

Abstract
Background and Aims:
Biliary disease is associated with a proliferative/fibrogenic ductular reaction (DR). p300 is an epigenetic regulator that acetylates lysine 27 on histone 3 (H3K27ac) and is activated during fibrosis. Long non-coding RNAs (lncRNAs) are aberrantly expressed in cholangiopathies, but little is known about how they recruit epigenetic complexes and regulate DR. We investigated epigenetic complexes, including transcription factors (TFs) and lncRNAs, contributing to p300-mediated transcription during fibrosis.
Methods:
We evaluated p300 in vivo using tamoxifen-inducible, cholangiocyte-selective, p300 knockout (KO) coupled with bile duct ligation (BDL) and Mdr KO mice treated with SGC-CBP30. Primary cholangiocytes and liver tissue were analyzed for expression of Acta2-as1 lncRNA by qPCR and RNA ISH. In vitro, we performed RNA-sequencing in human cholangiocytes with a p300 inhibitor. Cholangiocytes were exposed to lipopolysaccharide (LPS) as an injury model. We confirmed formation of a p300/ELK1 complex by immunoprecipitation (IP). RNA IP examined interactions between ACTA2-AS1 and p300. Chromatin IP assays evaluated p300/ELK1 occupancy and p300-mediated H3K27ac. Organoids were generated from ACTA2-AS1-depleted cholangiocytes.
Results:
BDL-induced DR and fibrosis were reduced in Krt19-CreERT/p300fl/fl mice. Similarly, Mdr KO mice were protected from DR and fibrosis after SGC-CBP30 treatment. In vitro, depletion of ACTA2-AS1 reduced expression of proliferative/fibrogenic markers, reduced LPS-induced cholangiocyte proliferation, and impaired organoid formation. ACTA2-AS1 regulated transcription by facilitating p300/ELK1 binding to the PDGFB promoter after LPS. Correspondingly, LPS-induced H3K27ac was mediated by p300/ELK1 and was reduced in ACTA2-AS1-depleted cholangiocytes.
Conclusion:
Cholangiocyte-selective p300 KO or p300 inhibition attenuate DR/fibrosis in mice. ACTA2-AS1 influences recruitment of p300/ELK1 to specific promoters to drive H3K27ac and epigenetic activation of proliferative/fibrogenic genes. This suggests that cooperation between epigenetic co-activators and lncRNAs facilitates DR/fibrosis in biliary diseases.
Keywords: ACTA2-AS1, p300, ELK1, ductular reaction, cholangiocyte
Graphical Abstract
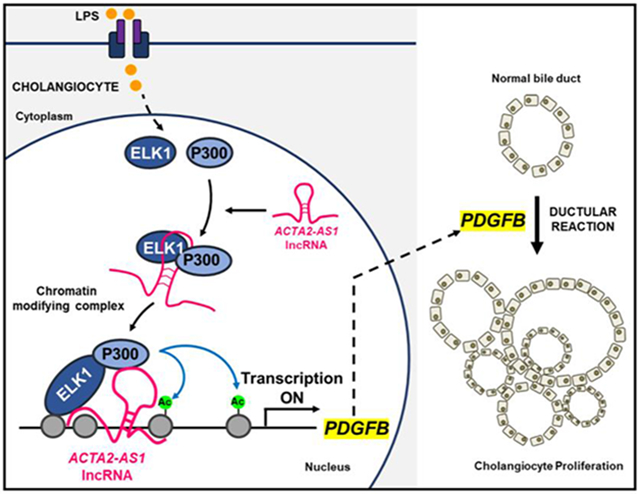
Lay Summary
We identified a three-part complex containing an RNA molecule, a TF, and an epigenetic enzyme. The complex is active in injured bile duct cells and contributes to activation of genes involved in proliferation and fibrosis.
Introduction
Cholangiocytes are epithelial cells lining the bile ducts and are the target cells of a diverse group of cholestatic liver diseases, termed cholangiopathies. Cholangiopathies are characterized by biliary fibrosis, which may progress to cirrhosis, leading to significant morbidity and mortality1. Progression towards end-stage disease is associated with a prominent ductular reaction (DR), a histologic feature characterized by persistent inflammatory response and ductular proliferation2. DR associates with activation and expansion of small, peri-portal ductular cells2. However, concepts regarding the origin and fate of ductular reactive cells are continuing to evolve. The DR may include both proliferation of biliary cells and transdifferentiation of mature hepatocytes3. However, the mechanisms regulating DR and portal fibrogenesis are poorly understood. Biliary fibrosis has complex pathobiology, including environmental exposures in genetically susceptible individuals, microbiome factors, and immune-mediated mechanisms4. These signals are likely modulating the epigenome, but few studies have addressed this5,6. Therefore, it is critical to elucidate signaling pathways and epigenetic events that coordinate DR and cholestatic fibrogenesis to identify new therapeutics.
Injured cholangiocytes acquire a proliferative and fibrogenic phenotype, including secretion of cytokines, growth factors, chemokines, and matrix molecules. These lead to proliferation, inflammation, and eventually, fibrosis7. Histone modification is an important mechanism for epigenetic regulation that is under-explored in cholestasis8. Lysine 27 on histone 3 (H3K27) is a key regulatory residue that is acetylated by the acetyltransferase p300. p300 is an enzyme that facilitates acetylation of H3K27 (H3K27ac), promoting transcription9. In cholangiocytes, p300 is implicated in the transition from repressive to permissive chromatin10. However, p300 does not bind to DNA, but requires the formation of epigenetic complexes with other molecules, including TFs and long non-coding RNAs (lncRNAs)11, guiding p300 to target genes in a tissue-specific manner.
LncRNAs are non-coding transcripts that exceed 200 nucleotides and lack protein coding capability. LncRNAs are implicated in regulation of gene expression by controlling chromatin remodeling, transcription initiation, and mRNA stability12. LncRNAs regulate cellular functions through interactions with proteins, TFs, and nucleic acids. They guide chromatin remodeling complexes to specific genetic loci resulting in regulation of transcription. They also act as scaffolds, linking protein complexes in a spatio-temporal fashion to coordinate activities. LncRNA biology represents an emerging but understudied field, particularly in the context of biliary disease. We understand the function of only a small number of lncRNAs in liver, including HULC and H1913, 14, and their expression typically correlates with disease progression.
The aim of this study was to identify epigenetic complexes including TFs and lncRNAs that contribute to pathological p300-mediated transcription within cholangiocytes and to define the mechanisms of their action. We show that ACTA2-AS1 recruits p300, mediates its binding to ELK1, and epigenetically activates transcription of proliferative and fibrogenic genes.
Materials and Methods
Cell Treatments
Human intrahepatic biliary epithelial cells (HiBEC) (ScienCell, #5100) and normal human cholangiocytes (NHC) were serum starved in DMEM containing 1%penicillin/streptomycin overnight before incubation with 0.5 μg/mL lipopolysaccharide (LPS;Millipore-Sigma, #L2880). For some experiments, cells were incubated with 10 μM SGC-CBP30 (Selleckchem, #S7256), dissolved in dimethyl sulfoxide (DMSO).
Constructs, RNA in vitro Transcription and Transfections
ACTA2-AS1 sense and antisense DNA constructs (Integrated DNA Technologies) were designed with the SP6 RNA promoter. Samples were resuspended in nuclease-free water (Invitrogen, #10977) to 20 ng/mL. Samples were heated at 50°C for 20 min and stored at −80°C. Transcription was performed using Megascript SP6 (Invitrogen, #AM1330)15. Constructs were transfected using Lipofectamine-RNAiMAX (Thermo Fisher Scientific, #M276S). Dilutions were performed in Opti-MEM (Thermo Fisher Scientific, #31985070). Cells were collected after 24 h. pCGN-ELK-1 and pCGN-ELK-l-S383A constructs were purchased from Addgene (#27156, #27160)16. Constructs were transfected using Lipofectamine-3000 (Thermo Fisher Scientific, #L3000008). Cells were collected after 48 h.
Animal Model
p300 floxed (p300fl/fl) mice17 (on a mixed background of C57BL/6J and 129) were crossed with mice harboring tamoxifen-inducible Cre-recombinase, driven by a cholangiocyte-selective promoter, Krt19-CreERT (provided by Dr. Stuart Forbes)18. Females and males (6-8 week-old) were used for experiments. Breeding was carried out with heterozygous animals to obtain homozygous littermates (5-7 mice/group). Cre-negative p300fl/fl mice were used as controls. Mice received intraperitoneal tamoxifen (75 mg/kg) for 5 days. 7 days after tamoxifen, bile duct ligation (BDL) was performed19. Briefly, two ties were placed on the common bile duct and the bile duct was transected between the two ties. Animals received humane care, were maintained on 12 h light/dark-cycle and had free access to water and food. After three weeks, fibrosis was analyzed by picro Sirius Red and Masson’s Trichrome stains, hydroxyproline analysis, qPCR, immunoblotting, and immunohistochemistry (IHC). Histology was analyzed by hematoxylin and eosin staining. Image J software was utilized to quantify positive stained areas (see supplementary information).
Eight-week-old Mdr2−/− mice (3-4 mice/group) were treated with 10 μg/g SGC-CBP30 or vehicle (2%DMSO, 30%PEG, 5%Tween80 and water) three times per week for 8 weeks by intraperitoneal injection. After 8 weeks, mice were sacrificed, and livers collected. Tissues were analyzed by picro Sirius Red and Masson’s Trichrome stains. DR was analyzed by IHC against CK19. Isolated mRNA was analyzed by RT-PCR. All animal experiments were approved by Mayo Clinic Institutional Animal Care and Use Committee.
Detailed information regarding mouse work is described following the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines20 (Supplementary Table 1).
Patient Samples
Liver tissues were obtained and analyzed under Mayo Clinic institutional review board–approved protocols. Detailed characteristics of patients are outlined in Supplementary Table 2.
Statistical Analysis
Analysis was performed using GraphPad Prism. Normal distribution was examined utilizing Shapiro-Wilk test. Student’s t-test and Analysis of variance (ANOVA) with a Bonferroni post-test were used to assess significance between normally-distributed groups. Mann-Whitney U and Kruskal-Wallis tests were used for non-normally distributed groups. The difference was considered significant for p values lower than 0.05. Data are from representative experiments reproduced at least three times. Data are presented as the mean ± standard error (SEM).
Data availability
All methods/protocols used were peer-reviewed and cited in the reference section. Please refer to supplementary information for additional material/methods. Reagents, antibodies and resources are listed in the CTAT table.
Results
Cholangiocyte-selective knockout of p300 attenuates ductular reaction in mice.
The acetyltransferase p300 mediates transcription through H3K27ac and is active in cholangiocytes10. To evaluate its role in cholangiocytes, we utilized a mouse model of tamoxifen-inducible, cholangiocyte-selective p300 knockout (KO) coupled with the BDL model of cholestatic fibrosis. p300fl/fl mice (control) and Krt19-CreERTLSLTdTomato/p300fl/fl mice (p300 KO) were injected with tamoxifen to induce Cre-mediated excision of p300 followed by sham or BDL surgery (Fig. S1A). We showed that tamoxifen-induced Cre-RFP was efficiently expressed in cholangiocytes using IHC against RFP (Fig. S1B). We also evaluated p300 mRNA expression in isolated cholangiocytes (Fig. S1C). We evaluated the role of p300 in DR using liver from control or p300 KO mice three weeks after surgery. IHC for CK19 demonstrated prominent DR induced by BDL that was attenuated upon p300 KO (Fig. 1A). Similar results were observed by mRNA and protein expression of CK19, a DR marker (Fig. S1D-E). We noted increased expression of the DR markers epithelial cell adhesion molecule (Epcam) and neural cell adhesion molecule 1 (Ncam1) in control mice that was attenuated in p300 KO (Fig. 1B, S1F-G). IHC for SRY-Box TF 9 (SOX9) also showed an increase in peri-portal accumulation in controls after BDL that was attenuated upon p300 KO (Fig. 1C). We evaluated expression/co-localization of CK7/Ki67 by immunofluorescence in tissue sections from P300 KO mice (Fig. S2A). Lastly, we measured Trop2 and AFP protein and mRNA to better reflect the diversity of cell types involved in DR (Fig. S2B-D). Overall, this data demonstrates impaired DR in mice lacking p300 within cholangiocytes.
Figure 1:

Cholangiocyte- selective knockout of p300 attenuates DR and biliary fibrosis. (A) Immunohistochemistry for CK19; (B) Immunoblotting for EpCAM; (C) Immunohistochemistry for SOX9; (D) Masson’s Trichrome staining; (E) Hydroxyproline assays; (F) Picro-Sirius red staining; and (G) Serum ALP in p300fl/fl and Krt19Cre/p300fl/fl animals with sham or BDL surgery. (n=5- 7 animals/group). Graph bars represent SEM. (A-D),(F-G) Mann-Whitney test *p<0.05,**p<0.01;(E) one-way ANOVA with Bonferroni comparison *p<0.05,****p<0.001.
We further evaluated the effect of p300 KO on biliary fibrosis. Control mice developed periductal fibrosis while p300 KO mice showed less accumulation of fibrosis (Fig. 1D). Increases in the fibrogenic markers, Fn1 and Serpine1 were less prominent after KO (Fig. S3A-B). Hydroxyproline assays confirmed less collagen deposition following BDL in animals lacking p300 (Fig. 1E). Piero Sirius red staining confirmed less peri-portal collagen in p300 KO animals after BDL (Fig. 1F). Cholestasis, assessed by serum alkaline phosphatase (ALP), was attenuated in p300 KO mice relative to controls (Fig. 1G). Alanine aminotransferase (ALT), total bilirubin levels (Fig. S3C-D) and hematoxylin and eosin staining (Fig. S3I) also suggested less inflammation and fibrosis in mice lacking p300. We noted residual elevations in BA levels and GGT activity (Figure S3F-H) in the p300 KO animals after BDL. Correspondingly, we observed a similar level of bile/bilirubin staining between the two BDL groups (Figure S3E). Overall, we find that proliferative and fibrogenic processes were attenuated in cholangiocyte-selective p300 KO mice after BDL.
To validate the role of p300 in vivo, we evaluated a second murine cholangiopathy model by treating Mdr2−/− mice with SGC-CBP30 for eight weeks. This data confirmed the protective role of p300 inhibition in terms of DR and hepatic fibrosis. Quantification of Masson’s trichrome staining shows a significant reduction in liver fibrosis after SGC-CBP30 treatment (Fig. 2A). We find reduced levels of cell adhesion molecules (Fig. 2B), cytokeratins (Fig. 2C-D), progenitor markers (Fig. 2E), and Ki-67 (Fig. 2F). We analyzed liver biochemistries including ALP, ALT, and total bilirubin (Figure S4A-C), as well as bile/bilirubin staining (Figure S4D), bile acid levels, and GGT activity (Figure S4E-F). Overall, the data confirms improved, but ongoing cholestasis after p300 inhibition. Liver/body weight ratio did not change after SGC-CBP30 administration (Fig. S4G).
Figure 2:

Inhibition of p300 attenuates DR and biliary fibrosis in Mdr−/− mice. (A) Masson’s Trichrome staining (Unpaired Student's t-test *p<0.05); (B-C) qRT-PCR for Epcam, Ncam, Krt19 and Krt7 (Unpaired Student's t-test *p<0.05,**p<0.01); (D) Immunohistochemistry for CK19 Mann-Whitney test *p<0.05); (E-F) qRT-PCR analysis for Afp, Trop2, and Ki-67 in liver from Mdr−/− mice treated with vehicle or SGC-CBP30 (Unpaired Student's t-test *p<0.05,**p<0.01). (n=3-4 animals/group). Graph bars represent SEM.
P300 regulates fibrogenic and proliferative genes in cholangiocytes.
In human PSC livers, we observed a significant increase in p300 expression as well as nuclear localization of H3K27ac within bile ducts (Fig. S5A-B). This increase was also observed in our control mice after BDL surgery (Fig. S5C-D). Given the effects of p300 knockout on DR and biliary fibrosis, we hypothesized that p300 modulates transcription of proliferative and fibrogenic genes in cholangiocytes. RNA-seq was performed in human intrahepatic biliary epithelial cells (HiBEC) incubated with SGC-CBP30 (CBP30), an inhibitor targeting p30021, once optimal concentration was determined (Fig. S6). Hierarchical clustering of differentially expressed genes after CBP30 revealed distinct expression profiles compared to controls (Fig. S7A). We found 274 genes decreased ≥2-fold after p300 inhibition (Fig. S7A). Ingenuity Pathway Analysis (IPA) demonstrated that genes downregulated by p300 inhibition were associated with fibrogenic processes, cell growth, and proliferation (Fig. 3A, S7B). This included genes relevant to cholestatic diseases, including matrix molecules (collagen isoforms), fibrogenic genes (ACTA2, FN1, GLI1, SERPINE1), proliferative factors (PDGFB), and genes such as VIM, CDH2, ITGA5 and WNT family members. Gene Set Enrichment Analysis (GSEA) showed that genes involved in “Extracellular matrix”, “KRAS signaling”, and “Epithelial/Mesenchymal Transition (EMT)” were downregulated by p300 inhibition (Fig. 3B, S7C-D). We validated down-regulation of several targets by qRT-PCR and WB in HiBEC cells (Fig. 3C-D), normal human cholangiocytes (NHC) (Fig. S7E), and cholangiocytes isolated from P300 KO mice (Figure S7F). This confirmed that p300 inhibition disrupts expression of DR and fibrosis related genes. To evaluate whether p300 inhibition impacts histone acetylation at the promoters, we performed ChIP assays for H3K27ac at representative genes. Indeed, p300 inhibition prevented H3K27ac at the promoters of PDGFRB, FN1, ACTA2 and PDGFB (Fig. 3E). The effect of p300 inhibitor was further analyzed in stimulated HiBEC, showing that LPS induced the expression of PDGFB and this effect was attenuated in P300 knock-down HiBEC (Fig. S7G). Overall, this data suggests that p300-mediated H3K27ac activates transcription in a subset of pathogenic genes in cholangiocytes.
Figure 3:

P300 activates transcription related to proliferation and fibrosis in cholangiocytes. (A) Pathways identified by IPA. (B) GSEA comparing control (DMSO) and P300-inhibited (CBP30) cells. (C) Validation by qRT- PCR. (D) Validation by WB. (E) ChIP-qPCR analysis in HiBEC treated with CBP30 or DMSO for H3K27ac at ACTA2, PDGFB, PDGFRB and FN1 promoters. (n=3). Graph bars represent SEM. Unpaired Student's t-test *p<0.05,**p<0.01,***p<0.005,****p<0.001 compared with DMSO group.
Inhibition of p300 in cholangiocytes reduces paracrine hepatic stellate cells (HSC) activation.
To evaluate mechanisms by which p300 mediates fibrosis, we examined its role in biliary senescence and in cholangiocyte-HSC cross-talk. Cholangiocytes were treated +/− CBP30 for 24 hours and expression of senescence markers, p16 and p21, were analyzed by qPCR (Fig. S8A). p300 inhibition did not affect p16 and p21. However, we observed a decrease in IL-6 mRNA in cells treated with CBP30 compared with controls, suggesting that p300 regulates the senescence-associated secretory phenotype (Figure S8A). P300 also modulates HiBEC proliferation since a reduction in Ki-67 was detected in CBP30-treated cells (Figure S8A). Next, cholangiocytes were treated with or without CBP30 for 24 hours and conditioned media was used to treat human HSC. HSC treated with media from cholangiocytes exposed to CBP30 were less activated than controls, as measured by α-SMA and FN expression (Figure S8B). This suggests that p300 mediates cross-talk between cholangiocytes and HSC, modulating paracrine HSC activation.
P300 binds to ELK1 promoting the expression of DR-related genes.
To define the epigenetic complex responsible for p300 effects, we aimed to identify relevant TFs. We studied putative motifs in the promoters of genes downregulated by p300 inhibition, noting motifs for ETS family members near the transcription start site of p300-dependent genes (Fig. 4A). The motifs included binding sites for ELK1 (CCGGAA), in the promoters of many genes including PDGFB (Fig. 4B). ELK1 is a ternary complex factor, a subclass of the ETS family22. In human liver tissue and in our animal model, ELK1 was increased in PSC patients as well as in mice that underwent BDL (Fig. S9A-B). In addition, ELK1 was an interesting candidate since it contributes to cell cycle control and is an LPS target23. LPS is commonly utilized as an in vitro model of cholangiocyte injury since translocation of endotoxin from gut to liver is of pathobiological importance in biliary disease23. qPCR and WB showed that ELK1 knockdown reduces expression of FN1, PDGFRB, α-SMA and PDGFB (Fig. 4C-D). LPS induced phosphorylation of ELK1 at 15 and 30 min whereas total ELK1 was unchanged (Fig. 4E). Nuclear fractionation showed that LPS-activated ELK1 localized to the nucleus (Fig. S9C). Similar results were obtained by immunofluorescence (Fig 4F). Furthermore, mutation of the main phosphorylation site of ELK1 (S383A) abolished LPS-induced effects observed in wild type ELK1-transfected NHC (Fig. S9D). We find that ELK1 mediates LPS-induced expression of α-SMA and PDGFB (Fig. 4G), cell proliferation (Fig. S9E), and ELK1 binds the promoter of PDGFB (Fig. 4H). Immunoprecipitation confirmed that LPS induces formation of a p300/p-ELK1 complex in cholangiocytes following LPS (Fig. 4I). This complex was also observed in mice after BDL (Fig. S9F). These results demonstrate that LPS regulates gene expression in cholangiocytes through formation of a p300/p-ELK1 complex.
Figure 4:

ELK1 regulates proliferative and fibrogenic genes in cholangiocytes. (A) ETS motifs identified by HOMER. (B) ELK1 motif from Jaspar. (C) qRT- PCR for ELK1, FN1, PDGFRB, ACTA2, and PDGFB in NHC transfected with control or ELK1 siRNA (Unpaired Student's t-test *p<0.05,**p<0.01 compared with control siRNA group). (D) WB for ELK1, FN1, PDGFRB, α-SMA and PDGFB in NHC transfected with control or ELK1 siRNA (Unpaired Student's t-test *p<0.05 compared with control siRNA group). (E) WB for p-ELK1, ELK1, p-ERK and ERK in NHC treated with LPS. (F) IF for p-ELK1 in NHC treated with LPS. (G) WB for ELK1, α-SMA and PDGFB in NHC transfected with control or ELK1 siRNA +/− LPS (one-way ANOVA with Bonferroni comparison *p<0.05,**p<0.01). (H) ChIP-qPCR in NHC treated with LPS. (I) WB on p-ELK1 and IgG immunoprecipitates from NHC +/− LPS (Unpaired Student’s t-test *p<0.05 compared with control group).(n=3). Graph bars represent SEM.
ACTA2-AS1 lncRNA is regulated by p300 and is overexpressed in PSC.
P300 and ELK1 are broadly active in transcription in various tissues and cell types. However, lncRNAs are generally implicated in tissue-specific regulation of more focused gene networks. Importantly, they often function as scaffolds that tether proteins into chromatin modifying complexes24. Therefore, we hypothesized that a lncRNA may interact with p300 and/or p-ELK1. Thus, we interrogated our RNA-seq data for changes in lncRNAs. We identified 21 antisense lncRNAs regulated by p300 (Fig.5A), including changes in ACTA2-AS1, PAPPA-AS1, and VCAN-AS1. Changes were validated by qRT-PCR (Fig. 5B-C). We analyzed expression of these lncRNAs in liver from PSC subjects versus healthy subjects (Supplementary Table 2) and found that PAPPA-AS1 and ACTA2-AS1 were increased in PSC (Fig. 5D). Fractionation of cholangiocytes and qRT-PCR showed ACTA2-AS1 and VCAN-AS1 in the nucleus whereas PAPPA-AS1 was located in cytoplasm (Fig.5E). We selected ACTA2-AS1 for further study because: 1) its expression was higher compared with PAPPA-AS1 and VCAN-AS1; 2) it was increased in human cholestatic liver disease; and 3) nuclear localization suggested a role in transcription. To evaluate cell-type specificity, we analyzed RNA-seq data from cholangiocyte and hepatocyte cells lines. This showed that cholangiocytes express ACTA2-AS1 whereas hepatocytes do not (Fig. S10A). We validated this by qRT-PCR in the cell lines (Fig. S10B) and in isolated mouse cholangiocytes and hepatocytes (Fig. S10C-E). We performed RNA in situ hybridization studies in our animal model (Fig. S10F). This confirmed the presence of ACTA2-AS1 in cholangiocytes with the expected reduction in staining after p300 knockout.
Figure 5:

ACTA2-AS1 is regulated by p300 and overexpressed in PSC. (A) Heatmap shows expression of lncRNAs in HiBEC treated with SGC-CBP30 or DMSO. (B, C) Validation by qRT-PCR. (D) qRT-PCR for PAPPA-AS1, ACTA2-AS1, and VCAN-AS1 in liver from healthy and PSC patients. (E) Percentage of RNA in the nuclear and cytoplasmic fractions by qRT-PCR. MALAT1 lncRNA and HPRT1 as nuclear and cytoplasmic controls.(n=3). Graph bars represent SEM. Unpaired Student's t-test *p<0.05,**p<0.01,****p<0.001 compared with control group.
ACTA2-AS1 promotes transcription of proliferative and fibrogenic genes.
ACTA2-AS1 is located on human chromosome 10 (10q23.31) and generates a 2,043-bp RNA (Fig. S11A) without protein-coding capacity (Fig. S11B). Gene Ontology analyses found associations between ACTA2-AS1 and “extracellular matrix assembly”, “focal adhesion assembly”, and “regulation of epithelial proliferation” (Fig. 6A), which are relevant in cholestatic liver disease. Antisense oligonucleotides (ASO) targeting ACTA2-AS1 were transfected into cholangiocytes, suppressing endogenous levels of ACTA2-AS1 (Fig.6B). Gene expression was analyzed by qPCR array and a scatterplot was generated (Fig.6C). ACTA2-AS1 silencing attenuated mRNA of fibrogenic genes (FN1, TGFB1, TGFB2), integrins (ITGB1), cadherins (CDH2), proliferative genes (PDGFRB), WNT-related genes (CTNNB1, TCF4, WNT5A, WNT5B), and TFs (STAT3, TWIST1, ZEB1) among others (Fig.6C). qRT-PCR and WB validated changes in FN1, PDGFRB, α-SMA, and PDGFB (Fig. 6D-E). We also generated epithelial organoids from cholangiocytes with and without ACTA2-AS1 knockdown and observed organoid size, and measured expression of proliferative and profibrogenic factors (Fig. S11C-E), noting reductions after ACTA2-AS1 knockdown.
Figure 6:
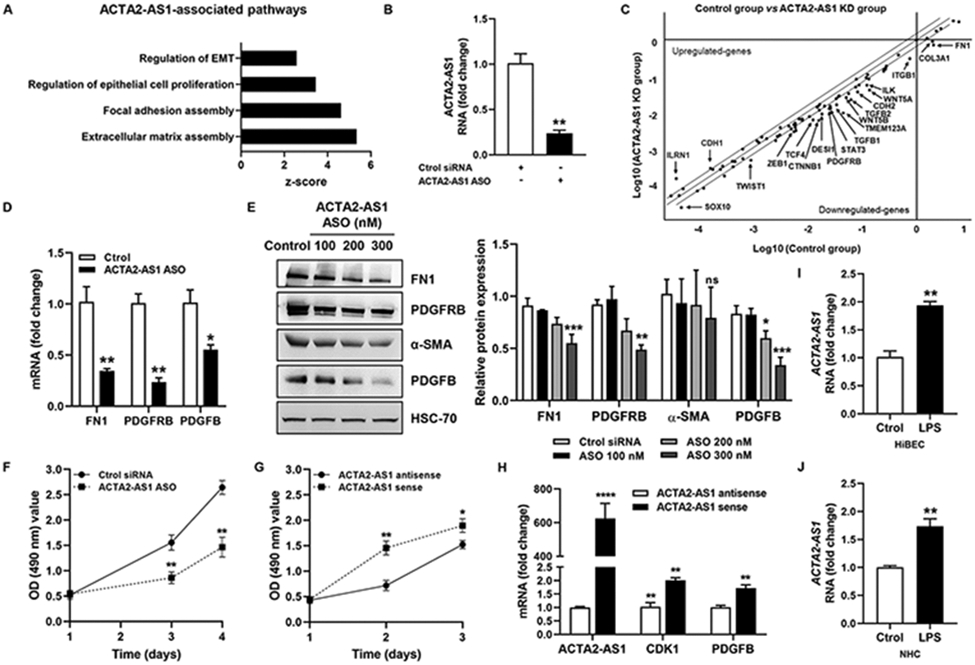
ACTA2-AS1 regulates proliferative and fibrogenic genes in cholangiocytes. (A) lncHUB shows pathways associated with ACTA2-AS1. (B) Analysis of ACTA2-AS1 expression in HiBEC transfected with control or ACTA2-AS1 antisense oligonucleotide (ASO). (C) Scatterplot showing gene changes in HiBEC transfected with control or ACTA2-AS1 ASO. (D) Validation by qPCR. (E) WB for FN1, PDGFRB, α-SMA and PDGFB. (F) Proliferation of control and ACTA2-AS1 knocked-down cells. (G) Proliferation of cells transfected with ACTA2-AS1 antisense and cells transfected with ACTA2-AS1 sense. (H) qRT- PCR for CDK1 and PDGFB in cells transfected with ACTA2-AS1 sense or antisense. (I, J) qRT- PCR for ACTA2-AS1 in HiBEC and NHC +/− LPS. (n=3). Graph bars represent SEM. Unpaired Student's t-test *p<0.05,**p<0.01,***p<0.005,****p<0.001 compared with control group.
We performed proliferation assays with loss- or gain-of-function of ACTA2-AS1 (Fig. 6F-G). ACTA2-AS1 knockdown impaired proliferation compared with controls (Fig. 6F). Correspondingly, cells overexpressing ACTA2-AS1 showed more proliferation (Fig. 6G). We further observed that ACTA2-AS1 overexpression increased mRNA of cyclin-dependent kinase 1 (CDK1) which is required for proliferation25, as well as the proliferative factor, PDGFB (Fig. 6H). Finally, we confirmed that ACTA2-AS1 expression is induced in response to LPS (Fig. 6I-J). Collectively, these observations point to ACTA2-AS1 as a lncRNA involved in pathological fibrosis and proliferation of cholangiocytes.
ACTA2-AS1 binds p300 and mediates p300-dependent transcription.
We next studied whether ACTA2-AS1 can bind p300 and promote transcription through H3K27ac. We performed RIP assays with an antibody against p300 using LPS-treated cholangiocytes. We observed enrichment of ACTA2-AS1 with the p300 antibody in response to LPS compared with controls (Fig. 7A). We performed ChIP assays to determine whether p300 binds the promoter of PDGFB in response to LPS and whether ACTA2-AS1 silencing affects binding. We observed enrichment of PDGFB promoter fragments with the p300 antibody in response to LPS (Fig. 7B). However, less PDGFB DNA bound to p300 in ACTA2-AS1 knocked-down cells (Fig. 7B). There was also a decrease in H3K27ac in the PDGFB promoter after ACTA2-AS1 knockdown (Fig. 7C), corresponding with decreased PDGFB protein (Fig. 7D). These results suggest that transcription of PDGFB is dependent upon ACTA2-AS1 for recruitment of p300 and subsequent H3K27ac.
Figure 7:
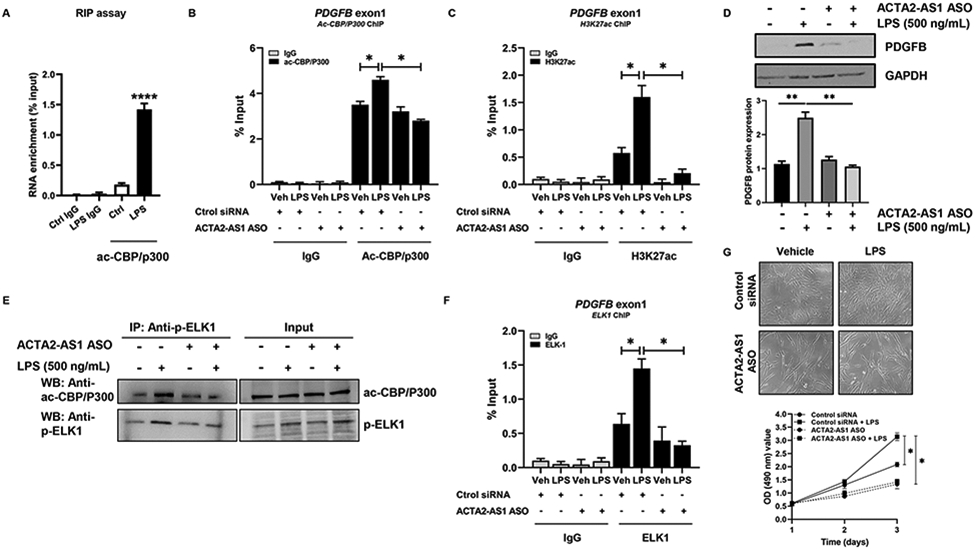
ACTA2-AS1 mediates P300- and ELK1-dependent transcription. (A) RIP of ac-CBP/P300-bound ACTA2-AS1 in NHC +/−LPS (Unpaired Student's t-test ****p<0.001 compared with control group). (B, C) NHC transfected with control or ACTA2-AS1 ASO +/− LPS analyzed by ChIP for ac-CBP/P300 and H3K27ac on the PDGFB promoter. (D) WB for PDGFB in cells transfected with ACTA2-AS1 ASO following LPS. (E) WB on ac-CBP/P300, p-ELK1 and IgG immunoprecipitates from HiBEC transfected with ACTA2-AS1 ASO +/− LPS. (F) Cholangiocytes transfected with control or ACTA2-AS1 ASO +/− LPS analyzed by ChIP for ELK1 on the PDGFB promoter. (G) Proliferation of cells transfected with ACTA2-AS1 ASO +/− LPS. (B-D),(F-G) one-way ANOVA with Bonferroni comparison *p<0.05,**p<0.01.(n=3). Graph bars represent SEM.
LncRNAs often act as scaffolds that facilitate epigenetic complexes containing TFs26. To evaluate whether ACTA2-AS1 mediates interaction between p300 and p-ELK1, we knocked-down ACTA2-AS1 and analyzed complex formation by IP. Knockdown of ACTA2-AS1 prevented p300/p-ELK1 complex formation after LPS (Fig. 7E). We also evaluated complex formation after SGC-CBP30 (Fig. S12A) and noted similar findings. ChIP assays determined that LPS induced enrichment of PDGFB promoter fragments with ELK1 antibody and ACTA2-AS1 silencing prevented ELK1 binding to the PDGFB promoter (Fig. 7F). Proliferation assays using ACTA2-AS1-silenced cholangiocytes demonstrated that ACTA2-AS1 knockdown impairs LPS-induced proliferation (Fig. 7G). These experiments confirm that cholangiocyte proliferation is dependent upon ACTA2-AS1 and its ability to recruit p300/p-ELK1 to proliferative and fibrogenic promoters, such as PDGFB.
Discussion
p300 is a histone acetyltransferase that activates transcription by mediating deposition of H3K27ac. We aimed to define the effects of cholangiocyte-selective loss of H3K27ac by deleting p300 from cholangiocytes during DR and biliary fibrosis in vivo. Furthermore, we sought to define the epigenetic complex through which p300 exerts proliferative and fibrogenic effects. We report several novel mechanistic insights into the epigenetic activation of DR and biliary fibrosis. We find that cholangiocyte-selective KO or pharmacologic inhibition of p300 is protective against surgically- or genetically-induced DR in mice and impairs the associated fibrosis. We identified proliferative and fibrogenic genes in cholangiocytes that are dependent upon p300 using RNA-seq and bioinformatics. Mechanistically, we defined a tripartite complex involving p300, a scaffolding lncRNA (ACTA2-AS1), and a TF (ELK1) that drives pathological gene expression. Thus, we identified an epigenetic complex that mediates proliferative and fibrogenic signals within cholangiocytes. Although ACTA2-AS1 appears to be cell type selective within liver epithelia, further profiling would be needed to assess its expression and complex formation outside of the liver.
p300 is the paralog of cyclic-AMP response element binding protein (CBP). CBP and p300 function as critical epigenetic co-activators in a variety of tissues and cell types27. Through acetylation of H3K27, they are involved in transcriptional activation of normal and pathological cellular processes. In liver, p300 is implicated in gene expression in hepatocytes, HSC, and endothelial cells, controlling diverse pathways including glucose homeostasis, cell cycle control, fibrosis, and inflammation28. In cholangiocytes, p300 promotes resistance to apoptosis through interaction with ETS110. Significant progress has been made in the development of specific p300 inhibitors29. Most target either the histone acetyltransferase (HAT) domain or the bromodomain (BRD). The HAT domain is responsible for transfer of acetyl groups to histones while the BRD domain allows for recognition of acetyl-lysines on other proteins27. Given the broad spectrum of p300 activity, within and outside of liver, inhibition of p300 in a clinical context may have off-target effects. Indeed, global KO of p300 results in embryonic lethality30. This led us to consider cell type-specific and context-dependent regulation of p300 including epigenetic complexes involving lncRNAs, which are often expressed in a tissue-specific manner31.
lncRNAs are increasingly recognized in liver for their roles in development, regeneration, fibrosis, and neoplasia32. However, lncRNAs are understudied in cholangiocytes and little is known about their role in biliary disease. Previous studies have identified roles for H1933 and HULC34 in proliferation and carcinogenesis. Our study is the first in cholangiocytes to implicate a lncRNA in epigenetic complex formation, acetyltransferase activity, and activation of pathological transcription. We identified three lncRNAs, ACTA2-AS1, PAPPA-AS1, and VCAN-AS1, that may be involved in p300-dependent transcription. We focused on ACTA2-AS1 since its expression is induced by p300, it is increased in human disease, and it has nuclear localization, consistent with an epigenetic role. Most lncRNAs do not have sequence homology across species35. However, we identified the mouse homologue of ACTA2-AS1, uc008hgf.1, and noted that it was specifically expressed in bile ducts. We also noted that it was markedly increased after BDL and that this effect was lost after genetic p300 KO. This supports the importance of this lncRNA in liver since it appears to be relevant in multiple species. We show that ACTA2-AS1 can bind p300 and induce accumulation of H3K27ac to drive PDGFB transcription.
Several lncRNAs (e.g. NEAT1)36 epigenetically regulate gene expression, but only a few, such as SATB2-AS137 and ZEB1-AS138, bind p300. Many lncRNAs function in cis and regulate expression of their corresponding sense genes39. Indeed, we find that ACTA2 expression is decreased in ACTA2-AS1-silenced cholangiocytes. Similar to other lncRNAs, such as TGFB2-AS140 and VIM-AS141, ACTA2-AS1 might facilitate accessibility of transcriptional regulators through R loops41. We show that ACTA2-AS1 acts as a scaffold, allowing formation of a complex including p300 and ELK1 downstream of LPS. The trio specifically targets certain gene promoters involved in proliferation and fibrosis and activates transcription through the acetyltransferase activity of p300.
In summary, cholangiocyte-selective p300 knockout or pharmacologic inhibition in mice is protective against BDL-induced DR and fibrosis. We defined a p300-dependent gene network in cholangiocytes that supports proliferation and fibrogenesis and mechanistically depends on interactions between p300, ELK1, and ACTA2-AS1. Therefore, this triumvirate is an interesting target for epigenetic treatment of cholangiopathies since its activity is responsible for a pathogenic subset of p300-dependent genes. This is the first study to link a tripartite acetyltransferase complex with biliary fibrosis. The mechanistic insights gained from this study may contribute to development of epigenetic pharmacology targeting DR and biliary fibrosis.
Supplementary Material
Highlights:
P300 knockout is protective against ductular reaction and biliary fibrosis in vivo
P300 regulates fibrogenic and proliferative gene networks in cholangiocytes
A P300/ELK1 complex epigenetically activates transcription downstream of LPS
ACTA2-AS1 lncRNA is a scaffold required for P300/ELK1 complex formation
Financial Support:
This publication was supported by the National Institutes of Health under Award Numbers DK057993 (NFL), DK084567 (NFL), DK059615 (VHS), DK113339 (RCH), and DK117861 (RCH), and the American Association for the Study of Liver Diseases (NJS). JMB was funded by the Spanish Carlos III Health Institute, Fondo Europeo de Desarrollo Regional, Euskadi RIS3, Department of Industry of the Basque Country, and Fundación Científica de la Asociación Española Contra el Cáncer.
Abbreviations:
- H3K27ac
acetylation of lysine 27 on histone 3
- BDL
bile duct ligation
- BRD
bromodomain
- ChIP
chromatin immunoprecipitation
- DR
ductular reaction
- Epcam
epithelial cell adhesion molecule
- ETS
E26 transformation-specific
- HAT
histone acetyltransferase
- HiBEC
human intrahepatic biliary epithelial cells
- LPS
lipopolysaccharide
- lncRNAs
long non-coding RNAs
- H3K27
lysine 27 on histone 3
- NHC
normal human cholangiocytes
- PSC
primary sclerosing cholangitis
- TFs
transcription factors
Footnotes
Conflict of Interest: The authors declare no conflicts of interest relevant to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Banales JM, Huebert RC, Karlsen T, Strazzabosco M, LaRusso NF, Gores GJ. Cholangiocyte pathobiology. Nat Rev Gastroenterol Hepatol 2019;16:269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sato K, Marzioni M, Meng F, Francis H, Glaser S, Alpini G. Ductular Reaction in Liver Diseases: Pathological Mechanisms and Translational Significances. Hepatology 2019;69:420–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ko S, Russell JO, Molina LM, Monga SP. Liver Progenitors and Adult Cell Plasticity in Hepatic Injury and Repair: Knowns and Unknowns. Annual review of pathology 2020;15:23–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lazaridis KN, LaRusso NF. Primary Sclerosing Cholangitis. N Engl J Med 2016;375:1161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jalan-Sakrikar N, De Assuncao TM, Shi G, Aseem SO, Chi C, Shah VH, et al. Proteasomal Degradation of Enhancer of Zeste Homologue 2 in Cholangiocytes Promotes Biliary Fibrosis. Hepatology 2019;70:1674–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aseem SO, Jalan-Sakrikar N, Chi C, Navarro-Corcuera A, De Assuncao TM, Hamdanet FH, et al. Epigenomic Evaluation of Cholangiocyte Transforming Growth Factor-beta Signaling Identifies a Selective Role for Histone 3 Lysine 9 Acetylation in Biliary Fibrosis. Gastroenterology 2021;160:889–905 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guicciardi ME, Trussoni CE, LaRusso NF, Gores GJ. The Spectrum of Reactive Cholangiocytes in Primary Sclerosing Cholangitis. Hepatology 2020;71:741–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mann DA. Epigenetics in liver disease. Hepatology 2014;60:1418–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dou C, Liu Z, Tu K, Zhang H, Chen C, Yaqoob U, et al. P300 Acetyltransferase Mediates Stiffness-Induced Activation of Hepatic Stellate Cells Into Tumor-Promoting Myofibroblasts. Gastroenterology 2018;154:2209–2221 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Hara SP, Splinter PL, Trussoni CE, Guicciardi ME, Splinter NP, Al Suraih MS, et al. The transcription factor ETS1 promotes apoptosis resistance of senescent cholangiocytes by epigenetically up-regulating the apoptosis suppressor BCL2L1. J Biol Chem 2019;294:18698–18713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bose DA, Donahue G, Reinberg D, Shiekhattar R, Bonasio R, Berger SL. RNA Binding to CBP Stimulates Histone Acetylation and Transcription. Cell 2017;168:135–149.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kopp F, Mendell JT. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018;172:393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li X, Liu R, Huang Z, Gurley EC, Wang X, Wang J, et al. Cholangiocyte-derived exosomal long noncoding RNA H19 promotes cholestatic liver injury in mouse and humans. Hepatology 2018;68:599–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang WT, Ye H, Wei PP, Han BW, He B, Chen ZH, et al. LncRNAs H19 and HULC, activated by oxidative stress, promote cell migration and invasion in cholangiocarcinoma through a ceRNA manner. Journal of Hematology & Oncology 2016;9:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tossberg JT, Esmond TM, Aune TM. A simplified method to produce mRNAs and functional proteins from synthetic double-stranded DNA templates. Biotechniques 2020;69:281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee SM, Vasishtha M, Prywes R. Activation and repression of cellular immediate early genes by serum response factor cofactors. J Biol Chem 2010;285:22036–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L, Koutelou E, Hirsch C, McCarthy R, Schibler A, Lin K, et al. GCN5 Regulates FGF Signaling and Activates Selective MYC Target Genes during Early Embryoid Body Differentiation. Stem Cell Reports 2018;10:287–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guest RV, Boulter L, Kendall TJ, Minnis-Lyons SE, Walker R, Wigmore SJ, et al. Cell lineage tracing reveals a biliary origin of intrahepatic cholangiocarcinoma. Cancer Res 2014;74:1005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang A, Okabe H, Popovic B, Preziosi ME, Pradhan-Sundd T, Poddar M, et al. Loss of Wnt Secretion by Macrophages Promotes Hepatobiliary Injury after Administration of 3,5-Diethoxycarbonyl-1, 4-Dihydrocollidine Diet. Am J Pathol 2019;189:590–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, et al. The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLOS Biology 2020;18:e3000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hammitzsch A, Tallant C, Fedorov O, O'Mahony A, Brennan PE, Hay DA, et al. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proceedings of the National Academy of Sciences 2015;112:10768–10773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buchwalter G, Gross C, Wasylyk B. Ets ternary complex transcription factors. Gene 2004;324:1–14. [DOI] [PubMed] [Google Scholar]
- 23.Park J, Gores GJ, Patel T. Lipopolysaccharide induces cholangiocyte proliferation via an interleukin-6-mediated activation of p44/p42 mitogen-activated protein kinase. Hepatology 1999;29:1037–43. [DOI] [PubMed] [Google Scholar]
- 24.Marchese FP, Huarte M. Long non-coding RNAs and chromatin modifiers: their place in the epigenetic code. Epigenetics 2014;9:21–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diril MK, Ratnacaram CK, Padmakumar VC, Du T, Wasser M, Coppola V, et al. Cyclin-dependent kinase 1 (Cdk1) is essential for cell division and suppression of DNA re-replication but not for liver regeneration. Proceedings of the National Academy of Sciences 2012;109:3826–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Molecular cell 2011;43:904–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He ZX, Wei BF, Zhang X, Gong YP, Ma LY, Zhao W, et al. Current development of CBP/p300 inhibitors in the last decade. Eur J Med Chem 2021;209:112861. [DOI] [PubMed] [Google Scholar]
- 28.Gao J, Wei B, Liu M, Hirsova P, Sehrawat TS, Cao S, et al. Endothelial p300 promotes portal hypertension and hepatic fibrosis through CCL2-mediated angiocrine signaling. Hepatology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lasko LM, Jakob CG, Edalji RP, Qiu W, Montgomery D, Digiammarino EL, et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017;550:128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kasper LH, Fukuyama T, Biesen MA, Boussouar F, Tong C, de Pauw A, et al. Conditional knockout mice reveal distinct functions for the global transcriptional coactivators CBP and p300 in T-cell development. Mol Cell Biol 2006;26:789–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou Q, Wan Q, Jiang Y, Liu J, Qiang L, Sun L. A Landscape of Murine Long Non-Coding RNAs Reveals the Leading Transcriptome Alterations in Adipose Tissue during Aging. Cell Reports 2020;31:107694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sato K, Glaser S, Francis H, Alpini G. Concise Review: Functional Roles and Therapeutic Potentials of Long Non-coding RNAs in Cholangiopathies. Frontiers in Medicine 2020;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiao Y, Liu R, Li X, Gurley EC, Hylemon PB, Lu Y, et al. Long Noncoding RNA H19 Contributes to Cholangiocyte Proliferation and Cholestatic Liver Fibrosis in Biliary Atresia. Hepatology 2019;70:1658–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang WT, Ye H, Wei PP, Han BW, He B, Chen ZH, et al. LncRNAs H19 and HULC, activated by oxidative stress, promote cell migration and invasion in cholangiocarcinoma through a ceRNA manner. J Hematol Oncol 2016;9:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahpour A, Mullen AC. Our emerging understanding of the roles of long non-coding RNAs in normal liver function, disease, and malignancy. JHEP Rep 2021;3:100177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Q, Cai J, Wang Q, Wang Y, Liu M, Yang J, et al. Long Noncoding RNA NEAT1, Regulated by the EGFR Pathway, Contributes to Glioblastoma Progression Through the WNT/beta-Catenin Pathway by Scaffolding EZH2. Clin Cancer Res 2018;24:684–695. [DOI] [PubMed] [Google Scholar]
- 37.Wang YQ, Jiang DM, Hu SS, Zhao L, Wang L, Yang MH, et al. SATB2-AS1 Suppresses Colorectal Carcinoma Aggressiveness by Inhibiting SATB2-Dependent Snail Transcription and Epithelial-Mesenchymal Transition. Cancer Res 2019;79:3542–3556. [DOI] [PubMed] [Google Scholar]
- 38.Liu C, Lin J. Long noncoding RNA ZEB1-AS1 acts as an oncogene in osteosarcoma by epigenetically activating ZEB1. Am J Transl Res 2016;8:4095–4105. [PMC free article] [PubMed] [Google Scholar]
- 39.Pelechano V, Steinmetz LM. Gene regulation by antisense transcription. Nat Rev Genet 2013;14:880–93. [DOI] [PubMed] [Google Scholar]
- 40.Papoutsoglou P, Tsubakihara Y, Caja L, Morén A, Pallis P, Ameur A, et al. The TGFB2-AS1 lncRNA Regulates TGF-beta Signaling by Modulating Corepressor Activity. Cell Rep 2019;28:3182–3198 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boque-Sastre R, Soler M, Oliveira-Mateos C, Portela A, Moutinho C, Sayols S, et al. Head-to-head antisense transcription and R-loop formation promotes transcriptional activation. Proc Natl Acad Sci U S A 2015;112:5785–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All methods/protocols used were peer-reviewed and cited in the reference section. Please refer to supplementary information for additional material/methods. Reagents, antibodies and resources are listed in the CTAT table.