

Summary
Targeting protein quality control pathways using proteasome or p97/VCP inhibition can effectively treat blood tumors. However, in solid tumors, only p97/VCP inhibitors are effective. To probe this difference in efficacy, we tracked HCT116 colon cancer cells using temporal proteomics to define the cellular and molecular responses to proteasome and p97 inhibition. Proteins involved in general protein quality control pathways were similarly upregulated by both treatments, suggesting that the proteotoxic stress caused by inhibitors does not explain the differential therapeutic effectiveness. Unexpectedly, proteins specifically dysregulated by two p97 inhibitors are involved in cell cycle control. Indeed, eleven cell cycle proteins were downregulated by p97 inhibition but not by proteasome inhibition. Western blot analysis validated degradation of Cyclin D1 and Securin depends on proteasome but not on p97. Differing regulation of cell cycle proteins by p97 and the proteasome may therefore explain the therapeutic efficacy of p97 inhibitors in colon cancer.
Keywords: p97 inhibitor, proteasome inhibitor, E2F1, Cyclin D1, cell cycle, anticancer, proteomic
Graphical Abstract
eTOC Blurb
Wang et al. perform temporal proteomics of p97 and proteasome inhibition to reveal the differential mechanism of action and the distinct regulation of cell cycle oncoproteins. This work provides a systematic view of protein markers that are common as well as different caused by compounds targeting potential anticancer targets.
Introduction
Protein homeostasis depends on regulation of protein degradation through the ubiquitin-proteasome system (UPS) and autophagy. Dysregulation of protein homeostasis is associated with cancer and the development of neurodegenerative disease (Cheng et al., 2018). Imbalances in protein synthesis caused by mutations in protein coding sequences and aneuploidy drive cancer cells toward stronger reliance on protein quality control (PQC) mechanisms. Interfering with PQC through proteasome inhibition has proven successful as an anticancer treatment, as indicated by FDA approval of two proteasome inhibitors (PI), bortezomib and carfilzomib (Deshaies, 2014). In addition to the proteasome, p97 is another essential component of the protein homeostasis regulatory network and is implicated in several PQC pathways. p97 is a strong candidate as an alternative anticancer drug target in the PQC pathway (Ye et al., 2017). p97 is an ATPase that removes misfolded proteins from the endoplasmic reticulum (ER) membrane for proteasomal degradation (Ye et al., 2004, Wu and Rapoport, 2018) and also facilitates degradation of substrates embedded in other large structures, including mitochondria and chromosomes (Wolf and Stolz, 2012, van den Boom and Meyer, 2018, Ravanelli et al., 2020). In addition to its role in regulating protein homeostasis, p97 is also involved in non-degradative pathways including Golgi and nuclear envelope reassembly and endosomal trafficking (Meyer, 2005, Ramanathan and Ye, 2012, Kaneko et al., 2021, Meyer et al., 2002). Recently, p97 is shown to require to clear damaged lysosomes (Papadopoulos et al., 2020, Papadopoulos et al., 2017) and maintain lysosomal homeostasis (Arhzaouy et al., 2019).
To assess whether p97 is a potential anticancer drug target, groups, including ours, have performed chemical screens in search of compounds that directly inhibit the ATPase activity of p97. Such screens identified a reversible ATP competitive inhibitor, DBeQ, (Chou et al., 2011) and two different allosteric p97 inhibitors, NMS-873 (Magnaghi et al., 2013) and UPCDC-30245 (Alverez et al., 2015, LaPorte et al., 2018, Wang et al., 2020). Structure-activity-relationship (SAR) studies of DBeQ led to a collection of more potent and specific p97 inhibitors, including ML240 (Chou et al., 2013) and CB-5083 (Anderson et al., 2015, Zhou et al., 2015). CB-5083 strongly inhibited cancer cell proliferation, and was also efficacious in inhibiting tumor growth in mouse xenograft models implanted with HCT116 tumor cells (Anderson et al., 2015). CB-5083 entered phase I clinical trials for multiple myeloma and advanced solid tumors in 2015, highlighting p97 as a potential drug target in oncology. However clinical development was halted due to off-target effects (Huryn et al., 2020). p97 inhibitors remain a promising avenue however, and Cleave Therapeutics recently initiated a Phase 1 clinical trial of CB-5339 in patients with acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS). The National Cancer Institute (NCI) is also evaluating CB-5339 for patients with solid tumors and lymphomas (https://clinicaltrials.gov/ct2/show/NCT04372641 and NCT04402541).
Current data indicate that p97 inhibitors are promising treatments in solid tumors where proteasome inhibitors are ineffective (Yang et al., 2006, Engel et al., 2007, Schmid et al., 2008, Trinh et al., 2012). To facilitate the development of p97 inhibitors as potential therapeutic agents and help define their clinical application, it is necessary to dissect the mechanism of action (MOA) of p97 inhibitors and compare them with proteasome inhibitors (PIs). Moreover, identifying specific cellular markers is critical for both the drug discovery and development process, to help validate candidate drugs and quantify their effect. In this study, we performed proteomic analysis and systematically compared p97 shRNA knockdown (KD), and three p97 inhibitors (CB-5083, NMS-873 and UPCDC-30245), with the proteasome inhibitor MG132 in HCT116 colon cancer cells. Through proteomic analyses we uncover the specific molecular mechanism by which p97 inhibition is distinct from proteasome inhibition, potentially relevant to understanding the efficacious effects of the former in solid tumors.
Results
Proteomic profiling and analysis of p97 inhibition and knockdown.
The first orally bioavailable p97 inhibitor, CB-5083, was originally developed using xenografts-derived from HCT116 colon cancer cells (Anderson et al., 2015). We therefore chose HCT116 cells to evaluate the effect of p97 inhibition, and used proteomic analysis with label-free quantification (LFQ), which provides the deepest coverage of unperturbed proteomes (Li et al., 2012), to comprehensively measure the functional impact of p97 knockdown. From a 2h gradient run on nano-LC coupled with Eclipse mass spectrometry (MS), 6884 proteins were identified and quantified (Table S1). Principal component analysis (PCA) showed that replicate samples had similar principal component scores (Fig. S1a–b), indicating that our biological replicates correlated well with each other. By performing differential expression (DE) analysis, we have identified 410 proteins that were significantly upregulated, and 356 proteins that were significantly downregulated (p-value<0.05) in p97 KD HCT116 cells (Table S1). DE analysis confirmed that p97 was downregulated by 78% and that ATF3 and DDIT3 (CHOP) were upregulated more than 10-fold (Fig. 1a). The change in these proteins was consistent with western blot analysis (Fig. S1c–d).
Fig. 1. Proteomic analysis of p97 inhibition with shRNA knockdown and pharmacological inhibitors in HCT116 cells.

(a) Volcano plot displaying the proteomic changes following p97 knockdown, Log2FC indicates the logarithm to the base 2 of fold change, n=3. (b) Venn diagram displaying UPR proteins that are DE caused by p97 KD. (c) Representative KEGG or Reactome pathways affected by p97 KD. (d) Heatmap showing fold change in proteasome proteins, cell cycle related proteins and E2 ubiquitin-conjugating enzymes which are significantly dysregulated by p97 KD. (e) The percentage of overlapping DE proteins following p97 KD and p97 inhibitor treatment increases time-dependently. This percentage was calculated by dividing the number of overlapping proteins by the sum of all DE proteins identified by p97 KD and/or inhibitor treatment. HCT116 cells were treated with 4 μM UPCDC-30245, 2 μM CB-5083 or 4 μM of NMS-874 for the indicated time points, n=2. (f) Functional enrichment analysis on proteins affected by both p97 KD and p97 inhibitors. (g) Heatmap displays the fold change of DE proteins participating in the four cellular pathways affected by p97 inhibition. (See Table S1–2).
We compared our identified DE proteins to components of the three key unfolded protein response (UPR) pathways (Table S1), PERK, ATF6 and IRE1a. p97 knockdown leads to DE of 5 proteins in the PERK pathway, 4 proteins in ATF6 pathway and 12 proteins in IRE1α pathway. These proteins were all upregulated in p97 KD HCT116 cells, suggesting p97 depletion in HCT116 cells activates all three UPR pathways (Fig. 1b). Functional enrichment analysis on all DE proteins revealed that multiple well-known p97-associated functions were affected by p97 KD, such as PQC, cell proliferation, and transport (Fig. 1c, Table S1). In addition, our data showed that the Asparagine N-linked glycosylation pathway was affected by p97 KD. Asparagine N-linked glycosylation is an important form of protein post-translational modification (PTM) and is dependent on ER quality control (ERQC) and ER-to-Golgi transport (Shrimal et al., 2019, Dall’Olio et al., 2011). The dysregulation of this pathway indicates that p97 may participate in maintaining PTMs. Multiple proteins in cellular pathways linked to p97 function were upregulated upon p97 KD, including proteasome components and E2 ubiquitin-conjugating enzymes (E2s; Fig. 1d). The upregulation of proteasome components and E2s may compensate for reduced proteolytic activity due to p97 KD. We detected a significant increase in nuclear levels of active TCF11 in p97 KD cells (Fig. S1d). This change may contribute to transcriptional upregulation of proteasome components, consistent with previous reports (Steffen et al., 2010, Radhakrishnan et al., 2014). The majority of cell cycle related proteins were downregulated by p97 KD, including the MCM complex (Fig. 1d). This finding is consistent with the lower proliferation rate of p97 KD cells and with the essential role of p97 in DNA replication (Akopian and Rape, 2017, Li and Xu, 2019).
To explore the temporal proteomic profiles of p97 inhibition, we treated HCT116 cells with DMSO, and p97 inhibitors (CB-5083, NMS-873 or UPCDC-30245) for 2h, 6h, 10h, 18h, or 24h in duplicate. To allow consistent comparison between inhibitors, we chose a treatment concentration that is 4-fold of the IC50 concentration, to aim for a similar cellular potency and avoid indirect effects caused by cell death (Fig. S2a). We identified 7703 proteins from 40 samples (Table S2). PCA on the temporal proteomics profiles showed that 2h and 6h treatment of CB-5083 and the NMS-873 treatment samples grouped in one cluster while 8h, 18h and 24h grouped into another cluster (Fig. S2b). Therefore, 6h and 24h likely represent reasonable timepoints to probe early and late responses to p97 inhibition. To compare the proteomic profile of p97 shRNA KD to that of different p97 inhibitors, we compared the DE proteins identified through p97 KD with those identified under different inhibitor treatments (p<0.05) at each time point. With the exception of UPCDC-30245, the percentage of overlapping DE proteins increased with treatment duration (Fig. 1e). The proteomic profile of p97 KD was most similar to later timepoints following pharmacological p97 inhibition since p97 depletion by shRNA takes 72h. To reveal the cellular effects common to both p97 KD and p97 inhibitors, we combined overlapping DE proteins across different timepoints for each inhibitor treatment. Functional enrichment analysis on each of these overlapping protein sets revealed that overlapping DE proteins from CB-5083 and NMS-873 treatment are linked to the same cellular pathways affected by p97 KD (Fig. 1f). Interestingly, we found that DE proteins that are affected by both p97 KD and UPCDC-30245 are not linked to pathways typically impacted by p97 inhibition, such as protein processing in the ER, UPR and asparagine N-linked glycosylation (Fig. 1f). UPR proteins and those linked to protein processing in the ER were all upregulated by CB-5083, NMS-873 and p97 KD, but not by UPCDC-30245 (Fig. 1g). These data suggest that the MOA of CB-5083 and NMS-873 in HCT116 cells is similar to p97 KD but is distinct from the MOA of UPCDC-30245.
Comparative proteomic analysis of p97 and proteasome inhibition reveals differential molecular mechanisms.
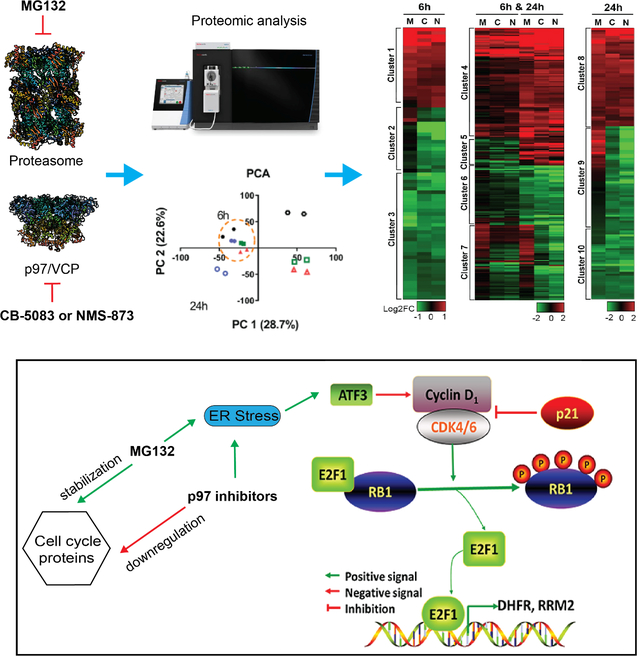
As a step toward understanding the differential effects of inhibiting p97 and the proteasome in cancer treatment, we compared the effect of inhibiting p97 and the proteasome using tandem mass tag (TMT) 16-plexed labelling. This technique allowed us to capture alterations in the proteome after exposure to MG132 (proteasome inhibitor) and p97 inhibitors (CB-5083 and NMS-873). Considering our previous results, we chose 6h and 24h treatments as representative of early (6h) and late (24h) responses to inhibitors. Samples were pooled together, fractionated into 8 fractions using high pH reverse chromatography, and analyzed using the highly accurate RTS-SPS-MS3 method (Schweppe et al., 2020, Li et al., 2012). We identified 7942 proteins, of which 6956 proteins were quantified across all 16 samples (Table S3). PCA showed 6h samples clustered separately from 24h samples, consistent with LFQ results, and confirmed that treatment time contributed to large changes in protein expression (Fig. 2a).
Fig. 2. Comparison of the temporal proteomic profile resulting from treatment with p97 inhibitors and treatment with MG132.

(a) PCA of TMT-labeled proteomic data. Cells were treated with 1 μM MG132, 2 μM CB-5083 or 4 μM of NMS-874 for 6h or 24h, n=2. (b) Venn diagram displays the number of proteins that were dysregulated by NMS-873 and CB-5083 treatment. We found that the DE proteins regulated by both p97 inhibitors can be separated into 3 groups. Group 1 contains 233 DE proteins that were only differentially expressed at 6h (6h, black rectangle), group 2 contains 349 DE proteins at both 6h and 24h (6h & 24h, blue rectangle), and group 3 contains 302 DE proteins differentially expressed only at 24h (24h, red rectangle). (c) Hierarchical clustering of proteins regulated by both NMS-873 and CB-5083. M, C and N represents MG132, CB-5083 and NMS-873 respectively. (d) Pathway analysis on proteins regulated both by MG132 and p97 inhibitors. (e) Pathway analysis on proteins specifically downregulated by p97 inhibitors. (f) Venn diagram of the proteins upregulated by MG132, NMS-873 and CB-5083. (g) Pathway analysis on proteins specifically upregulated by p97 inhibitors. (h) Log2FC of four XBP1 activates genes which were significantly upregulated by CB-5083 (CB) and NMS-873 (NMS) after 24h of treatment (p<0.05), but not by MG132 (MG). (See Table S2).
To identify the proteomic changes common to the different p97 inhibitors, we performed DE analysis (Table S3) and identified DE proteins (p<0.05) affected in both CB-5083 and NMS-873 treated samples (Fig. 2b). As shown in Fig. 2b, we divided the overlapping DE proteins into 3 groups (6h, 6h & 24h or 24h) and performed hierarchical clustering on those proteins which were significantly dysregulated by p97 inhibitors in respective samples with a threshold of |log2FC|>0.5 (Fig. 2c). Clusters 1, 4, and 8 contained proteins upregulated by both p97 inhibitors and MG132. Clusters 3, 6, and 10 contained proteins downregulated by both p97 inhibitors and MG132. Clusters 2, 7, and 9 contained proteins downregulated by p97 inhibitors but not MG132. Since p97 acts upstream of the proteasome and plays an important role in UPS, as expected the majority of DE proteins affected by p97 inhibitors were regulated similarly by MG132 (clusters 1, 4, 8, 3, 6 and 10). Functional enrichment analysis on the DE proteins dysregulated by both p97 inhibitors and MG132 identified pathways related to UPS, such as ERQC, protein processing in ER and UPR (Fig. 2d).
Next, we examined the proteins downregulated by p97 inhibitors but not by MG132 (clusters 2, 7, and 9). Using hierarchical clustering, we identified 121 proteins that were specifically downregulated by p97 inhibitors (Table S3). These proteins are involved in the p53 signaling pathway, cell cycle, virus infection and RHO GTPase-related pathways (Fig. 2e). To compare the cell cycle effects induced by a p97 inhibitor to that induced by a proteasome inhibitor, the DNA content was measured in both HCT116 cells after treatments with CB-5083 or MG132 for 24h (Fig. S3a) and showed a reduced distribution in the G0/G1 phase when treated with CB-5083 or MG132. Compared to MG132, CB-5083 is less potent in reducing the distribution of cells in G0/G1 phase and in increasing the distribution of cells in G2/M phase.
Moreover, we found that the autophagy related proteins BCL2L1, GABARAPL2 (ATG8C), TAX1BP1 and p62 (SQSTM1) were downregulated at 6h and upregulated at 24h by p97 inhibitors in cluster 5. Both TAX1BP1 and p62 were upregulated by MG132 at 6h and 24h (Fig. S3b). We failed to find any proteins specifically upregulated by p97 inhibitors using hierarchical clustering. Therefore, we used a Venn diagram to examine upregulated proteins (p<0.05, |log2FC|>0.3) identified from MG132, NMS-873 and CB-5083 treatment to identify proteins that were specifically upregulated by p97 inhibitors (Fig. 2f). From the Venn diagram, we have identified 210 DE proteins which were specifically upregulated by p97 inhibitors but not by MG132 after combining 6h and 24h data (Fig. 2f, Table S3). Functional enrichment analysis on these proteins revealed that p97 inhibition had a different effect on protein processing in the ER and UPR relative to MG132 treatment. In particular, p97 inhibition showed higher potency in activating the IREα and XBP1 pathways (Fig. 2g). Four XBP1 activated proteins, DNAJB11, SRPR, SRPRB and SYVN1 were significantly upregulated by p97 inhibitors but not by MG132 after 24h treatment (Fig. 2h). The upregulation of these four proteins was further validated by western blot (Fig S3c). Our results are consistent with reported findings that CB-5083 is more potent at activating the XBP1 pathway than PIs in models of multiple myeloma (Le Moigne et al., 2017). Taken together, these results validate that our comparative proteomic analysis can reveal the differential effect of p97 versus proteasome inhibition.
Overall, these results suggest that p97 inhibition impacts specific cellular pathways that are unaffected by proteasome inhibition.
Identifying specific protein markers of p97 inhibition.
In order to validate the 121 downregulated proteins that are specifically affected by p97 inhibitors (Table S3), we compared these with the proteins that were downregulated by p97 KD and found 33 overlapping proteins (Fig. 3a). Comparing our data to two published studies in HEK293 and U2OS cells revealed that the 33 proteins are also downregulated by p97 KD and NMS-873 treatment in other datasets (Fig. 3b) (Heidelberger et al., 2018, Xue et al., 2016). This comparison demonstrated these 33 protein levels are regulated similarly by p97 inhibition in HCT116, HEK293, and U2OS cells. We further analyzed the 210 upregulated proteins (Table S3) and found that the 21 most significantly upregulated proteins were affected by p97 inhibitors but not by MG132 (Fig. 3c). We further compared the fold change calculated from TMT experiments with that from LFQ results for the 21 upregulated and 33 downregulated proteins and in samples treated with p97 inhibitors or MG132. Six out of the 54 proteins were also identified in our LFQ runs and showed time-dependent changes in response to p97 inhibitors (Fig. 3d). Of the six proteins, the regulation of CCNB1, PAID4 and HMMR were validated through western blot analysis (Fig. S3d) and Benjamini-Hochberg adjusted p-values for FDR less than 0.05 at least in one condition for all six proteins (Table S3). Our results indicate that these 6 proteins are affected by p97 but not proteasome inhibition, and are potential markers that distinguish these two treatments.
Fig. 3. Identifying specific markers of p97 inhibition.

(a-b) Heatmap of proteins specifically downregulated by p97 inhibitors in HCT116 cells (a), in HEK293 and U2OS cells (b). The log2 fold change data of these proteins in HEK293 and U2OS cells was obtained from published results. Protein indicates proteomic data and RNA indicates RNAseq data. HEK293 cells were treated with p97 shRNA and NMS-873, U2OS cells were treated with NMS-873 and MG132. Blank indicates the protein was not detected. (c) Heatmap showing proteins upregulated by both CB-5083 and NMS-873, but not by MG132, after 6h or 24h of treatment. Of the 210 upregulated overlapping DE proteins (p<0.05) (Fig. 2f, SI table 8), the heatmap displays those which had log2FC>0.5 when treated by p97 inhibitors and log2FC<0.3 when treated by MG132. (d) Temporal proteomic profile of 6 potential p97 inhibition specific markers. Samples were treated with CB-5083 (green), NMS-873 (red) and MG132 (black) and data was collected from LFQ results. For Fig. 3a–c, M, C and N indicates MG132, CB-5083 and NMS-873 treatment respectively, shRNA represents p97 KD. (See Table S3, BH adjusted p-values for FDR less than 0.05 highlighted yellow).
p97 inhibition blocks E2F1-mediated transcription via downregulation of the CCND-CDK4/6 complex.
To further examine mechanisms specific to p97, rather than proteasome, inhibition we focused on the proteins specifically downregulated by the former treatment. To investigate whether the protein translation was inhibited by p97 or proteasome inhibitor, we examined the phosphorylation state of eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1). Phosphorylation of 4E-BP1 serves to increase total protein translation (Walejko et al., 2021). After 6h of treatments, both MG132 and p97 inhibitors inhibit global protein translation, to a greater extent in MG132 treated cells (Fig. S4a). 20 of the 33 proteins we identified are downregulated by NMS-873 at RNA levels in U2OS cells (Fig. 3b) in a previous dataset which sought to identify general p97 substrates (Heidelberger et al., 2018). The effect of p97 inhibition on specific protein levels likely arises from a change in transcript levels. To examine how transcript levels might be affected, we examined potential transcription factor binding sites (TFBSs) in the promoters of these 33 genes in the TRANSFAC database. This search revealed putative binding sites for two transcription factors, E2F1 and MYB (Fig. 4a). By examining protein-protein interaction networks related to E2F1 and MYB in the STRING database (https://string-db.org/), we found 11 E2F1 interactors (Fig. S3b) and 11 MYB interactors (Fig. S3c). Two proteins, Cyclin A2 (CCNA2) and CDC6, are E2F1 interactors and are also significantly dysregulated in our TMT proteomic analysis following proteasome and p97 inhibition (Fig. S3d, Table S4). In addition, some of the proteins downregulated by p97 inhibition (Fig. 4a), such as DHFR, TK1 and RRM2, have previously been experimentally validated as E2F1-specific target genes (Wells et al., 2002, Chen et al., 2012, Fang et al., 2015). We therefore decided to further investigate the role of E2F1, rather than MYB, in cells responding to p97 and proteasome inhibition.
Fig. 4. p97 inhibition impairs the transcriptional activity of E2F1 by downregulating the CCND1/CDK complex.

(a) The putative transcription factor binding sites (TFBSs) of the 33 proteins which were specifically downregulated by p97 inhibition. Data was analyzed using the g:Profiler. Blue indicates the gene is a potential target of transcription factor c-Myb or E2F1. Blank indicates it is not a target gene. (b) Depiction of the known regulatory network of the CCND1/CDK/RB1/E2F1 pathway. (c) Log2 fold change of the proteins regulating E2F1 function which were significantly affected by MG, CB and NMS in our TMT results. (d) The dysregulation of E2F1 related proteins as confirmed by western blot. HCT116 cells were treated with 1 μM MG132, 2 μM CB-5083 or 4 μM of NMS-874 for 6h. (e) The transcriptional activity of E2F1 was measured using E2F1 reporter assay. HCT116 cells were transfected with pGL2-AN plasmid for 24h. Then cells were plated in 384 well plate and incubated for 16h. The luminescence were detected after 8h treatment with MG132, CB-5083 or NMS-873. Data are shown as mean ± SD, n=3. (f) qRT-PCR analysis of ATF3, DHFR and CCND1 RNAs following treatment with MG132, CB-5083 or NMS-874. HCT116 cells were treated for 2, 6 and 24h using the same concentration as proteomic and western blot assay (n=4). (See Fig. S4 and Table S4).
To determine if the decreased expression of E2F1 target genes was caused by the downregulation of E2F1 levels due to ER stress (Pagliarini et al., 2015), we used qPCR and western blot to evaluate E2F1 levels. E2F1 RNA levels were strongly downregulated by MG132 and slightly downregulated by p97 inhibitors (Fig. S3e), consistent with the fact that ER stress induces the downregulation of E2F1. However, the protein level of E2F1 was not significantly affected by p97 inhibitors and was increased by MG132 (Fig. S3f). The transcriptional activity of E2F1 can also be repressed through the formation of a complex with the tumor suppressor protein retinoblastoma (RB1) (Fig. 4b) (McNair et al., 2018). Hyperphosphorylation of RB1 by cyclin-D–cyclin-dependent kinase 4/6 complex (CCND1-CDK4/6) inactivates RB1 and induces the release of the E2F1 from the complex (Burkhart and Sage, 2008). The newly released E2F1 leads to transcriptional activation of target genes (Lange and Yee, 2011). From our proteomic analysis, CCND1 (cyclin D1) and CDK4/6 were all significantly downregulated by p97 inhibitors but upregulated by MG132 (Fig. 4c). This downregulation of CCND1 and CDK4 was further confirmed by western blot (Fig. 4d). By performing a published E2F1 reporter assay (Neuman et al., 1994), we found both MG132 and p97 inhibitors reduce the transcriptional activity of E2F1, and the IC50 of p97 inhibitors is 2-fold lower than that of MG132 (Fig. 4e). Additionally, qPCR analysis indicates that mRNA levels of the E2F1 target gene DHFR are also downregulated by p97 inhibitors (Fig. 4f). Our data suggests the transcriptional activity of E2F1 is decreased by p97 inhibition and leading to downregulation of E2F1 target genes. Furthermore, the downregulation of CCND1 occurred after the upregulation of ATF3 (Fig. 4f). ATF3 is a stress-inducible gene, which binds to the cyclin D1 promoter and represses its transcription (Lu et al., 2006). Therefore, the upregulation of ATF3 potentially contribute to the downregulation of CCND1.
We hypothesized that CCND1 and CDK4/6 upregulation by MG132 promotes RB1 hyperphosphorylation. However, the western blots showed that MG132 treatment instead reduced phosphorylation of RB1 in addition to reducing the mRNA level of DHFR (Fig. 4e and f), indicating that the transcriptional activity of E2F1 was also inhibited by MG132. To reveal why the upregulated CCND1-CDK4/6 complex does not promote phosphorylation of RB1 (Fig. 4b and c), we focused on the CKI (cyclin-dependent kinase inhibitor) and found that p21 (CDKN1A) was significantly upregulated by MG132 but not by p97 inhibitors (Fig. 4c and d). This suggests that upregulation of p21 can block the increased activity of CCND1-CDK4/6 complex in MG132 treated cells and reduce RB1 phosphorylation.
Taken together, our results suggest that HCT116 cells respond to proteasome and p97 inhibition by upregulating ATF3 mRNA and protein levels (Fig. 4c, d and e) leading to a subsequent reduction in CCND1 mRNA (Fig. 4e). Unlike proteasome inhibition which increases cyclin D1 and CDK4/6, what distinguishes p97 inhibition is the reduction of all three of these oncoproteins at the protein level. Our results indicate that both cyclin D1 and p21 play an important role in regulating the E2F1 pathway in cells treated with p97 inhibitors or MG132. Overall, both MG132 and p97 inhibitors lead to the reduction of unphosphorylated RB1 which, as a result, sequesters E2F1 and blocks its transcriptional activity.
p97 inhibition promotes the downregulation of cell cycle oncoproteins.
Functional enrichment analysis on proteins specifically downregulated by p97 inhibitors revealed that cell cycle factors are highly associated with p97 inhibition (Fig. 2e). To follow up on this effect, we examined TMT experiments for cell cycle proteins that are significantly differentially expressed (Table S4) with Benjamini-Hochberg adjusted p-values for FDR less than 0.05 highlighted yellow. We found that eleven cell cycle proteins – Securin (PTTG1), Cyclin D1 (CCND1), MYC, Survivin (BIRC5), Emi1 (FBXO5), CDC20, Bub1, CDC25B, ORC6, GMNN, RRM2– are specifically downregulated after 6h and 24h treatment with p97 inhibitors only (Table 1). To exclude cell-specific effects, we detected the representative cell cycle proteins, Cyclin D1, Myc, Securin, Survivin and CDC20 in HT29 cells treated with p97 inhibitors or MG132, and found all the five proteins displayed similar changes as observed in HCT116 cells (Fig. S5a). To probe the effect at the transcriptional level, we determined the RNA levels of Myc, Securin, Emi1 and CDC20 after 6h treatment (Fig. 5a) and found only CDC20 was downregulated by CB-5083. We focused on the most affected two proteins, cyclin D1 and Securin, to investigate how protein levels are affected by both proteasome and p97 inhibition.
Table 1.
Cell cycle proteins specifically downregulated by p97 inhibitors.
| Description | Gene name | Log2FC |
Cell cycle | |||||
|---|---|---|---|---|---|---|---|---|
| 6h | 24h | |||||||
|
| ||||||||
| MG132 | CB-5083 | NMS-873 | MG132 | CB-5083 | NMS-873 | |||
|
| ||||||||
| Securin | PTTG1 | 1.4 | −0.9 | −1.2 | −0.1 | −1.9 | −2.9 | M |
| G1/S-specific cyclin-D1 | CCND1 | 0.2 | −1.4 | −1.8 | 0.4 | −2.0 | −3.2 | G1/S |
| Myc proto-oncogene protein | MYC | 1.2 | −0.5 | −1.1 | 0.2 | −0.7 | −1.1 | G1/S |
| Baculoviral IAP repeat-containing protein 5 | BIRC5 | 0.6 | −0.5 | −0.5 | 1.0 | −0.8 | −2.0 | M/check point |
| F-box only protein 5 | FBXO5 | 0.2 | −0.8 | −1.1 | 1.5 | −1.0 | −2.4 | M/G1 |
| Cell division cycle protein 20 homolog | CDC20 | 0.8 | −0.4 | −0.6 | 1.1 | −1.5 | −2.8 | M/check point |
| Mitotic checkpoint serine/threonine-protein kinase BUB1 | BUB1 | 1.2 | −0.3 | −0.4 | 1.4 | −1.1 | −2.1 | M/check point |
| M-phase inducer phosphatase 2 | CDC25B | 0.3 | −1.1 | −1.6 | −0.2 | −1.6 | −2.2 | S/G2/M |
| Origin recognition complex subunit 6 | ORC6 | 0.3 | −0.4 | −0.5 | 1.0 | −0.3 | −0.6 | G1/S/ch eckpoint |
| Geminin | GMNN | 0.9 | −0.4 | −0.6 | 1.4 | −1.1 | −1.8 | G1/S |
| Ribonucleoside-diphosphate reductase subunit M2 | RRM2 | 0.6 | −0.2 | −0.3 | 1.2 | −1.1 | −2.7 | G1 |
TMT label proteomic results for 6h and 24h treatment. Log2FC indicates log2 fold change (compound vs DMSO). BH adjusted p-values with FDR less than 0.05 see Table S4.
Fig. 5. p97 promotes the stability of cell-cycle oncoproteins.

(a) qRT-PCR analysis of Myc, Securin, Emi and CDC20 mRNA levels. HCT116 cells were treated with 1 μM of MG132 or 2 μM of CB-5083 for 6h, n=4. (b-c) MG132 rescued CB-5083 mediated cyclin D1 downregulation at the protein level (b) but not at the mRNA level (c) while Baf A1 had no effect on cyclin D1. The concentration of MG132, CB-5083 and Baf A1 was 1 μM, 2 μM and 10 μM respectively. Cells were treated for 6 hours, n=4, **** indicates p<0.001. (d-e) The half-life of Securin in HCT116 cells. HCT116 cells were treated with 1 μM of MG132, 2 μM of CB-5083, 1 μM of MG132 plus 2 μM of CB-5083 or DMSO. 50 μM of CHX was added immediately after compounds treatment, n=3, * indicates p<0.05. (f-g) The degradation of Securin was detected in the total lysate of HCT116 and HT29 cells. Cells were pretreated with 2 μM of MG132 for 1h. Then harvest the cells as 0 minute samples, or replace the culture media with fresh media and add DMSO, 2 μM of CB-5083 or 1 μM of MG132 together with 50 μM of CHX for 60, 90, 120,180 minutes, n=3, * indicates p<0.05, ** p < 0.01. For fig. 5b–g, D, B, M, C and N represents DMSO, Baf A1, MG132, CB-5083 and NMS-873 respectively. Data are shown as mean ± SD. Statistical analysis was performed using one-way ANOVA. (See Fig. S5).
Since p97 is involved in both the proteasomal and autophagy degradation pathways, we next tested which process is linked to the changes seen in cyclin D1 levels upon p97 inhibition. We co-treated cells with CB-5083 and either the proteasome inhibitor MG132 or the autophagy inhibitor Bafilomycin A1 (Baf A1). Our results showed that MG132 treatment rescued CB-5083 mediated cyclin D1 downregulation at the protein level but not at the mRNA level while Baf A1 had no effect on cyclin D1 levels (Fig. 5b and c). In addition, there was a greater reduction in cyclin D1 protein in samples co-treated with CB-5083 and MG132 than those treated with MG132 treatment alone. This result indicates that CB-5083 may promote cyclin D1 degradation in HCT116 cells. We further examined cyclin D1 degradation in the cytosol and nucleus (Fig. S5b) and found that in both the cytosol and nucleus, cyclin D1 turnover was blocked by MG132. Due to the short half-life of cyclin D1 (less than 30 minutes), we found that CB-5083 only slightly accelerated nuclear, but not cytosolic, cyclin D1 degradation in HCT116 cells (Fig. S5c).
We also examined Securin protein degradation from the cytosol and nucleus and found that it was blocked by MG132 (Fig. 5d, Fig. S5b). In addition, CB-5083 and MG132 co-treatment reduced the stabilization effect of MG132 on Securin (Fig. 5d and e), suggesting that the degradation of Securin does not depend on p97. When the cells were pretreated with MG132 for 1h to buildup Securin, CB-5083 slightly accelerated the degradation of Securin in both HCT116 and HT29 cells (Fig. 5f and g). We confirmed that cyclin D1 and Securin are proteasome substrates and do not require p97 for degradation by proteasome. Overall, p97 inhibitors did not increase the protein level of these cell cycle oncoproteins, and instead downregulated protein levels. These results distinguish the role of p97 from that of the proteasome in regulating cell cycle proteins.
Discussion
While previous studies have used proteomics to examine potential p97 partners and substrates in HEK293, and 293T cells, none of these have obtained temporal profiles and examined mechanisms that are potentially linked to the specific therapeutic effects of p97 inhibition (Alexandru et al., 2008, Raman et al., 2015, Rosenow, 1990). Two studies used proteomic profiling in U2OS cell (Heidelberger et al., 2018) and HEK293 cells (Xue et al., 2016). One study performed proteomic profiling of NMS-873 after 6 h treatment in HCT116 cells (Her et al., 2016). These studies identified interesting p97 functions and substrates that are degraded in a p97-dependent manner. Here, however, we have conducted a systematic analysis of p97 inhibition over time, using genetic knockdown and three small molecule inhibitors with different binding modes. Previous studies have also shown that p97/VCP is involved in multiple cellular processes (Stach and Freemont, 2017) and that p97 inhibition is a potentially promising therapeutic strategy for treating cancers, neurodegenerative disease and virus infection (Huryn et al., 2020). However, most published studies on the cellular effects of p97 inhibition have focused on one or a few aspects of p97 functions, such as p97-adaptor binding (Xue et al., 2016, Her et al., 2016) and the degradation of ubiquitinated proteins through endoplasmic-reticulum-associated protein degradation (ERAD) (Braun and Zischka, 2008). Here we used mass spectrometry-based quantitative proteomics to reveal a comprehensive picture of the proteomic and cellular pathways altered in HCT116 cells in response to both genetic and pharmacological p97 inhibition. We then compared the proteomic profiles of p97 inhibition to those of proteasome inhibition at several time points.
Our data reveal the overall influence of p97 inhibition on cellular functions (Fig. 1c and f), and argue that p97 KD upregulates formation of proteasome as well as E2 ubiquitin-conjugating enzyme complexes to compensate for reduced proteolytic activity (Fig. 1d). In addition, we compared the proteomic changes induced by p97 shRNA KD to those induced by three p97 inhibitors and found that inhibition by CB-5083 and NMS-873 is more similar to p97 KD than UPCDC-30245. Specifically, UPR and factors associated with protein processing in the ER pathway were upregulated by both CB-5083 and NMS-873 as well as p97 KD (as previously reported), but not by UPCDC-30245 (Fig. 1g) (Magnaghi et al., 2013, Her et al., 2016, Anderson et al., 2015, Bastola et al., 2016, Zhao et al., 2020, Garrison and Bangs, 2020, Wang et al., 2020). This result is consistent with our previous study as demonstrated by western blot analysis of two UPR markers (CHOP and ATF4) and two autophagy markers (p62 and LC3) (Wang et al., 2020). It is possible that UPCDC-30245 is inhibiting distinct p97 complexes or inhibiting other proteins that directly affect the autophagy machinery. Further studies will reveal whether these possibilities are the case (Chou and Deshaies, 2011).
The cell fate decisions under ER stress could be divided into two stages, the adaptive response phase (early stage) and apoptotic phase (late stage) (Hetz, 2012, Corazzari et al., 2017). During the early stage, global protein translation is inhibited by PERK (Harding et al., 2000) and selective degradation of mRNA (Hollien et al., 2009, Hollien and Weissman, 2006) and autophagy is activated (Kroemer et al., 2010) to reduce the influx of proteins into the ER and reestablish homeostasis. Prolonged stress conditions and CHOP activation results in pro-death signaling overweighing pro-survival signaling, which leads to apoptosis (Corazzari et al., 2017, Hetz and Papa, 2018). We used temporal proteomics to identify representative time points for the different cell fate decisions induced by p97 inhibition. We found that 6h and 24h provide representative time points that reveal early and late proteomic changes in response to p97 inhibition (Fig. S2b).
We next compared the proteomic profile of p97 inhibition to the proteomic profile of proteasome inhibition. Proteasome inhibition and p97 inhibition had a similar effect on some protein clusters (Fig. 2c). However, p97 inhibitors activated the XBP1 pathway more potently than MG132 (Fig. 2g and h), consistent with what has been reported in models of multiple myeloma (Le Moigne et al., 2017). In addition, both TAX1BP1 and p62 were downregulated by p97 inhibitors but upregulated by MG132 after 6h treatment, revealing differential effects of p97 and proteasome inhibition on autophagy (Fig. S3b). This supports the notion that upregulation of autophagy is a possible mechanism by which resistance to proteasome inhibition emerges. In addition, this reveals that p97 inhibition blocks two major proteasome degradation pathways (Chou et al., 2011). TMBIM6 (Bax inhibitor 1) enhances autophagy via regulating lysosomal calcium and accelerates p62 degradation (Kim et al., 2020). The rapid upregulation of TMBIM6 by p97 inhibitors, but not by MG132, may explain the downregulation of p62 at 6h (Fig. 3a and d).
The most striking difference between p97 and proteasome inhibition is that many cell cycle proteins were specifically downregulated by p97 inhibition. Since the discovery of yeast p97 via isolation of temperature-sensitive Cdc48 mutants (Moir et al., 1982), the role of p97 in regulating the cell cycle has focused on its ability to extract cell cycle proteins from complexes and deliver them to the proteasome for degradation. Based on this, p97 and the proteasome could be considered to regulate the cell cycle as part of the same pathway. For example, they are both recruited to K11/K48 ubiquitinylated H2B, promoting its degradation and maintaining cell identify during cell division (Oh et al., 2020). Studies have reported both p97 inhibition and MG132 treatment lead to HCT116 cells arresting in the G2-M phase (Magnaghi et al., 2013, Kim et al., 2004). However, we have here identified two major differences between the effect of p97 inhibition and proteasome inhibition on cell cycle regulation:
The mechanism underlying E2F1 inhibition by p97 inhibitors is different from MG132. p97 ATPase activity is required to maintain levels of the CCND1-CDK4/6 complex, whereas proteasome activity degrades p21 protein, and therefore promotes active CCND1-CDK4/6 complex. The overall effect is the same for both inhibitors, reduced Rb1 phosphorylation and E2F1 sequestration and thus reduced mRNA levels of the E2F1 target genes (Fig. 4b–d).
That p97 inhibitors and proteasome inhibitors have important opposing effects on the protein levels of several cell cycle that are also oncoproteins (Table 1). Although both MG132 and p97 inhibitors can affect the cell cycle (Fig. S3a), our proteomics data suggest that the molecular mechanism by which they affect cell cycle is different. We therefore selected two proteins, cyclin D1 and Securin, and determined their half lives in the presence of p97 or proteasome inhibitors. While we expected to see stabilization of cyclin D1 and Securin with MG132 as both are proteasome substrates, we also found that p97 inhibitors do not stabilize degradation of cyclin D1 and Securin. Congruently, Parisi et al. showed that p97 and yeast Cdc48 prevent degradation of ubiquitinated cyclin D1 and that the first generation p97 inhibitor, DBeQ, promotes cyclin D1 degradation (Parisi et al., 2018).
One of the major hallmarks of cancer is upregulation of cell cycle oncoproteins. The CCND-CDK4/6-INK4-Rb pathway is more frequently dysregulated in solid tumors and plays a central role in tumorigenesis and progression (Schettini et al., 2018). Of the eleven cell-cycle proteins specifically downregulated by p97, ten are significantly upregulated in TCGA-COAD patient tumors compared to normal matched tissue and most are upregulated in other GI cancers as well. (Fig. S6). Overexpression of some these proteins, such as Cyclin D1 is an unfavorable prognostic factor and is associated with tumor size and metastasis (Qie and Diehl, 2016). Mutations that directly perturb the degradation of Cyclin D1 and its nuclear export are also frequently observed in esophageal and uterine cancers (Benzeno et al., 2006). Therefore, targeting the Cyclin D1-CDK4/6-INK4-Rb pathway represents a valid treatment in a broad spectrum of solid tumors and numerous inhibitors of CDK4/6 are currently under development and in clinical trials (Qie and Diehl, 2016). Inhibition of p97 impairs the Cyclin D1-CDK4/6-INK4-Rb pathway and the transcriptional activity of E2F1. That leads to the direct downregulation of cell cycle proteins and the deficit cell cycle. The deficit cell cycle may further amplify the downregulation of cell cycle proteins. Taken together, we propose that the down regulation of cell cycle proteins potentially explains why p97 inhibitors were effective in solid tumor models (Anderson et al., 2015) while proteasome inhibitors, which stabilizes cell cycle oncoproteins, are largely ineffective.
A better understanding of the diversity and complexity of ubiquitin signaling in cell cycle regulation will shed new light on the precise control of cell cycle progression and guide anticancer drug development (Dang et al., 2021). Our findings demonstrated the value of chemical tools in combination with accurate temporal proteomic measurement. Our results also provide strong evidence for p97’s critical role in regulating many cell cycle oncoproteins, solidifying its potential as an excellent drug target in cancers that exhibit upregulated cell cycle oncoproteins.
Significance
Small molecule inhibitors of p97/VCP are being developed as anticancer drug. Our study aimed to identify the distinct mechanism of action of three classes of p97 inhibitors by comparing with genetic knockdown of the p97 and with a proteasome inhibitor. To reveal early and late responders, we used label free proteomics to compare 5 time points for 4 compounds. To quantify the differential expressed proteins more precisely, we used TMT labeling to compare 6h and 24h. We used qPCR and western blot to reveal the unique mechanism regulated by p97 inhibition. We demonstrated that p97 inhibition blocks E2F1-mediated transcription via downregulation of the CCND-CDK4/6 complex and promotes the downregulation of cell cycle oncoproteins.
STAR★Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Tsui-Fen Chou (tfchou@caltech.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
All relevant data generated during this study are included in the article and the supplementary Information. The mass spectrometry raw data related to Fig. 1a–d are deposited to the ProteomeXchance Consortium (https://www.ebi.ac.uk/pride/) via the PRIDE repository with the dataset identifier PXD025094 and 10.6019/PXD025094”. The mass spectrometry raw data related to Figs. 1e–g with dataset identifier PXD025167 and 10.6019/PXD025167”. The mass spectrometry raw data related to Fig. 2 with dataset identifier PXD025225 and 10.6019/PXD025225”. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Cell lines
The human colon cancer cell lines HCT116 (ATCC, cat# CCL-247) was established from a tumor sample collected from an adult male with colorectal carcinoma, and HT29 (ATCC, cat# HTB-38) was isolated from a primary adenocarcinoma of the colon from a 44 year old female donor. Both cell lines were grown in RPMI1640 medium (Corning, Cat# 10–040-CM) supplemented with 10% FBS (R&D, Cat# S11150) and 1% penicillin-streptomycin (Gibco, Cat# 15140–122), and cultured at 37 °C in a 5% CO2 incubator. Cells were tested for mycoplasma routinely using MycoAlert PLUS Mycoplasma Detection Kit (Lonza, cat# LT07–710).
Cell samples prepared for mass spec, qPCR, and western blot experiments were plated in 6-well plates at a density of 1.5 × 106/well in RPMI1640 medium supplemented with 5% of FBS and 1% of penicillin-streptomycin. Twenty-four hours after seeding, the cell plates were changed with 2 mL fresh medium per well. Then 10 μL of compound prepared in DMSO was added into each well, 10 μL DMSO was added to the vehicle control well. Plates were incubated at 37 °C in a 5% CO2 incubator until harvesting for experiments.
Stable HCT116 cell lines expressing doxycycline-inducible shRNA against non-targeting control or p97 were generated through the TripZ lent-viral shRNA system (Thermo Fisher Scientific) as described previously (Radhakrishnan et al., 2014). The targeted sequence for p97 is 5’-AAACAGCCATTCTCAAACAGAA-3’. The nontargeting control shRNA comes directly from Thermo Fisher Scientific. The control or p97 shRNA plasmid and lentivirus packaging plasmids (pHDM-G, CAG4-RTR2 and CAGGHIVgpco) were transiently co-transfected into 293T cells with BioT reagent. Then, the supernatant containing lentivirus particles was harvested at 24 and 48 hours post transfection, and was filtered through 0.45 μM filter. Next, HCT116 cells were seeded and transduced with lentivirus in the presence of 8 μg/mL Polybrene, and 5 μg/mL Puromycin was used to generate stable cell lines. HCT116 cells transfected with doxycycline-inducible expressing control shRNA or p97 shRNA cells were cultured in the presence of 1 μg/ml doxycycline for 72 hours, and cell pellets were harvested and further prepared for mass spec, qPCR, and western blot.
Method details
Quantitative real-time PCR
After harvesting cells, pellets were re-suspended in DPBS/TRIzol-LS mixture (v/v 1:3). Total RNA was extracted from the TRIzol-LS mixture using Direct-zol RNA MiniPrep plus kit (Zymo Research, cat# R2072) according to the manufacturer’s instructions. RNA concentrations were measured using a NanoDrop Lite UV visible spectrophotometer (Thermo Scientific, cat# S/N 2361). 1 μg total RNA was reverse transcribed into complementary DNA using SensiFAST™ cDNA Synthesis Kit (Bioline, cat# BIO-65054). qRT-PCR reactions were performed using primers and PowerUp SYBR Green Master Mix (Thermo Scientific, cat# A25742), or qPCR probes and TaqMan Universal Master Mix II no UNG (Thermo Scientific, cat# 4440040) on the QuantStudio™ 5 Real-Time PCR System (Thermo Scientific, cat# A28140). 2ˆ(−ΔCT) values were calculated after normalizing to GAPDH levels.
Western blot
After harvesting, cell pellets were re-suspended in 150 μL lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% Triton X-100 with protease inhibitor tablet, 50 μM MG132, and 50 mM NEM), and incubated on ice for 10 min with occasional vortex. Samples were then centrifuged at 15000 rpm at 4 °C for 10 min, and 120 μL of the supernatant was transferred into a new 1.5 mL tube. Total soluble protein concentrations were measured using the Bradford reagent (Bio-Rad, cat# 5000006). After that, 40 μL of 4 × Laemmli sample buffer (Bio-Rad, cat# 161–0774) containing 0.1 M DTT (Cytiva, cat#17–1318-02) was mixed with the samples and heated for 5 min at 95 °C. Equal amounts of protein samples were loaded and separated using 4–20% Mini-PROTEAN TGX precast gels (Bio-Rad, cat# 456–1096) and transferred to nitrocellulose membranes using Trans-Blot Turbo Transfer System (Bio-Rad, cat# 170–4155). Membranes were blocked with 5% w/v nonfat milk prepared in TBST buffer, incubated with primary antibodies overnight at 4 °C, washed with 5% milk (10 min × 3 times), incubated with proper secondary antibodies for 2 h at room temperature, and washed with TBST buffer (5 min × 3 times). The blots were imaged using ECL reagent (MilliporeSigma, cat# WBKLS0500) and the ChemiDoc MP Imaging System (Bio-Rad). Blot densities were analyzed using Image Lab 6.0.1 software (Bio-Rad).
Flow cytometric analysis of DNA content
After 24h of treatments with DMSO, 2 μM of CB-5083 or 1 μM of MG132, cells were detached with TrypLE™ Select Enzyme (Gibco, 50–591-420). The analysis of DNA content assay was performed using Cell Cycle Analysis Kit (Sigma-Aldrich, MAK344) following the manufacturer’s instructions.
E2F1 reporter assay
HCT116 cells (2*106) were transfected with 2 μg pGL2-AN plasmid (Addgene: 20950) in one well of 6 well plate using BioT transfection reagent (B01–01, Bioland Scientific). After 24h, transfected cells were re-plated in 384 well plate (1*10^4 cell per well in 30 μL media) and incubated for 16h. Compounds (0–42 μM, 3-fold serial dilution for MG132, CB-5083 or NMS-873) were added to cell in triplicate. 10 μL of luciferin (2 mg/mL in DPBS) was add to each well 8h after the treatments. The luminescence was detected with Synergy Neo2 (BioTek) after 10 minutes incubation at room temperature.
Label-free proteomics
Cells were harvested and spun down to remove the supernatant. LC-MS samples were prepared using the Thermo EasyPep Mini MS Sample Prep Kit (Thermo Scientific, cat# A4006) according to the manufacturer’s instructions. Samples were then re-suspended in 0.1% formic acid (Thermo, cat# 85178) solution, and peptide concentration was tested using the Pierce Quantitative Fluorometric Peptide Assay (cat# 23290).
LC-MS/MS experiments were performed by loading 1 μg sample onto an EASY-nLC 1000 (ThermoFisher Scientific, San Jose, CA) connected to an Orbitrap Eclipse Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA). Peptides were separated on an Aurora UHPLC Column (25 cm × 75 μm, 1.6 μm C18, AUR2–25075C18A, Ion Opticks) with a flow rate of 0.4 μL/min and for a total duration of 131 min. The gradient was composed of 3% Solvent B for 1 min, 3–19% B for 72 min, 19–29% B for 28 min, 29–41% B for 20 min, 41–95% B for 3 min, and 95–98% B for 7 min. Solvent A consists of 97.8% H2O, 2% ACN, and 0.2% formic acid, and solvent B of 19.8% H2O, 80% ACN, and 0.2% formic acid. MS1 scans were acquired with a range of 400–1600 m/z in the Orbitrap at 120k resolution. The maximum injection time was 50 ms, and the AGC target was 2 × 105. MS2 scans were acquired using quadrupole isolation mode and higher-energy collisional dissociation (HCD) activation type in the Iontrap. The isolation window was 1.4 m/z, collision energy was 35%, maximum injection time was 35 ms, and the AGC target was 1×104. Other global settings were set to the following: ion source type, NSI; spray voltage, 2500 V; ion transfer tube temperature, 300 °C. Method modification and data collection were performed using Xcalibur software (Thermo Scientific).
TMT label proteomics
Sample preparation and determination of peptide concentrations were performed as described in “Label-free proteomics”. 20 μg of peptide from each sample was transferred into a new 1.5 mL tube and dried using a vacuum centrifuge. 40 μL of 100 mM TEAB (Sigma, cat# 17902) was added to dissolve the sample. TMTpro 16plex reagents (Thermo, cat# A44522) were dissolved with anhydrous acetonitrile (Sigma, cat# 900644), added 10x by weight to each sample and incubated for 2 hours at room temperature. The reaction was quenched by adding 2 μL of 0.5% hydroxylamine (Thermo, cat# 90115) and incubated for 15 min at room temperature. Samples were combined and dried using vacuum centrifugation. Samples were then re-suspended with 0.1% TFA (Thermo, cat# 85183), and separated into 8 fractions using the High pH reversed-phase peptide Fractionation Kit (Thermo, cat# 84868). After that, the fractions were dissolved with 0.1% FA and peptide concentrations were tested through Pierce Quantitative Colorimetric Peptide Assay (cat# 23275).
TMT labeling LC-MS/MS experiments were performed using an EASY-nLC 1000 connected to an Orbitrap Eclipse Tribrid mass spectrometer. 1 μg of sample was loaded onto an Aurora UHPLC Column and separated over 136 min at a flow rate of 0.4 μL/min with the following gradient: 2–6% Solvent B (7.5 min), 6–25% B (82.5 min), 25–40% B (30 min), 40–95% B (1 min), and 95% B (15 min). Solvent A consisted of 97.8% H2O, 2% ACN, and 0.2% formic acid, and solvent B consisted of 19.8% H2O, 80% ACN, and 0.2% formic acid. An MS1 scan was acquired in the Orbitrap at 120k resolution with a scan range of 350–1600 m/z. The AGC target was 8 × 105, and the maximum injection time was 50 ms. Dynamic exclusion was set to exclude features after 1 time for 60 s with a 5-ppm mass tolerance. MS2 scans were acquired with collision-induced dissociation (CID) activation type with the Iontrap. The isolation window was 0.4 m/z, the collision energy was 35%, the maximum injection time was 45 ms and the AGC target was 104. MS3 scans were acquired with higher-energy collisional dissociation (HCD) activation type in the Orbitrap at 50k resolution with a scan range of 100–500 m/z. The isolation window was 0.7 m/z, the collision energy was 55%, the maximum injection time was 86 ms and the AGC target was 2.5 × 105. Ion source settings were as follows: ion source type, NSI; spray voltage, 2200 V; ion transfer tube temperature, 300 °C. System control and data collection were performed by Xcalibur software.
Quantification and statistical analysis
Proteomic analysis was performed using Proteome Discoverer 2.4 (PD 2.4, Thermo Scientific) software, the Uniprot human database and SequestHT with Percolator validation. Percolator FDRs were set at 0.001 (strict) and 0.01 (relaxed). Peptide FDRs was set at 0.001 (strict) and 0.01 (relaxed), with medium confidence and a minimum peptide length of 6. Carbamidomethyl (C) was set as a static modification; oxidation (M) was set as a dynamic modification; acetyl (protein N-term), Met-loss (Protein N-term M) and Met-loss+acetyl (Protein N-term M) were set as dynamic N-Terminal modifications. For TMT label proteomic data analysis, TMTpro (Any N-Terminus) was set as a static N-Terminal Modification; TMTpro (K) and carbamidomethyl (C) were set as static modifications; oxidation (M) was set as a dynamic modification; acetyl (protein N-term), Met-loss (Protein N-term M) and Met-loss+acetyl (Protein N-term M) were set as dynamic N-Terminal modifications.
Protein abundance normalization was performed relative to the total peptide amount. DE analysis was performed with limma using R studio following the user guide 7 or one-tailed Student’s t-test with PD 2.4. PCA analyses were conducted with PD 2.4 and plotted using GraphPad Prism 7.0. The volcano and heatmap figures were plotted by Prism 7.0. The Venn diagram was plotted using Origin 2019b. Gene ontology analysis was performed using g:Profiler (website (https://biit.cs.ut.ee/gprofiler/gost). Other statistical analyses were carried out by one-tailed Student’s t-test or one-way ANOVA using Prism 7.0. Three independent biological replicates were used. P-values < 0.05 are reported as statistically significant and are depicted as follows throughout the manuscript: * p < 0.05, ** p < 0.01, **** p < 0.0001.
Supplementary Material
Table S1. Proteomic profiling of p97 knockdown in HCT116 cells, related to Figure 1
Table S2. Temporal proteomics of HCT116 cells treated with DMSO or inhibitors, related to Figure 1
Table S3. Proteomic data of p97 inhibitors and MG132 detected by TMTPro16 label, related to Figure 2–3
Table S4. E2F1 network proteins and cell cycle proteins list from Reactome database, related to Table 1, Figure 4
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| Anti- Cyclin D1 | Santa Cruz | Cat# sc-8396; AB_627344 |
| Anti-DHFR | Thermo Fisher | Cat# PA5–30992; AB_2548466 |
| Anti-p62 | MBL | Cat# M162–3; AB_1279299 |
| Anti-ATF3 | CST | Cat# 18665; AB_2827506 |
| Anti-p21 | BD | Cat# 610233; AB_397628 |
| Anti-CDK4 | Santa Cruz | Cat# sc-23896; AB_627239 |
| Anti-pRB | Santa Cruz | Cat# sc-377528 |
| Anti-E2F1 | Santa Cruz | Cat# sc-251; AB_627476 |
| Anti-Securin | Abcam | Cat# Ab3305; AB_2173413 |
| Anti-GAPDH | CST | Cat# 2118; AB_561053 |
| Anti-Lamin B1 | Proteintech | Cat# 66095–1-Ig; AB_11232208 |
| Anti-k48 | Boston Biochem | Cat# A-101; AB_10699867 |
| Anti-TCF11 | Santa Cruz | Cat# sc-13031; AB_2298615 |
| Anti-p97 | Thermo Fisher | Cat# MA3–004; AB_2214638 |
| p-4E-BP1 | Santa Cruz | Cat# sc-293124 |
| Anti-DNAJB11 | Santa Cruz | Cat# sc-271240; AB_10608210 |
| Anti-SYVN1 | Thermo Fisher | Cat# 13473–1-AP; AB_2287023 |
| Anti-SRPRB | Santa Cruz | Cat# sc-376723 |
| Anti-SRPR | Thermo Fisher | Cat# A305–140A; AB_2631535 |
| Anti-CCNB1 | Millipore | Cat# 05–373; AB_309701 |
| Anti-HMMR | Santa Cruz | Cat# sc-515221 |
| Anti-PDIA4 | Santa Cruz | Cat# sc-376230; AB_10988045 |
| Goat Anti-Rabbit IgG-HRP Conjugate | BIO-RAD | Cat# 170–6515; AB_11125142 |
| Goat Anti-Mouse IgG-HRP Conjugate | BIO-RAD | Cat# 170–6516; AB_11125547 |
| Chemicals, peptides, and recombinant proteins | ||
| CB-5083 | MedKoo Biosciences | Cat# 206489 |
| NMS-873 | MedKoo Biosciences | Cat# 406458 |
| UPCDC-30245 | Sigma | Cat# SML1674 |
| MG132 | Selleckchem | Cat# S2619 |
| Baf-A1 | Selleckchem | Cat# S1413 |
| RPMI-1640 medium | Corning, | Cat# 10–040-CM |
| Heat-inactivated fetal bovine serum (FBS) | R&D, | Cat# S11150 |
| penicillin-streptomycin (100x) | Gibco | Cat# 15140–122 |
| TrypLE™ Select Enzyme | Gibco | Cat# 50–591-420 |
| Dulbecco's PBS (DPBS) | Sigma | Cat# D8537–1 |
| 10xTBST | Bioland | Cat# TBST01–03 |
| 4–20% Mini-PROTEAN TGX precast gels | Bio-Rad | Cat# 456–1096 |
| ECL reagent | MilliporeSigma | Cat# WBKLS0500 |
| DTT | Cytiva | Cat# 17–1318-02 |
| UltraPure™ DNase/RNase-Free distilled water | Thermo Scientific | Cat#10977035 |
| TRIzol™ LS Reagent | Thermo Scientific | Cat# 10296010 |
| Direct-zol RNA MiniPrep plus kit | Zymo Research | Cat# R2072 |
| SensiFAST™ cDNA Synthesis Kit | Bioline | Cat# BIO-65054 |
| PowerUp SYBR Green Master Mix | Thermo Scientific | Cat# A25742 |
| TaqMan Universal Master Mix II no UNG | Thermo Scientific | Cat# 4440040 |
| DMSO | Fisher Scientific | Cat# BP231–1 |
| Absolute Ethanol (200 proof) | Fisher Scientific | Cat# BP2816–500 |
| Triethylammonium bicarbonate buffer (TEAB) | Sigma | Cat# 17902 |
| Thermo EasyPep Mini MS Sample Prep Kit | Thermo Scientific | Cat# A4006 |
| High pH reversed-phase peptide Fractionation Kit | Thermo Scientific | Cat# 84868 |
| Hydroxylamine | Thermo Scientific | Cat# 90115 |
| TMTpro 16plex reagents | Thermo Scientific | Cat# A44522 |
| LC-MS grade formic acid (FA) | Thermo Scientific | Cat# 85178 |
| Water, Optima™ LC/MS Grade | Fisher Scientific | Cat# W6500 |
| Acetonitrile (ACN), LC-MS Grade | Fisher Scientific | Cat# PI51101 |
| Trifluoroacetic acid (TFA) | Thermo Scientific | Cat# 85183 |
| BioT transfection reagent | Bioland Scientific | Cat# B01–01 |
| D-Luciferin | Medchemexpress | Cat# HY-12591A |
| Polybrene Infection / Transfection Reagent | Sigma | Cat# TR-1003 |
| Puromycin | Thermo Scientific | Cat# A1113802 |
| N-Ethylmaleimide (NEM) | Medchemexpress | Cat# HY-D0843 |
| Sodium Chloride (NaCl) | Fisher Scientific | Cat# S271–10 |
| 4 × Laemmli sample buffer | Bio-Rad | Cat# 161–0774 |
| TaqMan qPCR probe_GAPDH | Thermo Fisher | Hs02786624_g1 |
| TaqMan qPCR probe_p97 | Thermo Fisher | Hs00997642_m1 |
| TaqMan qPCR probe_CHOP | Thermo Fisher | Hs00358796_g1 |
| TaqMan qPCR probe_ATF3 | Thermo Fisher | Hs00231069_m1 |
| Critical commercial assays | ||
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat# G7573 |
| Pierce Quantitative Fluorometric Peptide Assay | Thermo Fisher | Cat# 23290 |
| Pierce Quantitative Colorimetric Peptide Assay | Thermo Fisher | Cat# 23275 |
| MycoAlert PLUS Mycoplasma Detection Kit | Lonza | Cat# LT07–710 |
| Cell Cycle Analysis Kit | Sigma-Aldrich | Cat# MAK344 |
| Deposited data | ||
| p97 KD proteomic raw data | This paper | PRIDE: 10.6019/PXD025094 |
| p97 inhibitors and MG132 proteomic raw data | This paper | PRIDE: 10.6019/PXD025167 |
| TMT labeling proteomic raw data | This paper | PRIDE: 10.6019/PXD025225 |
| Experimental models: Cell lines | ||
| HCT116 cell line | ATCC | Cat# CCL-247 |
| HT29 cell line | ATCC | Cat# HTB-38 |
| Oligonucleotides | ||
| See Table S10 | This paper | N/A |
| Recombinant DNA | ||
| TripZ lent-viral shRNA system | Thermo Fisher Scientific | N/A |
| The nontargeting control shRNA | Thermo Fisher Scientific | N/A |
| pGL2-AN plasmid | Neuman et al., 1994 | Addgene 20950 |
| Software and algorithms | ||
| FlowJo | BD Biosciences | https://www.flowjo.com/ |
| Proteome Discoverer 2.4 | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home.html |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| ImageLab™ software | BioRad | https://www.bio-rad.com/ |
| Xcalibur v.4.0 | Thermo Scientific | https://www.thermofisher.com/us/en/home.html |
| Sequest HT | Thermo Scientific | https://www.thermofisher.com/us/en/home.html |
| RStudio v.1.2.5033 | RStudio | https://www.rstudio.com |
| Limma package | Ritchie et al. (2015) | https://bioconductor.org/packages/release/bioc/html/limma.html |
| R v.3.6.3 | The R Foundation | https://www.r-project.org/ |
Highlights.
Proteomics of p97 and proteasome inhibition reveals distinct molecular mechanisms.
p97 Inhibition activates the IREα and XBP1 pathways more than that of proteasome.
p97 Inhibition blocks E2F1-mediated transcription via reducing CCND-CDK4/6 levels.
Inhibition of p97 promotes the downregulation of cell cycle oncoproteins.
Acknowledgements
This work was supported in part with funds from the National Institute of Neurological Disorders and Stroke, R01NS100815 and R01NS102279.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- AKOPIAN D & RAPE M 2017. Conducting the finale of DNA replication. Genes Dev, 31, 226–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALEXANDRU G, GRAUMANN J, SMITH GT, KOLAWA NJ, FANG R & DESHAIES RJ 2008. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell, 134, 804–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALVEREZ C, ARKIN MR, BULFER SL, COLOMBO R, KOVALIOV M, LAPORTE MG, LIM C, LIANG M, MOORE WJ, NEITZ RJ, YAN Y, YUE Z, HURYN DM & WIPF P 2015. Structure-Activity Study of Bioisosteric Trifluoromethyl and Pentafluorosulfanyl Indole Inhibitors of the AAA ATPase p97. ACS Med Chem Lett, 6, 1225–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ANDERSON DJ, LE MOIGNE R, DJAKOVIC S, KUMAR B, RICE J, WONG S, WANG J, YAO B, VALLE E, KISS VON SOLY S, MADRIAGA A, SORIANO F, MENON MK, WU ZY, KAMPMANN M, CHEN Y, WEISSMAN JS, AFTAB BT, YAKES FM, SHAWVER L, ZHOU HJ, WUSTROW D & ROLFE M 2015. Targeting the AAA ATPase p97 as an Approach to Treat Cancer through Disruption of Protein Homeostasis. Cancer Cell, 28, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ARHZAOUY K, PAPADOPOULOS C, SCHULZE N, PITTMAN SK, MEYER H & WEIHL CC 2019. VCP maintains lysosomal homeostasis and TFEB activity in differentiated skeletal muscle. Autophagy, 15, 1082–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BASTOLA P, NEUMS L, SCHOENEN FJ & CHIEN J 2016. VCP inhibitors induce endoplasmic reticulum stress, cause cell cycle arrest, trigger caspase-mediated cell death and synergistically kill ovarian cancer cells in combination with Salubrinal. Mol Oncol, 10, 1559–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENZENO S, LU F, GUO M, BARBASH O, ZHANG F, HERMAN JG, KLEIN PS, RUSTGI A & DIEHL JA 2006. Identification of mutations that disrupt phosphorylation-dependent nuclear export of cyclin D1. Oncogene, 25, 6291–6303. [DOI] [PubMed] [Google Scholar]
- BRAUN RJ & ZISCHKA H 2008. Mechanisms of Cdc48/VCP-mediated cell death: from yeast apoptosis to human disease. Biochim Biophys Acta, 1783, 1418–35. [DOI] [PubMed] [Google Scholar]
- BURKHART DL & SAGE J 2008. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat Rev Cancer, 8, 671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN T, XUE L, NIU J, MA L, LI N, CAO X, LI Q, WANG M, ZHAO W, LI G, WANG J & TONG T 2012. The retinoblastoma protein selectively represses E2F1 targets via a TAAC DNA element during cellular senescence. J Biol Chem, 287, 37540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHENG J, NORTH BJ, ZHANG T, DAI X, TAO K, GUO J & WEI W 2018. The emerging roles of protein homeostasis-governing pathways in Alzheimer’s disease. Aging Cell, 17, e12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOU TF, BROWN SJ, MINOND D, NORDIN BE, LI K, JONES AC, CHASE P, PORUBSKY PR, STOLTZ BM, SCHOENEN FJ, PATRICELLI MP, HODDER P, ROSEN H & DESHAIES RJ 2011. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc Natl Acad Sci U S A, 108, 4834–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOU TF & DESHAIES RJ 2011. Development of p97 AAA ATPase inhibitors. Autophagy, 7, 1091–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOU TF, LI K, FRANKOWSKI KJ, SCHOENEN FJ & DESHAIES RJ 2013. Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase. ChemMedChem, 8, 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CORAZZARI M, GAGLIARDI M, FIMIA GM & PIACENTINI M 2017. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front Oncol, 7, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DALL’OLIO GM, JASSAL B, MONTANUCCI L, GAGNEUX P, BERTRANPETIT J & LAAYOUNI H 2011. The annotation of the asparagine N-linked glycosylation pathway in the Reactome database. Glycobiology, 21, 1395–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DANG F, NIE L & WEI W 2021. Ubiquitin signaling in cell cycle control and tumorigenesis. Cell Death Differ, 28, 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DESHAIES RJ 2014. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol, 12, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENGEL RH, BROWN JA, VON ROENN JH, O’REGAN RM, BERGAN R, BADVE S, RADEMAKER A & GRADISHAR WJ 2007. A phase II study of single agent bortezomib in patients with metastatic breast cancer: a single institution experience. Cancer Invest, 25, 733–7. [DOI] [PubMed] [Google Scholar]
- FANG Z, GONG C, LIU H, ZHANG X, MEI L, SONG M, QIU L, LUO S, ZHU Z, ZHANG R, GU H & CHEN X 2015. E2F1 promote the aggressiveness of human colorectal cancer by activating the ribonucleotide reductase small subunit M2. Biochem Biophys Res Commun, 464, 407–15. [DOI] [PubMed] [Google Scholar]
- GARRISON P & BANGS JD 2020. p97 Inhibitor CB-5083 Blocks ERAD in Trypanosoma brucei. Mol Biochem Parasitol, 239, 111313. [DOI] [PubMed] [Google Scholar]
- HARDING HP, NOVOA I, ZHANG Y, ZENG H, WEK R, SCHAPIRA M & RON D 2000. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Molecular Cell, 6, 1099–1108. [DOI] [PubMed] [Google Scholar]
- HEIDELBERGER JB, VOIGT A, BORISOVA ME, PETROSINO G, RUF S, WAGNER SA & BELI P 2018. Proteomic profiling of VCP substrates links VCP to K6-linked ubiquitylation and c-Myc function. EMBO Rep, 19, e44754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HER NG, TOTH JI, MA CT, WEI Y, MOTAMEDCHABOKI K, SERGIENKO E & PETROSKI MD 2016. p97 Composition Changes Caused by Allosteric Inhibition Are Suppressed by an On-Target Mechanism that Increases the Enzyme’s ATPase Activity. Cell Chem Biol, 23, 517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HETZ C 2012. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol, 13, 89–102. [DOI] [PubMed] [Google Scholar]
- HETZ C & PAPA FR 2018. The Unfolded Protein Response and Cell Fate Control. Mol Cell, 69, 169–181. [DOI] [PubMed] [Google Scholar]
- HOLLIEN J, LIN JH, LI H, STEVENS N, WALTER P & WEISSMAN JS 2009. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J Cell Biol, 186, 323–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLLIEN J & WEISSMAN JS 2006. Decay of endoplasmic reticulum-localized mRNAs during the unfolded protein response. Science, 313, 104–7. [DOI] [PubMed] [Google Scholar]
- HURYN DM, KORNFILT DJP & WIPF P 2020. p97: An Emerging Target for Cancer, Neurodegenerative Diseases, and Viral Infections. J Med Chem, 63, 1892–1907. [DOI] [PubMed] [Google Scholar]
- KANEKO Y, SHIMODA K, AYALA R, GOTO Y, PANICO S, ZHANG X & KONDO H 2021. p97 and p47 function in membrane tethering in cooperation with FTCD during mitotic Golgi reassembly. EMBO J, e105853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM HK, LEE GH, BHATTARAI KR, LEE MS, BACK SH, KIM HR & CHAE HJ 2020. TMBIM6 (transmembrane BAX inhibitor motif containing 6) enhances autophagy through regulation of lysosomal calcium. Autophagy, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM OH, LIM JH, WOO KJ, KIM Y-H, JIN I-N, HAN ST, PARK J-W & KWON TK 2004. Influence of p53 and p21Waf1 expression on G2/M phase arrest of colorectal carcinoma HCT116 cells to proteasome inhibitors. International journal of oncology, 24, 935–941. [PubMed] [Google Scholar]
- KROEMER G, MARINO G & LEVINE B 2010. Autophagy and the integrated stress response. Mol Cell, 40, 280–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANGE CA & YEE D 2011. Killing the second messenger: targeting loss of cell cycle control in endocrine-resistant breast cancer. Endocr Relat Cancer, 18, C19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAPORTE MG, BURNETT JC, COLOMBO R, BULFER SL, ALVEREZ C, CHOU TF, NEITZ RJ, GREEN N, MOORE WJ, YUE Z, LI S, ARKIN MR, WIPF P & HURYN DM 2018. Optimization of Phenyl Indole Inhibitors of the AAA+ ATPase p97. ACS Med Chem Lett, 9, 1075–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LE MOIGNE R, AFTAB BT, DJAKOVIC S, DHIMOLEA E, VALLE E, MURNANE M, KING EM, SORIANO F, MENON MK, WU ZY, WONG ST, LEE GJ, YAO B, WIITA AP, LAM C, RICE J, WANG J, CHESI M, BERGSAGEL PL, KRAUS M, DRIESSEN C, KISS VON SOLY S, YAKES FM, WUSTROW D, SHAWVER L, ZHOU HJ, MARTIN TG 3RD, WOLF JL, MITSIADES CS, ANDERSON DJ & ROLFE M 2017. The p97 Inhibitor CB-5083 Is a Unique Disrupter of Protein Homeostasis in Models of Multiple Myeloma. Mol Cancer Ther, 16, 2375–2386. [DOI] [PubMed] [Google Scholar]
- LI Z, ADAMS RM, CHOUREY K, HURST GB, HETTICH RL & PAN C 2012. Systematic comparison of label-free, metabolic labeling, and isobaric chemical labeling for quantitative proteomics on LTQ Orbitrap Velos. J Proteome Res, 11, 1582–90. [DOI] [PubMed] [Google Scholar]
- LI Z & XU X 2019. Post-Translational Modifications of the Mini-Chromosome Maintenance Proteins in DNA Replication. Genes (Basel), 10, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LU D, WOLFGANG CD & HAI T 2006. Activating transcription factor 3, a stress-inducible gene, suppresses Ras-stimulated tumorigenesis. J Biol Chem, 281, 10473–81. [DOI] [PubMed] [Google Scholar]
- MAGNAGHI P, D’ALESSIO R, VALSASINA B, AVANZI N, RIZZI S, ASA D, GASPARRI F, COZZI L, CUCCHI U, ORRENIUS C, POLUCCI P, BALLINARI D, PERRERA C, LEONE A, CERVI G, CASALE E, XIAO Y, WONG C, ANDERSON DJ, GALVANI A, DONATI D, O’BRIEN T, JACKSON PK & ISACCHI A 2013. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat Chem Biol, 9, 548–56. [DOI] [PubMed] [Google Scholar]
- MCNAIR C, XU K, MANDIGO AC, BENELLI M, LEIBY B, RODRIGUES D, LINDBERG J, GRONBERG H, CRESPO M, DE LAERE B, DIRIX L, VISAKORPI T, LI F, FENG FY, DE BONO J, DEMICHELIS F, RUBIN MA, BROWN M & KNUDSEN KE 2018. Differential impact of RB status on E2F1 reprogramming in human cancer. J Clin Invest, 128, 341–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MEYER HH 2005. Golgi reassembly after mitosis: the AAA family meets the ubiquitin family. Biochim Biophys Acta, 1744, 108–19. [DOI] [PubMed] [Google Scholar]
- MEYER HH, WANG Y & WARREN G 2002. Direct binding of ubiquitin conjugates by the mammalian p97 adaptor complexes, p47 and Ufd1-Npl4. EMBO J, 21, 5645–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MOIR D, STEWART SE, OSMOND BC & BOTSTEIN D 1982. Cold-sensitive cell-division-cycle mutants of yeast: isolation, properties, and pseudoreversion studies. Genetics, 100, 547–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEUMAN E, FLEMINGTON EK, SELLERS WR & KAELIN WG JR. 1994. Transcription of the E2F-1 gene is rendered cell cycle dependent by E2F DNA-binding sites within its promoter. Mol Cell Biol, 14, 6607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OH E, MARK KG, MOCCIARO A, WATSON ER, PRABU JR, CHA DD, KAMPMANN M, GAMARRA N, ZHOU CY & RAPE M 2020. Gene expression and cell identity controlled by anaphase-promoting complex. Nature, 579, 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAGLIARINI V, GIGLIO P, BERNARDONI P, DE ZIO D, FIMIA GM, PIACENTINI M & CORAZZARI M 2015. Downregulation of E2F1 during ER stress is required to induce apoptosis. J Cell Sci, 128, 1166–79. [DOI] [PubMed] [Google Scholar]
- PAPADOPOULOS C, KIRCHNER P, BUG M, GRUM D, KOERVER L, SCHULZE N, POEHLER R, DRESSLER A, FENGLER S, ARHZAOUY K, LUX V, EHRMANN M, WEIHL CC & MEYER H 2017. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J, 36, 135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAPADOPOULOS C, KRAVIC B & MEYER H 2020. Repair or Lysophagy: Dealing with Damaged Lysosomes. J Mol Biol, 432, 231–239. [DOI] [PubMed] [Google Scholar]
- PARISI E, YAHYA G, FLORES A & ALDEA M 2018. Cdc48/p97 segregase is modulated by cyclin-dependent kinase to determine cyclin fate during G1 progression. EMBO J, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QIE S & DIEHL JA 2016. Cyclin D1, cancer progression, and opportunities in cancer treatment. J Mol Med (Berl), 94, 1313–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RADHAKRISHNAN SK, DEN BESTEN W & DESHAIES RJ 2014. p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. Elife, 3, e01856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAMAN M, SERGEEV M, GARNAAS M, LYDEARD JR, HUTTLIN EL, GOESSLING W, SHAH JV & HARPER JW 2015. Systematic proteomics of the VCP-UBXD adaptor network identifies a role for UBXN10 in regulating ciliogenesis. Nat Cell Biol, 17, 1356–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAMANATHAN HN & YE Y 2012. The p97 ATPase associates with EEA1 to regulate the size of early endosomes. Cell Res, 22, 346–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAVANELLI S, DEN BRAVE F & HOPPE T 2020. Mitochondrial Quality Control Governed by Ubiquitin. Front Cell Dev Biol, 8, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSENOW EC 3RD 1990. Diffuse pulmonary infiltrates in the immunocompromised host. Clin Chest Med, 11, 55–64. [PubMed] [Google Scholar]
- SCHETTINI F, DE SANTO I, REA CG, DE PLACIDO P, FORMISANO L, GIULIANO M, ARPINO G, DE LAURENTIIS M, PUGLISI F, DE PLACIDO S & DEL MASTRO L 2018. CDK 4/6 Inhibitors as Single Agent in Advanced Solid Tumors. Front Oncol, 8, 608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHMID P, KUHNHARDT D, KIEWE P, LEHENBAUER-DEHM S, SCHIPPINGER W, GREIL R, LANGE W, PREISS J, NIEDERLE N, BROSSART P, FREIER W, KUMMEL S, VAN DE VELDE H, REGIERER A & POSSINGER K 2008. A phase I/II study of bortezomib and capecitabine in patients with metastatic breast cancer previously treated with taxanes and/or anthracyclines. Ann Oncol, 19, 871–6. [DOI] [PubMed] [Google Scholar]
- SCHWEPPE DK, ENG JK, YU Q, BAILEY D, RAD R, NAVARRETE-PEREA J, HUTTLIN EL, ERICKSON BK, PAULO JA & GYGI SP 2020. Full-Featured, Real-Time Database Searching Platform Enables Fast and Accurate Multiplexed Quantitative Proteomics. J Proteome Res, 19, 2026–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHRIMAL S, CHEREPANOVA NA, MANDON EC, VENEV SV & GILMORE R 2019. Asparagine-linked glycosylation is not directly coupled to protein translocation across the endoplasmic reticulum in Saccharomyces cerevisiae. Mol Biol Cell, 30, 2626–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STACH L & FREEMONT PS 2017. The AAA+ ATPase p97, a cellular multitool. Biochem J, 474, 2953–2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STEFFEN J, SEEGER M, KOCH A & KRUGER E 2010. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol Cell, 40, 147–58. [DOI] [PubMed] [Google Scholar]
- TRINH XB, SAS L, VAN LAERE SJ, PROVE A, DELEU I, RASSCHAERT M, VAN DE VELDE H, VINKEN P, VERMEULEN PB, VAN DAM PA, WOJTASIK A, DE MESMAEKER P, TJALMA WA & DIRIX LY 2012. A phase II study of the combination of endocrine treatment and bortezomib in patients with endocrine-resistant metastatic breast cancer. Oncol Rep, 27, 657–63. [DOI] [PubMed] [Google Scholar]
- VAN DEN BOOM J & MEYER H 2018. VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol Cell, 69, 182–194. [DOI] [PubMed] [Google Scholar]
- WALEJKO JM, CHRISTOPHER BA, CROWN SB, ZHANG GF, PICKAR-OLIVER A, YONESHIRO T, FOSTER MW, PAGE S, VAN VLIET S, ILKAYEVA O, MUEHLBAUER MJ, CARSON MW, BROZINICK JT, HAMMOND CD, GIMENO RE, MOSELEY MA, KAJIMURA S, GERSBACH CA, NEWGARD CB, WHITE PJ & MCGARRAH RW 2021. Branched-chain alpha-ketoacids are preferentially reaminated and activate protein synthesis in the heart. Nat Commun, 12, 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG F, LI S, GAN T, STOTT GM, FLINT A & CHOU TF 2020. Allosteric p97 Inhibitors Can Overcome Resistance to ATP-Competitive p97 Inhibitors for Potential Anticancer Therapy. ChemMedChem, 15, 685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WELLS J, GRAVEEL CR, BARTLEY SM, MADORE SJ & FARNHAM PJ 2002. The identification of E2F1-specific target genes. Proc Natl Acad Sci U S A, 99, 3890–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOLF DH & STOLZ A 2012. The Cdc48 machine in endoplasmic reticulum associated protein degradation. Biochim Biophys Acta, 1823, 117–24. [DOI] [PubMed] [Google Scholar]
- WU X & RAPOPORT TA 2018. Mechanistic insights into ER-associated protein degradation. Curr Opin Cell Biol, 53, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XUE L, BLYTHE EE, FREIBERGER EC, MAMROSH JL, HEBERT AS, REITSMA JM, HESS S, COON JJ & DESHAIES RJ 2016. Valosin-containing protein (VCP)-Adaptor Interactions are Exceptionally Dynamic and Subject to Differential Modulation by a VCP Inhibitor. Mol Cell Proteomics, 15, 2970–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG CH, GONZALEZ-ANGULO AM, REUBEN JM, BOOSER DJ, PUSZTAI L, KRISHNAMURTHY S, ESSELTINE D, STEC J, BROGLIO KR, ISLAM R, HORTOBAGYI GN & CRISTOFANILLI M 2006. Bortezomib (VELCADE) in metastatic breast cancer: pharmacodynamics, biological effects, and prediction of clinical benefits. Ann Oncol, 17, 813–7. [DOI] [PubMed] [Google Scholar]
- YE Y, SHIBATA Y, YUN C, RON D & RAPOPORT TA 2004. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature, 429, 841–7. [DOI] [PubMed] [Google Scholar]
- YE Y, TANG WK, ZHANG T & XIA D 2017. A Mighty “Protein Extractor” of the Cell: Structure and Function of the p97/CDC48 ATPase. Front Mol Biosci, 4, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO Z, WU M, ZHANG X, JIN Q, WANG Y, ZOU C, HUANG G, YIN J, XIE X & SHEN J 2020. CB-5083, an inhibitor of P97, suppresses osteosarcoma growth and stem cell properties by altering protein homeostasis. Am J Transl Res, 12, 2956–2967. [PMC free article] [PubMed] [Google Scholar]
- ZHOU HJ, WANG J, YAO B, WONG S, DJAKOVIC S, KUMAR B, RICE J, VALLE E, SORIANO F, MENON MK, MADRIAGA A, KISS VON SOLY S, KUMAR A, PARLATI F, YAKES FM, SHAWVER L, LE MOIGNE R, ANDERSON DJ, ROLFE M & WUSTROW D 2015. Discovery of a First-in-Class, Potent, Selective, and Orally Bioavailable Inhibitor of the p97 AAA ATPase (CB-5083). J Med Chem, 58, 9480–97. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Proteomic profiling of p97 knockdown in HCT116 cells, related to Figure 1
Table S2. Temporal proteomics of HCT116 cells treated with DMSO or inhibitors, related to Figure 1
Table S3. Proteomic data of p97 inhibitors and MG132 detected by TMTPro16 label, related to Figure 2–3
Table S4. E2F1 network proteins and cell cycle proteins list from Reactome database, related to Table 1, Figure 4
Data Availability Statement
All relevant data generated during this study are included in the article and the supplementary Information. The mass spectrometry raw data related to Fig. 1a–d are deposited to the ProteomeXchance Consortium (https://www.ebi.ac.uk/pride/) via the PRIDE repository with the dataset identifier PXD025094 and 10.6019/PXD025094”. The mass spectrometry raw data related to Figs. 1e–g with dataset identifier PXD025167 and 10.6019/PXD025167”. The mass spectrometry raw data related to Fig. 2 with dataset identifier PXD025225 and 10.6019/PXD025225”. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.