21mer-T3 system, we pre-paired the target with miRNA while in real structure the target may not perfectly pair with miRNA due to excessive tension and cannot pull the miRNA from the PAZ domain. Validation of the release of 3′ tail by experiment will be the focus of our follow-up research.
Distinct pairing patterns can cause different conformational dynamics of AGO2–RNA complex. By comparing the conformational dynamics between the seed-pairing-only and seed-plus-supplementary pairing systems, we confirmed the structural model that the PAZ domain shifted away from the central cleft for the opening of supplementary chamber to form additional supplementary pairing between the target RNA and miRNA. Moreover, results indicated that in addition to the supplementary guide-target interaction, supplementary pairing could increase target affinity by promoting the proximity of the PAZ and PIWI domains to wrap target RNA more tightly.
We investigated how miRNA lengths influence target affinity. Previous studies speculated that miRNA extensions increased target affinity by releasing tension in miRNA 3′ tail to promote the supplementary interaction between guide and target. Consistent with this speculation, we found that miRNA extensions could promote pairing at the edge of supplementary region. However, the sum of hydrogen bonds formed between target and guide was similar with different miRNA lengths which can be ascribed to the compensation of inner stronger pairing. This result explained and accorded with the fact that mismatches at the edge of supplementary pairing had a minor effect on target binding. Based on this, we assumed that the enhancement of edge pairing of supplementary region was not the key factor causing the difference in target affinity with miRNA extensions. Furthermore, we put forward that miRNA extensions could induce conformational transition from “departing” to “approaching” with the proximity of the PAZ, PIWI and N domains. This conformational transition could tighten the mouth of the supplementary chamber, which in turn prevented the escape of target RNA from the complex and shielded it from solvent water attack. This conformational change created by the miRNA extensions can be ascribed to the enhanced domain connectivity between the PAZ, PIWI and N domains and the increased plasticity of the PAZ domain.
In view of the wide experimental application of miRNA and the great potential of miRNA therapy in the treatment of tumor, cardiovascular disease, endocrine and metabolic disease, the design of miRNA mimics and anti-miRNAs has become the focus of research [11], [81], [82]. Much guidance on miRNA mimics and anti-miRNAs design has been established [83], [84]. However, the structural basis for the assembly of AGO–RNA complex needs to be further investigated for the development of new design strategies. Our study illuminated the structural basis of pairing pattern and miRNA lengths’ effect on target RNA recognition, providing insights on control of pairing patterns and miRNA lengths on the design of miRNA mimics and anti-miRNAs. For the potential engineering of AGO proteins as gene silencing and even editing tools [85], our study may provide a useful guidance to tune AGO proteins for new gene regulation tools.
5. Conclusion
In summary, the collective sampling of 90 μs MD simulations revealed the effect of pairing patterns and miRNA lengths on the conformational dynamics and the assembly of AGO–RNA complexes. Compared to the seed-paring-only systems, the supplementary pairing can strengthen target-guide interactions and promote the proximity of the PAZ and PIWI domains, which can tighten the mouth of supplementary chamber to protect the target RNA from dissociation. Further, in the seed-plus-supplementary pairing systems, the extension of miRNA lengths could induce the increased plasticity of the PAZ domain and enhance domain connectivity between the PAZ, PIWI and N domains, and thereby promote the proximity of these domains to tighten the mouth of supplementary chamber, which in turn protected the target RNA from water solvent attacking and hindered target RNA’s escaping from the AGO–RNA complexes through the mouth of supplementary chamber. Moreover, our results showed that miRNA extensions could strengthen the target-guide pairing at the edge of supplementary region while the sum of hydrogen bonds formed between target and guide RNAs were similar with different miRNA lengths and the inner stronger matching can compensate for edge weaker matching. The proximity of the PAZ, PIWI and N domains rather than the enhanced edge pairing is the key factor for the increased target affinity with the miRNA extensions. These results shed light on the structural details of the assembly of AGO–RNA complex with the supplementary pairing, which were instructive to the design of miRNA mimics and anti-miRNA for clinical and experimental use.
6. Declarations
Ethics approval and consent to participate.
Not applicable.
7. Consent for publication
All the authors have approved and agreed to publish this manuscript.
8. Availability of data and material
All data generated and analysed to support the conclusion of this study is included in this published manuscript and its additional supporting files.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
This work was supported by the National Natural Science Foundation of China (82172846 and 81901255).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2022.03.001.
Contributor Information
Shaoyong Lu, Email: lushaoyong@sjtu.edu.cn.
Xiaolong Li, Email: lixl868@163.com.
Zongtao Chai, Email: runout@163.com.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Höck J., Meister G. The Argonaute protein family. Genome Biol. 2008;9:210. doi: 10.1186/gb-2008-9-2-210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meister G. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet. 2013;14:447–459. doi: 10.1038/nrg3462. [DOI] [PubMed] [Google Scholar]
- 3.Wu J., Yang J., Cho W.C., Zheng Y. Argonaute proteins: structural features, functions and emerging roles. J Adv Res. 2020;24:317–324. doi: 10.1016/j.jare.2020.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hammond S.M. An overview of microRNAs. Adv Drug Deliv Rev. 2015;87:3–14. doi: 10.1016/j.addr.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gebert L.F.R., MacRae I.J. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol. 2019;20:21–27. doi: 10.1038/s41580-018-0045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q., Ye Y., Lin R., Weng S., Cai F., Zou M., et al. Analysis of the expression, function, prognosis and co-expression genes of DDX20 in gastric cancer. Comput Struct Biotechnol J. 2020;18:2453–2462. doi: 10.1016/j.csbj.2020.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruiz-Saavedra S., García-González H., Arboleya S., Salazar N., Emilio Labra-Gayo J., Díaz I., et al. Intestinal microbiota alterations by dietary exposure to chemicals from food cooking and processing. Application of data science for risk prediction. Comput Struct Biotechnol J. 2021;19:1081–1091. doi: 10.1016/j.csbj.2021.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 9.Elbashir S.M., Harborth J., Lendeckel W., Yalcin A., Weber K., Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:694–698. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 10.Hammond S.M., Bernstein E., Beach D., Hannon G.J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature. 2000;404:293–296. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- 11.Rupaimoole R., Slack F.J. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discovery. 2017;16:203–222. doi: 10.1038/nrd.2016.246. [DOI] [PubMed] [Google Scholar]
- 12.Ganju A., Khan S., Hafeez B.B., Behrman S.W., Yallapu M.M., Chauhan S.C., et al. miRNA nanotherapeutics for cancer. Drug Discov Today. 2017;22:424–432. doi: 10.1016/j.drudis.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu T.X., Rothenberg M.E. MicroRNA. J Allergy Clin Immunol. 2018;141:1202–1207. doi: 10.1016/j.jaci.2017.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez-Ledesma E., Flores D., Trevino V. Computational methods for detecting cancer hotspots. Comput Struct Biotechnol J. 2020;18:3567–3576. doi: 10.1016/j.csbj.2020.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu J., Carmell M.A., Rivas F.V., Marsden C.G., Thomson J.M., Song J.J., et al. Argonaute2 is the catalytic engine of mammalian RNAi. Science. 2004;305:1437–1441. doi: 10.1126/science.1102513. [DOI] [PubMed] [Google Scholar]
- 16.Schirle N.T., MacRae I.J. The crystal structure of human argonaute2. Science. 2012;336:1037–1040. doi: 10.1126/science.1221551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elkayam E., Kuhn C.D., Tocilj A., Haase A.D., Greene E.M., Hannon G.J., et al. The structure of human argonaute-2 in complex with miR-20a. Cell. 2012;150:100–110. doi: 10.1016/j.cell.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wee L.M., Flores-Jasso C.F., Salomon W.E., Zamore P.D. Argonaute divides Its RNA guide into domains with distinct functions and RNA-binding properties. Cell. 2012;151:1055–1067. doi: 10.1016/j.cell.2012.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schirle N.T., Sheu-Gruttadauria J., MacRae I.J. Structural basis for microRNA targeting. Science. 2014;346:608–613. doi: 10.1126/science.1258040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brennecke J., Stark A., Russell R.B., Cohen S.M. Principles of microRNA-target recognition. PLoS Biol. 2005;3 doi: 10.1371/journal.pbio.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grosswendt S., Filipchyk A., Manzano M., Klironomos F., Schilling M., Herzog M., et al. Unambiguous identification of miRNA: target site interactions by different types of ligation reactions. Mol Cell. 2014;54:1042–1054. doi: 10.1016/j.molcel.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedman R.C., Farh K.K.H., Burge C.B., Bartel D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grimson A., Farh K.K.H., Johnston W.K., Garrett-Engele P., Lim L.P., Bartel D.P. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Broughton J.P., Lovci M.T., Huang J.L., Yeo G.W., Pasquinelli A.E. Pairing beyond the seed supports MicroRNA targeting specificity. Mol Cell. 2016;64:320–333. doi: 10.1016/j.molcel.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Helwak A., Kudla G., Dudnakova T., Tollervey D. Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell. 2013;153:654–665. doi: 10.1016/j.cell.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brancati G., Großhans H. An interplay of miRNA abundance and target site architecture determines miRNA activity and specificity. Nucleic Acids Res. 2018;46:3259–3269. doi: 10.1093/nar/gky201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao Y., Macrae I.J. Robust differential microRNA targeting driven by supplementary interactions in vitro. RNA. 2020;26:162–174. doi: 10.1261/rna.072264.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salomon W.E., Jolly S.M., Moore M.J., Zamore P.D., Serebrov V. Single-molecule imaging reveals that argonaute reshapes the binding properties of its nucleic acid guides. Cell. 2015;162:84–95. doi: 10.1016/j.cell.2015.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sheu-Gruttadauria J., Xiao Y., Gebert L., MacRae I. Beyond the seed: structural basis for supplementary microRNA targeting. EMBO J. 2018;38 doi: 10.15252/embj.2018101153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sheu-Gruttadauria J., Pawlica P., Klum S.M., Wang S., Yario T.A., Schirle Oakdale N.T., et al. Structural basis for target-directed MicroRNA degradation. Mol Cell. 2019;75:1243–1255. doi: 10.1016/j.molcel.2019.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Park J.H., Shin S.Y., Shin C. Non-canonical targets destabilize microRNAs in human Argonautes. Nucleic Acids Res. 2017;45:1569–1583. doi: 10.1093/nar/gkx029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi CY, Kingston ER, Kleaveland B, Lin DH, Stubna MW, Bartel DP. The ZSWIM8 ubiquitin ligase mediates target-directed microRNA degradation. Science 2020;370:eabc9359. [DOI] [PMC free article] [PubMed]
- 33.Ameres S.L., Horwich M.D., Hung J.H., Xu J., Ghildiyal M., Weng Z., et al. Target RNA-directed trimming and tailing of small silencing RNAs. Science. 2010;328:1534–1539. doi: 10.1126/science.1187058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Machlin E.S., Sarnow P., Sagan S.M. Masking the 5′ terminal nucleotides of the hepatitis C virus genome by an unconventional microRNA-target RNA complex. PNAS. 2011;108:3193–3198. doi: 10.1073/pnas.1012464108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wyman S.K., Knouf E.C., Parkin R.K., Fritz B.R., Lin D.W., Dennis L.M., et al. Post-transcriptional generation of miRNA variants by multiple nucleotidyl transferases contributes to miRNA transcriptome complexity. Genome Res. 2011;21:1450–1461. doi: 10.1101/gr.118059.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fernandez-Valverde S.L., Taft R.J., Mattick J.S. Dynamic isomiR regulation in Drosophila development. RNA. 2010;16:1881–1888. doi: 10.1261/rna.2379610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gonzalez-Rivera J.C., Orr A.A., Engels S.M., Jakubowski J.M., Sherman M.W., O’Connor K.N., et al. Computational evolution of an RNA-binding protein towards enhanced oxidized-RNA binding. Comput Struct Biotechnol J. 2020;18:137–152. doi: 10.1016/j.csbj.2019.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan F., Gao F. A systematic strategy for the investigation of vaccines and drugs targeting bacteria. Comput Struct Biotechnol J. 2020;18:1525–1538. doi: 10.1016/j.csbj.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang K.C., der Wen J. Programmed −1 ribosomal frameshifting from the perspective of the conformational dynamics of mRNA and ribosomes. Comput Struct Biotechnol J. 2021;19:3580–3588. doi: 10.1016/j.csbj.2021.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khan A., Tahir Khan M., Saleem S., Junaid M., Ali A., Shujait Ali S., et al. Structural insights into the mechanism of RNA recognition by the N-terminal RNA-binding domain of the SARS-CoV-2 nucleocapsid phosphoprotein. Comput Struct Biotechnol J. 2020;18:2174–2184. doi: 10.1016/j.csbj.2020.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mahalapbutr P., Kongtaworn N., Rungrotmongkol T. Structural insight into the recognition of S-adenosyl-L-homocysteine and sinefungin in SARS-CoV-2 Nsp16/Nsp10 RNA cap 2′-O-Methyltransferase. Comput Struct Biotechnol J. 2020;18:2757–2765. doi: 10.1016/j.csbj.2020.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Long C., Romero M.E., la Rocco D., Yu J. Dissecting nucleotide selectivity in viral RNA polymerases. Comput Struct Biotechnol J. 2021;19:3339–3348. doi: 10.1016/j.csbj.2021.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Joseph R.E., Amatya N., Fulton D.B., Engen J.R., Wales T.E., Andreotti A.H. Differential impact of BTK active site inhibitors on the conformational state of full-length BTK. ELife. 2020;9 doi: 10.7554/eLife.60470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan J., Liu Y., Kong R., Ni D., Yu Z., Lu S., et al. Harnessing reversed allosteric communication: a novel strategy for allosteric drug discovery. J Med Chem. 2021;64:17728–17743. doi: 10.1021/acs.jmedchem.1c01695. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Q., Chen Y., Ni D., Huang Z., Wei J., Feng L., et al. Targeting a cryptic allosteric site of SIRT6 with small-molecule inhibitors that inhibit the migration of pancreatic cancer cells. Acta Pharm Sin B. 2022;12:876–889. doi: 10.1016/j.apsb.2021.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu S., Chen Y., Wei J., Zhao M., Ni D., He X., et al. Mechanism of allosteric activation of SIRT6 revealed by the action of rationally designed activators. Acta Pharm Sin B. 2021;11:1355–1361. doi: 10.1016/j.apsb.2020.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu S., He X., Yang Z., Chai Z., Zhou S., Wang J., et al. Activation pathway of a G protein-coupled receptor uncovers conformational intermediates as targets for allosteric drug design. Nat Commun. 2021;12:4721. doi: 10.1038/s41467-021-25020-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li X., Dai J., Ni D., He X., Zhang H., Zhang J., et al. Insight into the mechanism of allosteric activation of PI3Kα by oncoprotein K-Ras4B. Int J Biol Macromol. 2020;144:643–655. doi: 10.1016/j.ijbiomac.2019.12.020. [DOI] [PubMed] [Google Scholar]
- 49.Feng L., Lu S., Zheng Z., Chen Y., Zhao Y., Song K., et al. Identification of an allosteric hotspot for additive activation of PPARγ in antidiabetic effects. Sci Bull. 2021;66:1559–1570. doi: 10.1016/j.scib.2021.01.023. [DOI] [PubMed] [Google Scholar]
- 50.Wang Y., Ji D., Lei C., Chen Y., Qiu Y., Li X., et al. Mechanistic insights into the effect of phosphorylation on Ras conformational dynamics and its interactions with cell signaling proteins. Comput Struct Biotechnol J. 2021;19:1184–1199. doi: 10.1016/j.csbj.2021.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li X., Wang C., Peng T., Chai Z., Ni D., Liu Y., et al. Atomic-scale insights into allosteric inhibition and evolutional rescue mechanism of Streptococcus thermophilus Cas9 by the anti-CRISPR protein AcrIIA6. Comput Struct Biotechnol J. 2021;19:6108–6124. doi: 10.1016/j.csbj.2021.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang Y., Li M., Liang W., Shi X., Fan J., Kong R., et al. Delineating the activation mechanism and conformational landscape of a class B G protein-coupled receptor glucagon receptor. Comput Struct Biotechnol J. 2022;20:628–639. doi: 10.1016/j.csbj.2022.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qiu Y., Yin X., Li X., Wang Y., Fu Q., Huang R., et al. Untangling dual-targeting therapeutic mechanism of epidermal growth factor receptor (EGFR) based on reversed allosteric communication. Pharmaceutics. 2021;13:747. doi: 10.3390/pharmaceutics13050747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li X., Qi Z., Ni D., Lu S., Chen L., Chen X. Markov state models and molecular dynamics simulations provide understanding of the nucleotide-dependent dimerization-based activation of lrrk2 roc domain. Molecules. 2021;26:5647. doi: 10.3390/molecules26185647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu S., Shen Q., Zhang J. Allosteric methods and their applications: facilitating the discovery of allosteric drugs and the investigation of allosteric mechanisms. Acc Chem Res. 2019;52:492–500. doi: 10.1021/acs.accounts.8b00570. [DOI] [PubMed] [Google Scholar]
- 56.Lu S., Zhang J. Small molecule allosteric modulators of G-protein-coupled receptors: drug-target interactions. J Med Chem. 2019;62:24–45. doi: 10.1021/acs.jmedchem.7b01844. [DOI] [PubMed] [Google Scholar]
- 57.Lu S., He X., Ni D., Zhang J. Allosteric modulator discovery: from serendipity to structure-based design. J Med Chem. 2019;62:6405–6421. doi: 10.1021/acs.jmedchem.8b01749. [DOI] [PubMed] [Google Scholar]
- 58.Anand R., Sanjeev B.S. Insights into the effect of molecular crowding on the structure, interactions and functions of siRNA–PAZ complex through molecular dynamics studies. J Biomol Struct Dyn. 2019;37:2745–2752. doi: 10.1080/07391102.2018.1498391. [DOI] [PubMed] [Google Scholar]
- 59.Jiang H., Sheong F.K., Zhu L., Gao X., Bernauer J., Huang X. Markov state models reveal a two-step mechanism of miRNA loading into the human argonaute protein: selective binding followed by structural re-arrangement. PLoS Comput Biol. 2015;11 doi: 10.1371/journal.pcbi.1004404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harikrishna S., Pradeepkumar P.I. Probing the binding interactions between chemically modified siRNAs and human argonaute 2 using microsecond molecular dynamics simulations. J Chem Inf Model. 2017;57:883–896. doi: 10.1021/acs.jcim.6b00773. [DOI] [PubMed] [Google Scholar]
- 61.Lei J., Sheng G., Cheung P.P.H., Wang S., Li Y., Gao X., et al. Two symmetric arginine residues play distinct roles in thermus thermophilus argonaute DNA guide strand-mediated DNA target cleavage. PNAS. 2019;116:845–853. doi: 10.1073/pnas.1817041116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schäfer M., Ciaudo C. Prediction of the miRNA interactome – established methods and upcoming perspectives. Comput Struct Biotechnol J. 2020;18:548–557. doi: 10.1016/j.csbj.2020.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Quillet A., Anouar Y., Lecroq T., Dubessy C. Prediction methods for microRNA targets in bilaterian animals: toward a better understanding by biologists. Comput Struct Biotechnol J. 2021;19:5811–5825. doi: 10.1016/j.csbj.2021.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schirle N.T., Sheu-Gruttadauria J., Chandradoss S.D., Joo C., MacRae I.J. Water-mediated recognition of t1-adenosine anchors Argonaute2 to microRNA targets. ELife. 2015;4 doi: 10.7554/eLife.07646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zgarbová M, Otyepka M, Šponer J, Mládek A, Banáš P, Cheatham TE, et al. Refinement of the Cornell et al. Nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. Journal of Chemical Theory and Computation 2011;7:2886-2902. [DOI] [PMC free article] [PubMed]
- 66.Pérez A., Marchán I., Svozil D., Sponer J., Cheatham T.E., Laughton C.A., et al. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maier J.A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K.E., Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926. [Google Scholar]
- 69.York DM, Wlodawer A, Pedersen LG, Darden TA. Atomic-level accuracy in simulations of large protein crystals. Proceedings of the National Academy of Sciences of the United States of America 1994;91. doi:10.1073/pnas.91.18.8715. [DOI] [PMC free article] [PubMed]
- 70.Hassan N.Z.A., Mokhtar N.M., Sin T.K., Rose I.M., Sagap I., Harun R., et al. Integrated analysis of copy number variation and genome-wide expression profiling in colorectal cancer tissues. PLoS ONE. 2014;9 doi: 10.1371/journal.pone.0092553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Daidone I., Amadei A. Essential dynamics: foundation and applications. Wiley Interdisc Rev: Comput Mol Sci. 2012;2 [Google Scholar]
- 72.Lange O.F., Grubmüller H. Generalized correlation for biomolecular dynamics. Proteins Struct Funct Genet. 2006;62:1053–1061. doi: 10.1002/prot.20784. [DOI] [PubMed] [Google Scholar]
- 73.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J Mol Model. 2001;7:306–317. [Google Scholar]
- 74.Eargle J., Luthey-Schulten Z. Network view: 3D display and analysis of protein ·rNA interaction networks. Bioinformatics. 2012;28:3000–3001. doi: 10.1093/bioinformatics/bts546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Girvan M., Newman M.E.J. Community structure in social and biological networks. PNAS. 2002;99:7821–7826. doi: 10.1073/pnas.122653799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Husic B.E., Pande V.S. Markov state models: from an art to a science. J Am Chem Soc. 2018;140:2386–2396. doi: 10.1021/jacs.7b12191. [DOI] [PubMed] [Google Scholar]
- 77.Jiang Y., Wang D., Wang W., Xu D. Computational methods for protein localization prediction. Comput Struct Biotechnol J. 2021;19:5834–5844. doi: 10.1016/j.csbj.2021.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang C., Chowdhury D., Zhang Z., Cheung W.K., Lu A., Bian Z., et al. A review of computational tools for generating metagenome-assembled genomes from metagenomic sequencing data. Comput Struct Biotechnol J. 2021;19:6301–6314. doi: 10.1016/j.csbj.2021.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ni D., Wei J., He X., Rehman A.U., Li X., Qiu Y., et al. Discovery of cryptic allosteric sites using reversed allosteric communication by a combined computational and experimental strategy. Chem Sci. 2021;12:464–476. doi: 10.1039/d0sc05131d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lu S., Ni D., Wang C., He X., Lin H., Wang Z., et al. Deactivation pathway of Ras GTPase underlies conformational substates as targets for drug design. ACS Catal. 2019;9:7188–7196. [Google Scholar]
- 81.Mellis D., Caporali A. MicroRNA-based therapeutics in cardiovascular disease: screening and delivery to the target. Biochem Soc Trans. 2018;46:11–21. doi: 10.1042/BST20170037. [DOI] [PubMed] [Google Scholar]
- 82.van Rooij E., Purcell A.L., Levin A.A. Developing MicroRNA therapeutics. Circ Res. 2012;110:496–507. doi: 10.1161/CIRCRESAHA.111.247916. [DOI] [PubMed] [Google Scholar]
- 83.Lima J.F., Cerqueira L., Figueiredo C., Oliveira C., Azevedo N.F. Anti-miRNA oligonucleotides: a comprehensive guide for design. RNA Biol. 2018;15:338–352. doi: 10.1080/15476286.2018.1445959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bofill-De Ros X., Gu S. Guidelines for the optimal design of miRNA-based shRNAs. Methods. 2016;103:157–166. doi: 10.1016/j.ymeth.2016.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang X., Jiang J., Li Z., Liang J., Xiang Y. Strategies for mitochondrial gene editing. Comput Struct Biotechnol J. 2021;19:3319–3329. doi: 10.1016/j.csbj.2021.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated and analysed to support the conclusion of this study is included in this published manuscript and its additional supporting files.