

Abstract
The use of light-responsive proteins to control both living or synthetic cells, is at the core of the expanding fields of optogenetics and synthetic biology. It is thus apparent that a richer reaction toolbox for the preparation of such systems is of fundamental importance. Here, we provide a proof-of-principle demonstration that Morita-Baylis-Hillman adducts can be employed to perform a facile site-specific, irreversible and diastereoselective click-functionalization of a lysine residue buried into a lipophilic binding pocket and yielding an unnatural chromophore with an extended π-system. In doing so we effectively open the path to the in vitro preparation of a library of synthetic proteins structurally reminiscent of xanthopsin eubacterial photoreceptors. We argue that such library, made of variable unnatural chromophores inserted in an easy to mutate and crystallize retinoic acid transporter, significantly expand the scope of the recently introduced rhodopsin mimics as both optogenetic and “lab-on-a-molecule” tools.
Keywords: Morita-Baylis-Hillman adducts, synthetic Xanthopsin-like proteins, site-specific reaction, light-sensitive proteins, PYP-like chromophores
Graphical Abstract

We demonstrate that Morita-Baylis-Hillman adducts can be employed in the irreversible site-specific and diastereoselective click-functionalization of a lysine residue buried into a lipophilic binding pocket. This research open the path to the preparation of synthetic proteins structurally reminiscent of xanthopsin eubacterial photoreceptors potentially useful for both optogenetic and “lab-on-a-molecule” tools.
1. Introduction
In recent years, much interest has been focused on synthetic light-responsive proteins (LRPs) capable to perform light-driven biomimetic functions such as light color sensing, fluorescence, structural changes, pKa changes, ion channeling and pumping/transport.[1] The preparation of diverse LRPs is important for the rapidly expanding fields of synthetic biology[2,3] and optogenetics.[4] At a basic level, the accomplishment of such task requires the site-specific functionalization of an amino acid residue, ultimately producing an unnatural and covalently-bounded prosthetic group capable to respond to a light stimulus. It is apparent, that the preparation of such a system in vitro requires a reaction selectivity well beyond the discrimination of organic functions.
In addition to the required selectivity, it is desirable to perform such a preparation via “click-like chemistries” and starting from a library of precursors with spectroscopic properties different from those of the target prosthetic groups. In this way, the precursor-protein coupling can be easily followed, with obvious advantages. Ideally, one is looking for prosthetic groups with a significantly red-shifted absorption with respect to the precursor.
Click-like chemistries with the properties discussed above exist in Nature. The canonical example is that of the coupling between retinal and opsin proteins.[5–7] Such coupling exploits a specific lysine residue strategically located in the opsin hydrophobic pocket. The environment surrounding the lysine side-chain, not only increases its nucleophilic character but results in a stabilization of the produced Schiff base linkage via protonation. This avoids the thermodynamic instability of Schiff base linkages in aqueous media. Most relevantly, retinal absorbs maximally around 380 nm in methanol while the resulting retinal protonated Schiff base absorbs at 480 nm in the same solvent and at a range of longer wavelengths in the opsin environment.[7] Remarkably, once inserted in the opsin cavity, the same chromophore displays further red-shifted wavelengths which can go beyond 600 nm, and are modulated by variations in the opsin sequence.[7,8]
A variety of reactions[9,10] and methods[11] have been considered for the development of site-specific protein functionalizations. In order not to alter the protein integrity, these must be compatible with the aqueous media and mild reaction conditions.[11] Thanks to their nucleophilicity and relative abundance, lysine and cysteine residues have been frequently targeted.[12] N-hydroxysuccinimide (NHS) esters,[13] isocyanate (or thioisocyanate),[14] and aldehydes for reductive amination[15] are the electrophiles attacked by the −NH2 group of lysine, while maleimides[16] and α-halocarbonyls[17] react with the −SH group of cysteine.[18]
We shall also mention that genetic manipulation techniques have been leveraged to introduce natural (cysteine,[19] tryptophan,[20] tyrosine,[21] and histidine[22]) ) or unnatural (e. g. photo-Lys,[23] pTyr,[24] or AmAzZLys[25]) ) residues and obtain functionalized sites. These have also been exploited by selective synthetic procedures involving metal-catalyzed or metal-directed reactions.[26] However, these methods often require the preliminary synthesis of diverse unnatural amino acids. Therefore, the possibility of a more practical site-specific functionalization of native protein structures remains highly attractive.[27–29]
In this paper we report, as a proof of principle, that Morita-Baylis-Hillman adducts (MBHAs) can functionalize a lysine residue strategically located in the cavity of a human cellular retinoic acid binding protein II (hCRABPII) mutant.[30] We will also show that, due to their expanded π-system, the resulting naphthalene-, phenyl- and p-hydroxyphenyl-acrylate prosthetic groups formed via click-functionalization at room temperature in an aqueous buffer, have red-shifted absorption maxima with respect to their MBHA precursors.
In the past MBHAs have been employed as reagents in imidazole modification.[31–33] Some of these reagents were prepared[34,35] and characterized[36,37] in our laboratory (i. e. 1a-c and 2a-e, Fig. 1A). However, only functionalization of nucleophilic residues (i.e. histidine) belonging to a peptide chain and/or residing on protein tails were targeted. On the other hand, the photophysical features displayed by the products of their conjugation with imidazole or N-acetylhexahistidine suggested that MBHAs could be employed to build, via side-chain modification (Fig. 1B), a chromophore group even deep inside a protein cavity. As evident from inspection of the figure, the functionalization reaction can be described as a Michael reaction immediately followed by a E1CB elimination that provides the driving force to achieve the product.
Figure 1.
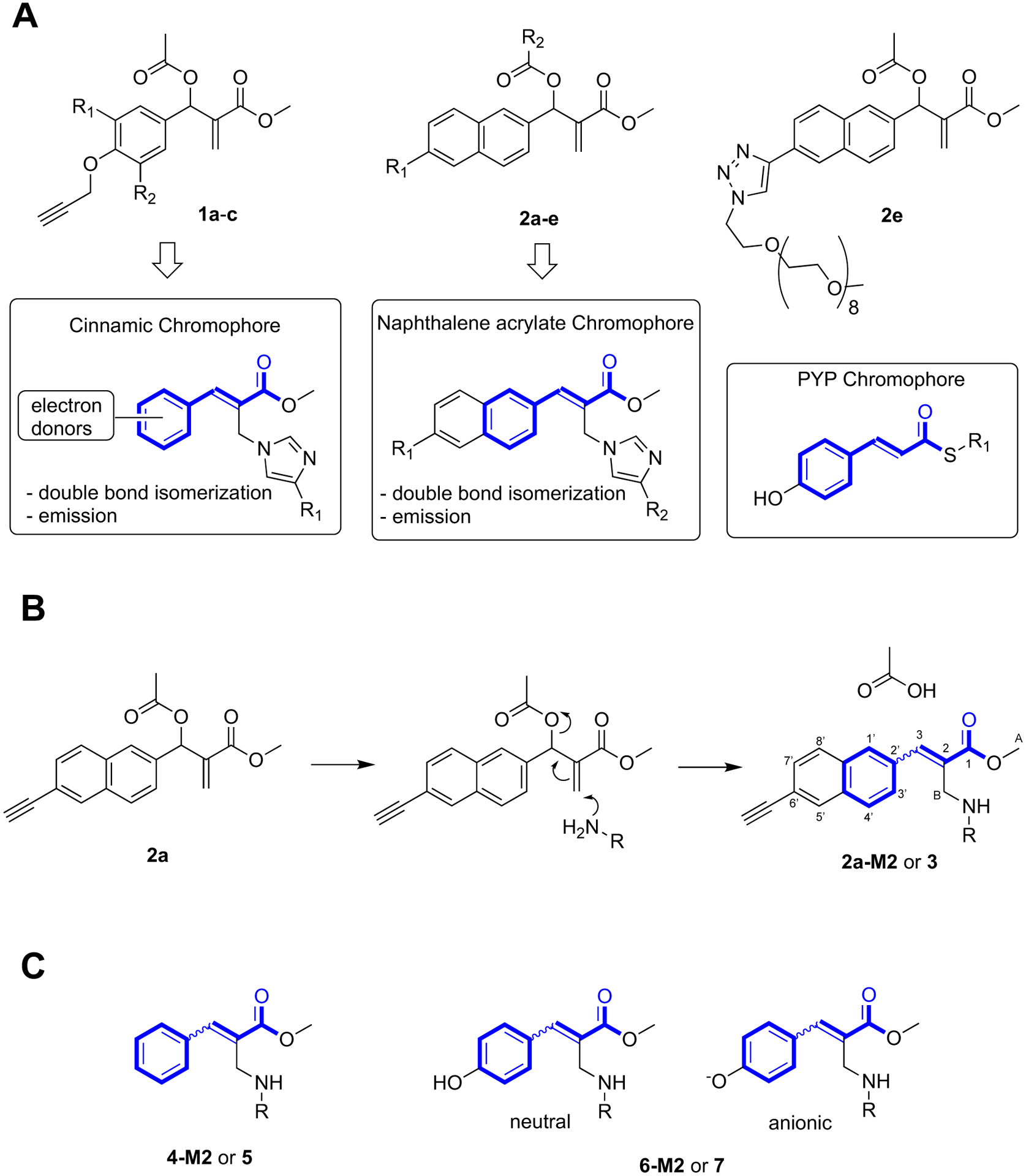
MBHAs reactivity. A. Structures of protein-reactive MBHAs 1a-c (1a: R1 = H, R2 = H; 1b: R1 = H R2 = OMe, 1c: R1 = OMe, R2 = OMe) and 2a-e (2a: R1 = C≡CH, R2 = OAc; 2b: R1 = OCH2C≡CH, R2 = OAc, 2c: R1 = OCH2C≡CH, R2 = COCH2CH2(OCH2CH2)11OMe, 2d: R1 = C≡CH, R2 = COCH2CH2(OCH2CH2)11OMe) and of their products of conjugation with proteins compared (blue-bold) with the PYP chromophore. B. Reaction of MBHA 2a with a primary amino group leading to 2a-M2 (R = side-chain of Lys111) and 3 (R = n-butyl, reagent n-butylamine, CHCl3). C. Phenyl-acrylate and p-hydroxyphenyl-acrylate protein constructs and corresponding models. 4-M2 (R = side-chain of Lys111) and 5 (R = n-butyl). 6-M2 (R = side-chain of Lys111) and 7 (R = n-butyl).
As shown in bold in Fig. 1A and 1B, the achieved functionalization would generate unnatural prosthetic groups incorporating the framework of natural chromophores such as that of the photoactive yellow protein (PYP),[35] the most thoroughly characterized blue-light eubacterial photoreceptor belonging to the xanthopsin family. This observation together with the fact that certain PYP constructs have, for instance, been employed to modulate the genetic expression via light irradiation,[38] provides a realistic route to flexible syntheses of novel optogenetic tools as well as to the development of a “lab-on-a-molecule” concept: a prospective tool for the systematic study of the effects of the position and orientation of amino acid residues on synthetic chromophores.
Our study starts by describing the conjugation of the sterically demanding MBHA 2a to a selected hCRABPII mutant (hereafter called M2) and the structural characterization of the resulting 2a-M2 conjugate. It then follows by looking at the consequences of the steric reduction (i.e. from 2a-M2 to 4-M2) and increase in polarity (i.e. from 4-M2 to 6-M2) of the employed MBHA, ultimately achieving a hCRABPII-based construct structurally resembling PYP.
2. Results and discussion
The results of MBHA conjugation studies[37] with an antibody bearing a hexahistidine tag and other nucleophiles including several lysine residues, suggested that 2e reacts with the residues of the peptide tag with a partial selectivity. In fact, digestion studies identified both arginine and lysine residues as reacting nucleophiles.[37] This result provided the motivation for testing the conjugation of MBHAs with proteins offering one lysine site embedded in a molecular cavity. The hope was that the lipophilic cavity environment could make possible the systematic construction of novel synthetic LRPs selectively, irreversibly and in large quantity.
Our study was conducted by using, as a lab model, the previously reported hCRABPII hexa-mutant (R111K:R134F:T54V:R132Q:P39Y:R59Y) variant M2, whose conjugation derivative with a retinal chromophore precursor had been shown to crystallize easily.[30] M2 was reacted with equimolar amounts of the MBHA 2a, and the reaction kinetic followed by performing electrospray ionization mass spectrometry (ESI-MS) measurements at regular time intervals. Accordingly, 2a was dissolved in DMSO and the solution mixed with a buffer containing the protein (see the details in the Supporting Information). We assumed that the conjugation between M2 and 2a occurs in two sequential steps: (i) formation of a non-covalent complex 2a@M2 between the MBHA and the protein and (ii) an addition-elimination reaction (Fig. 1B) leading to release of an acetate molecule and formation of a 2a-M2 conjugate.
ESI-MS measurements, which were used to investigate the kinetics of the process, confirmed that 2a reacts with M2. In fact, 24 hours after the mixing, the mass spectrum showed the presence of mono- and di-conjugates along with unreacted M2 and small amounts of tri- and tetra-conjugates (see Fig. S1–S4 in Supporting Information). The M2 sequence displays 14 lysine and 4 arginine residues plus a single histidine residue located near the N-terminus. The previously reported reactivity of 2a with imidazole nucleophiles,[36] suggested formation of mono- and di-conjugates on the imidazole ring of the histidine residue. To investigate such hypothesis, M2 and 2a-M2 were crystallized and submitted to X-ray diffraction analysis (see supporting information for the details). The structure of M2 was obtained at 2.40 Å resolution. The overall fold of the protein consists of two five-stranded β-sheets (β1 – β5 and β6 – β10) forming an orthogonal barrel and two α-helices (α1 and α2) that connect β1 and β2 (Fig. 2).
Figure 2.

X-ray crystallographic structure of M2 (PDB: 6Z2U). Cartoon representation of the M2 tertiary structure. α-helices are colored liliac-violet, β strands in magenta and loops in black. Histidine (orange carbon atoms), arginine (green carbon atoms) and lysine (yellow carbon atoms) are displayed in sticks. A label marks the reactive Lys111 site.
The binding pocket, defined by the two β-sheets, is about 13.0 Å deep, 10.0 Å by 10.0 Å wide and partially solvent exposed. The construct resulted to be highly similar to that of a retinal-M2 conjugate reported in literature (see Fig. S5).[30] Being located in the N-terminus, the single histidine residue is solvent-exposed (Fig. 2) and characterized by positional disorder that prevents modeling of its side-chain. The structure also shows that all arginine and almost all lysine residues are located on the solvent-exposed protein surface. The only exception is Lys111 that is located inside the binding pocket (Fig. 2).
Despite the inhomogeneity of the species in solution, the crystallization of the reaction mixture at 24 hours gave us an interesting result. The structure of the 2a-M2 conjugate (Fig. 1B, 3B) was determined at 2.55 Å resolution. A single protein chain populates the crystal asymmetric unit, displaying a conserved structure with respect to the unreacted protein. Remarkably, the structure shows that the electrophilic dangling double bond of 2a was attacked by the amine nitrogen of Lys111 forming a synthetic prosthetic group with an extended π-system and, therefore, functioning as a chromophore. It also shows that the conjugate displays a C2=C3 bond with a Z configuration (this may be seen as counterintuitive given the specific design of the protein cavity to host an all-E retinoic acid guest). The obtained chromophore, which is reminiscent of the neutral (protonated) thiol-cinnamate chromophore of PYP (Fig. 1A), corresponds to a naphthalene-acrylate moiety forming a bond angle between the Lys111 Cε-N bond and the CB carbon of 117.5° ± 6.8°. No crystallographic water molecules were seen inside the cavity. Notice that, the occupancy of the protein cavity estimated on the basis of the refined atomic displacement parameters (see details in the Supporting Information) was found to be 70% ± 9% indicating, on the one hand, the possible selection of mono-functionalized derivatives during the crystallization process, and on the other hand, that 2a can reacts preferentially with the internal Lys111 residue rather than with the external M2 nucleophilic sites with the internal functionalization present also in the multi-derivatized species.
Figure 3.

Binding pocket structure. A. M2 pocket with its hydrophobic residues displayed in sticks (carbon atoms in green). B. 2a-M2 pocket (PDB: 6Z2Z). The chromophore and surrounding amino acid residues are shown as sticks (orange and green colors indicate carbon atoms. Oxygen, nitrogen, and sulphur atoms are colored in red, blue, and yellow, respectively). C. Superposition of the binding pockets of 2a-M2 and retinal-M2 rhodopsin mimics (PDB: 4YFQ) featuring the all-trans retinal protonated Schiff base chromophore in cyan. Residues are color coded as the respective chromophores. Oxygen and nitrogen atoms are colored red and blue, respectively. D. The same comparison with the 15-cis retinal protonated Schiff base (PDB: 4YFP).
We hypothesize that the observed Z configuration of C2=C3 is the result of a diastereoselective attack driven by the asymmetric M2 cavity and due to stabilizing van der Waals interactions of the naphthalene moiety with Leu19, Tyr39, Val54, Val76, Trp109 and Met123 (Fig. 3B). This is supported by the observation that the reaction of 2a with n-butylamine in chloroform is not diastereoselective affording the model compound 3 (Fig. 1B) with an E/Z ratio of 7/3 suggesting that C2-C3 single bond twisting was necessary to stabilize a specific 2a conformer in 2a@M2 and achieve a fully diastereoselective nucleophilic attack ultimately leading to 2a-M2.
The described 2a-M2 structure parallels that reported for the above mentioned retinal-M2 rhodopsin mimic.[30] This is a synthetic LRP obtained employing the natural click-like chemistry to conjugate all-trans retinal and M2 rather than to an opsin. The resulting conjugate mimics, structurally, the members of a vast family of photoreceptors called rhodopsins, which include visual pigments.[7] From inspection of Fig. 3C it is apparent that, in spite of substantial structural differences, 2a replaces all-trans-retinal in the interaction with the M2 pocket indicating, when considering the modest reactivity of MBHAs with nucleophilic residues, a diffusion-controlled process leading to 2a@M2 formation. Thus, the presence of small amounts of di-substituted, as well as tri- and tetra-substituted, 2a-M2 adducts in the ESI-MS spectrum obtained at long incubation times (e. g. after 24 h) can be explained by unfavored attacks to the external nucleophilic residues competing with 2a@M2 formation. 2a-M2 formation has also been tentatively followed via UV-vis absorption spectroscopy (see Fig. S8 and related text in the Supporting Information).
2a-M2 is not a close PYP mimics as it does not incorporate the less sterically demanding and more polar p-hydroxyphenyl-thioacrylate (p-hydroxy-thiocinnamate) chromophore (Fig. 1A), which could be accommodated into the cavity better than the ethynylnaphthyl-acrylate moiety of 2a-M2. Therefore, in order to develop closer mimics showing hopefully higher selectivity, we leveraged the MBHA preparation flexibility to achieve the unsubstituted phenyl-acrylate (cinnamate) (see structure 4-M2 in Fig. 1C) and the p-hydroxy-phenyl-acrylate chromophores (see structure 6-M2 in Fig. 1C). Accordingly, we prepared MBHA derivative 4 (see Fig. 4A, top), which was then conjugated to M2 (see the Supporting Information for details) successfully using the same incubation and crystallization procedure used previously. X-ray crystallographic analysis on the obtained crystals indicated that the resulting conjugate (see Fig. 4B) incorporates the wanted chromophore on the Lys111 with a 80% ± 10% occupancy (estimated on the basis of the refined atomic displacement parameters, as detailed in the Supporting Information). Moreover, the structure confirms that the Z-diastereoselectivity of the conjugation reaction is conserved. Interestingly, it is apparent that the reduced steric hindrance of the 4-M2 chromophore leads to a different cavity localization with respect to the naphthyl-acrylate chromophore of 2a-M2 (see Fig. 4B), possibly as a consequence of favorable π-stacking interaction with Trp109 or, from another point of view, a higher fit into the small binding cavity between Trp109 and Val76.
Figure 4.
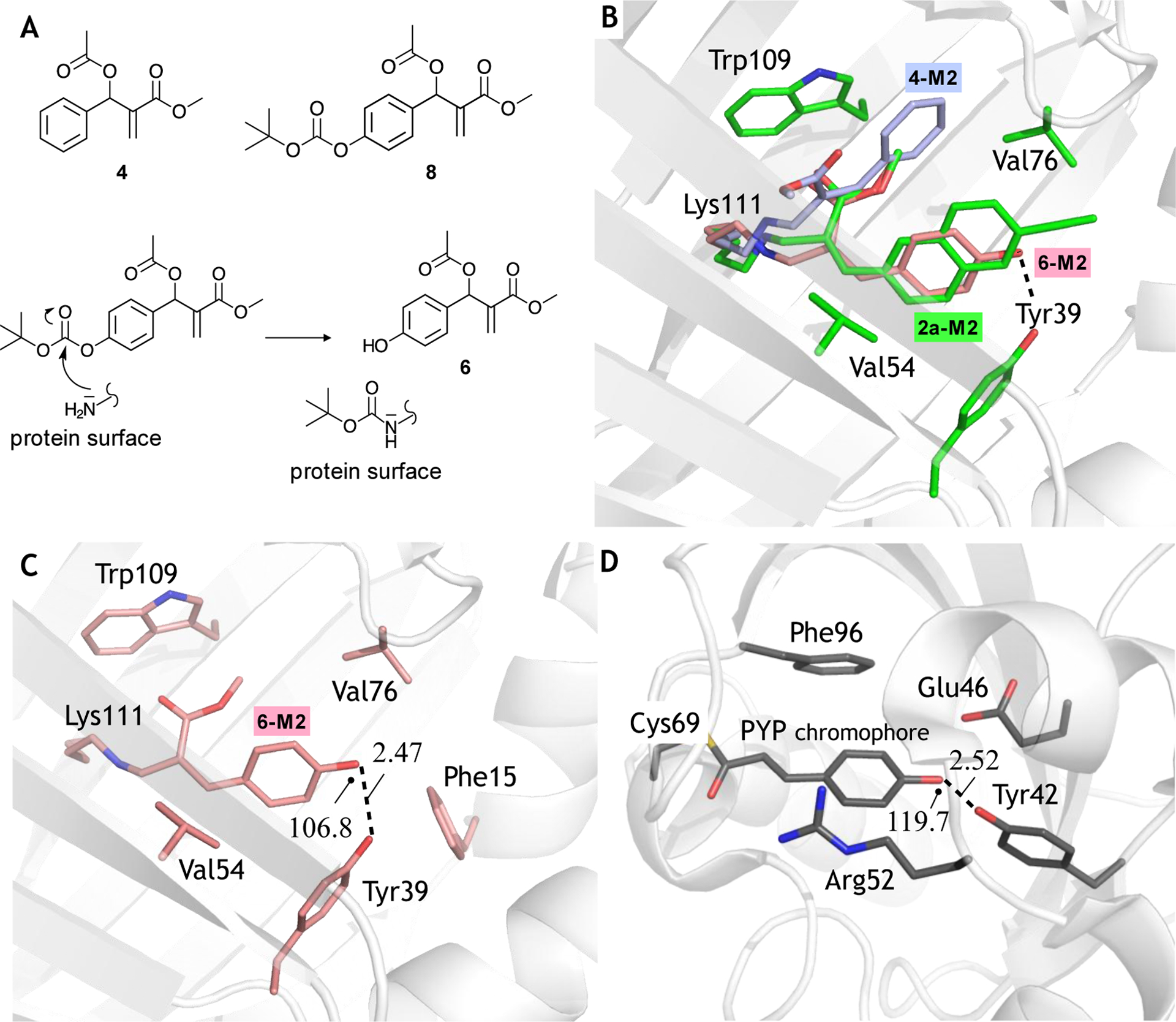
Structural characterization of Xanthopsin-like systems. A. Top. MBHAs used to prepare the conjugates 4-M2 and 6-M2 (see Fig. 1). Bottom. Possible mechanism for the in situ production of the MBHA 6. B. Chromophore-binding cavity view of the superimposition between 2a-M2 construct (PDB id: 6Z2Z) (white cartoon and green carbons) with the phenolic chromophore of 6-M2 (PDB id: 6ZSW) (in stick pink carbons) and phenolic chromophore of 4-M2 (PDB id: 6ZSX) (in stick light blue carbons). Residues are color coded as 2a carbons. In the figure, oxygen and nitrogen atoms are colored red and blue, respectively. C. Chromophore-binding cavity view of 6-M2 construct. White color was used in the cartoon for the M2 portion, while the generated chromophore and the amino acid surroundings are shown as stick pink carbons. D. Chromophore-binding cavity view of the PYP X-ray crystallographic structure (PDB id: 2ZOI). White color was used in the cartoon for the protein portion, while its p-hydroxythiocinnamate chromophore and the surroundings residues are shown as stick grey carbons. Oxygen atoms are colored in red, nitrogen atoms in blue, and the sulfur atom in yellow. In panels C and D the marked (see dashed line) hydrogen bond lengths is given Å and the relative angles in degrees.
Inspection of the same crystallographic structure revealed that the ability of the Tyr39 residue to form hydrogen bonds may be exploited to re-orient the phenyl-acrylate chromophore. More specifically, we hypothesized that a suitable substituent in the p-position of the chromophore phenyl moiety would provide a hydrogen bond acceptor (or donor) site possibly binding to Tyr39. More relevantly for the design of a PYP mimic, the resulting p-hydroxyphenyl moiety would be the same found in the PYP chromophore. We therefore attempted the synthesis of a suitable MBHA which we hoped could yield the biomimetic 6-M2 conjugate of Fig. 1C. Owing to the reactivity of the phenolic function, we prepared the MBHA 8 (see Experimental Section) featuring a tert-butyloxycarbonyl (BOC) protected phenolic hydroxyl function with the hope that the resulting conjugate could then be deprotected in physiological conditions. Remarkably, the X-ray crystallographic analysis carried out on the crystals obtained from the reaction mixture of 8 and M2 incubated for 24 hours showed a free phenol function pointing to a “spontaneous” deprotection event during the conjugation or crystallization steps and yielding the 6-M2 target (see Fig. 4B and Fig. 4C). In both covalent adducts 6-M2 and 4-M2 the angle between the Lys111 Cε-N bond and the chromophore CB carbon is of 116.9° (± 3.2°), coherently with the 2a-M2 adduct. ESI-MS measurements performed on the mixture of the conjugation reaction between 8 and M2 showed the presence, along with the expected conjugate MS peak at around 16162 m/z, of an intense peak at 16262.5 m/z (see Fig. S6, Supporting Information), which suggested that the BOC group was still present in a significant amount of 6-M2 after conjugation step. In order to rationalize this apparent discrepancy with respect to the results obtained by crystallography, we took into consideration also the hypothesis that the BOC could be covalently linked to one or different mobile protein fragment located in the protein surface. As schematically illustrated in Fig. 4A bottom, and supported by ref. [39], this would be indeed possible via a nucleophilic attack of an M2 external lysine (or any other external nucleophilic residue) to the BOC carbonyl of 8, ultimately leading to formation of the desired MBHA 6 that then fully penetrates in the protein cavity to form the 6@M2 complex and react with the Lys111 residue. Such externally linked BOC would not be detectable in the crystallographic analysis presumably due to its high mobility. On the other hand, a significant portion of the BOC protecting group appeared be lost during the crystallization step as suggested by ESI-MS measurements performed on the crystals used in the crystallographic studies (see the peak at 16163.2 in Fig. S7, Supporting Information).
As apparent from inspection of the X-ray crystallographic structure of Fig. 4B and, in spite of a steric encumbrance similar to that of the phenyl-acrylate hosted in 4-M2, the 6-M2 chromophore has recovered the cavity orientation of the naphthalene-acrylate and retinal chromophores found in ionic 2a-M2 and retinal-M2 respectively. The same structure supports the conclusion that a hydrogen bond connecting the Tyr39 residue with the phenolic hydroxyl group of the chromophore play a crucial role in determining both the chromophore re-orientation and the high selectivity of the interaction/reaction (as indicated by a 80% ± 10% occupancy of the protein cavity estimated on the basis of the refined atomic displacement parameters). Furthermore, the comparison of Fig. 4C and 4D shows that a similar hydrogen bonded structure is found in the PYP X-ray crystallographic structure suggesting that 6-M2 incorporates a biomimetic feature known to be critical for the PYP function.[40,41] In PYP the hydroxyl moiety of the chromophore is positioned within H-bond distance from both Tyr42 hydroxyl and Glu46 carboxylate (2.52 ± 0.08 Å and 2.56 ± 0.08 Å, respectively). On the other hand, in 6-M2 the chromophore hydroxyl entails an H-bond only with Tyr39 (2.47 ± 0.24 Å), structurally matching Tyr42 of PYP, whereas hydrophobic residues are present on the facing wall of the cavity, not able to replace the function of the PYP glutamate. However, the 6-M2 and PYP chromophores and cavities show remarkable differences. Firstly, while the PYP chromophore features an isomerizable double-bond incorporated in an all-trans π-framework, in 6-M2 chromophore the π-framework displays a reactive Z (i. e. cis) C2=C3 double bond. Secondly, the hydrogen bond connecting the phenolic hydroxyl group of the chromophore to the −OH group of Tyr39 is less linear (i. e. 106.8° ± 3.2° in terms of C-O…..O angle) and, presumably, weaker than its PYP homologue (119.7° ± 1.1°). Thirdly, in PYP one has a positively charged Arg52 stabilizing a deprotonated (i.e. anionic) form of the chromophore, while the lack of such interaction suggests that 6-M2 incorporates a protonated (i. e. neutral) chromophore more similar to the transient intermediate generated during the PYP photocycle.[40]
While the present report focuses on the preparation and structural characterization of 6-M2 and homologues, these studies were integrated by spectroscopic and photochemical characterizations. To do so, we started by looking at compound 7 as a model of the 6-M2 chromophore in solution while the naphthyl and phenyl models (3 and 5 respectively) of 2a-M2 and 4-M2 were only partially characterized for comparative purposes (see Fig. S8–S10 and related text in the Supporting Information). Accordingly, the UV-visible absorption spectra of the pure E diastereoisomer of the model compound 7 (E-7) were measured in aqueous buffer (Tris) at pH 8 and after the addition of a KOH excess. As shown in Fig. 5A, they are characterized by an intense absorption at 308 nm with a shoulder at ca. 357 nm, which increases in intensity after the KOH addition.
Figure 5.

UV-Vis absorption spectra. A. Comparison of the normalized absorption spectra of M2, 7 (pure E diastereoisomer), and 8 in tris buffer solution and anionic form of 7 obtained by addition of KOH. B. Absorption spectra of M2, of the M2+8 mixture registered after 24 hours in tris buffer solution at concentration of 2.6 × 10−5 M and differential spectrum obtained by subtracting the spectrum of the first one from that of the second.
In contrast, the corresponding MBHA derivative 8 shows only negligible absorption in this region and therefore could be easily differentiated. Similarly, the native M2 protein show only weak absorption in the relevant 300–400 nm region. Interestingly, it was found that after conjugation (see Fig. 5B) only quantitative changes are observed in the 6-M2 spectrum compared to that of M2. In particular, the differential spectrum obtained subtracting the spectrum of M2 from that of 6-M2 reveals an increased absorption peaking around 280 nm. Accordingly, the absorption of the chromophore of 6-M2 would remaining hidden behind the M2 absorption band and only contribute to an intensity increase. These findings are interpreted by hypothesizing that the Z-diastereoisomer of 6-M2 (see Fig. 4C) has a blue shifted absorption with respect to the corresponding E-diastereoisomer of E-7 displaying, in water, 308 and 357 nm peaks. Such hypothesis is supported by literature data where a blue-shifted absorption for a Z form in water has been reported for many chromophore analogues including cinnamic, coumarin, ferulic, and caffeic acid.[42] Furthermore, a similar effect is demonstrated in PYP upon chromophore conversion from the trans to the cis form.[43] On the other hand, the shoulder around 310 nm shown in the same difference spectrum can be assigned to a minor protein fraction containing the E-diastereoisomer of the chromophore, most likely bounded to the protein surface.
The ability of the biomimetic chromophore to photoisomerize was investigated by focusing on the neutral form of 7 in methanol solution (see Fig. S11). In fact, in methanol (i) the E-7 stereochemistry seen in the X-ray crystallographic analysis (see Fig. S12) of the synthesized compound does not undergo thermal C2=C3 isomerization in the chosen experimental conditions and (ii) the absorption maximum value of the anionic form of E-7 (i. e. after addition of KOH or NaOH), is largely red-shifted with respect to the neutral form. Properties i-ii facilitate the interpretation of the outcome of irradiation experiments and provide evidence that a neutral chromophore is present in the M2 cavity as no red-shifted (ca. 350 nm) band is seen in Fig. 5B.
The photochemical reactivity of the biomimetic p-hydroxy-phenyl chromophore of 7 and 6-M2 (and also 3, see Fig. S13) is supported by the comparison of the ground (S0) and excited (S1) state structures of Z-7 (and Z-3 in Fig. S13) computed via multiconfigurational second-order perturbation theory quantum chemical calculations (see the Supporting Information for details). As illustrated in Fig. 6A, we found that, for neutral Z-7 (and for Z-3, the Supporting Information) model in isolated conditions, the first allowed transition (S0→S2) may trigger an excited state relaxation along stretching and torsional modes and leads to S1 excited state equilibrium geometries with inverted bond orders and increased or, in the case of Z-7, complete torsional deformation indicating facile C2=C3 twisting and isomerization (see the Supporting Information for an analysis of the electronic transitions). The corresponding S0 and S1 electronic structures indicate that the relaxation is accompanied by partial charge separation leading to a negatively charged acrylate and p-hydroxyphenyl (for Z-7) or naphthyl (for Z-3) moieties. These will therefore behave like photo-bases not releasing but, rather, conserving the protonation state during the excited state relaxation.
Figure 6.

Photoisomerization of the neutral form of 7. A. Excitation energies (kcal/mol) of the S0 and S1 equilibrium (eq.) structures of the neutral form of Z-7. The relaxation from the allowed S2 state (exp. 318 nm in solution vs. comp. 257 nm (#303 nm after correcting the geometry for electron correlation effects) in vacuo may involve a S2/S1 conical intersection. The relevant bond lengths (Å) and torsion (degrees) of the same structures are also given. Red (negative) and blue (positive) balloons represent charge magnitudes (positive electron units). B. 1H NMR spectra of E-7 in methanol-d4. C. 1H NMR spectra of the E-7 and Z-7 mixture obtained after irradiating E-7 for two hours with a UV-B light lamp. D. The absorption spectra of neutral E-7 in methanol (black line) and of its E/Z mixture (red line) obtained after continuous irradiation at 313 nm for 18 minutes is consistent with a blue-shifted absorption for Z-7 with respect to E-7.
When ca. 2 mg of E-7 were dissolved in deuterated methanol and irradiated with UV-B light for two hours, the 1H NMR spectrum (compare Fig. 6B and Fig. 6C) showed the presence of the Z-7 (50%) mixed with the E-7 (50%) indicating that the 6-M2 chromophore is, potentially, photochemically reactive. The irradiated solution was stored at room temperature in the dark for a few days without displaying a significant change in E/Z composition. The absorption spectra recorded before and after the irradiation shows, as hypothesized above, that the blue-shifted absorption of Z-7 would be hidden (see above and Fig. 5B) by the protein absorption in 6-M2. However, the supported effectiveness of the photoisomerization of the biomimetic p-hydroxy-phenyl chromophore could be largely modulated by the M2 cavity. Thus, photoisomerization experiments were performed with 6-M2 in Tris buffer at different pH values.
The irradiation of 6-M2 at 280 nm at pH 8, and 10.5 (the pH 10.5 spectra are displayed in Figure S14) produced only negligible effects in the UV-visible absorption spectrum (e. i. Fig. 7A), suggesting that the photoisomerization of the Z form of the chromophore is hampered by the interactions with the protein cavity, which mainly conserves the native folding as suggested by the Circular Dichroism (CD) spectra (see Fig. S15B). On the other hand, after the protein de-folding observed at pH 12.7 (see CD spectra of M2 and 6-M2 samples in Fig. S15), no spectral variations have been detected in the dark. In contrast, irradiation at 280 nm induce an increase of a broad band around 350 nm. These facts are consistent with the photoinduced generation of the E-form of the chromophore. In fact, the raising absorption band of Fig. 7B, is in good agreement with the one observed in the E-7 absorption spectrum recorded in buffer in the presence of a KOH excess (see Fig. 5A).
Figure 7.

Kinetic isomerization of 6-M2 monitored by UV-vis spectra registered under continuous irradiation at 280 nm in tris buffer solution pH 8 (A) and in tris buffer solution added with NaOH until pH 12.7 (B). In panel B, the arrows indicate the progressive increase or decrease of the spectrum as a consequence of the irradiation.
The results above support the effectiveness of the photoisomerization process when the chromophore is located in the, presumably, looser surrounding generated by protein de-folding. Moreover, they confirmed the selective interaction/reaction of MBHA derivative 6 with M2 binding pocket displayed by the 6-M2 crystallographic structure.
3. Conclusion
We have demonstrated the feasibility of a site-specific click-functionalization of a lysine residue buried into a lipophilic binding sites and leading to the production of three different xanthopsin-like structures. This result opens the path to the preparation of entire libraries of novel synthetic LRPs through the design of diverse MBHAs whose reactivity with M2 may be computationally screened via docking and, as done for rhodopsin mimics,[44–47] studied via quantum chemical calculations (see ref. [46]) and X-ray crystallographic analysis. For instance, such libraries can be prepared by: (i) designing MBHAs yielding sets of diversely substituted p-hydroxycinnamate (p-coumarate) derivatives that would then yield M2-based xanthopsin mimics with different spectroscopic and reactivity properties, (ii) engineering M2 variants to be conjugated with a specific MBHA derivative and useful for tuning/controlling, via the protein environment, the chromophore photochemistry and (iii) combining concepts i-ii to engineer systems where both the chromophore and protein are modified, yielding a plethora of M2-based xanthopsin-like LRPs to be used as a “lab-on-a-molecule”: a tool that would allow the systematic modification of a reference LRP and, as a consequence, the discovery or verification of engineering principles (e.g. for the tuning of light absorption, emission and photochemical reactivity). An application of such a tool in the investigation of the photoisomerization mechanism of green fluorescent protein variants has been recently reported.[48] Future more technical studies will be instead devoted to quantify the selectivity towards the protein pocket. This could involve digestion studies of 6-M2 coupled with mass spectroscopic analysis. Furthermore, using protein enriched with nitrogen-15 and/or ligand bearing a carbon-13 label it will be possible to carry out structural studies such as 1H−15N HSQC and 1H-13C HSQC aimed at detecting the chromophores outside the site of interest. These methods have already been used by the authors in past studies.[16,37]
The research activity outlined above will be continued in our laboratories. One target will be learning how to design an LPR photoisomerization function mimicking that of natural systems such as PYP with obvious applications in optogenetics and synthetic biology. A second objective will be related to the ongoing developments in time-resolved X-ray and ultrafast electron diffraction techniques that are beginning to allow the imaging of the nuclear and electronic response to light irradiation in systems ranging from organic chromophores to PYP[49] and rhodopsins.[50–52] In this context we believe that the development of the presented MBHAs-based technology will allow the preparation of robust and easy to crystallize model systems for systematic time-resolved structural studies.
4. Experimental Section/Methods
Materials and Methods.
Chemistry.
Yields refer to purified products and are not optimized. Merck silica gel 60 (230–400 mesh) was used for column chromatography. Merck TLC plates, silica gel 60 F254 were used for TLC. NMR spectra were recorded with either a Bruker DRX-400 AVANCE, a Bruker AMX-600 AVANCE, Varian Mercury 300, or a Varian INOVA 500 spectrometer operating at 400, 600, 300, and 500 MHz respectively, in the indicated solvents (TMS as internal standard); the values of the chemical shifts (δ) are expressed in ppm and the coupling constants (J) in Hz. Mass spectra were recorded on an Agilent 1100 LC/MSD. The absorption spectra were recorded with a PerkinElmer Lambda 40.
MBHA derivatives 2a and 4 were synthesized as reported in the literature, (ref. [35] and [31]). MBHA derivative 8 was prepared by modifying a previously published procedure (Scheme 1).
Scheme 1.
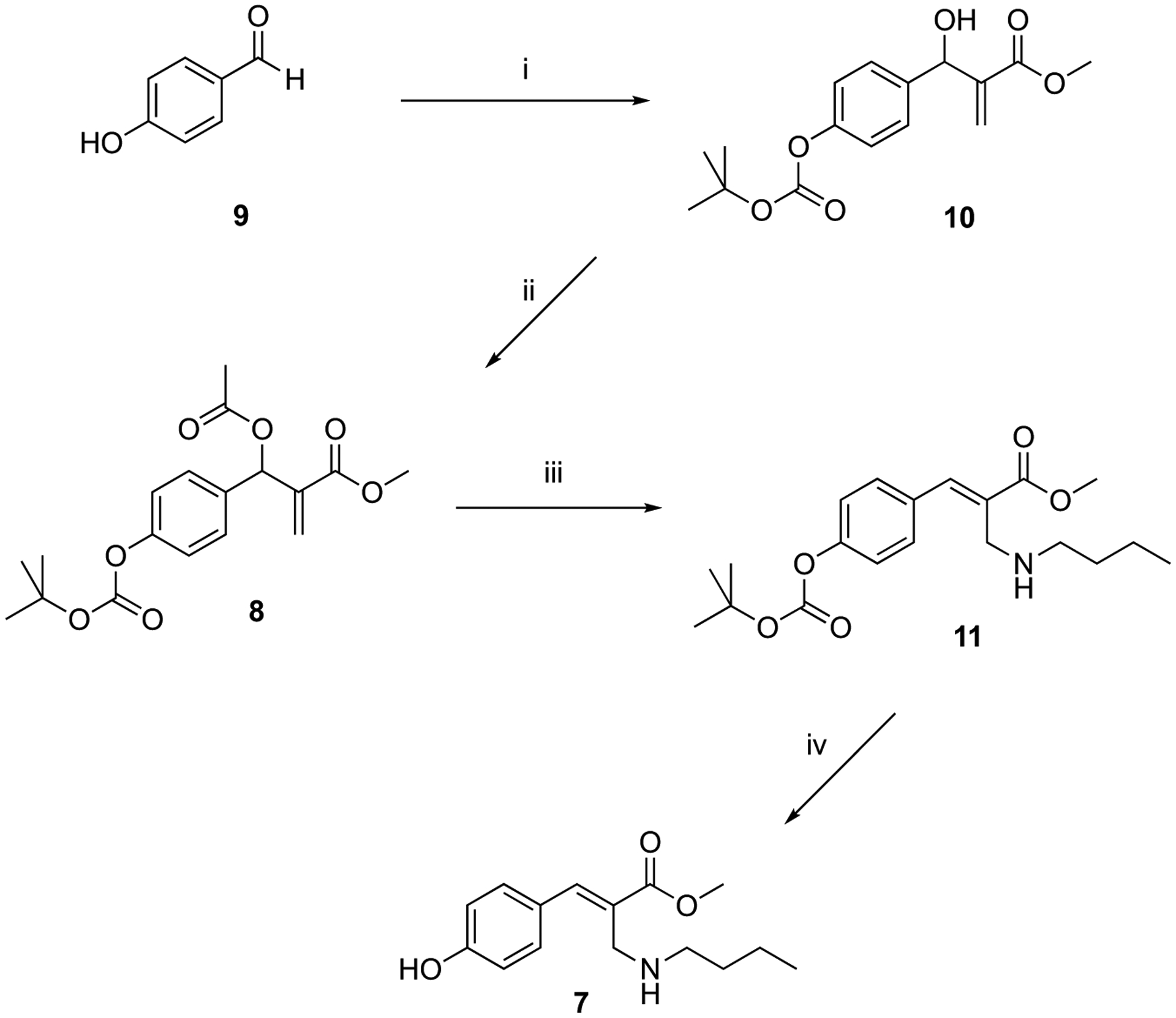
Synthesis of MBHA derivative 8 and its reaction with n-butylamine.
Reagents: (i) BOC anhydride, methyl acrylate, DABCO, CH3OH, THF; (ii) CH3COCl, TEA, CH2Cl2; (iii) n-butylamine, THF-H2O (5:1); (iv) CF3COOH, CH2Cl2.
Methyl (E)-2-[(butylamino)methyl]-3-(6-ethynylnaphthalen-2-yl)acrylate (3).
A mixture of MBHA acetate 2a[35] (53 mg, 0.172 mmol) in chloroform (5.0 mL) containing n-butylamine (0.080 mL, 0.859 mmol) was stirred at room temperature for 2 h. The reaction mixture was concentrated under reduced pressure and the resulting residue was purified by flash chromatography with petroleum ether-ethyl acetate (8:2) as the eluent to obtain pure amine derivative 3 as a pale-yellow oil (38 mg, yield 69%). 1H NMR (400 MHz, CD3OD): 0.84 (t, J = 7.3, 3H), 1.20–1.36 (m, 2H), 1.37–1.46 (m, 2H), 2.57 (t, J = 7.4, 2H), 3.60 (s, 1H), 3.71 (s, 2H), 3.85 (s, 3H), 7.53 (d, J = 8.4, 1H), 7.57 (d, J = 8.5, 1H), 7.88 (m, 2H), 7.95 (s, 1H), 8.00 (s, 1H), 8.02 (s, 1H). 13C NMR (75 MHz, CD3OD): 15.1, 22.3, 33.3, 47.0, 50.7, 53.6, 80.7, 83.4, 123.1, 129.4, 130.0, 130.5, 130.9, 131.1, 133.0, 133.6, 134.0, 134.9, 135.7, 143.7, 170.5. HRMS (ESI) m/z: [M + H]+ Calcd for C21H24NO2 322.1802; Found 322.1800.
Methyl (E)-2-[(butylamino)methyl]-3-phenylacrylate (5).
MBHA acetate 4[31] (0.21 g, 0.897 mmol) was dissolved in 10 mL of THF-H2O (5:1 v/v) containing n-butylamine (0.13 mL, 1.36 mmol). The resulting mixture was stirred at reflux temperature for 1 h and then concentrated under reduced pressure. The residue was purified by flash chromatography with petroleum ether-ethyl acetate (1:1) as the eluent to obtain a pale-yellow oil consisting of amine derivative 5 as mixture (7:3) of E and Z isomers (0.11 g, yield 51%). 1H NMR (400 MHz, CDCl3): 0.78 (t, J = 7.3, 3H Z), 0.89 (t, J = 7.3, 3H E), 1.10–1.52 (m, 4H Z, 4H E), 2.37 (t, J = 7.3, 2H Z), 2.60 (t, J = 7.0, 2H E), 3.45 (s, 2H Z), 3.58 (s, 2H E), 3.71 (s, 3H Z), 3.82 (s, 3H E), 7.28–7.44 (m, 3H Z, 3H E), 7.47 (d, J = 7.5, 2H E), 7.54 (m, 2H Z), 7.76 (s, 1H Z), 7.80 (s, 1H E). Pure E isomer was obtained as pale-yellow oil by further flash chromatography purification using petroleum ether-ethyl acetate (7:3) as the eluent. 1H NMR (600 MHz, CD3OD): 0.89 (t, J = 7.4, 3H), 1.30 (m, 2H), 1.40 (m, 2H), 2.57 (t, J = 7.4, 2H), 3.67 (s, 2H), 3.84 (s, 3H), 7.41–7.47 (m, 5H), 7.89 (s, 1H). 13C NMR (150 MHz, CD3OD): 15.1, 22.2, 32.9, 46.8, 50.5, 55.0, 130.6, 131.0, 131.3, 131.7, 137.1, 144.7, 170.4. HRMS (ESI) m/z: [M + H]+ Calcd for C15H22NO2 248.1645; Found 248.1647.
Methyl 2-[[4-[(tert-butoxycarbonyl)oxy]phenyl](hydroxy)methyl]acrylate (10).
A mixture of aldehyde 9 (1.0 g, 8.19 mmol), THF (5.0 mL) containing di-tert-butyl dicarbonate (BOC anhydride) (1.94 g, 8.19 mmol), was stirred at room temperature for 15 min. Then, ethyl acrylate (10 mL), methanol (10 mL) and 1,4-diazabicyclo[2.2.2]octane (DABCO) (0.92 g, 8.19 mmol) were added and the resulting mixture was stirred at room temperature for 48 h in the darkness, and then concentrated under reduced pressure. The resulting residue was dissolved in dichloromethane and washed six times with a solution of NH4Cl. The organic layer was dried over sodium sulfate and concentrated under reduced pressure and purified by flash chromatography with dichloromethane as the eluent to obtain compound 10 (1.08 g, yield 43%) as a white solid melting at 71–72 °C. 1H NMR (400 MHz, CDCl3): 1.53 (s, 9H), 3.01 (d, J = 5.4, 1H), 3.72 (s, 3H), 5.55 (d, J = 5.4, 1H), 5.82 (s, 1H), 6.32 (s, 1H), 7.14 (d, J = 8.4, 2H), 7.38 (d, J = 8.3, 2H). 13C NMR (125 MHz, CDCl3): 27.7, 52.0, 72.6, 83.6, 121.2, 126.1, 127.7, 138.8, 141.8, 150.5, 151.8, 166.6. HRMS (ESI) m/z: [M + Na]+ Calcd for C16H20NaO6 331.1152; Found 331.1158.
Methyl 2-[acetoxy[4-[(tert-butoxycarbonyl)oxy]phenyl]methyl]acrylate (8).
To a solution of the alcohol 10 (0.10 g, 0.324 mmol), in dry dichloromethane (10 mL) containing TEA (0.11 mL, 0.811 mmol), acetyl chloride (0.046 mL, 0.648 mmol) was added dropwise. After stirring at room temperature for 1 h, the reaction mixture was washed with water. The organic layer was dried over sodium sulfate and concentrated under reduced pressure. The residue was purified by flash chromatography with petroleum ether-ethyl acetate (8:2) as the eluent to afford the corresponding acetate 8 (0.102 g, yield 90%) as a colorless oil, which crystallized on standing (mp 77–78 °C). 1H NMR (400 MHz, CDCl3): 1.53 (s, 9H), 2.08 (s, 3H,), 3.68 (s, 3H), 5.86 (s, 1H), 6.38 (s, 1H), 6.67 (s, 1H), 7.13 (d, J = 8.3, 2H), 7.37 (d, J = 8.3, 2H). 13C NMR (125 MHz, CDCl3): 21.0, 27.7, 52.0, 72.4, 83.7, 121.3, 125.7, 128.8, 135.3, 139.5, 150.9, 151.7, 165.2, 169.3. HRMS (ESI) m/z: [M + Na]+ Calcd for C18H22NaO7 373.1258; Found 373.1260.
Methyl (E)-3-[4-[(tert-butoxycarbonyl)oxy]phenyl]-2-[(butylamino)methyl]acrylate (11).
MBHA acetate 8 (0.15 g, 0.43 mmol) was dissolved in 10 mL of a mixture of THF-H2O (5:1 v/v) containing n-butylamine (0.066 mL, 0.64 mmol) and stirred at reflux temperature for 2 h. The reaction mixture was concentrated under reduced pressure and the resulting residue was purified by flash chromatography with petroleum ether-ethyl acetate (8:2) as the eluent to obtain pure amine derivative 11 as a pale-yellow oil (0.10 g, yield 64%). 1H NMR (400 MHz, CDCl3): 0.90 (t, J = 7.2, 3H), 1.20–1.36 (m, 2H), 1.37–1.46 (m, 2H), 1.56 (s, 9H), 2.61 (t, J = 7.0, 2H), 3.54 (s, 2H), 3.60 (s, 3H), 7.20 (d, J = 8.6, 2H), 7.53 (d, J = 8.6, 2H), 7.75 (s, 1H). HRMS (ESI) m/z: [M + H]+ Calcd for C20H30NO5 364.2118; Found 364.2122.
Methyl (E)-2-[(butylamino)methyl]-3-(4-hydroxyphenyl)acrylate (7).
To a solution of 11 (80 mg, 0.22 mmol), in dry dichloromethane (10 mL) trifluoroacetic acid (TFA) (0.025 mL, 0.33 mmol) was added dropwise. After stirring at room temperature for 1 h, the reaction mixture was concentrated under reduced pressure. The residue was purified by crystallization from ethyl acetate to afford the trifluoroacetate salt of compound 7 (47 mg, yield 63%) as a colorless solid (mp 141–142 °C). 1H NMR (400 MHz, CD3OD): 0.93 (t, J = 7.4, 3H), 1.35 (m, 2H), 1.60 (m, 2H), 3.0 (t, J = 8.0, 2H), 3.87 (s, 3H), 4.16 (s, 2H), 6.89 (d, J = 8.6, 2H), 7.35 (d, J = 8.6, 2H), 8.04 (s, 1H). 13C NMR (125 MHz, CDCl3): 13.8, 20.3, 31.2, 45.3, 48.7, 52.1, 115.9, 125.5, 126.0, 131.2, 143.6, 158.6, 168.7. HRMS (ESI) m/z: [M + H]+ Calcd for C15H22NO3 264.1594; Found 264.1596.
Supplementary Material
Acknowledgements
The authors are grateful to Tatiana Domratcheva for helpful discussions. This work was supported, in part, by the Italian MIUR for funding (PRIN 2015) and, in part, by the National Institute of Health under grant no. R15GM126627. The authors are also grateful to MIUR for a grant “Dipartimento di Eccellenza, 2018–2022”. The authors would like to thank Diamond Light Source for beamtime (proposal mx15832 and mx21741), and the staff of beamlines I03 and I04 for assistance with crystal testing and data collection.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
[CCDC 2071148 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.]
References
- [1].Rodriguez EA, Campbell RE, Lin JY, Lin MZ, Miyawaki A, Palmer AE, Shu X, Zhang J, Tsien RY, Trends Biochem. Sci 2017, 42, 111–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Schmidt D, Cho YK, Trends Biotechnol. 2015, 33, 80–91. [DOI] [PubMed] [Google Scholar]
- [3].Stano P, Mavelli F, Life 2015, 5, 1700–1702. [Google Scholar]
- [4].Zhang F, Vierock J, Yizhar O, Fenno LE, Tsunoda S, Kianianmomeni A, Prigge M, Berndt A, Cushman J, Polle J, Magnuson J, Hegemann P, Deisseroth K, Cell 2011, 147, 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kandori H, Bull. Chem. Soc. Jpn 2019, 93, 76–85. [Google Scholar]
- [6].Gozem S, Luk HL, Schapiro I, Olivucci M, Chem. Rev 2017, 117, 13502–13565. [DOI] [PubMed] [Google Scholar]
- [7].Ernst OP, Lodowski DT, Elstner M, Hegemann P, Brown LS, Kandori H, Chem. Rev 2014, 114, 126–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bedbrook CN, Yang KK, Robinson JE, Mackey ED, Gradinaru V, Arnold FH, Nat. Methods 2019, 16, 1176–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hoyt EA, Cal PMSD, Oliveira BL, Bernardes GJL, Nat. Rev. Chem 2019, 3, 147–171. [Google Scholar]
- [10].Devaraj NK, ACS Cent. Sci 2018, 4, 952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Shadish JA, DeForest CA, Matter 2020, 2, 50–77. [Google Scholar]
- [12].Spicer CD, Davis BG, Nat. Commun 2014, 5, 4740. [DOI] [PubMed] [Google Scholar]
- [13].Ward CC, Kleinman JI, Nomura DK, ACS Chem. Biol 2017, 12, 1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rosa-Neto P, Wängler B, Iovkova L, Boening G, Reader A, Jurkschat K, Schirrmacher E, ChemBioChem 2009, 10, 1321–1324. [DOI] [PubMed] [Google Scholar]
- [15].Raindlová V, Pohl R, Hocek M, Chem. – A Eur. J 2012, 18, 4080–4087. [DOI] [PubMed] [Google Scholar]
- [16].Pagano K, Paolino M, Fusi S, Zanirato V, Trapella C, Giuliani G, Cappelli A, Zanzoni S, Molinari H, Ragona L, Olivucci M, J. Phys. Chem. Lett 2019, 10, 2235–2243. [DOI] [PubMed] [Google Scholar]
- [17].Smith BD, Higgin JJ, Raines RT, Bioorg. Med. Chem. Lett 2011, 21, 5029–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gunnoo SB, Madder A, ChemBioChem 2016, 17, 529–553. [DOI] [PubMed] [Google Scholar]
- [19].Krall N, da Cruz FP, Boutureira O, Bernardes GJL, Nat. Chem 2016, 8, 103–113. [DOI] [PubMed] [Google Scholar]
- [20].Hansen MB, Hubálek F, Skrydstrup T, Hoeg-Jensen T, Chem. – A Eur. J 2016, 22, 1572–1576. [DOI] [PubMed] [Google Scholar]
- [21].Wang L, Gruzdys V, Pang N, Meng F, Sun X-L, RSC Adv. 2014, 4, 39446–39452. [Google Scholar]
- [22].Solomatina AI, Chelushkin PS, V Krupenya D, Podkorytov IS, Artamonova TO, V Sizov V, Melnikov AS, V Gurzhiy V, Koshel EI, Shcheslavskiy VI, Tunik SP, Bioconjug. Chem 2017, 28, 426–437. [DOI] [PubMed] [Google Scholar]
- [23].Yang T, Li X-M, Bao X, Fung YME, Li XD, Nat. Chem. Biol 2016, 12, 70–72. [DOI] [PubMed] [Google Scholar]
- [24].Hoppmann C, Wong A, Yang B, Li S, Hunter T, Shokat KM, Wang L, Nat. Chem. Biol 2017, 13, 842–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yamaguchi A, Matsuda T, Ohtake K, Yanagisawa T, Yokoyama S, Fujiwara Y, Watanabe T, Hohsaka T, Sakamoto K, Bioconjug. Chem 2016, 27, 198–206. [DOI] [PubMed] [Google Scholar]
- [26].Isenegger PG, Davis BG, J. Am. Chem. Soc 2019, 141, 8005–8013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Walsh SJ, Omarjee S, Galloway WRJD, Kwan TT-L, Sore HF, Parker JS, Hyvönen M, Carroll JS, Spring DR, Chem. Sci 2019, 10, 694–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chen X, Muthoosamy K, Pfisterer A, Neumann B, Weil T, Bioconjug. Chem 2012, 23, 500–508. [DOI] [PubMed] [Google Scholar]
- [29].Matos MJ, Oliveira BL, Martínez-Sáez N, Guerreiro A, Cal PMSD, Bertoldo J, Maneiro M, Perkins E, Howard J, Deery MJ, Chalker JM, Corzana F, Jimenez-Oses G, Bernardes GJL, J. Am. Chem. Soc 2018, 140, 4004–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nosrati M, Berbasova T, Vasileiou C, Borhan B, Geiger JH, J. Am. Chem. Soc 2016, 138, 8802–8808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li J, Wang X, Zhang Y, Tetrahedron Lett. 2005, 46, 5233–5237. [Google Scholar]
- [32].Liu XX, Melman A, Chem. Commun 2013, 49, 9042–9044. [DOI] [PubMed] [Google Scholar]
- [33].Rodrigues MT, Santos MS, Santos H, Coelho F, Tetrahedron Lett. 2014, 55, 180–183. [Google Scholar]
- [34].Razzano V, Paolino M, Reale A, Giuliani G, Artusi R, Caselli G, Visintin M, Makovec F, Donati A, Villafiorita-Monteleone F, Botta C, Cappelli A, ACS Omega 2017, 2, 5453–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Razzano V, Paolino M, Reale A, Giuliani G, Donati A, Giorgi G, Artusi R, Caselli G, Visintin M, Makovec F, Battiato S, Samperi F, Villafiorita-Monteleone F, Botta C, Cappelli A, RSC Adv. 2018, 8, 8638–8656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Paolino M, Reale A, Razzano V, Giuliani G, Donati A, Bonechi C, Caselli G, Visintin M, Makovec F, Scialabba C, Licciardi M, Paccagnini E, Gentile M, Salvini L, Tavanti F, Menziani MC, Cappelli A, New J Chem. 2019, 43, 6834–6837. [Google Scholar]
- [37].Paolino M, Visintin M, Margotti E, Visentini M, Salvini L, Reale A, Razzano V, Giuliani G, Caselli G, Tavanti F, Menziani MC, Cappelli A, New J Chem. 2019, 43, 17946–17953. [Google Scholar]
- [38].Ali AM, Reis JM, Xia Y, Rashid AJ, Mercaldo V, Walters BJ, Brechun KE, Borisenko V, Josselyn SA, Karanicolas J, Wolley GA, Chem. Biol 2015, 22, 1531–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pittelkow M, Lewinsky R, Christensen JB, Synthesis (Stuttg). 2002, 2002, 2195–2202. [Google Scholar]
- [40].Larsen DS, van Grondelle R, Hellingwerf KJ, in Ultrashort Laser Pulses Biol. Med (Eds.: Braun M, Gilch P, Zinth W), Springer, 2008, pp. 165–199. [Google Scholar]
- [41].Hellingwerf KJ, Hendriks J, Gensch T, J. Phys. Chem. A 2003, 107, 1082–1094. [Google Scholar]
- [42].Salum ML, Arroyo Mañez P, Luque FJ, Erra-Balsells R, J. Photochem. Photobiol. B Biol 2015, 148, 128–135. [DOI] [PubMed] [Google Scholar]
- [43].Rubinstenn G, Vuister GW, Mulder FAA, Düx PE, Boelens R, Hellingwerf KJ, Kaptein R, Nat. Struct. Biol 1998, 5, 568–570. [DOI] [PubMed] [Google Scholar]
- [44].Huntress MM, Gozem S, Malley KR, Jailaubekov AE, Vasileiou C, Vengris M, Geiger JH, Borhan B, Schapiro I, Larsen DS, Olivucci M, J. Phys. Chem. B 2013, 117, 10053–10070. [DOI] [PubMed] [Google Scholar]
- [45].Gueye M, Manathunga M, Agathangelou D, Orozco Y, Paolino M, Fusi S, Haacke S, Olivucci M, Léonard J, Nat. Commun 2018, 9, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Manathunga M, Jenkins AJ, Orozco-Gonzalez Y, Ghanbarpour A, Borhan B, Geiger JH, Larsen DS, Olivucci M, J. Phys. Chem. Lett 2020, 11, 4245–4252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Schapiro I, Gueye M, Paolino M, Fusi S, Marchand G, Haacke S, Martin ME, Huntress M, Vysotskiy VP, Veryazov V, Leonard J, Olivucci M, Photochem. Photobiol. Sci 2019, 18, 2259–2269. [DOI] [PubMed] [Google Scholar]
- [48].Romei MG, Lin C-Y, Mathews II, Boxer SG, Science (80-. ) 2020, 367, 76–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Pande K, Hutchison CDM, Groenhof G, Aquila A, Robinson JS, Tenboer J, Basu S, Boutet S, DePonte DP, Liang M, White TA, Zatsepin NA, Yefanov O, Morozov D, Oberthuer D, Gati C, Subramanian G, James D, Zhao Y, Koralek J, Brayshaw J, Kupitz C, Conrad C, Roy-Chowdhury S, Coe JD, Metz M, Xavier PL, Grant TD, Koglin JE, Ketawala G, Fromme R, Srajer V, Henning R, Spence JCH, Ourmazd A, Schwander P, Weierstall U, Frank M, Fromme P, Barty A, Chapman HN, Moffat K, van Thor JJ, Schmidt M, Science (80-. ) 2016, 352, 725–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Nogly P, Weinert T, James D, Carbajo S, Ozerov D, Furrer A, Gashi D, Borin V, Skopintsev P, Jaeger K, Nass K, Båth P, Bosman R, Koglin J, Seaberg M, Lane T, Kekilli D, Brünle S, Tanaka T, Wu W, Milne C, White T, Barty A, Weierstall U, Panneels V, Nango E, Iwata S, Hunter M, Schapiro I, Schertler G, Neutze R, Standfuss J, Science (80-. ). 2018, 361, aat0094. [Google Scholar]
- [51].Nass Kovacs G, Colletier J-P, Grünbein ML, Yang Y, Stensitzki T, Batyuk A, Carbajo S, Doak RB, Ehrenberg D, Foucar L, Gasper R, Gorel A, Hilpert M, Kloos M, Koglin JE, Reinstein J, Roome CM, Schlesinger R, Seaberg M, Shoeman RL, Stricker M, Boutet S, Haacke S, Heberle J, Heyne K, Domratcheva T, Barends TRM, Schlichting I Nat. Commun 2019, 10, 3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Skopintsev P, Ehrenberg D, Weinert T, James D, Kar RK, Johnson PJM, Ozerov D, Furrer A, Martiel I, Dworkowski F, Nass K, Knopp G, Cirelli C, Arrell C, Gashi D, Mous S, Wranik M, Gruhl T, Kekilli D, Brünle S, Deupi X, Schertler GFX, Benoit RM, Panneels V, Nogly P, Schapiro I, Milne C, Heberle J, Standfuss J, Nature 2020, 583, 314–318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.