

Abstract
Background and Aims:
A leading cause of hepatocellular carcinoma (HCC) is non-alcoholic steatohepatitis (NASH), but mechanisms linking NASH to eventual tumor formation remain poorly understood. Here we investigate the role of TAZ/WWTR1, which is induced in hepatocytes in NASH, in the progression of NASH to HCC.
Methods:
The roles of hepatocyte TAZ and its downstream targets were investigated in diet-induced and genetic models of NASH-HCC using gene-targeting, AAV8-H1-mediated gene silencing, or AAV8-TBG-mediated gene expression. The biochemical signature of the newly elucidated pathway was probed in liver specimens from humans with NASH-HCC.
Results:
When hepatocyte-TAZ was silenced in pre-tumor NASH mice using AAV8-H1-shTaz, subsequent HCC tumor development was suppressed. In this setting, this tumor-suppressing effect of shTaz was not dependent of TAZ silencing in the tumors themselves and could be dissociated from the NASH-suppressing effects of shTaz. The mechanism linking pre-tumor hepatocyte-TAZ to eventual tumor formation involved TAZ-mediated induction of the NOX2-encoding gene Cybb, which led to NADPH-mediated oxidative DNA damage. As evidence, DNA damage and tumor formation could be suppressed by treatment of pre-tumor NASH mice with AAV8-H1-shCybb; AAV8-TBG-OGG1, encoding the oxidative DNA-repair enzyme 8-oxoguanine glycosylase; or AAV8-TBG-NHEJ1, encoding the dsDNA repair enzyme non-homologous end-joining factor 1. In tumor-surrounding tissue in human NASH-HCC liver, there were strong correlations among TAZ, NOX2, oxidative DNA damage.
Conclusions:
TAZ in pre-tumor NASH-hepatocytes, via induction of Cybb and NOX2-mediated DNA damage, contributes to subsequent HCC tumor development. These findings illustrate how NASH provides a unique window into the early molecular events that can lead to tumor formation and suggest that NASH therapies targeting TAZ might also prevent NASH-HCC.
Keywords: nonalcoholic steatohepatitis (NASH), hepatocellular carcinoma (HCC), TAZ/WWTR1, NOX2/Cybb, oxidative DNA damage
Graphical Abstract
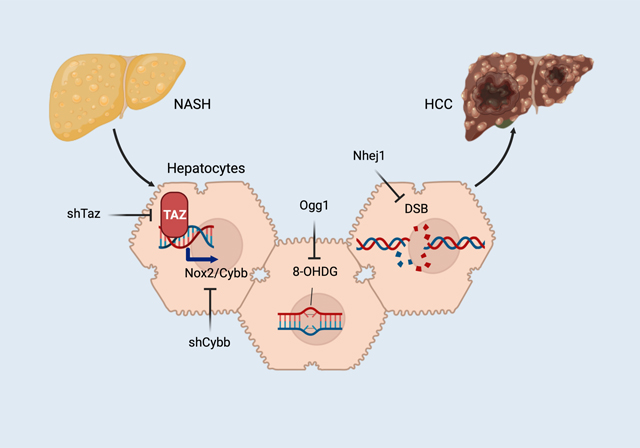
Lay summary
Nonalcoholic steatohepatitis (NASH) is emerging as the leading cause of a type of liver cancer called hepatocellular carcinoma (HCC), but molecular events in pre-tumor NASH hepatocytes leading to HCC remain largely unknown. Our study shows that a protein called TAZ in pre-tumor NASH-hepatocytes promotes damage to the DNA of hepatocytes and thereby contributes to eventual HCC. This study reveals a very early event in HCC that is induced in pre-tumor NASH, and the findings suggest that NASH therapies targeting TAZ might also prevent NASH-HCC.
INTRODUCTION
Nonalcoholic steatohepatitis (NASH) is emerging as the leading cause of both liver disease1–3 and hepatocellular carcinoma (HCC)3,4. NASH-HCC can develop in the absence of cirrhosis5–7, suggesting that NASH-mediated signals within hepatocytes may drive carcinogenesis before the carcinogenic effects of cirrhosis are present. Indeed, the prolonged pre-cancer stage of NASH provides a unique opportunity to address a major challenge in cancer, namely, identifying very early changes in non-cancer cells that can subsequently lead to tumor formation. However, the mechanisms of how NASH predisposes to eventual HCC tumor formation remains largely unknown.
To address this challenge, we investigated three features in common between NASH hepatocytes and HCC tumor cells, namely, TAZ/WWTR1, oxidative stress, and DNA damage8–11. The gene regulator TAZ/WWTR1 is increased in mouse and human hepatocytes as hepatosteatosis progresses to NASH8,12–14 and promotes NASH by inducing the secretory protein Indian hedgehog (Ihh)8. Oxidative stress occurs in NASH hepatocytes and can cause double-stranded DNA breaks (DSB) and chromosome instability, which, by causing mutations in tumor-suppressor genes, can induce HCC when other “hits” are present10,11,15,16. Although TAZ can promote tumor growth and spread, including in liver cancer17–19, we hypothesized that it might have an independent role in activating molecular events in pre-tumor NASH hepatocytes that could eventually lead to HCC. We now present evidence that TAZ, by inducing the pro-oxidant gene Cybb, promotes oxidative DNA damage in pre-tumor NASH, leading to eventual HCC tumor formation. This conclusion is supported by molecular-genetic causation data in experimental NASH-HCC, and the biochemical signature of the pathway is present in human NASH-HCC.
MATERIALS AND METHODS
Animal Studies
Male wild-type C57BL/6J mice (#000664, 9–10 weeks/old), Cybbfl/fl mice (#031777), and RosaNICD mice (#008159) were from Jackson Laboratory (Bar Harbor, ME) and allowed to adapt in the animal facility for 1 week prior to random assignment to experimental cohorts. Wwtr1fl/fl mice20, backcrossed to C57BL/6J, were provided by Dr. Eric Olson (University of Texas Southwestern). The mice were fed a diet containing sugar water (23.1 g fructose/L and 18.9 g glucose/L), palmitate, and 1.25% cholesterol (“NASH diet”; Teklad, TD.160785 PWD), which induces NASH after 16 weeks8. All AAV8-viruses were injected by tail vein (2×1011 genome copies/mouse) as indicated in the figure legends. For the DMBA model, 50 μl of 0.5% DMBA (7,12-dimethylbenz [a]anthracene, Sigma) in acetone was administered to the dorsal surface on postnatal day 4–521; the NASH diet was begun at weaning (3 wks/o). Animals were housed in standard cages at 22°C in a 12–12-hour light-dark cycle in a barrier facility. For mouse HCC, the predetermined endpoint was tumor weight estimated to be <10% of body weight. All animal experiments were performed in accordance with institutional guidelines and regulations and approved by the Institutional Animal Care and Use Committee at Columbia University.
RESULTS
Silencing hepatocyte TAZ in pre-tumor NASH mice suppresses HCC tumor development
We began our investigation with a well-characterized and validated NASH model that uses a diet rich in fructose, palmitate, and cholesterol8,14,22–24. In this model, early fibrosis occurs after 16 weeks on diet, but we extended the feeding period to 15 months to look for HCC. All of the mice developed tumors showing features of HCC, including far fewer portal tracts in the tumors than in the surrounding liver, reticulin staining showing expanded hepatocyte cords, and positive glypican-3 staining (Figure 1A). In another cohort, we administered AAV8-H1-shTaz or AAV8-H1-scrambled RNA (Scr) at the 8-month time point, which is before tumors develop, and analyzed the mice at 13 months (Figure 1B). AAV8-shTaz potently lowers TAZ specifically in hepatocytes8, and we documented TAZ silencing in the livers of the 13-month NASH diet-fed mice (Figure 1C). We found that hepatocyte-TAZ silencing completely prevented tumor development (Figures 1D). The percentage of Ki67+ HNF4α+ liver cells in non-tumor tissue was also decreased in the shTaz cohort (Figures 1E). For a second model, we treated newborn mice with the mutagen DMBA and then placed them on the NASH diet from 1–9 months of age. DMBA alone does not cause tumors in this timeframe21, but the combination of DMBA and the NASH diet led to the development of numerous tumor (Figure 1F). DMBA/NASH diet-treated Wwtr1fl/fl mice were treated with AAV8-TBG-Cre to delete hepatocyte-TAZ, or AAV8-TBG-LacZ control, at the 5-month time point, which is before tumors form (Figures 1G–1H). Deletion of hepatocyte-TAZ markedly decreased tumor number and size at 10 months (Figures 1I and S1A). The percentage of Ki67+HNF4α+ cells in non-tumor tissue was also decreased by hepatocyte-TAZ deletion (Figures 1J). As a third model, we activated hepatocyte Notch by treating RosaNICD mice with AAV8-TBG-Cre and then fed the NASH diet, which leads to NASH features after 2 months and NASH diet-dependent HCC tumor formation by 3–4 months23. We verified that tumors formed at 4 months (Figure 1K) and then used RosaNICD Wwtr1fl/fl to test our hypothesis. The experimental group was administered AAV8-TBG-Cre to enable both Notch activation and TAZ deletion in hepatocytes, with Cre-injected RosaNICD mice serving as the intact-TAZ control cohort (Figures 1L–1M). The mice were then fed the NASH diet for 4 months. Deletion of hepatocyte TAZ lowered tumor number and size and the percentage of Ki67+HNF4α+ cells in non-tumor tissue (Figures 1N–1O and S1B). Thus, in three separate models of NASH diet-dependent HCC, silencing or deleting TAZ in hepatocytes before tumors form suppresses the eventual formation of HCC tumors.
Figure 1. Silencing hepatocyte TAZ in pre-tumor NASH mice suppresses the development of HCC tumors.
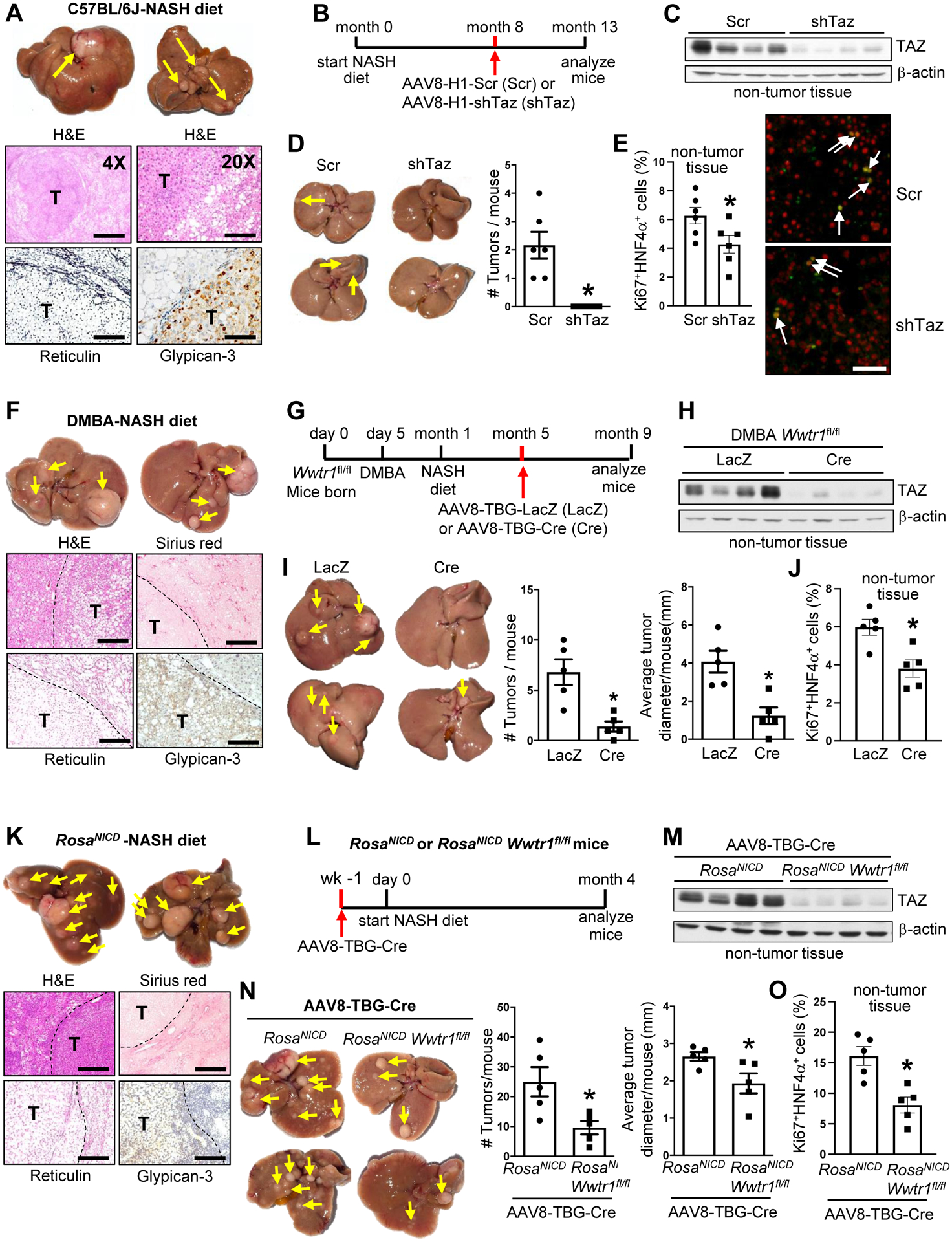
(A) Livers (arrows, tumors) and liver sections of mice fed the NASH diet for 15 months. The sections were stained with H&E (imaged at 4X and 20X; bars, 1 mm and 200 μm, respectively; T, tumor) and with reticulin and ant-iglypican-3 (bars, 100 μm). (B-E) Mice were fed the NASH diet for 13 months, with AAV8-H1-shTaz (shTaz) or control vector (Scr) administered at 8 monts. (B) Experimental scheme. (C) TAZ immunoblot from non-tumor liver tissue. (D) Livers (arrows, tumors) and tumor numbers/mouse. (E) Liver sections from non-tumor areas stained for Ki67 (green) and HNF4α (red) and quantified for the percent Ki67+HNF4α+ cells (arrows, Ki67+HNF4α+ cells; bar, 100 μm). For D-E, n = 6 mice/group; means ± SEM; *p < 0.05 by Student’s t-test. (F) Livers (arrows, tumors) and liver sections of mice that were administered DMBA post-natal day 5; placed on NASH diet at 1 month of age, and analyzed at 9 months. The sections were stained with H&E, Sirius red (bars, 500 μm; T, tumor), reticulin, and anti-glypican-3 (bars, 200 μm). (G-J) Wwtr1fl/fl male mice were administered DMBA on postnatal day 5; placed on the NASH diet at 1 month; injected with AAV8-TBG-LacZ or AAV8-TBG-Cre at 5 months; and analyzed at 9 months. (G) Experimental scheme. (H) TAZ immunoblot from non-tumor liver tissue. (I) Livers (arrows, tumors) and tumor numbers and average diameter. (J) Percent Ki67+HNF4α+ cells in non-tumor areas. For I-J, n = 5 mice/group; means ± SEM; *p < 0.05 by Student’s t-test. (K) Livers (arrows, tumors) and liver sections of RosaNICD mice injected with AAV8-TBG-Cre to activate hepatocyte Notch; started on the NASH diet 1 week later; and analyed after 4 months on diet. The sections were stained with H&E, Sirius red (bars, 500 μm), reticulin, and anti-glypican-3 (bars, 200 μm). (L-O) RosaNICD or RosaNICD Wwtr1fl/fl mice were treated with AAV8-TBG-Cre; placed on NASH diet 1 week later; and analyzed 4 months later. (L) Experimental scheme. (M) TAZ immunoblot from non-tumor liver tissue. (N) Livers (arrows, tumors) and tumor numbers and average diameter. (O) Percent Ki67+HNF4α+ cells in non-tumor areas. For N-O, n = 5 mice/group; means ± SEM; *p < 0.05 by Student’s t-test.
HCC tumor suppression by hepatocyte-TAZ silencing is not dependent of TAZ silencing in tumors
As with other types of HCC25,26, TAZ was expressed in human and mouse NASH-HCC tumors (Figures S1C–S1G), and we found that TAZ deletion using the cre-lox method, i.e., AAV8-TBG-Cre in the DMBA-Wwtr1fl/fl and RosaNICD Wwtr1fl/fl models, lowered tumor TAZ (Figure S1H–S1I). Thus, it was possible that silencing of TAZ in tumor cells was responsible for tumor suppression. In contrast, episomally expressed AAV8-shTaz becomes diluted as cells divide, resulting in eventual elimination of gene silencing in tumors. Thus, the tumor-preventative effect of AAV8-H1-shTaz in the 13-month NASH-diet model (Figures 1B–E) suggests a pre-tumor effect. To test this principle in a more robust model, we turned to the Notch-NASH diet model. Three months after Notch activation and start of the NASH diet, mice were injected with AAV8-H1-shTaz or AAV8-H1-Scr and analyzed 2 months later (Figure 2A). As designed, shTaz silenced TAZ in surrounding NASH tissue but not in the tumor tissue (Figure 2B). We found that tumor number and size were decreased by shTaz treatment (Figure 2C and S1J), as was the percentage of Ki67+HNF4α+ cells in surrounding tissue (Figure 2D). Note that shTaz did not alter the expression of the Notch downstream gene Hes1 (Figure S1K), indicating lack of interference with Notch function itself. These data suggest the TAZ in pre-tumor hepatocytes contributes to molecular events that can eventually lead to tumor formation.
Figure 2. The suppression of HCC tumor formation by hepatocyte-TAZ silencing is not dependent of tumor-TAZ silencing.
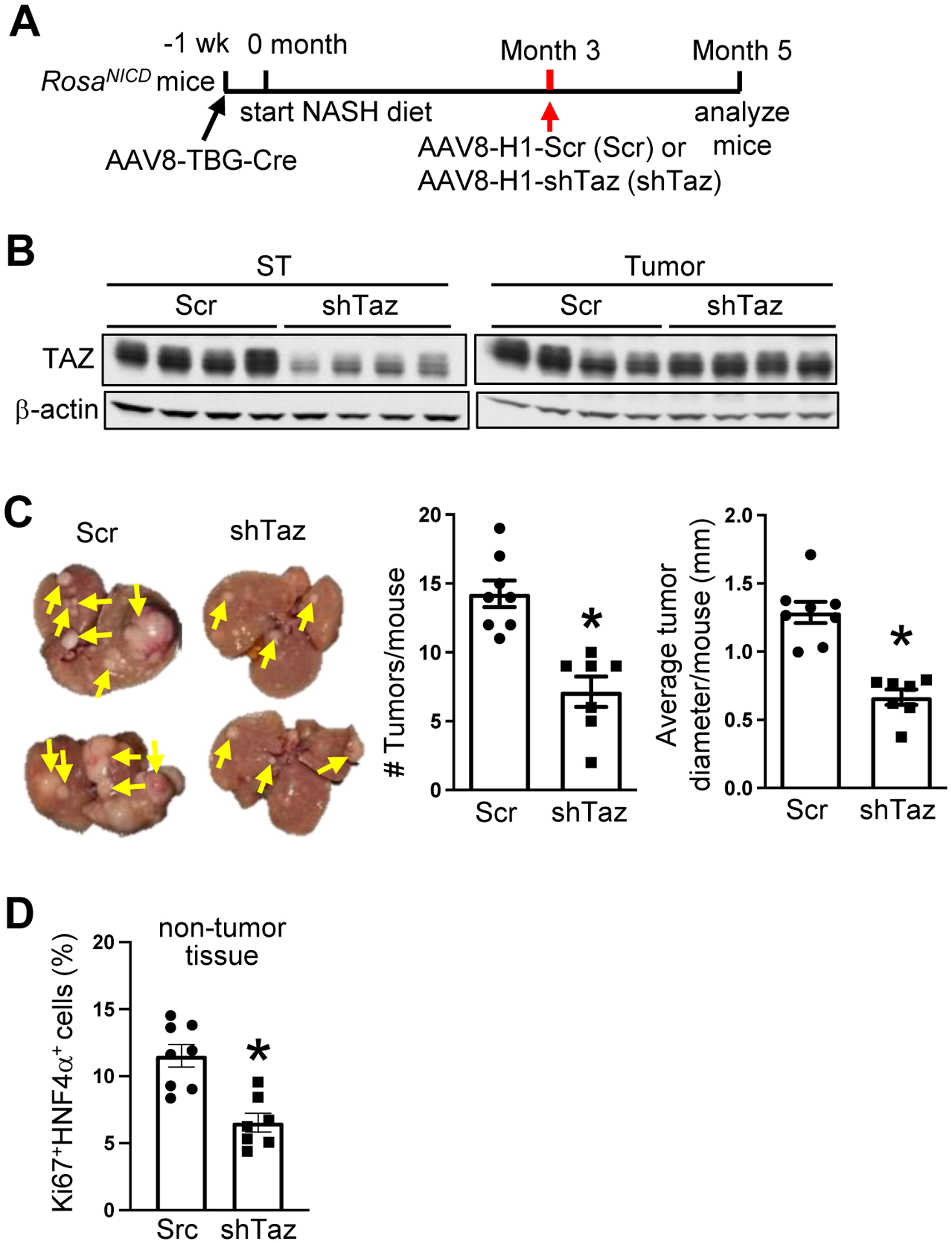
(A-D) AAV8-TBG-Cre-treated RosaNICD mice were fed the NASH diet and, 3 months later, injected with AAV8-H1-scrambled RNA or AAV8-H1-shTaz. The mice were analyzed at month 5. (A) Experimental scheme. (B) TAZ immunoblot from surrounding tissue (ST) and tumor tissue. (C) Livers (arrows, tumors) and tumor numbers and average diameter. (D) Quantification of the percentage of Ki67+HNF4α+ cells in liver sections from non-tumor areas. For C-D, n = 7–8 mice/group; means ± SEM; *p < 0.05 by Student’s t-test.
The tumor-suppressing and NASH-suppressing effects of shTaz can be dissociated in experimental NASH-HCC
As expected from our previous work8, hepatocyte-TAZ deletion lowered liver inflammation, fibrosis, and cell death and plasma ALT in the models studied in Figures 1 and 2 (Figure S2A–S2T). Thus the anti-tumor-suppressing effect of hepatocyte-TAZ silencing in our models could be secondary to suppressing the NASH niche, which can in its advanced form contribute to HCC development27,28. However, NASH is relatively low-grade in our models, suggesting that anti-tumor effect of shTaz might be independent of its NASH-suppression effect. To test this idea, we examined the role of Indian hedgehog (Ihh) in NASH-HCC, as Ihh is the major gene target of TAZ/TEAD responsible for TAZ-induced NASH liver inflammation, fibrosis, and cell death8. Using the Notch (NICD)/NASH-diet model, mice were treated with AAV8-H1-shTaz plus either AAV8-TBG-Ihh or control AAV8-TBG-LacZ at the 2-month timepoint and then examined at 4 months (Figure 3A). In a parallel experiment, we showed that shTaz in this 2-month → 4-month protocol suppressed tumors (Figure S3A–S3B), lowered TAZ and Ihh expression only in non-tumor tissue (Figures S3C), and decreased NASH endpoints without affecting body weight or fasting plasma glucose (Figure S3D–S3F). As designed, treatment of TAZ-silenced mice with AAV8-TBG-Ihh increased Ihh in non-tumor-bearing NASH liver but not in the tumors themselves (Figure 3B), and, consistent with our previous data8, Ihh restored NASH features in the TAZ-silenced mice (Figures 3C–3E) without affecting body weight or fasting plasma glucose (Figures S3G–S3H). Most importantly, AAV8-TBG-Ihh did not increase tumor number or size in the TAZ-silenced mice (Figure 3F and S3I). Next, we directly silenced Ihh in this model (Figure 3G), which resulted in lower Ihh in non-tumor tissue but not tumor tissue (Figure 3H). As expected, this intervention lowered liver inflammation, fibrosis, and TUNEL+ cells in the liver and plasma ALT (Figure 3I–3K) without affecting body weight or fasting plasma glucose (Figures S3J–S3K). Most importantly, shIhh did not lower HCC development (Figure 3L and S3L). These combined data dissociate the tumor-suppressing effect of shTaz from its NASH-suppressing effects in this model. Moreover, while Ihh is a key TAZ gene target that contributes to NASH progression, Ihh does not appear to be involved in TAZ-induced HCC.
Figure 3. The tumor-suppressing effect of shTaz in experimental NASH-HCC can be dissociated from its NASH-suppressing effects.
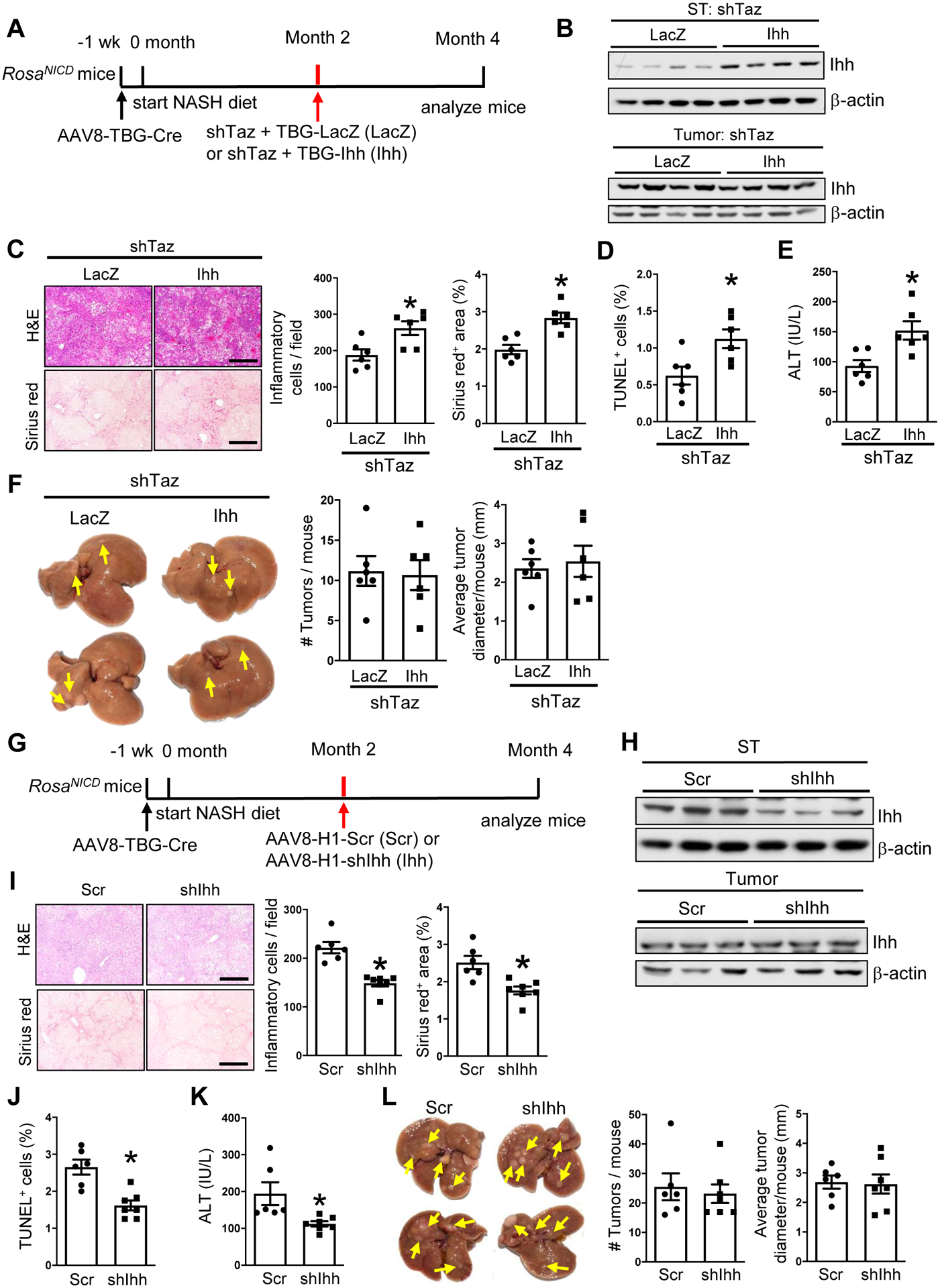
(A-F) AAV8-TBG-Cre-treated RosaNICD mice were fed the NASH diet and, 2 months later, injected with AAV8-H1-shTaz and either AAV8-TBG-LacZ or AAV8-TBG-Ihh. The mice were analyzed at month 4. (A) Experimental scheme. (B) Ihh immunoblot from surrounding tissue (ST) and tumor tissue. (C) Liver sections stained with H&E (upper images) and Sirius red (lower images), with quantification of inflammatory cells and percent Sirius red-positive area. Bars, 200 μm. (D) Percent TUNEL+ cells from non-tumor areas. (E) Plasma ALT. (F) Livers (arrows, tumors) and tumor numbers and diameter. For C-F, n = 6 mice/group; means ± SEM; *p < 0.05 by Student’s t-test. (G-L) AAV8-TBG-Cre-treated RosaNICD mice were fed the NASH diet and, 2 months later, injected with AAV8-H1-Scr or AAV8-H1-shIhh. The mice were analyzed at month 4. (G) Experimental scheme. (H) Ihh immunoblot from surrounding tissue (ST) and tumor tissue. (I) Liver sections were stained with H&E (upper images) and Sirius red (lower images), with quantification of inflammatory cells and percent Sirius red-positive area. Bars, 200 μm. (J) Percent TUNEL+ cells from non-tumor areas. (K) Plasma ALT. (L) Livers (arrows, tumors) and tumor numbers and diameter. For I-L, n = 6–7 mice/group; means ± SEM; *p < 0.05 by Student’s t-test.
TAZ-mediated oxidative DNA damage in pre-tumor NASH is linked to tumor formation
In our search for TAZ-mediated process in NASH that might contribute to the eventual tumor formation, we investigated a key process in HCC, namely, DNA damage9,10. First, we found that a marker of double-stranded DNA (dsDNA) damage, γH2AX (phospho-H2AX), was increased in the livers of humans and mice with non-tumor NASH (Figure S4A–S4B). Next, we found that treatment with AAV8-H1-shTaz eliminated the increase in γH2AX in NASH mice (Figure 4A). Although the decrease in the γHA2X signal by shTaz could have resulted from the suppression of hepatocyte proliferation10, shTaz did not affect the percentage of Ki67+HNF4α+ cells in these non-HCC NASH livers (Figure S4C). Moreover, this finding shows that TAZ promotes DNA damage in hepatocytes before proliferation occurs. One mechanism of dsDNA damage in HCC is oxidative DNA damage, and there is evidence that this process is relevant to NASH-HCC in humans11. Using the livers of mice fed the NASH diet for 8 weeks (steatosis) or 16 weeks (early NASH), we used immunofluorescence microscopy to detect hepatocytes expressing a marker of oxidative DNA damage, 8-oxo-2′-deoxyguanosine (8-OHDG). The percent of 8-OHDG+ hepatocytes increased during the period of steatosis-to-NASH progression (Figures 4B and S4D), and the increase at 16 weeks was diminished in mice by AAV8-H1-shTaz treatment (Figure 4C).
Figure 4. TAZ promotes hepatocyte oxidative DNA damage in NASH, and its suppression by OGG1 supresses tumor formation in NASH-HCC mice.
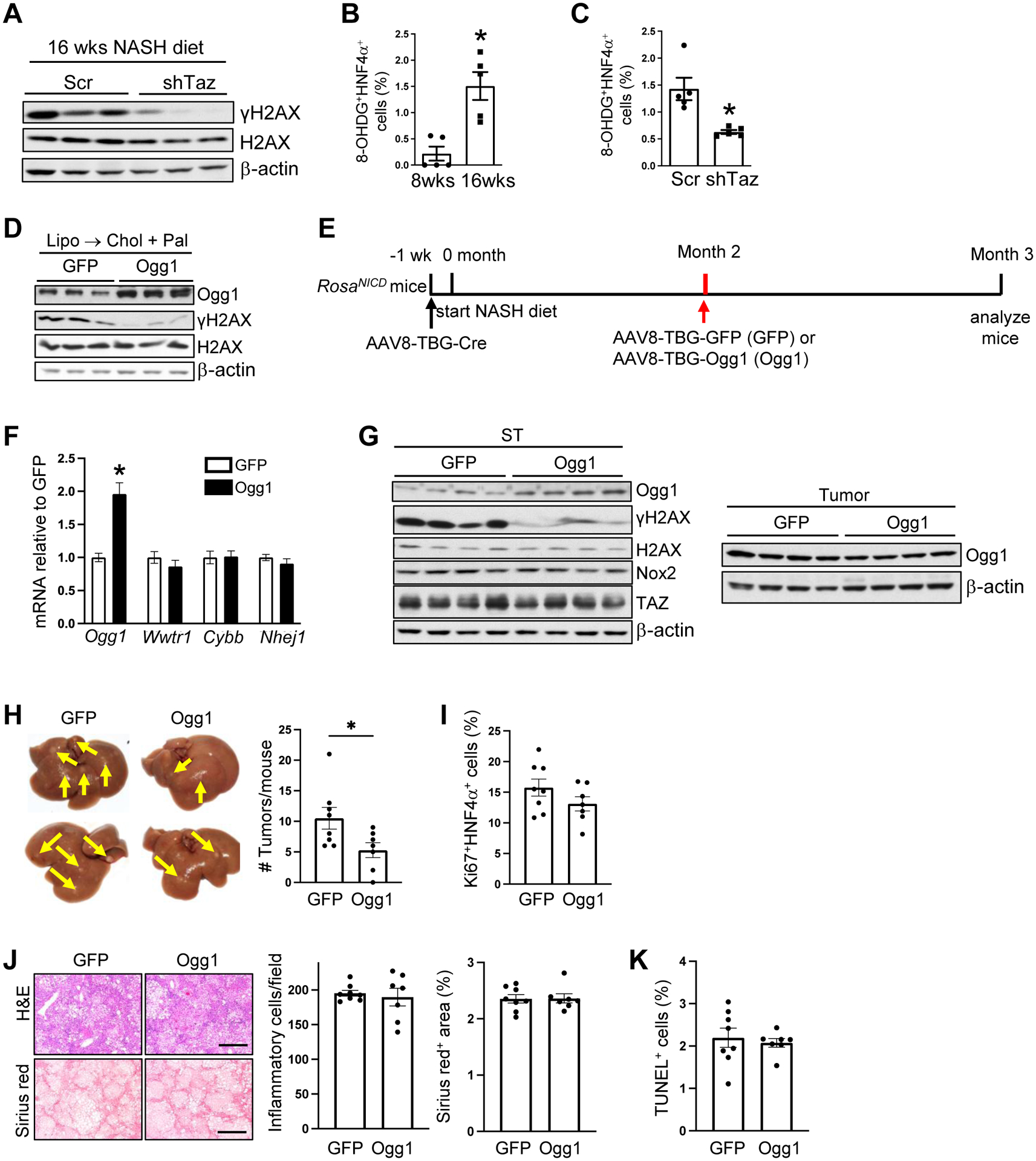
(A) γH2AX and H2AX immunoblots from liver of mice injected with AAV8-H1-Scr or AAV8-H1-shTaz and fed the NASH diet for 16 weeks. (B) Percent 8-OHDG+ HNF4α+ cells in the livers of mice fed the NASH diet for 8 or 16 weeks (n = 5 mice/group; means ± SEM; **p < 0.01 by Student’s t-test). (C) Percent 8-OHDG+ HNF4α+ cells in livers of mice treated like those in panel A (n = 5 mice/group; means ± SEM; **p < 0.01 by Student’s t-test). (D) Ogg1, γH2AX, and H2AX immunoblots from AML12 cells transfected with GFP or Ogg1 plasmids and then incubated for 24 h with liposomes to deplete cholesterol (Lipo) and then 16 h with cholesterol-rich liposomes and palmitate (Lipo → Chol + Pal). (E-K) AAV8-TBG-Cre-treated RosaNICD mice were fed the NASH diet and, 2 months later, injected with AAV8-TBG-GFP or AAV8-TBG-Ogg1. The mice were analyzed at month 4. (n = 7–8 mice/group; means ± SEM) (E) Experimental scheme. (F) Liver Ogg1, Wwtr1, Cybb and Nhej1 mRNA. (*p < 0.05 by two-way ANOVA/Sidak’s post-hoc analysis). (G) Immunoblots of the indicated proteins in the surrounding tumor tissue (ST) and tumor tissue from the livers of the two groups of mice. (H) Livers (arrows, tumors) and tumor numbers/mouse. (*p < 0.05 by Student’s t-test) (I) Percent Ki67+HNF4α+ cells in non-tumor tissue. (J) Liver sections were stained with H&E (upper images) and Sirius red (lower images), with quantification of inflammatory cells and percent Sirius red-positive area. Bars, 500 μm. (K) Percent TUNEL+ cells in non-tumor areas.
We next sought direct evidence that oxidative DNA damage was involved in NASH-HCC by testing the effect if 8-oxoguanine glycosylase (OGG1), which mediates base excision repair of oxidatively damaged DNA29. First, transfection of AML12 cells with Ogg1 prevented cholesterol/palmitate-induced DNA damage as assessed by γH2AX immunoblot (Figure 4D). Next, we administered AAV8-TBG-Ogg1 or control virus (AAV8-TBG-GFP) 2 months after the start of the NASH diet in Cre-treated RosaNICD mice and then analyzed the mice 1 month later (Figure 4E). AAV8-TBG-Ogg1 successfully increased liver Ogg1 mRNA without affecting Wwtr1 (TAZ); increased OGG1 protein in surrounding tissue but not tumors; and decreased γH2AX1 in surrounding tissue (Figures 4F–G). Most importantly, OGG1 decreased the number of tumors that developed in these mice (Figure 4H) without affecting the percentage of Ki67+HNF4α+ cells in the liver (Figure 4I) or systemic or NASH parameters (Figure 4J–K and S4E–F). These combined data show causative links among between TAZ and DNA damage in pre-tumor NASH and eventual tumor formation.
TAZ-induced Cybb/NOX2 contributes to oxidative DNA damage in NASH and to NASH-HCC tumor formation
We surveyed 12 mRNAs that encode oxidant-related proteins in pre-tumor NASH vs. control liver and found that Cybb, which encodes the NOX2 (gp91) subunit of the pro-oxidant protein complex NADPH oxidase, was increased in NASH (Figure 5A). Cybb mRNA was also elevated in both tumor tissue and non-tumor surrounding tissue in the livers of the 13-month NASH-diet HCC model (Figure S4G). Further, NOX2 protein was increased in mouse and human NASH liver (Figure 5B and 5C, top); in surrounding tissue and tumor tissue of two of our mouse NASH-HCC models; and in human NASH-HCC tumors (Figure S4H–J). We also found strong correlations between TAZ and NOX2, NOX2 and γH2AX, and TAZ and γH2AX and between the percent of 8-OHDG+ cells and NOX2, TAZ, and γH2AX in non-tumor tissue of human NASH-HCC liver (Figure S4K–N). Immunostaining using a validated anti-NOX2 antibody (Figure S4O) showed a strong NOX2 signal in hepatocytes in mouse and human NASH liver (Figure 5B–5C, images). Although NOX2 staining was also seen in liver macrophages, there was a far greater number of NOX2-positive hepatocytes (Figure S4P). Further, neutrophils were not a major sources of NOX2 in human NASH liver, as their numbers were very low (Figure S4Q). Thus, hepatocytes contributed to most of the NOX2 signal in NASH liver (Figure 5B–5C, graphs). Most importantly, silencing hepatocyte TAZ in NASH mice led to a substantial decrease in hepatic Cybb and NOX2 expression (Figure 5D). We next conducted anti-TAZ ChIP analysis and showed that TAZ was enriched on a TAZ/TEAD binding sequence in a Cybb promoter in liver extracts from NASH diet-fed mice compared with either control liver extracts or liver extracts from NASH diet-fed mice that had been treated with AAV8-H1-shTaz (Figure 5E).
Figure 5. Cybb is a TAZ gene target that contributes to hepatocyte oxidative DNA damage in NASH.
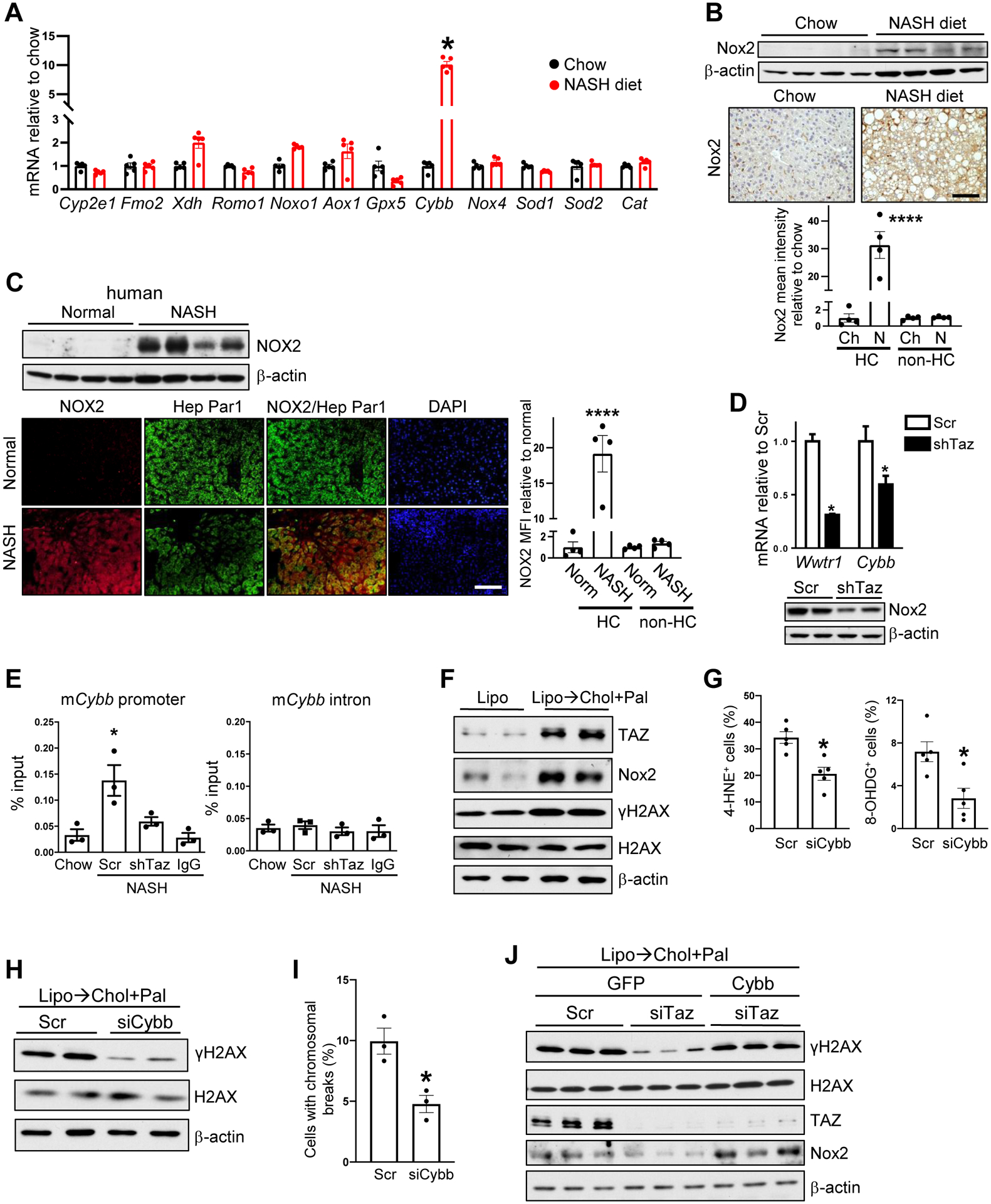
(A) Quantification of pro-oxidant genes in the livers of mice fed chow or the NASH diet for 16 weeks NASH (n = 5 mice/group; means ± SEM; *p < 0.0001 by two-way ANOVA with Sidak’s post-hoc analysis). (B) Immunoblot and immunohistochemical staining of Nox2 in the livers of mice fed chow diet (Ch) or NASH diet (N) for 28 weeks. Bar, 100 μM, with quantification of Nox2 MFI in hepatocytes (HC) and non-hepatocytes (non-HC) (n = 4 mice/group; means ± SEM; ****p < 0.0001 by two-way ANOVA/Sidak’s post-hoc analysis). (C) Immunoblot of NOX2 and immunofluorescence of NOX2 and Hep-Par1 (hepatocytes) in normal human livers (Norm) or livers from subjects with NASH. Bar, 100 μM, with quantification of NOX2 MFI in hepatocytes (HC) and non-hepatocytes (non-HC) (n = 4 specimens/group; means ± SEM; ****p < 0.0001 by two-way ANOVA/Sidak’s post-hoc analysis). (D) Wwtr1 and Cybb mRNA and Nox2 immunoblot in the livers of mice injected with AAV8-H1-Scr or AAV8-H1-shTaz and then fed the NASH diet for 16 weeks (n = 5 mice/group; means ± SEM; *p < 0.05 by two-way ANOVA/Sidak’s post-hoc analysis). (E) The livers of chow-fed mice or mice injected with AAV8-H1-Scr or AAV8-H1-shTaz and then fed the NASH diet for 16 weeks were subjected to TAZ ChIP, followed by qPCR of the precipitated DNA for a TAZ/TEAD binding sequence in a Cybb promoter or a non-consensus sequence in a Cybb intron. IgG served as the antibody control; the data were normalized to the values obtained from input DNA (n = 3 mice/group; means ± SEM; *p < 0.05 by one-way ANOVA/Tukey’s post-hoc analysis). (F) Immunoblots of TAZ, Nox2, γH2AX, and H2AX in AML12 cells incubated for 40 h with liposomes (Lipo) to deplete cholesterol (Lipo) or for 24 h with liposomes and then 16 h with cholesterol-rich liposomes and palmitate (Lipo → Chol + Pal). (G) Percent 4-HNE+ and 8-OHDG+ cells among scrambled RNA- or siCybb-treated AML12 cells that were incubated for 24 h with liposomes and then 16 h with liposomal-cholesterol and palmitate (n = 5 biological replicates/group; means ± SEM; *p < 0.05 by Student’s t-test). (H) γH2AX and H2AX immunoblots from the AML12 cells in panel J. (I) Chromosome spread assay of the AML12 cells in panel J, with quantification of percent cells with chromosomal breaks (n = 3 biological replicates/group; means ± SEM; *p < 0.05 by Student’s t-test). (J) γH2AX, H2AX, TAZ, and Nox2 immunoblots from AML12 cells transfected with Scr or siTaz and with GFP control or Cybb, and then incubated for 24 h with liposomes and then 16 h with liposomal-cholesterol and palmitate..
To document cell-autonomous links among TAZ, Cybb/NOX2, and dsDNA damage, we turned to a NASH-relevant model in which AML12 hepatocytes are first depleted of cholesterol using phospholipid liposomes and then loaded with cholesterol using cholesterol-loaded liposomes. This treatment induces TAZ by the same mechanism that occurs in NASH hepatocytes in vivo14. We also added palmitate to the incubation medium to induce NASH-relevant lipid stress. We found that AML12 cells treated in this manner had increases in TAZ, NOX2, and γH2AX compared with control AML12 cells (Figure 5F). Further, using a chromosomal spread assay, we observed an increase in chromosomal breaks in the treated cells but not in the control cells (Figure S4R). Most importantly, siCybb lowered 4-hydroxynonenal (4-HNE), a marker of oxidative stress; 8-OHDG; γH2AX; and chromosomal breaks (Figures 5G–5I). Moreover, siTaz treatment lowered γH2AX and NOX2 by approximately 50%, and genetic restoration of NOX2 in the siTaz-treated hepatocytes abrogated the decrease in γH2AX (Figure 5J). These combined data show that the increase in hepatocyte TAZ in NASH leads to the induction of Cybb, which results in NOX2-mediated oxidative dsDNA damage.
We reasoned that expressing a dsDNA-repair enzyme in hepatocytes in pre-tumor NASH HCC might provide a causal link between DNA damage in NASH and eventual tumor formation. Based on a screen of mRNAs encoding dsDNA-damage repair enzymes, we chose non-homologous end joining factor 1 (NHEJ1; also known as XRCC4-like factor [XLF]). Nhej1 was uniquely decreased in NASH versus control liver (Figure S5A). Nhej1 was also decreased in cholesterol/palmitate-treated versus untreated AML12 cells (Figure S5B), and transfection of these cells with Nhej1 lowered γH2AX (Figure S5C). For the in-vivo test, the Notch (NICD)/NASH-diet model was transduced with AAV8-TBG-Nhej1 between months 2 and 3 (Figure S5D). As planned, the vector increased liver Nhej1 expression without affecting Wwtr1 (TAZ) or Cybb; increased NHEJ1 protein in surround tissue but not tumors; and decreased γH2AX1 in non-tumor tissue (Figures S5E–S5F). Most importantly, NHEJ1 decreased the number of tumors in these mice (Figure S5G), without affecting the percentage of Ki67+HNF4α+ cells (Figure S5H) or systemic or NASH parameters (Figures S5I–S5L).
We next tested the role of Cybb in NASH-HCC by treating Notch (NICD)/NASH-diet mice with AAV8-H1-shCybb or AAV8-H1-Scr between months 2–4 (Figure 6A). NOX2 was decreased in non-tumor tissue but not the tumors, and this was accompanied by a decrease in γH2AX in the non-tumor tissue (Figure 6B and Figure S6A). Most importantly, shCybb lowered tumor numbers in these mice (Figure 6C) without affecting systemic or NASH endpoints (Figures 6D–6F and S6B–S6C). Interestingly, neither the average size of the tumors nor the percentage of Ki67+HNF4α+ cells (Figure 6G and S6D) was altered, suggesting that hepatocyte-Cybb contributed to an early, pre-proliferative molecular process that can eventually lead to new tumor formation.
Figure 6. TAZ-induced Cybb/NOX2 contributes to the development of NASH-HCC tumors.
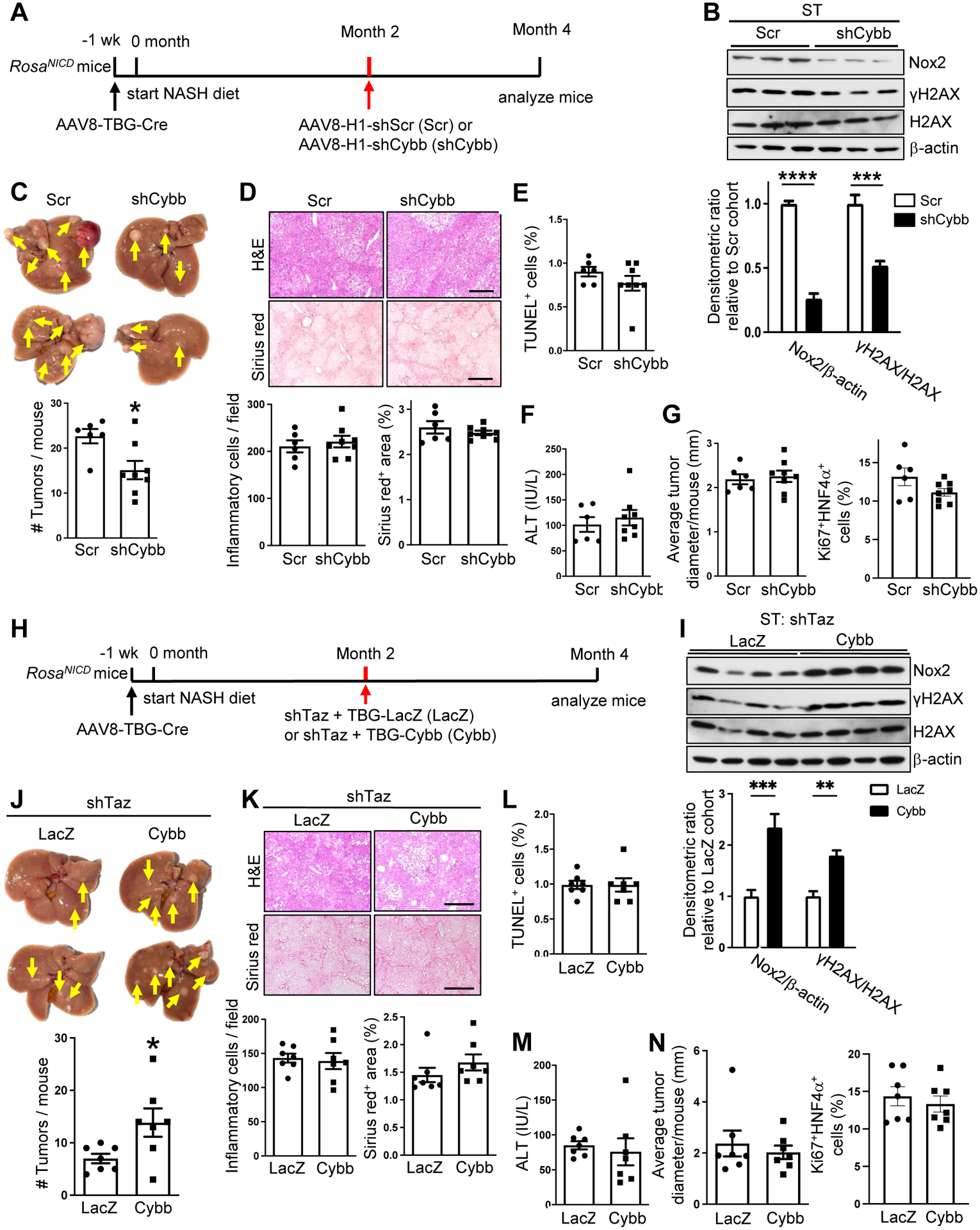
(A-G) AAV8-TBG-Cre-treated RosaNICD mice were fed the NASH diet and, 2 months later, injected with AAV8-H1-Scr or AAV8-H1-shCybb. The mice were analyzed at month 4. (A) Experimental scheme. (B) Nox2, γH2AX, and H2AX immunoblots from surrounding tissue (ST), with quantification (n = 3; means ± SEM; ***p<0.001, ****p<0.0001 by two-way ANOVA/Sidak’s post-hoc analysis). (C) Livers (arrows, tumors) and tumor numbers/mouse. (D) Liver sections were stained with H&E (upper images) and Sirius red (lower images) and quantified for the number of inflammatory cells and the percent Sirius red-positive area. Bars, 200 μm. (E) Percent TUNEL+ cells in non-tumor areas. (F) Plasma ALT. (G) Average tumor diameter and percent Ki67+HNF4α+ cells in non-tumor areas. For C-G, n = 6–8 mice/group; means ± SEM; *p < 0.05 by Student’s t-test. (H-N) AAV8-TBG-Cre-treated RosaNICD mice were fed the NASH diet and, 2 months later, injected with AAV8-H1-shTaz and either AAV8-TBG-LacZ or AAV8-TBG-Cybb. The mice were analyzed at month 4. (H) Experimental scheme. (I) Nox2, γH2AX, and H2AX immunoblots from ST, with quantification (n = 4; means ± SEM; **p<0.01, ***p<0.001 by two-way ANOVA/Sidak’s post-hoc analysis). (J) Livers (arrows, umors) and tumor numbers/mouse. (K) Liver sections were stained with H&E (upper images) and Sirius red (lower images), with quantification of inflammatory cells and percent Sirius red-positive area. Bars, 200 μm. (L) Percent TUNEL+ cells in non-tumor areas. (M) Plasma ALT. (N) Average tumor diameter and percent Ki67+HNF4α+ cells. For J-N, n = 7 mice/group; means ± SEM; *p < 0.05 by Student’s t-test.
Finally, we asked whether genetic restoration of Cybb in shTaz-treated mice could restore dsDNA damage and tumor development. Accordingly, Notch (NICD)/NASH-diet mice were treated with AAV8-H1-shTaz plus either AAV8-TBG-Cybb or AAV8-TBG-LacZ control (Figure 6H). In the AAV8-TBG-Cybb mice, NOX2 and γH2AX were restored in non-tumor liver tissue but not tumors (Figure 6I and Figure S6E). Most importantly, NOX2 restoration increased average tumor number to the value that is typically observed in control Notch/NASH-diet mice (Figure 6J) without affecting systemic or NASH endpoints (Figures 6K–6M and S6F–S6G). Consistent with the shCybb data above, neither average tumor size nor the percentage of Ki67+HNF4α+ cells was affected (Figure 6N and S6H). When combined with the previous data, these findings suggest NASH-mediated induction of TAZ in hepatocytes, by inducing Cybb and NOX2-mediated oxidative DNA damage, promotes an early, pre-proliferative process that contributes to eventual HCC tumor formation.
DISCUSSION
In NASH-induced HCC, hepatocytes undergo biological changes over a prolonged period of time prior to the formation of tumors, providing a unique window into the earliest molecular-cellular processes of tumor formation. Here we show the importance of a TAZ-Cybb-oxidative dsDNA damage pathway. Future studies will be needed to elucidate the molecular-genetic links among dsDNA damage, additional carcinogenic hits, and eventual tumor formation in this setting. Multiple mechanisms are possible for DNA damage, including the inactivation of tumor-suppresor genes30. With regard to additional hits, the NASH niche is likely important27,28, and TAZ itself may play and addition role to promote hepatocyte proliferation17–19.
The pathway described here focuses specifically on NOX-induced oxidative DNA damage. Oxidative stress is a well-known inducer of DNA damage and cancer-causing mutations, and it is associated with NASH-HCC10,15. More specifically, the formation of 8-OHDG is linked to epigenetic instability in human HCC11 and has been identified as a risk factor for HCC in chronic hepatitis C infection16. Moreover, NOX2-mediated superoxide generation has been implicated previously in certain non-liver cancers31, and several studies have shown correlations between the expression of various NOX proteins and HCC in cell lines, mouse models of HCC, and human HCC liver specimens32. However, direct in vivo causation studies and mechanistic links to NASH-HCC were previously lacking.
Most therapeutic efforts in HCC focus on arresting tumor growth or promoting tumor regression after the diagnosis of HCC in patients with cirrhosis. However, in the case of NASH, HCC can develop before frank cirrhosis occurs5–7. Moreover, pre-tumor NASH requires treament in its own right, i.e., to prevent liver failure. The fact that TAZ is induced in hepatocytes in NASH and contributes to both NASH and HCC provides a strong rationale for TAZ-based therapy in patients with NASH. For example, GalNAc-siTAZ, which is based on a platform currently in human use, can lower hepatocyte-TAZ to its healthy-liver level and block or reverse progression to fibrosis in experimental NASH22. Based on the pathway revealed here, we suggest that hepatocyte-targeted siTaz therapy would also block NASH-to-HCC progression.
Supplementary Material
Highlights.
Silencing hepatocyte TAZ in pre-tumor NASH suppresses subsequent HCC
Cybb is the key TAZ-induced gene in NASH hepatocytes that triggers tumor formation
Cybb encodes NOX2, which promotes HCC by inducing oxidative DNA damage
Silencing hepatocyte Cybb in pre-tumor NASH, or blocking DNA damage, suppresses HCC
TAZ, NOX2, oxidative DNA damage are strongly correlated in human NASH-HCC liver
Acknowledgements
We thank Dr. Eric Olson (University of Texas Southwestern) for providing the Wwtr1fl/fl mice; Drs. Anwesha Dey and Philamer Calses (Genentech) and Dr. Vivette D’Agati (CUIMC) for help with the chromosomal spread assay; and Dr. Ricard Masia (MGH) for advice on mouse HCC tumor characterization. The graphical abstract was created with BioRender.com.
Financial support
This work was supported by an American Liver Foundation Liver Scholar Award (to X.W.); NIH grants DK103818 and DK119767 (to U.P.); R01CA190844 and R01CA228483 (to R.F.S.); and R01DK116620 (to R.F.S. and I.T). Human liver samples were obtained from the Liver Tissue Cell Distribution System (University of Minnesota), which was funded by NIH contract HHSN276201200017C. Samples for histological analysis were prepared in the Molecular Pathology Shared Resource of the Herbert Irving Comprehensive Cancer Center at Columbia University, supported by NIH/NCI grant #P30 CA013696.
Conflict of interest
Dr. Tabas received an academic research grant from Takeda Pharmaceuticals to study the therapeutic potential of silencing TAZ in NASH.
Abbreviations:
- 8-OHDG
8-Oxo-2′-deoxyguanosine (8-Oxo-dG)
- AAV
Adeno-associated virus
- DMBA
7,12-dimethylbenz [a]anthracene
- FPC
fructose-palmitate-cholesterol
- HCC
hepatocellular carcinoma
- NASH
non-alcoholic steatohepatitis
- Nhej1
non-homologous end-joining factor
- Nox2
NADPH oxidase-2
- Ogg1
8-oxoguanine DNA glycosylase
- TAZ/Wwtr1)
WW domain-containing transcription regulator-1
- TBG
thyroxine-binding globulin promoter
- TUNEL
terminal deoxynucleotidyl transferase dUTP-nick-end labeling
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability statement
Data are available from the corresponding author upon reasonable request.
References
- [1].Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 2013;10:686–690. [DOI] [PubMed] [Google Scholar]
- [2].Corey KE, Kaplan LM. Obesity and liver disease: the epidemic of the twenty-first century. Clin Liver Dis 2014;18:1–18. [DOI] [PubMed] [Google Scholar]
- [3].Michelotti GA, Machado MV, Diehl AM. NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 2013;10:656–665. [DOI] [PubMed] [Google Scholar]
- [4].Huang DQ, El-Serag HB, Loomba R. Global epidemiology of NAFLD-related HCC: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 2021;18:223–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mittal S, El-Serag HB, Sada YH, Kanwal F, Duan Z, Temple S, et al. Hepatocellular carcinoma in the absence of cirrhosis in united states veterans is associated with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2016;14:124–131 e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Perumpail RB, Wong RJ, Ahmed A, Harrison SA. Hepatocellular carcinoma in the setting of non-cirrhotic nonalcoholic fatty liver disease and the metabolic syndrome: us experience. Dig Dis Sci 2015;60:3142–3148. [DOI] [PubMed] [Google Scholar]
- [7].Paradis V, Zalinski S, Chelbi E, Guedj N, Degos F, Vilgrain V, et al. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: a pathological analysis. Hepatology 2009;49:851–859. [DOI] [PubMed] [Google Scholar]
- [8].Wang X, Zheng Z, Caviglia JM, Corey KE, Herfel TM, Cai B, et al. Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab 2016;24:848–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Linhart KB, Glassen K, Peccerella T, Waldherr R, Linhart H, Bartsch H, et al. The generation of carcinogenic etheno-DNA adducts in the liver of patients with nonalcoholic fatty liver disease. Hepatobiliary Surg Nutr 2015;4:117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Boege Y, Malehmir M, Healy ME, Bettermann K, Lorentzen A, Vucur M, et al. A dual role of caspase-8 in triggering and sensing proliferation-associated DNA damage, a key determinant of liver cancer development. Cancer Cell 2017;32:342–359.e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kakehashi A, Suzuki S, Ishii N, Okuno T, Kuwae Y, Fujioka M, et al. Accumulation of 8-hydroxydeoxyguanosine, L-arginine and glucose metabolites by liver tumor cells are the important characteristic features of metabolic syndrome and non-alcoholic steatohepatitis-associated hepatocarcinogenesis. Int J Mol Sci 2020;21:7746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yang X, Sheng S, Du X, Su W, Tian J, Zhao X. Hepatocyte-specific TAZ deletion downregulates p62/ Sqstm1 expression in nonalcoholic steatohepatitis. Biochem Biophys Res Commun 2021;535:60–65. [DOI] [PubMed] [Google Scholar]
- [13].Khajehahmadi Z, Mohagheghi S, Nikeghbalian S, Geramizadeh B, Khodadadi I, Karimi J, et al. Downregulation of hedgehog ligands in human simple steatosis may protect against nonalcoholic steatohepatitis: Is TAZ a crucial regulator? IUBMB Life 2019;71:1382–1390. [DOI] [PubMed] [Google Scholar]
- [14].Wang X, Cai B, Yang X, Sonubi OO, Zheng Z, Ramakrishnan R, et al. Cholesterol stabilizes TAZ in hepatocytes to promote experimental non-alcoholic steatohepatitis. Cell Metab 2020;31:969–986 e967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pinyol R, Torrecilla S, Wang H, Montironi C, Pique-Gili M, Torres-Martin M, et al. Molecular characterisation of hepatocellular carcinoma in patients with non-alcoholic steatohepatitis. J Hepatol 2021;75:865–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chuma M, Hige S, Nakanishi M, Ogawa K, Natsuizaka M, Yamamoto Y, et al. 8-Hydroxy-2′-deoxy-guanosine is a risk factor for development of hepatocellular carcinoma in patients with chronic hepatitis C virus infection. J Gastroenterol Hepatol 2008;23:1431–1436. [DOI] [PubMed] [Google Scholar]
- [17].Hagenbeek TJ, Webster JD, Kljavin NM, Chang MT, Pham T, Lee HJ, et al. The Hippo pathway effector TAZ induces TEAD-dependent liver inflammation and tumors. Sci Signal 2018;11:eaaj1757. [DOI] [PubMed] [Google Scholar]
- [18].Hayashi H, Higashi T, Yokoyama N, Kaida T, Sakamoto K, Fukushima Y, et al. An imbalance in TAZ and YAP expression in hepatocellular carcinoma confers cancer stem cell-like behaviors contributing to disease progression. Cancer Res 2015;75:4985–4997. [DOI] [PubMed] [Google Scholar]
- [19].Wang H, Wang J, Zhang S, Jia J, Liu X, Zhang J, et al. Distinct and overlapping roles of hippo effectors YAP and TAZ during human and mouse hepatocarcinogenesis. Cell Mol Gastroenterol Hepatol 2021;11:1095–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Xin M, Kim Y, Sutherland LB, Murakami M, Qi X, McAnally J, et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc Natl Acad Sci U S A 2013;110:13839–13844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013;499:97–101. [DOI] [PubMed] [Google Scholar]
- [22].Wang X, Sommerfeld MR, Jahn-Hofmann K, Cai B, Filliol A, Remotti HE, et al. A therapeutic silencing RNA targeting hepatocyte TAZ prevents and reverses fibrosis in nonalcoholic steatohepatitis in mice. Hepatol Commun 2019;3:1221–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhu C, Ho YJ, Salomao MA, Dapito DH, Bartolome A, Schwabe RF, et al. Notch activity characterizes a common hepatocellular carcinoma subtype with unique molecular and clinicopathologic features. J Hepatol 2021;74:613–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhu C, Kim K, Wang X, Bartolome A, Salomao M, Dongiovanni P, et al. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci Transl Med 2018;10:eaat0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Moon H, Cho K, Shin S, Kim DY, Han KH, Ro SW. High risk of hepatocellular carcinoma development in fibrotic liver: Role of the Hippo-YAP/TAZ signaling pathway. Int J Mol Sci 2019;20:581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhang S, Zhou D. Role of the transcriptional coactivators YAP/TAZ in liver cancer. Curr Opin Cell Biol 2019;61:64–71. [DOI] [PubMed] [Google Scholar]
- [27].Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M. From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol 2019;16:411–428. [DOI] [PubMed] [Google Scholar]
- [28].Peiseler M, Tacke F. Inflammatory mechanisms underlying nonalcoholic steatohepatitis and the transition to hepatocellular carcinoma. Cancers (Basel) 2021;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nishimura S Mammalian Ogg1/Mmh gene plays a major role in repair of the 8-hydroxyguanine lesion in DNA. Prog Nucleic Acid Res Mol Biol 2001;68:107–123. [DOI] [PubMed] [Google Scholar]
- [30].Schumacher B, Pothof J, Vijg J, Hoeijmakers JHJ. The central role of DNA damage in the ageing process. Nature 2021;592:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Okada F, Kobayashi M, Tanaka H, Kobayashi T, Tazawa H, Iuchi Y, et al. The role of nicotinamide adenine dinucleotide phosphate oxidase-derived reactive oxygen species in the acquisition of metastatic ability of tumor cells. Am J Pathol 2006;169:294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Choi J, Corder NL, Koduru B, Wang Y. Oxidative stress and hepatic Nox proteins in chronic hepatitis C and hepatocellular carcinoma. Free Radic Biol Med 2014;72:267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available from the corresponding author upon reasonable request.