

Abstract
Ribosome rescue pathways recycle stalled ribosomes and target problematic mRNAs and aborted proteins for degradation1,2. In bacteria, it remains unclear how rescue pathways distinguish ribosomes stalled in the middle of a transcript from actively translating ribosomes3–6. In a genetic screen in E. coli, we discovered a novel rescue factor that has endonuclease activity. SmrB cleaves mRNAs upstream of stalled ribosomes, allowing the ribosome rescue factor tmRNA (which acts on truncated mRNAs3) to rescue upstream ribosomes. SmrB is recruited to ribosomes and activated by collisions; cryo-EM structures of collided disomes from E. coli and B. subtilis reveal a distinct and conserved arrangement of the individual ribosomes and the composite SmrB binding site. These findings reveal the underlying mechanisms by which ribosome collisions trigger ribosome rescue in bacteria.
The synthesis of roughly 1 out of every 250 proteins in E. coli ends in failure7. Problems arise in many ways. Ribosomes arrest at the 3’-end of transcripts lacking a stop codon, for example, due to premature transcriptional termination or mRNA decay8. Ribosomes also stall on chemically damaged messages8,9, codons that are decoded slowly10, and nascent peptides that inhibit their own translation11,12. Prolonged stalling traps ribosomes in inactive complexes and produces incomplete proteins that can be toxic. Ribosome rescue pathways selectively recognize stalled ribosomes, rescue the ribosomal subunits, and target problematic mRNAs and nascent proteins for degradation1.
How are stalled ribosomes recognized? tmRNA, the main rescue factor in bacteria, enters ribosomes and encodes a short peptide tag to target the nascent peptide for proteolysis2. Because it binds in the mRNA channel, tmRNA’s activity is inhibited by mRNA downstream of the stall site1,3,13. In cases where translation stalls on intact messages, the current model is that mRNA cleavage yields truncated mRNAs that are good substrates for tmRNA4. Yet the signal that triggers mRNA cleavage and the nuclease have remained elusive.
Recent work has revealed insights into how eukaryotic cells recognize stalled ribosomes. When an upstream ribosome collides with a stalled ribosome, Hel2 is recruited to the interface formed between the two small ribosomal subunits14,15 and adds ubiquitin to specific ribosomal proteins16. Through the activity of Hel2, ribosome collisions trigger subunit splitting, mRNA decay, and degradation of the nascent polypeptide17. In the absence of ubiquitin and Hel2 homologs, however, it has been unclear whether collisions play a similar role in bacteria. Here we report that ribosome collisions in E. coli recruit SmrB, triggering mRNA cleavage and ribosome rescue. Moreover, we present cryo-EM structures that reveal the architecture of collided ribosomes in E. coli and B. subtilis and the composite binding site that explains how SmrB is activated by ribosome collisions.
A selection for novel rescue factors
We performed a genetic selection for mutations that allow ribosomes to translate through a strong stalling motif. Our selection uses a reporter with a fusion of NanoLuc and the bleomycin resistance protein (Ble) (Fig. 1a). Cells expressing a control construct with a stop codon between the genes (Stop) produce NanoLuc alone and are sensitive to the antibiotic phleomycin (Fig. 1b). In contrast, cells expressing a second construct without any intervening stalling motif (Non-stall) produce full-length protein and are therefore resistant to phleomycin. In a third construct where the strong SecM stalling motif is inserted between NanoLuc and Ble (SecM), ribosomes arrest at the SecM motif and fail to complete translation of Ble. As expected, wild-type cells expressing the SecM reporter are phleomycin sensitive (Fig. 1b). We confirmed that the SecM reporter induces stalling and ribosome rescue11: although little full-length protein is produced, truncated protein can be detected in cells lacking tmRNA (Extended Data Fig. 1a).
Figure 1.
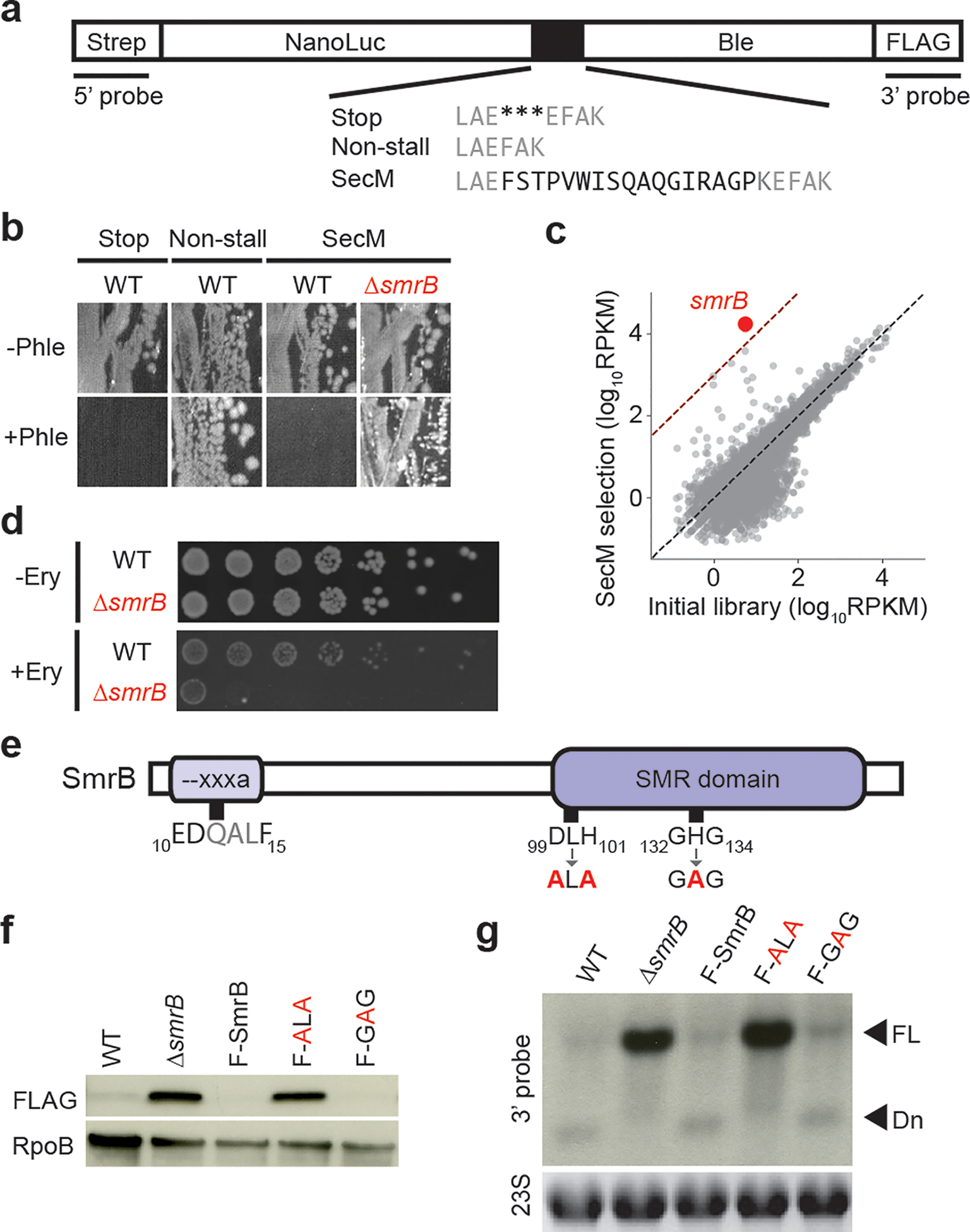
SmrB is a ribosome rescue factor. a, Between NanoLuc and the bleomycin resistance gene, we inserted stop codons, no added sequence, or the SecM stalling motif (black). b, Sections of plates with or without 50 μg/mL phleomycin showing growth of wild-type and ΔsmrB strains with reporters. c, The results of Tn-seq showing the number of transposon insertions for each gene (rpkm). d, Serial dilutions of wild-type and ΔsmrB cultures on plates with and without 200 μg/mL erythromycin. e, Domain organization of E. coli SmrB. Mutations at the conserved DxH and GxG motifs are indicated. f, Full-length SecM reporter protein was detected with anti-FLAG antibodies. Loading control = RpoB. F-SmrB, F-ALA, and F-GAG represent endogenously FLAG-tagged SmrB (not shown). g, SecM reporter mRNA was detected using a 3’-probe. Loading control = 23S rRNA. FL = full-length and Dn = downstream mRNA fragment.
Using a Tn-seq approach18, we created a knock-out library of 5 million colonies through random insertion of Tn5 transposase into the E. coli genome. After introducing the SecM reporter plasmid into the library and harvesting phleomycin-resistant colonies, we tallied the number of transposon insertions per gene (Fig. 1c). Following the selection, the smrB gene showed the strongest enrichment of transposon insertions (3100-fold). Deletion of smrB yields higher levels of full-length reporter protein (Fig. 1f) and makes cells phleomycin resistant (Fig. 1b). Furthermore, ΔsmrB cells show increased expression of reporters with other stalling motifs, such as Glu-Pro-stop (EP*) (Extended Data Fig. 1b,c).
We next asked if ΔsmrB cells are hypersensitive to conditions that induce widespread ribosome stalling on endogenous mRNAs. The ΔsmrB strain shows increased sensitivity to erythromycin, an antibiotic that stalls elongating ribosomes at specific sequences19,20 (Fig. 1d), but not to other antibiotics that target elongation more generally, including tetracycline, chloramphenicol, and kanamycin (not shown).
SmrB is a conserved nuclease
The E. coli SmrB protein contains a domain of the Small MutS Related (SMR) superfamily that possesses endonucleolytic RNase activity21,22. Indeed, we previously reported that an SMR protein, Cue2, cleaves mRNA on stalled ribosomes in yeast23. SMR domains are broadly conserved in bacteria, though notably underrepresented in the PVC group and the actinobacteria (Extended Data Fig. 2, 3a). They are found in all sampled eukaryotic lineages but are relatively uncommon in archaea.
Bacterial SMR domains cluster into three clades (Extended Data Fig. 3a,b). The first clade features SmrB from E. coli and other proteobacteria with an N-terminal extension containing a predicted helix (residues 9–20) with the conserved motif --xxxa (six-residues beginning with two acidic residues and ending with an aromatic amino acid) (Fig. 1e, Extended Data Fig. 3c). The second clade features the most common bacterial architecture typified by B. subtilis MutS2, which contains several domains found in the DNA mismatch repair factor MutS. The third clade is restricted to the Bacteroidetes lineage, with an N-terminal KOW domain.
An alignment of the SMR domain (residues 98–173) reveals that SmrB contains conserved residues corresponding to the DxH motif implicated in catalysis and the GxG motif predicted to interact with RNA substrates (Extended Data Fig. 3d)24,25. We mutated these motifs to ALA and GAG (Fig. 1e), respectively, at the endogenous smrB locus tagged with an N-terminal FLAG epitope. The FLAG tag does not affect SmrB activity (Fig. 1f). The ALA mutation increased abundance of full-length protein to a similar extent as deletion of smrB, whereas the GAG mutation had no discernible effect. These results suggest that only the DxH motif is critical for SmrB activity.
We followed SmrB activity using northern blots with a probe against the 3’-end of the reporter mRNA (Fig. 1g). In the wild-type strain, full-length mRNA is barely detectable and the strongest signal comes from the downstream fragment produced by cleavage near the SecM motif. In the ΔsmrB strain, the downstream fragment disappears and the levels of full-length mRNA are dramatically higher. As expected, the FLAG-tagged SmrB and the GAG mutant yield primarily cleaved mRNA, whereas the ALA mutant shows high levels of full-length mRNA. These results establish that SmrB cleavage is the dominant pathway that targets the reporter for decay.
To determine how ribosome rescue impacts mRNA decay, we used a probe against the 5’-end of the SecM and EP* reporters, revealing little full-length RNA in the wild-type strain (since it is cleaved and degraded) but detecting an upstream cleavage fragment in ΔssrA cells lacking tmRNA (Fig. 2a)4,11. While the upstream fragment is not detectable in the ΔsmrB strain, as expected, it is present for both reporters in the ΔssrA ΔsmrB strain (ΔΔ). This unexpected result reveals that the upstream fragment can be produced by additional pathways. It is unlikely that other endonucleases are responsible, given that the downstream fragment disappears in strains lacking SmrB (Fig. 2a). We speculate that in the absence of SmrB, the upstream fragment is generated by 3’−5’ exonucleolytic decay of the mRNA back to the stalled ribosome.
Figure 2.

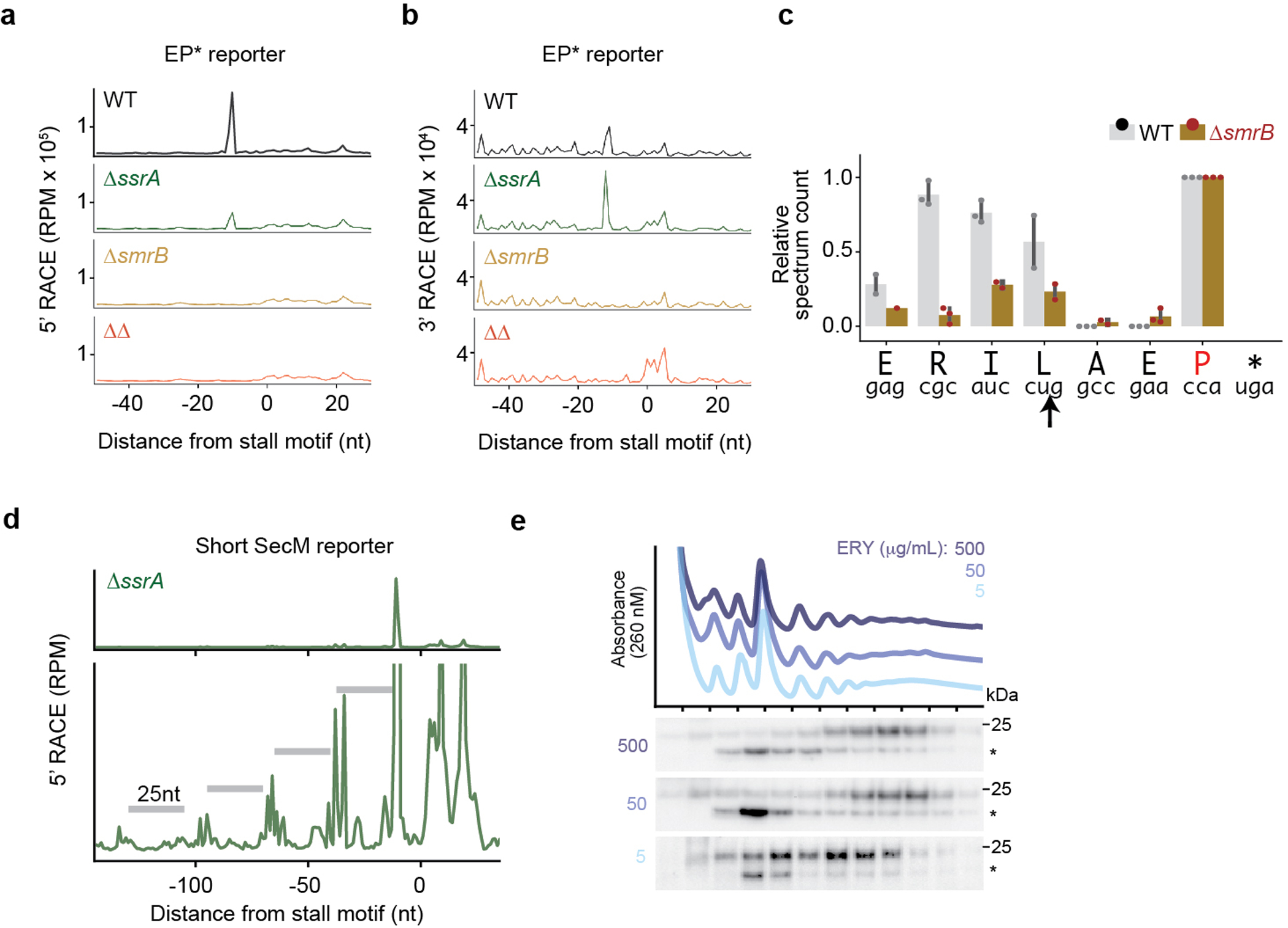
SmrB cleavage at the 5’ boundary of stalled ribosomes promotes ribosome rescue. a, Northern blots of reporter mRNA using the 5’-probe and the 3’-probe show full-length (FL) or truncated RNAs (upstream or downstream fragments). Loading control = 23S rRNA. b, 5’-RACE reveals the 5’-ends of downstream fragments in reads per million on the SecM reporter. The first nt in the A-site codon in the stall motif is 0. c, 3’-RACE reveals the 3’-ends of upstream fragments. d, tmRNA tagging sites on the Short SecM reporter in the wild-type and ΔsmrB strains were detected by targeted LC-MS-MS. The relative spectrum count is normalized by the count at the SecM stall site (red). The mean and standard deviation of three replicates is shown. The arrow indicates the SmrB cleavage site. e, A model for mRNA processing during ribosome rescue.
SmrB cleaves upstream of stalled ribosomes
We used RACE to identify the ends of the reporter mRNA fragments. 5’-RACE experiments reveal that the 5’-end of the downstream fragment is 11 nt upstream of the SecM stall site (Fig. 2b). This peak disappears in the ΔsmrB and the ΔsmrB ΔssrA strains. We see similar results from the EP* reporter (Extended Data Fig. 4a). Given that the ribosome protects roughly 12 nt of mRNA upstream of the A-site codon26, we conclude that SmrB cleaves mRNA at the 5’-boundary of the ribosome.
We also used 3’-RACE to determine the 3’-end of the upstream mRNA fragment. The data from these experiments are complex because multiple pathways generate truncated mRNAs. For the SecM reporter, in the wild-type and ΔssrA strains, the strongest peak is 11 nt upstream of the stall site (Fig. 2c); in the absence of SmrB, this peak disappears. This same phenomenon was observed in the EP* reporter (Extended Data Fig. 4b). This position corresponds perfectly with the site of SmrB cleavage identified for the downstream fragment by 5’-RACE.
In strains lacking both SmrB and tmRNA (ΔΔ) the 3’-RACE data provide additional information about other pathways that produce the upstream mRNA fragment. The strongest signal for the SecM reporter is 16 nt downstream of the stall site, corresponding to the 3’-boundary of the ribosome (Fig. 2c), likely the result of 3’−5’ exonuclease activity. In contrast, the strongest signal from the EP* reporter is at the A-site codon (Extended Data Fig. 4b). These results are broadly consistent with previous reports of mRNA cleavage at both the 5’- and 3’-boundaries of SecM-stalled ribosomes11 and A-site cleavage within EP*-stalled ribosomes4.
SmrB cleavage frees upstream ribosomes
To determine how SmrB cleavage affects tmRNA activity, we immunoprecipitated SecM reporter protein from the wild-type and ΔsmrB strains expressing a non-proteolytic tmRNA variant10, digested the protein, and subjected the resulting peptides to LC-MS-MS. In both the wild-type and ΔsmrB strains, the tmRNA tag is added to the SecM construct at the stall site, the second Gly residue (Fig. 2d). We also observe tagging four residues upstream (at the first Gly) in the wild-type strain. This position correlates with the SmrB cleavage site determined by RACE; tagging at this site is dramatically reduced in the ΔsmrB strain. Similar results were observed with the EP* reporter (Extended Data Fig. 4c).
These results lead us to propose the following model for ribosome rescue in E. coli (Fig. 2e). Ribosome stalling leads to endonucleolytic cleavage by SmrB at the 5’-boundary of the stalled ribosome (red). Upstream ribosomes (grey) translate to the end of the upstream mRNA fragment and arrest at the site of SmrB cleavage. tmRNA rescues these ribosomes, consistent with its well-characterized preference for truncated mRNAs3. tmRNA also rescues the first ribosome (red) at the initial stall site. Decay of the reporter mRNA back to the 3’-boundary of the ribosome (in the case of SecM) or the A-site codon (in the case of EP*) likely grants tmRNA access to these ribosomes4,6,11.
Collisions promote SmrB binding & activity
Close inspection of the 5’-RACE data reveals that SmrB also cleaves near ribosomes stacked behind the SecM-stalled ribosome (Extended Data Fig. 4d). To induce collisions throughout the transcriptome, we treated cells with the antibiotic mupirocin (MPC) which causes ribosome stalling at Ile codons26. In untreated cells, FLAG-SmrB is distributed across the sucrose gradient (Fig. 3a). After MPC treatment, FLAG-SmrB moves deeper into the polysomes, consistent with preferential binding to collided ribosomes on messages that are heavily translated. Furthermore, in lysates treated with RNase A to generate nuclease-resistant disomes, a hallmark of ribosome collisions15, SmrB is enriched in the disome peak in the MPC-treated sample (Fig. 3b).
Figure 3.

SmrB acts on collided ribosomes. a, The distribution of FLAG-SmrB in a sucrose gradient was observed with anti-FLAG antibodies in samples with and without mupirocin treatment (to induce pauses at Ile codons). A non-specific band is marked with *. b, The distribution of FLAG-SmrB in lysates treated with RNase A. c, Samples treated with four different tetracycline concentrations were analyzed as in a. d, Various lengths of the crp gene were fused to ble with the short SecM motif between them. N represents the number of nucleotides between the start codon and stall site. Reporter mRNA was detected using the 3’-probe. An arrow indicates the downstream fragment. Loading control = 16S rRNA.
To determine whether stalling or collisions recruit SmrB, we compared the effects of a high dose of a general elongation inhibitor (that stalls all ribosomes in place) to a lower dose (that induces collisions)17. While SmrB is enriched in polysomes in cells treated with 1.25 μg/mL tetracycline (TET), this effect is lost at 10- or 100-fold higher concentrations (Fig. 3c), arguing that SmrB specifically recognizes collided ribosomes. In contrast, ERY treatment enriches SmrB in polysomes even at high concentrations (Extended Data Fig. 4e) because it stalls ribosomes at specific sequences (e.g. RxR motifs19,20) rather than arresting all ribosomes, partially explaining the specific hypersensitivity of ΔsmrB cells to erythromycin (Fig. 1d).
We generated reporters to test whether ribosome collisions are required for mRNA cleavage by SmrB17. Different lengths of crp were fused upstream of the short SecM stalling motif (Fig. 3d). The closer the stalling motif is to the start codon, the less room there is for ribosomes to accumulate. The 3’-probe against the reporter mRNA reveals the downstream mRNA fragment and a strong reduction in full-length mRNA in the 111, 222, and 423 reporters (Fig. 3d). In contrast, for the 51 and 81 reporters where only one or two ribosomes can be loaded upstream of the SecM stalled ribosome, the downstream mRNA fragment is reduced and there is more full-length mRNA. In the ΔsmrB strain, all five reporters yield abundant full-length mRNA and no downstream fragment. Taken together, these results show that SmrB binding and activity is triggered by collisions.
The structure of collided ribosomes
In eukaryotes, collided ribosomes form interactions recognized by collision sensors like Hel214,15. To ask whether bacterial ribosomes display a similar behavior, we generated collided ribosomes by translating mRNAs encoding the arrest peptides MifM27 in B. subtilis extracts and VemP28 in the E. coli PURE system. Following separation on sucrose gradients, disome and trisome peaks were collected and subjected to structural analysis by cryo-EM.
The 3D reconstruction of E. coli disomes (Extended Data Fig. 5, Extended Data Table 1) revealed that the stalled ribosome resembles the VemP-stalled 70S in a non-rotated state29 whereas the collided ribosome is found in both the rotated and non-rotated states (Fig. 4). The stalled ribosome engages the collided ribosome in protein-protein and protein-rRNA interactions between the two juxtaposed 30S subunits. These interactions involve uS10, uS2, 16S rRNA helix h16, uS4, and 16S rRNA helices h5 and h17 in the collided ribosome interacting with uS9, uS2, 25S rRNA helix H78, uS11 and bS6, and bL9 in the stalled ribosome, respectively (Fig. 4c; Extended Data Fig. 6b). In addition, the L1 stalk of the stalled ribosome forms a bridge with the 30S subunit of the collided ribosome (Extended Data Fig. 6b).
Figure 4.

Cryo-EM structure of the E. coli disome. a, Cryo-EM density map of the E. coli disome with 50S subunits in grey, 30S subunits of the stalled and collided ribosome in yellow and orange, respectively, tRNAs in green, bS1 in purple, and bL9 in blue. b, Structural model of the E. coli disome. The bL9 proteins from stalled (bL9S) and collided (bL9C) ribosomes adopt different conformations. c, Interactions at the disome interface (opened up by rotation of the stalled and collided ribosomes); interacting partners are shown in matching colors.
There are substantial differences between the conformations of bL9 in the two ribosomes. In the collided ribosome, bL9 contacts uS6 and uL2 in the 30S subunit, as previously seen in individual 70S ribosomes. In contrast, in the stalled ribosome, the C-terminal domain of bL9 engages with helices h5 and h17 in the 30S subunit of the collided ribosome, forming a bridge between them (Extended Data Fig. 6b). Although bL9 also forms a bridge between 70S ribosomes in x-ray crystal structures30,31, that binding site does not overlap with the site observed in collided disomes. Binding of bL9 to the 30S subunit of the collided ribosome may block recruitment of factors like EF-G31, preventing attempts at translocation that may exert tension on the mRNA and induce frameshifting. Ribosomes lacking bL9 undergo frameshifting at very high rates32.
Notably, the largest protein of the 30S subunit, bS1, is missing in the stalled ribosome but present in the collided one (Fig. 4a,b; Extended Data Fig. 6a). Given that bS1 would sterically clash with the collided ribosome, we conclude that formation of the disome architecture requires dissociation of bS1 from the stalled ribosome. bS1 dissociation may serve as a checkpoint to discriminate between short-lived collisions in productive polysomes and longer-lasting, problematic stalling events.
In E. coli trisomes, the interface between the second and third ribosomes is essentially identical to the interface between the stalled and collided ribosomes (Extended Data Fig. 6c, 7). During long-lived stalling events, additional collisions may yield many such ribosome-ribosome interfaces. This observation may explain why SmrB primarily binds trisomes and heavier polysomes upon mupirocin treatment (Fig. 3a) and most efficiently cleaves stalling reporters with enough room for three or more ribosomes to collide (Fig. 3d).
Structures of MifM-stalled disomes from B. subtilis adopted an essentially identical conformation as observed in E. coli, including the bridges formed between the ribosomes by the L1 stalk and bL9 (Extended Data Fig. 6d,e, 7). This high degree of similarity indicates that dimerization is likely to be conserved in bacteria, with subtle differences at the disome interface. Notably, the architecture of these collided disomes is completely different from hibernating 100S disomes formed under stress conditions (Extended Data Fig. 6g,h)33,34.
The SmrB-disome structure
Next, we reconstituted mRNA cleavage on collided disomes in vitro. We incubated wild-type SmrB and the nuclease-deficient ALA mutant with E. coli disomes and analyzed the products on sucrose gradients (Fig. 5a). In a control reaction without SmrB we observed disomes, as expected, and 70S ribosomes arising from ribosome dissociation or background nucleolytic activity. In contrast, after incubation with wild-type but not mutant SmrB, the disome signal is substantially reduced and the 70S signal increases, consistent with SmrB cleavage between the ribosomes. This result argues that SmrB is sufficient to recognize disomes and cleave mRNA without additional factors.
Figure 5.
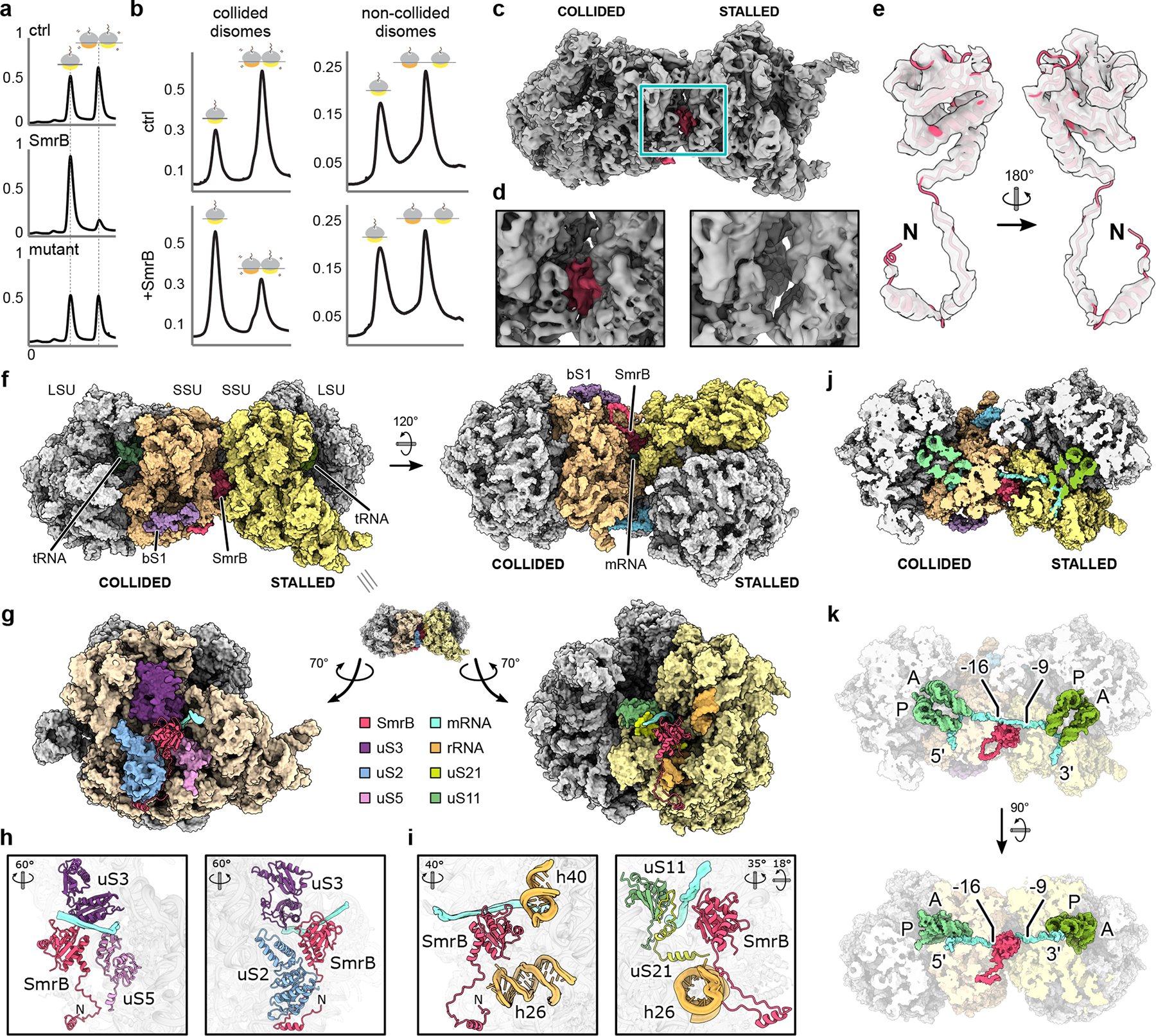
Cryo-EM structure of the SmrB-bound E. coli disome. a, Disome nuclease assay. Sucrose gradients showing VemP-stalled disomes alone (ctrl) and incubated with wild-type (SmrB) or nuclease- deficient SmrB (mutant). Y-axes are A260 nm. b, Sucrose gradients showing collided disomes (left) and non-collided disomes (right) alone and with SmrB. Y-axes are A260 nm. c, Cryo-EM density map of the SmrB-bound E. coli disome with SmrB in pink. d, Zoomed view of the disome interface comparing the SmrB bound (left) and unbound (right) maps. e, Isolated cryo-EM density with the fitted SmrB model. f, Structural model of SmrB bound to the E. coli disome. g, Interactions between SmrB and the stalled and collided ribosomes. The disome interface is opened up by rotation of the stalled and collided ribosomes. h & i, Interactions of SmrB with the collided ribosome (h) and the stalled ribosome (i). The orientations with respect to g are indicated. j, Cut view of the SmrB-bound disome showing the mRNA path. k, Interaction of SmrB with the mRNA. The orientation in the upper part of k corresponds to j. Approximately 8 nucleotides of the mRNA are exposed at the disome interface, in reach for SmrB cleavage. The nucleotides at the ribosome boundaries are indicated; the first nucleotide in the A site of the stalled ribosome is 0.
We also generated non-collided disomes through in vitro translation of an mRNA containing two short ORFs that each accommodate only one ribosome; these ribosomes were stalled with tetracycline (Extended Data Fig. 8a,b). Although addition of SmrB to collided disomes reduced the disome peak as expected, it had little or no effect on the non-collided disomes (Fig. 5b), arguing that SmrB exhibits little or no activity on single ribosomes or naked mRNA (Extended Data Fig. 8c,d).
Next, we added the nuclease-deficient SmrB mutant to E. coli disomes and subjected the complexes to structural analysis by cryo-EM (Extended Data Fig. 9, Extended Data Table 1). The reconstruction revealed extra density between the 30S subunits near the mRNA stretching between the ribosomes (Fig. 5c,d). The limited local resolution in this region prevented de novo model building for SmrB. However, the high-confidence prediction of the SmrB structure by AF235 corresponded well with the extra density and was used for rigid-body docking with only minor adjustments of the N-terminal region (Fig. 5e, Extended Data Fig. 10).
SmrB binds at the interface of the two ribosomes: on the collided ribosome, the SMR domain interacts with uS3, uS2, and uS5; on the stalled ribosome, it binds uS11, uS21, and 16S rRNA helices h40 and h26 (Fig. 5f–i). Notably, the genes encoding uS21 and SMR-domain proteins are linked in conserved operons in several bacteria, suggesting that this interaction may be conserved (Extended Data Fig. 11a). The N-terminal extension of SmrB forms a hook-like structure that wraps around uS2 of the collided ribosome (Fig. 5g). The N-terminal helix contains the --xxxa motif conserved in SMR-domain proteins in proteobacteria (Extended Data Fig. 3c). Although this helix also appears on the stalled ribosome, the SMR domain of this second copy of SmrB in the complex is not visible. We speculate that the N-terminal helix initially recruits SmrB to 70S ribosomes and that formation of the composite site between collided ribosomes stabilizes SMR domain binding and positions it for mRNA cleavage. Indeed, an SmrB mutant lacking the N-terminal extension does not bind ribosomes (Extended Data Fig. 11b). The active site of SmrB is poised to cleave the bridging mRNA between position −9 and −16, counting from the A-site codon in the stalled ribosome (Fig. 5j,k), in agreement with our biochemical data. Activation of SmrB on collided disomes is likely the result of precise positioning with respect to its substrate.
We asked if disruptions of the disome interface or SmrB binding pocket interfere with SmrB activity. SmrB efficiently cleaves reporter mRNA in a collision-dependent manner in a bL9 knockout strain (Extended Data Fig. 11c). Moreover, fusion of mCherry to the C-terminus of bL9 has no effect on SmrB activity. These results suggest that the bL9 bridge does not play an essential role in stabilizing the disome interaction. In contrast, fusion of MBP to the N-terminus of uS21 dramatically stabilizes full-length mRNA in the 111 and 222 reporters (Extended Data Fig. 11d). To a lesser extent, fusion of GFP to the C-terminus of S6 also stabilizes the 111 CRP reporter. These results are consistent with a reduction in SmrB activity due to a disruption of the SmrB binding pocket, validating our structural findings.
Conclusions
In this report, we show that SmrB recognizes ribosome collisions, cleaves mRNA, and triggers ribosome rescue. Our work reveals two striking parallels between ribosome rescue in bacteria and eukaryotes. First, ribosome collisions signal that a stalled ribosome requires rescue15,17. A role for collisions in E. coli is consistent with previous mathematical models of the effects of ribosome stalling on protein output5,36 and reports that collisions affect frameshifting37,38. The conservation of the disome interface between E. coli and B. subtilis argues that collisions play a conserved role in bacteria by providing a unique interface for recognition by rescue factors. Secondly, SMR-domain proteins cleave mRNA within collided ribosomes in yeast23; SMR-domain proteins were found in all but a few bacterial phyla, suggesting that their role in ribosome rescue may be widespread. These common features substantiate the universal significance of ribosome collisions and endonucleolytic cleavage in ribosome rescue.
ONLINE METHODS
Statistics and Reproducibility
Western blots (Fig. 1f & 3a–c and Extended Data Fig. 1, 4e, & 11b) and northern blots (Fig. 1g, 2a, & 3d and Extended Data Fig. 11c,d) were repeated at least three times. The MS experiments identifying the tmRNA tagging site were performed in triplicate (Fig. 2d and Extended Data Fig. 4c). The in vitro SmrB nuclease assays (Fig. 5a,b and Extended Data Fig. 8) were performed in triplicate. All of these attempts at replication were successful.
Bacterial strains and plasmids
A list of strains and plasmids and the details of their construction are given in Supplementary Table 1.
The L9 knockout strain (BW25113 rplI::kan) was obtained from the E. coli Genetic Stock Center. Strains QC101 and QC901 containing the L9-mCherry and S6-GFP, respectively, were gifts from Suparna Sanyal39. Additional knockout strains of MG1655 were constructed using one-step genomic replacement with a PCR fragment with λ Red recombinase40. Gene deletions and the endogenous epitope-tagged SmrB mutants were verified by PCR and sequencing.
The nanoLuc-ble reporter construct pKS-nonstall was expressed from plasmids containing an AmpR marker and a p15A origin of replication. DNA encoding various stalling motifs was inserted between the genes for nanoLuc and ble using Gibson assembly.
To construct the Crp collision reporters, the first 39, 69, 99, 109, and 411 bases of the crp coding region were amplified from MG1655 genomic DNA. The PCR also added the sequence encoding the short SecM ribosome stalling motif (GIRAGP) in-frame immediately downstream of the crp fragment. The PCR products were inserted into EcoRI and BglII digested pKS-nonstall to produce the pCRP51, pCRP81, pCRP111, pCRP121, and pCRP423 reporter plasmids using Gibson assembly. The names of the plasmids and numbers in the text represent the distance from the AUG to the stall site (the second Gly codon in GIRAGP) in each reporter.
To construct the plasmid pAC01 encoding tmRNA-DD, we switched the origin of replication of the pKW23 plasmid10 from p15A to pBR322 for compatibility. This was accomplished by Gibson assembly of two PCR products: 1) everything in pKW23 except the origin and 2) the pBR322 origin from pBAD-GFPuv41.
For overexpression and purification of SmrB, we amplified the smrB gene from genomic DNA from MG1655, adding a Twin-Strep tag, a TEV cleavage site, and a FLAG tag to the N-terminus of SmrB using nested PCR primers. This amplicon was inserted into pET24b cleaved with NdeI and BamHI using Gibson assembly.
Genetic screening
A library with random transposon insertions throughout the genome was constructed using the EZ-Tn5 <KAN-2>Tnp Transposome Kit (Lucigen). The EZ-Tn5 transposome was electroporated into the parental E. coli strain K12 MG1655 and the cells were plated on LB + kanamycin (50 mg/L). After one overnight incubation, approximately 5 million colonies were collected and stored at −80 °C in LB + 20% glycerol. Subsequently, the SecM reporter plasmid was electroporated into the random insertion library and transformants were plated on LB + ampicillin (50 mg/L) + phleomycin (50 mg/L). After one overnight incubation, phleomycin resistant colonies were collected and stored in at −80 °C in LB + 20% glycerol.
To quantify the number of transposon insertions in each gene throughout the genome, Tn-seq was performed on the random insertion library as well as the libraries of phleomycin-resistant colonies from the SecM screen. Sequencing libraries were prepared from genomic DNA from each library using the NEBNext Ultra II FS DNA Library Prep Kit for Illumina (NEB) following the manufacturer’s protocol. During PCR amplification of the adapter-ligated DNA, we used two PCR steps with primers that specifically bind to the mosaic end of EZ-Tn5 <KAN-2> Transposon to prepare libraries enriched in transposon insertion sites. Between the PCR steps, we included an additional enrichment step based on biotin-streptavidin purification. The two-PCR steps were performed using NEBNext Ultra II Q5 Master Mix (NEB) as follows: The 1st PCR enrichment was performed with primers KS_Tn5_1stPCR_F_biotin and KS_Tn5_1stPCR_R (Table_S1) with the following program: 30 s at 98 °C; 15 cycles of 10 s at 98 °C, 20 s at 59 °C, 60 s at 65 °C; and 5 min at 65 °C. The PCR products were purified first with DNA Clean & Concentrator-5 columns (Zymo Research) and then Dynabeads MyOne Streptavidin C1 beads (Thermo Fisher). The streptavidin beads were washed four times with binding and washing buffer (5 mM Tris-HCl pH 7.5, 0.5 mM EDTA pH 8.0, 1 M NaCl); the PCR products were added and incubated for 30 min at 25 °C; and the beads were washed twice with the binding and washing buffer and then twice by 0.1x TE buffer (1 mM Tris pH 8.0, 0.1 mM EDTA pH 8.0). The beads bound to the PCR products were used directly as the template for the 2nd PCR enrichment using KS_Tn_library_F and one of our custom Index primers for Tn-seq (Table S1) with the following program: 30 s at 98 °C; 7–9 cycles of 10 s at 98 °C, 20 s at 59 °C, 60 s at 65 °C; and 5 min at 65 °C. The products from the 2nd PCR were gel purified on a non-denaturing 5% TBE gel, analyzed on a BioAnalyzer high sensitivity DNA kit (Agilent), and sequenced on the NextSeq 500 instrument (Illumina).
The Tn-seq data were analyzed with custom scripts written in Python 2.7. The adaptor sequence AGATCGGAAGAGCACACGTC was removed from the 3’-ends of reads with Skewer version 0.2.242. Reads of interest contain the Tn5 transposase sequence at the 5’-end and genomic DNA sequence at the 3’-end. Reads lacking the Tn5 transposase sequence GGTTGAGATGTGTATAAGAGACAG at their 5’-ends were discarded using cutadapt version 1.1643. After trimming this sequence, the remaining reads were aligned to E. coli K12 MG1655 genome build NC_000913.2 using bowtie version 1.1.244. The site of the transposon insertion was assigned using the 5′-end of the aligned reads. The number of transposon insertion sites for each gene was counted and normalized as reads per kilobase per million mapped reads (RPKM), normalizing for both the sequencing depth for each library and the length of each gene.
Sequence Analyses
PSI-BLAST v2.1245 and JACKHMMER v3.3.246 were used to carry out iterative sequence profile searches to collect SMR domain-containing sequences. Proteins were clustered using BLASTCLUST (https://ftp.ncbi.nih.gov/blast/documents/blastclust.html) to identify shared domain architectural themes. Additional domains fused to the Smr domain were annotated using a database of domain sequence profiles including pfam A models47. For contextual analysis of prokaryotic gene neighborhoods, the GenBank genome files corresponding to unique GenBank genome assemblies (GCA ids) were used as starting material. Specific neighborhoods were extracted using a Perl script that reports upstream and downstream genes of the anchor SMR domain-containing gene. Proteins encoded by these genes were then clustered using BLASTCLUST to identify conserved gene neighborhoods based on conservation between different taxa. Additional filters outputted valid neighborhoods for further analysis: (1) nucleotide distance constraint (generally 50 nucleotides), (2) conservation of gene directionality within the neighborhood, and (3) presence in more than one phylum. Multiple sequence alignments were built using Kalign v2.0448, and manually improved based on the alignments outputted by sequence homology searches. Secondary structure prediction was done using JPred v3.049. Phylogenetic relationships were determined using an approximate maximum likelihood (ML) method as implemented in FastTree v2.1.1050: corresponding local support values were also estimated as implemented. To increase accuracy of topology, the rounds of minimum-evolution subtree-prune-regraft (SPR) moves were increased to 4 (-spr 4) and we utilized options -mlacc and -slownni to survey more exhaustively the ML nearest neighbor interchanges (NNIs).
Western blots
Cells were grown in LB + ampicillin (50 mg/L) to OD600 = 0.5, harvested by centrifugation, resuspended in 12.5 mM Tris pH 6.8 with 4% SDS, and lysed by heating to 90 °C for 10 min. 5x loading dye (250 mM Tris pH 6.8, 20% glycerol, 30% β-mercaptoethanol, 10% SDS, saturated bromophenol blue) was added and the lysate was denatured at 90 °C for 10 min. Protein was separated on a 4–12% Criterion XT Bis-Tris protein gel (Bio-Rad) using XT MES buffer and transferred to PVDF membrane using the Trans-Blot Turbo Transfer system (Bio-Rad). Membranes were blocked in 5% milk for 1 h at room temperature, washed, and then probed with antibodies diluted in TBS-tween. Antibodies dilutions were: anti-FLAG-HRP 1:10000 (Sigma); anti-Strep•Tag II-HRP 1:5000 (Millipore Sigma); anti-RpoB 1:1000 (BioLegend); anti-RpoC 1:1000 (BioLegend); and anti-mouse-HRP 1:2000 (Thermo Fisher). Chemiluminescent signals from HRP were detected using SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher) or SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher) and visualized on Amersham Hyperfilm ECL (GE).
Northern blots
Cells were grown in LB + ampicillin (50 mg/L) to OD600 = 0.5, harvested by centrifugation, and resuspended in 100 mM NaCl, 10 mM Tris pH 8.0, 1 mM EDTA pH 8.0, and 1% SDS. RNA was extracted twice by phenol, pH 4.5 (once at 65 °C and once at room temperature) followed by chloroform extraction. RNA in the aqueous layer was then precipitated by isopropanol and 0.3 M NaOAc (pH 5.5), washed with 80% ethanol, and resuspended in water. Purified RNA was separated on a 1.2% agarose-formaldehyde denaturing gel and transferred to a nylon membrane (Hybond-N+, Cytiva) in 10 × SSC buffer using a Model 785 Vacuum Blotter (Bio-Rad). RNA was crosslinked to the membrane with the Stratalinker UV crosslinker (Stratagene). Pre-hybridization and hybridization was performed in PerfectHyb Plus Hybridization Buffer (Milipore Sigma). RNA was probed with 50 nM 5’-digoxigenin labeled DNA oligos (IDT). Digoxigenin was detected with anti-Digoxigenin-AP antibodies diluted 1:1000 (Milipore Sigma). Chemiluminescent signals from alkaline phosphatase were detected with CDP-Star (Milipore Sigma) and visualized on Amersham Hyperfilm ECL (GE).
5’-RACE
RNA was extracted as described above. DNA contamination was depleted by treatment with RQ1 DNase (Promega). 5 μg of purified RNA was 5’-phosphorylated by incubating with T4 polynucleotide kinase (NEB) in 1 mM ATP at 37 °C for 30 min, after which PNK was denatured by heating to 75 °C for 10 min. The RNA adapter KS_5RACE_linker was ligated to the 5’-end of the RNA by incubating with T4 RNA ligase 1 (NEB) in 1 mM ATP and 15% PEG8000 at 25 °C for 3 h. Ligated samples were purified by 2.2x volume of RNAclean XP (Beckman). The 1st strand cDNA was synthesized using the KS_5RACE_RT primer and SuperScript III Reverse Transcriptase (Thermo Fisher) by incubating at 54 °C for 60 min after which RT was denatured by heating to 85 °C for 5 min. Denatured reverse transcription products were used directly in the 1st PCR reaction. The 1st PCR was performed with Phusion High-Fidelity DNA Polymerase (NEB) and primers KS_5RACE_F1 and KS_5RACE_R1 with the program: 30 s at 98 °C; 25 cycles of 10 s at 98 °C, 10 s at 65 °C, 60 s at 72 °C; and 5 min at 72 °C. The 1st PCR products were purified using DNA Clean & Concentrator-5 columns (Zymo Research). The 2nd PCR was performed with Phusion High-Fidelity DNA Polymerase (NEB) and primer NI-NI-2 and one of our custom Index primers for 5’-RACE (Table S1) with the program: 30 s at 98 °C; 25 cycles of 10 s at 98 °C, 10 s at 65 °C, 60 s at 72 °C; and 5 min at 72 °C. The 2nd PCR products were purified using DNA Clean & Concentrator-5 columns (Zymo Research), analyzed on a BioAnalyzer high sensitivity DNA kit (Agilent), and sequenced on the MiSeq Nano instrument (Illumina).
The 5’-RACE data were analyzed using custom scripts written in Python 2.7. The RNA adapter sequence TGCCCGAGTG was removed from the 5’-end of reads using cutadapt43. Reads without the RNA adapter sequence were discarded. The reverse primer sequence GCGGTCGAGTTCTGGACCGA from the 2nd PCR was removed from the 3’-ends of reads by cutadapt43. The processed reads were aligned to the SecM or EP* reporter plasmid sequences using bowtie44. The 5’ ends of mapped reads were counted and normalized as reads per million mapped reads (RPM), normalizing for the sequencing depth of each library.
3’-RACE
RNA was extracted as described above. DNA contamination was depleted by treatment with RQ1 DNase (Promega). 5 μg of purified RNA was 3’-dephosphorylated by incubating with T4 polynucleotide kinase (NEB) without ATP at 37 °C for 30 min, after which PNK was denatured by heating to 75 °C for 10 min. The 3’ DNA adapter was 5’ adenylated using the 5´ DNA Adenylation Kit (NEB) and purified with an Oligo Clean & Concentrator column (Zymo Research). This adaptor was ligated to 3’-end of dephosphorylated RNA by incubating with T4 RNA ligase 2 truncated (NEB) in 15% PEG8000 at 37 °C for 3 hours. Ligated samples were purified with RNAclean XP (Beckman). The 1st strand cDNA was synthesized with KS_3RACE_RT and SuperScript III Reverse Transcriptase (Thermo Fisher) by incubating at 54 °C for 60 min, after which RT was denatured by heating to 85 °C for 5 min. Denatured reverse transcription products were used directly in the 1st PCR reaction. The 1st PCR was performed with Phusion High-Fidelity DNA Polymerase (NEB) and primers KS_3RACE_F1 and KS_3RACE_RT with the program: 30 s at 98 °C; 9 cycles of 10 s at 98 °C, 10 s at 65 °C, 60 s at 72 °C; and 5 min at 72 °C. The PCR products were purified using DNA Clean & Concentrator-5 columns (Zymo Research). The 2nd PCR was performed with Phusion High-Fidelity DNA Polymerase (NEB) and one of our custom Index primers for 3’-RACE (Table S1) and KS_3RACE_R with the program: 30 s at 98 °C; 25 cycles of 10 s at 98 °C, 10 s at 65 °C, 60 s at 72 °C; and 5 min at 72 °C. The 2nd PCR products were purified by DNA Clean & Concentrator-5 columns (Zymo Research), analyzed on a BioAnalyzer high sensitivity DNA kit (Agilent), and sequenced on the MiSeq Nano (Illumina).
The 3’-RACE data were analyzed by custom scripts written in Python 2.7. The DNA adapter sequence TCCTTGGTGCCCGAGTGNNNNNN was removed from the 5’-end of reads using cutadapt43. Reads without the adapter sequence were discarded. The forward primer sequence AGATCGGAAGAGCACACGTC from the 2nd PCR was removed from 3’-end of reads using cutadapt43. Processed reads were aligned to the SecM or EP* reporter plasmid sequences using bowtie44. The 5’ ends of mapped reads were counted and normalized as reads per million mapped reads (RPM), normalizing for the sequencing depth of each library.
Mass spectrometric analysis of tmRNA tagging sites
Immunoprecipitation and processing of the reporter proteins
The EP* and the Short SecM (GIRAGP) reporters were expressed in both the wild-type MG1655 strain and the ΔsmrB strain. In addition, a modified tmRNA encoding ANDENYALDD was also expressed from the pAC01 plasmid to stabilize the products of tmRNA tagging. (pAC01 is a derivative of pKW2310 modified to contain a pBR322 origin). For each of these four samples, reporter protein was purified from three biological replicates as follows: 100 mL LB cultures were grown to OD600 = 0.5 and harvested by centrifugation. The pellet was frozen at −80 °C and thawed in 2x CellLytic B cell lysis reagent (Sigma) for 10 minutes. The lysate was clarified by centrifugation for 30 min at 20,000 × g. 50 μL Strep-tactin sepharose beads (IBA) were added to the supernatant and incubated at 4 °C for 1 h. The beads were washed with IP wash buffer (20 mM Tris pH 8.0, 100 mM NH4Cl, 0.4% Triton, 0.1% NP-40) for 5 min at 4 °C four times. Protein was eluted from the beads by shaking at 4 °C in elution buffer (20 mM Tris pH 8.0, 100 mM NH4Cl, 5 mM desthiobiotin) for 1 h. 36 μL of each immunoprecipitated sample was reduced with 1.5 mg/mL DTT in 50 μL of 50 mM tri-ethyl ammonium bicarbonate (TEAB) buffer at 57 °C for 60 min, then alkylated with 10 mg/mL iodoacetomide in 50 μL of 50 mM TEAB buffer in the dark at room temperature for 45 min. The samples were reconstituted in 36 μL of 50 mM HEPES pH 8.5 and digested with 2 ng/μL LysC at 37 °C overnight as described51. Peptides were desalted on Oasis u-HLB plates (Waters), eluted with 60% ACN / 0.1% TFA, dried, and reconstituted with 2% ACN / 0.1% formic acid.
LC/MS/MS analysis
Desalted peptides cleaved by LysC were analyzed by liquid chromatography/tandem mass spectrometry (LC/MS/MS). The peptides were separated by reverse-phase chromatography (2% - 90% acetonitrile / 0.1% formic acid gradient over 60 min at 300 nL/min) on an 75 μm × 150 mm ProntoSIL-120-5-C18 H column (Bischoff) using the nano-EasyLC 1200 system (Thermo). Eluting peptides were sprayed into an Orbitrap-Lumos_ETD mass spectrometer through a 1 μm emitter tip (New Objective) at 2.7 kV. Scans were acquired within 360–1700 Da m/z targeting the C-terminal SsrA fusion peptides with no dynamic exclusion. Precursor ions were individually isolated with 0.8 Da (no offset) and fragmented (MS/MS) using HCD activation collision energy 30. Precursor and the fragment ions were analyzed at resolution at 200Da 120,000 AGC target 1xe6, max IT 50ms and 60000, AGC target 1xe5, mx IT118ms, respectively, 3 cycles. Tandem MS/MS spectra were processed by Proteome Discoverer v2.4 (Thermo Fisher) and analyzed with Mascot v.2.6.2 (Matrix Science) using RefSeq2017_83Ecoli and a database with peptides from the nanoLuc-ble reporter protein. Peptide identifications from Mascot searches were processed within the Proteome Discoverer-Percolator to identify peptides with a confidence threshold of a 0.01% False Discovery Rate, based on a concatenated decoy database search to calculate the protein and peptide ratios. Only Peptide Rank 1 were considered.
Polysome profiling
Cells were cultured at 37 °C in 500 mL of LB (and antibiotics where appropriate) to OD600 = 0.5. For samples with antibiotic treatments, cells were cultured to OD600 = 0.45, then treated for 5 min by antibiotics at the concentrations indicated in the figures. Cells were then harvested by filtration using a Kontes 99 mm filtration apparatus with a 0.45 μm nitrocellulose filter (Whatman), and flash frozen in liquid nitrogen. Cells were lysed in lysis buffer (20 mM Tris pH 8.0, 10 mM MgCl2, 100 mM NH4Cl, 5 mM CaCl2, 100 U/mL DNase I, and 1 mM chloramphenicol) using a Spex 6870 freezer mill with 5 cycles of 1 min grinding at 5 Hz and 1 min cooling. Lysates were centrifuged at 20,000 × g for 30 min at 4 °C to pellet cell debris. 10–54% sucrose density gradients were prepared using the Gradient Master 108 (Biocomp) with gradient buffer (20 mM Tris pH 8.0, 10 mM MgCl2, 100 mM NH4Cl, and 2 mM DTT). 5–40 AU of E. coli lysate was loaded on top of sucrose gradient and centrifuged in a SW41 rotor at 35,000 rpm for 2.5 h at 4 °C. Fractionation was performed on a Piston Gradient Fractionator (Biocomp). To process each fraction for western blots, proteins were precipitated in 10% trichloroacetic acid (TCA) and the pellets were washed twice by ice-cold acetone, vacuum-dried briefly, resuspended in 5x loading dye, and neutralized with Tris-HCl pH 7.5.
Growth assays
Cells were grown overnight at 37 °C in liquid LB. Overnight cultures were diluted 100-fold in fresh LB and were cultured at 37 °C to log phase (OD600 at approximately 0.5). Cells were diluted to prepare 5-fold serial dilutions starting from OD600 = 0.005. 1.5 μL of diluted cultures were spotted on LB plates with or without erythromycin (200 mg/L). Plates were incubated at 37 °C.
Purification of E. coli SmrB and inactive SmrB mutant (99DLH101-ALA).
The plasmids pET24b coding for E. coli SmrB and the E. coli SmrB mutant with N-terminal TwinStrep-tag, TEV cleavage site and FLAG-tag were transformed in E. coli stain BL21 (DE3). Cells were grown in 3 L LB medium to mid-log phase (OD600 = 0.6) at 37 °C and induced with 1 mM IPTG at 18 °C for 20 h. Cells were harvested by centrifugation at 5,471 g and 4 °C for 8 min, resuspended with buffer A (25 mM HEPES/KOH pH 7.8, 300 mM KCl, 5 mM β-Mercaptoethanol, 1:1000 protease inhibitor (pill/mL), 10% glycerol) and lysed using a microfluidizer (Microfluidics M-110L). Cell debris was removed by centrifugation at 30,597 g and 4 °C for 20 min. The cleared lysate was then incubated with 5 mL of prewashed Strep-Tactin XT Superflow beads for 1 h. Afterwards the beads were washed with 10 column volumes (CVs) buffer B (25 mM HEPES/KOH pH 7.8, 1 M KCl, 5 mM β-mercaptoethanol, 10% glycerol), with CVs buffer C (25 mM HEPES/KOH pH 7.8, 300 mM KCl, 5 mM β-mercaptoethanol, 10% glycerol), and eluted with 1 CV Buffer D (25 mM HEPES/KOH pH 7.8, 300 mM KCl, 5 mM β-mercaptoethanol, 10% glycerol, 50 mM biotin).
275 μg of TEV protease were added to the elution fraction and incubated on a rotating wheel at 4 °C overnight. To remove the TEV protease, Dynabeads™ (Invitrogen, Thermo Fisher Scientific) were added and incubated for 30 minutes. The cleaved tag was removed by incubation with Strep-Tactin XT Superflow beads. SmrB was concentrated using an Amicon 10k MWCO and subjected to size-exclusion chromatography using a Superdex 75 in buffer C. SmrB-containing fractions were again concentrated and stored at −80 °C.
E. coli in vitro translation and isolation of disomes and trisomes
The mRNA used in the in vitro translation reaction was prepared as described before by PCR amplification, DNA purification, in vitro transcription and phenol/chloroform precipitation29. The VemP-encoding mRNA contains the VemP peptide without N-terminal signal sequence, a FLAG-tag and a cleavable His-tag. RNCs were generated with the PURExpress In Vitro Protein Synthesis Kit (New England Biolabs #E6800S, transcription and translation coupled) using 21 μg of mRNA per 25 μL reaction. 10 reactions were incubated at 30 °C for 35 min and subsequently loaded on sucrose density gradients (25 mM HEPES/KOH pH 7.5, 100 mM KOAc, 10 mM Mg(OAc)2, 0.01% DDM; 10–50% sucrose) and spun in a SW 40 Ti rotor (Beckman Coulter) at 54,322 g for 16 h at 4 °C. The gradient was fractionated at a BioComp Gradient Station ip using a Triax Flow Cell for UV measurement. The disome and trisomes peak fractions were collected and pelleted by centrifugation in a TLA110 rotor (Beckman Coulter) at 434,513 g for 2 h at 4 °C. After resuspension in RNC buffer (25 mM HEPES/KOH pH 7.5, 150 mM KOAc, 10 mM Mg(OAc)2, 2 mM DTT), samples were frozen in liquid nitrogen and stored at −80 °C.
The twin staller mRNA contains two short ORFs (Met-Lys-Lys-Stop) separated by a linker (Extended Data Fig. 8). The PURExpress In Vitro Protein Synthesis Kit (New England Biolabs #E6800S, transcription and translation coupled) was used to generate RNCs. 30 μg of mRNA were used per 25 μL reaction and additionally tetracycline was added to a final concentration of 125 μg/mL. A 250 μL reaction volume was incubated at 30 °C for 20 minutes. Disomes were separated and concentrated from other ribosomal fractions as described above.
B. subtilis in vitro translation and isolation of disomes
The MifM-encoding mRNA, which contains the MifM leader peptide with shortened C-terminus, a defined stalling site, the MifM N-terminal transmembrane segment (TM), a V5-tag and a cleavable His-tag, was prepared as described before by PCR amplification, DNA purification, in vitro transcription and phenol/chloroform precipitation52. The translation extract was prepared from the Bacillus subtilis strain 168 Δhpf ΔssrA ΔSAS1–253. Cells were grown in LB medium supplemented with 1% glucose, harvested at an OD600 between 0.6 and 0.8 and pelleted by centrifugation at 5,471 g and room temperature for 5 min. Afterwards, cells were resuspended in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH4PO4, pH 7.4), pelleted again by centrifugation at 5,471 g and 4 °C for 15 min and resuspended in as little as possible lysis buffer (10 mM HEPES pH 8.2, 60 mM K glutamate, 14 mM Mg(OAc)2). Cell lysis was performed using a microfluidizer (Microfluidics M-110L) and cell debris was removed by centrifugation at 30,597 g and 4 °C for 20 min. The extract was aliquoted and frozen in liquid nitrogen. Activity of the extract as well as Mg buffer concentration was determined using the Luciferase Assay System (Promega).
The in vitro translation reaction was performed in 4 × 500 μL reaction volume. 640 μL cell extract were mixed with energy buffer (final concentration in 2 mL: 2% PEG 8000, 50 mM HEPES/KOH pH 8.2, 10 mM NH4OAc, 130 mM KOAc, 30 mM Na-pyruvate, 4 mM Na-oxalate, 50 μg/ml tRNA (from E. coli; Sigma 10109541 001), 0.2 mg/ml folinic acid, 0.1 μg/ml creatine kinase, 20 mM creatine phosphate, 4 mM ATP, 3 mM GTP, 0.1 mM amino acid mix, 1 mM DTT, 0.08 U SUPERase•In™ RNase Inhibitor (Invitrogen), 15 mM Mg(OAc)2). After heating the mixture to 32 °C for 2 min, 50 μg of mRNA were added to each aliquot and the in vitro translation was incubated at 32 °C for 40 min while shaking at 900 rpm. For affinity purification of ribosome nascent chain complexes, the in vitro translation was incubated with 400 μL of prewashed TALON metal affinity resin for 45 min on a wheel. The flow-through was collected, beads were washed with 5 CVs buffer A (30 mM HEPES pH7.5/KOH, 250 mM KOAc, 25 mM Mg(OAc)2, 20 mM imidazole, 0.1% DDM) and eluted by incubation for 2 h with 1 CV buffer B (30 mM HEPES pH 7.5/KOH, 250 mM KOAc, 25 mM Mg(OAc)2, 0.1% DDM), 1.1 mg/mL 3C protease). The sample was loaded on a 10–40% sucrose gradient (30 mM HEPES pH 7.5/KOH, 250 mM KOAc, 25 mM Mg(OAc)2, 0.1% DDM, 10–40% (w/v) sucrose) and spun in a SW 40 Ti rotor (Beckman Coulter) at 54,322 g for 16 h at 4°C. The disome peak fractions were combined pelleted by centrifugation in a TLA110 rotor (Beckman Coulter) at 434,513 g for 2 h at 4 °C. The pellet was resuspended in buffer C (25 mM HEPES pH 7.5/KOH, 150 mM KOAc, 10 mM Mg(OAc)2, 2 mM DTT), frozen in liquid nitrogen and stored at −80 °C.
Nuclease Assays
Purified E. coli disomes were mixed with 10 times molar access of protein (SmrB wt or SmrB mut). As a control the same volume of buffer C (25 mM HEPES pH 7.5/KOH, 150 mM KOAc, 10 mM Mg(OAc)2, 2 mM DTT) was added to disomes. Samples were incubated at 30 °C for 3 h and then loaded on sucrose density gradients (25 mM pH 7.5 HEPES-KOH, 100 mM KOAc, 10 mM Mg(OAc)2, 0.01% DDM; 10–50% sucrose). The gradients were spun in a SW 40 Ti rotor (Beckman Coulter) at 54,322 g for 16 h at 4 °C. The gradient was fractionated at a BioComp Gradient Station ip using a Triax Flow Cell for UV measurement.
Purified collided disomes (VemP-stalled disomes) or non-collided disomes (twin staller) were mixed with 10 times molar access of SmrB or the same volume of buffer C (25 mM HEPES-KOH pH 7.5, 150 mM KOAc, 10 mM Mg(OAc)2, 2 mM DTT). All samples were incubated at 30 °C for 1 h and then loaded on sucrose density gradients (25 mM pH 7.5 HEPES-KOH, 100 mM KOAc, 10 mM Mg(OAc)2, 0.01% DDM; 10–50% sucrose). The gradients were spun in a SW 40 Ti rotor (Beckman Coulter) at 202,048 g for 3 h at 4 °C. The gradient was fractionated at a BioComp Gradient Station ip using a Triax Flow Cell for UV measurement. The quantification of the monosome and disome peaks was performed as described54. The relative SmrB activity was calculated as differences in relative disome peak area between reactions without and with SmrB.
Cryo-EM analysis
Data collection and processing of the E. coli disome and E. coli trisome.
A volume of 3.5 μL was applied to 2 nm pre-coated Quantifoil R3/3 holey carbon support grids and vitrified in liquid ethane using a Vitrobot mark IV (FEI Company, Netherlands) (wait time 45 s, blotting time 2 s). 2,437 and 14,849 movies were collected for the E. coli disome and trisome sample, respectively. Data were collected on a Titan Krios TEM using a Falcon II DED at 300 kV, with an electron dose of 2.5 e−/Å2 per frame for 16 frames (defocus range of 0.5 to 4 μm). The magnified pixel size was 1.09 Å/pixel. For the E. coli disome sample, frames were gain corrected, aligned and summed using MotionCor255 and CTF parameters were determined using CTFFIND56. Particles were picked using Gautomatch v0.56 (http://www.mrc-lmb.cam.ac.uk/kzhang/). The particles were extracted and processed following the standard workflow in RELION 3.157. The particles containing the stalled ribosome of the disomes were extracted with a box size of 380 pixel, imported to Cryosparc v3.2.058 and refined to a final resolution of 3.8 Å. After extension of the box and refinement of the full disome, the map was used to create templates for particle picking. 2D classes of disome particles were selected and then classified and refined to a final resolution of 4.3 Å (Extended Data Fig. 5). For the E. coli trisome, data were processed following the standard workflow in cryoSPARC v3.2.058 (Extended Data Fig. 7).
Data collection and processing of the B. subtilis disome
All samples were vitrified as described above. Two datasets of 8,842 and 19,354 movies were collected on a Titan Krios TEM using a Falcon II DED at 300 kV, with an electron dose of 2.5 e−/Å2 per frame for 16 frames (defocus range of 0.5 to 4 μm). The magnified pixel size was 1.084 Å/pixel. All frames were gain corrected and subsequently aligned and summed using MotionCor255. The data were processed following the standard workflow in cryoSPARC v3.2.058 (Extended Data Fig. 7).
Sample preparation, data collection and processing of the E. coli disome SmrB complex.
For reconstitution, disomes and SmrB mutant (99DLH101-ALA) were thawed on ice. Disomes were mixed with 10 times molar access of protein, incubated for 10 min at room temperature and subsequently analysed by cryo-EM. 8,350 movies were collected on a Titan Krios at 300 kV recorded on a K2 Summit direct electron detector (DED) with an electron dose of approx. 1.06 e−/Å2 per frame for 40 frames (defocus range of 0.5 to 3.5 μm). The magnified pixel size was 1.059 Å/pixel. All frames were gain corrected and subsequently aligned and summed using MotionCor255. The data were processed following the standard workflow in cryoSPARC v3.2.058. The processing scheme and final local resolution for SmrB are shown in Extended Data Fig. 9.
Model building and refinement
The E. coli disome model was prepared by rigid body docking of the model from Su et al. (PDB code 5NWY29). uL9 was taken from PDB-6WD159 and the N-terminal part (residues 1–52) and the C-terminal part (residues 53–149) were rigid body docked individually and rejoined to match the bridged conformation of uL9 in the stalled ribosome. uL1 and the L1-stalk rRNA (nucleotides 2099–2190) were taken from PDB-6WD1 and rigid body docked into the cryo-EM density. tRNA-Phe was taken from PDB-3L0U60 and rigid body docked into the A-sites of stalled and collided ribosomes although the identity of the tRNA in the A-site of the collided could not be determined. uS1 was taken from PDB-6BU861 and docked into the density map of the collided ribosome. The resolution of the mRNA between the two ribosomes was insufficient for modeling with nucleotide precision. Despite that, the mRNA of PDB-5NWY was extended at the 5’-end according to the sequence of the construct to provide an approximation of the number of nucleotides stretching from the exit of the stalled to the entry of the collided ribosome. The SmrB model was prepared using Alphafold 2 (AF2)35 predictions and Mmseqs262 for multiple sequence alignment of SmrB alone and of SmrB fused to the ribosomal interaction partner uS2 as described in Extended Data Fig. 10. AF2 predictions were performed using an API provided by the Söding lab. During preparation of this manuscript a model prediction for SmrB became available at the alphafold database at EMBL-EBI. It is in high agreement with our final adjusted model (Extended Data Fig. 10).
The E. coli trisome model was prepared by docking the two copies of the stalled 70S from the disome in the first and second ribosome and one copy of the collided 70S in the third ribosome. The B. subtilis disome model was prepared by docking two copies of the MifM-stalled ribosome complex (PDB 3J9W52) into the cryo-EM map.
All model adjustments were performed using coot v0.963. Given the comparably low resolution of the cryo-EM maps, our structural models should not be interpreted at the side-chain level. The models have the purpose of providing insights into the architecture of bacterial disomes and how SmrB is positioned at the composite interface of the disome to facilitate mRNA cleavage. For interpreting the atomic details of this process, higher resolution data is required. Structural figures were prepared using ChimeraX v1.364.
Extended Data
Extended Data Fig. 1:
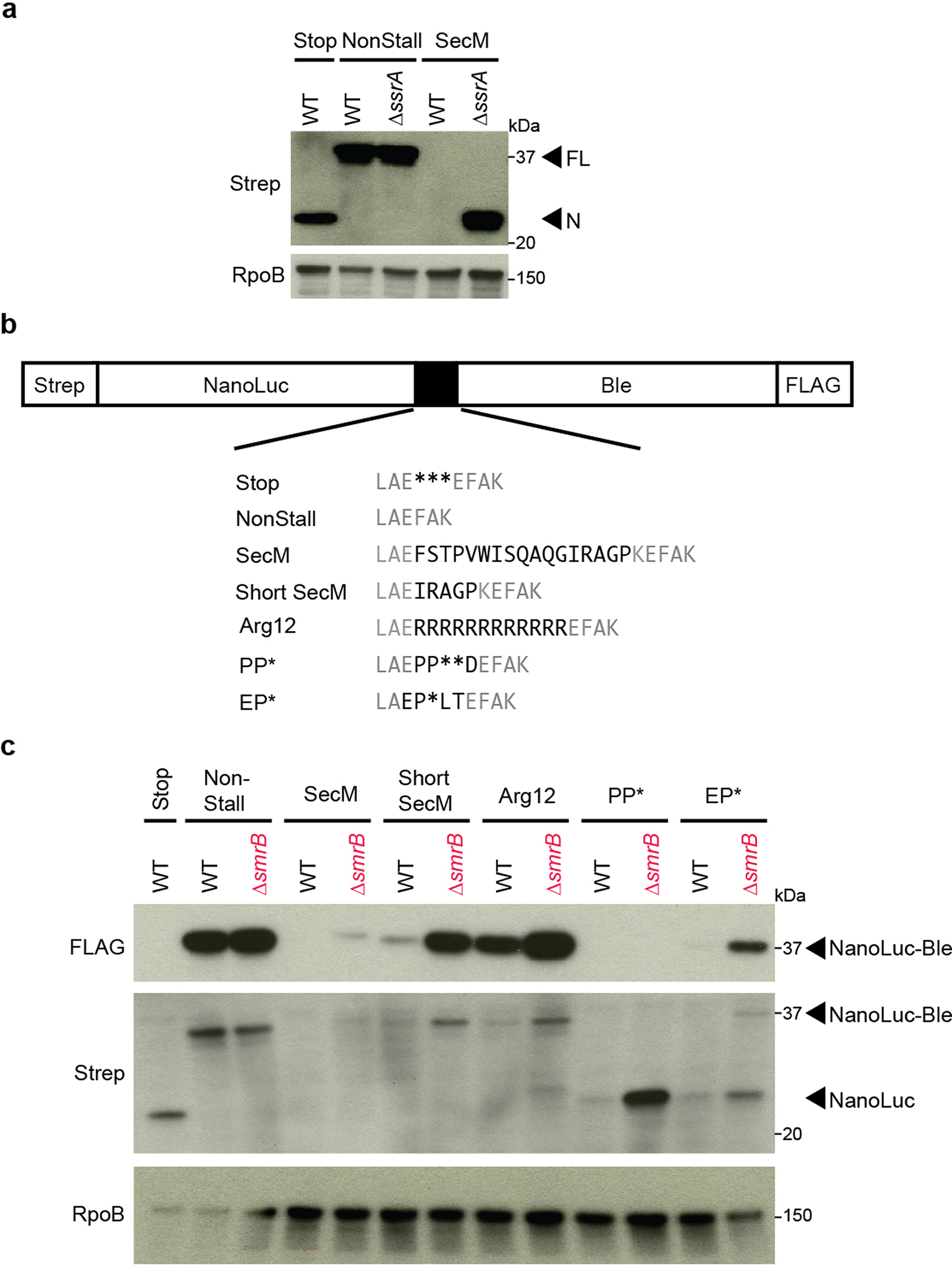
SmrB is a general ribosome rescue factor. a, Reporter protein from wild-type and ΔssrA strains was detected by antibodies against the N-terminal Strep-tag. Arrows indicate the full-length fusion protein (FL) and shorter NanoLuc protein (N). The RpoB protein serves as a loading control. b, Additional reporters to study ribosome rescue in E. coli with various stalling motifs. c, The expression of full-length NanoLuc-Ble protein was monitored with an anti-FLAG antibody; anti-Strep antibodies reveal both full-length NanoLuc-Ble and truncated NanoLuc protein. RpoB serves as a loading control.
Extended Data Fig. 2:

Phylogenetic tree of SMR domain proteins. Stylized phylogenetic tree depicting relationships between SMR domain clades. Clades with indicated bootstrap support are marked with circles. Clade names are given to the right of the tree. Dotted lines indicate positions with little or no bootstrap support.
Extended Data Fig. 3:
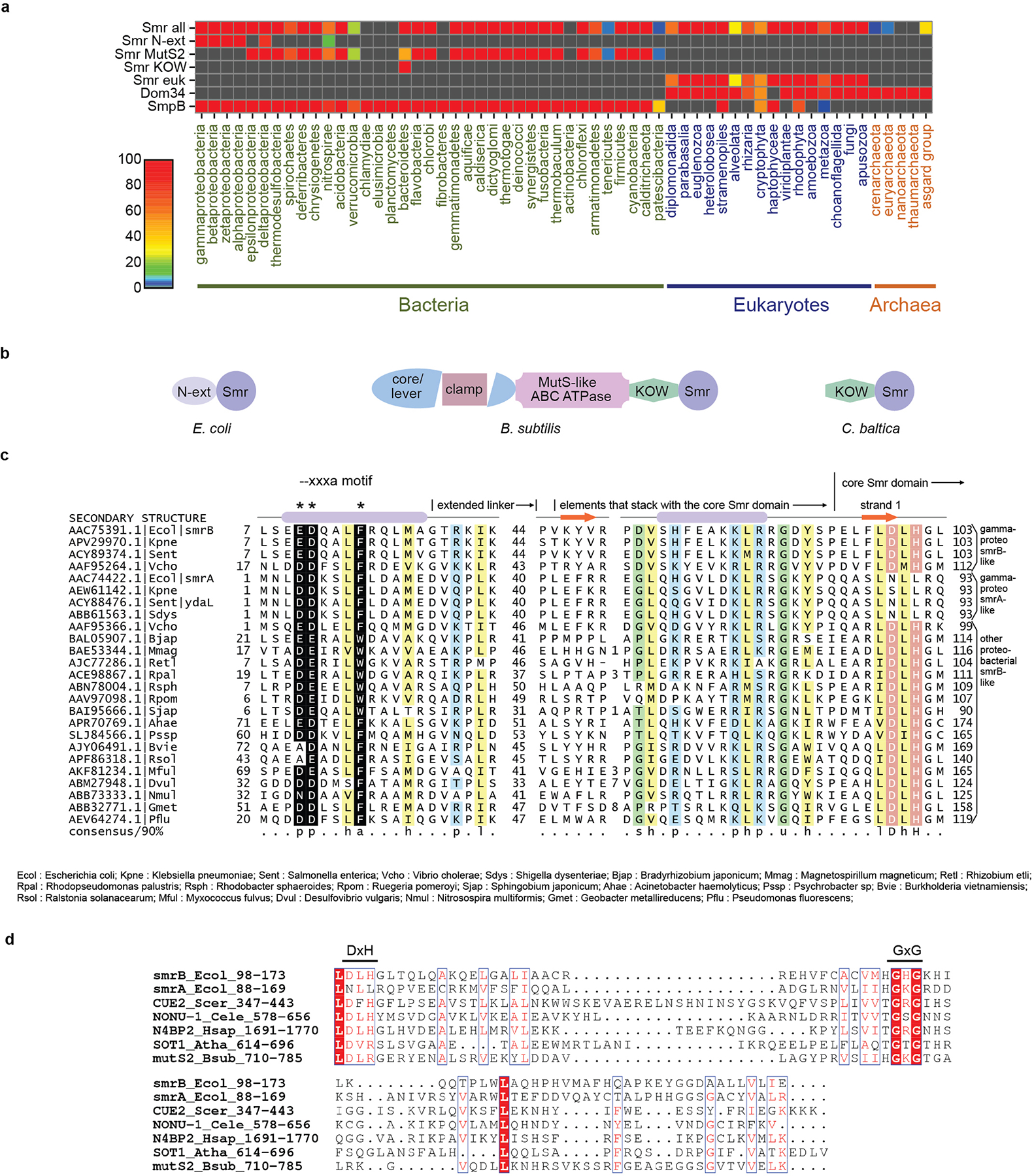
Distribution and architectures of SMR-domain proteins. a, Heat map demonstrating the conservation and distribution of SMR-domain proteins and other related translational quality control factors. Smr-all includes all types of SMR-domain proteins; Smr-euk includes only the eukaryotic branch. b, Domain organization of three representative bacterial proteins containing an SMR domain. c, Multiple alignment of the conserved regions in the N-terminal extension of SMR proteins from proteobacteria. Columns in the alignment are shaded and labeled according to biochemical character: -, negatively charged; h, hydrophobic in yellow; a, aromatic; p, polar in blue; l, aliphatic in yellow; s, small in green; u, tiny in green. Residue positions in the --xxxa motif are colored in white and shaded in black, marked by asterisks above the alignment. Residue positions forming part of the active site of the core SMR domain are colored in white and shaded in red. Sequences are labeled with NCBI accession number and organism abbreviation; abbreviations are provided below alignment. Secondary structure provided at top of alignment. Numbers to left and right of alignment denote positioning of the region. Internal numbers give the size of excised variable insert regions. d, Sequence alignment of SMR domains of representative proteins. Identical residues are shown in white with a red background; conserved residues are shown in red. The identity of each sequence is represented by the gene name, species name, and numbers indicating the beginning and the end of the residues used for the alignment. Ecol, Escherichia coli; Scer, Saccharomyces cerevisiae; Cele, Caenorhabditis elegans; Hsap, Homo sapiens; Atha, Arabidopsis thaliana; Bsub, Bacillus subtilis.
Extended Data Fig. 4:
SmrB cleavage, tmRNA tagging, and ribosome collisions. a, The results of 5’-RACE showing the 5’-ends of downstream fragments in reads per million on the EP* reporter. The first nt in the A site codon in the stall motif is designated as zero. b, The results of 3’-RACE showing the 3’-ends of upstream fragments. c, tmRNA tagging sites on the EP* reporter in the wild-type and ΔsmrB strains, corresponding to the residue immediately preceding the tmRNA tag in peptide sequences detected by targeted LC-MS-MS. The relative spectrum count is normalized by the count at the EP* stall site (red) where tmRNA tagging was expected to occur in both the wild-type and ΔsmrB strains. The spectrum count corresponds to the mean and the standard deviation of three replicates. The arrow indicates the SmrB cleavage site demonstrated by 5’-RACE. d, 5’-RACE data on the Short SecM reporter reveal the SmrB cleavage sites as in Fig. 2b, zoomed in to show smaller peaks upstream. e, The distribution of FLAG-SmrB in cells treated with 5, 50, or 500 μg/mL erythromycin (ERY) was determined by fractionation over a sucrose gradient and detected with an anti-FLAG antibody.
Extended Data Fig. 5:

Cryo-EM data processing for the E. coli disome sample. Shown are the classification scheme, representative micrographs (the scale bar is 500 Å), 2D class averages and the Gold standard Fourier Shell Correlation (GSFSC) curve for the volume containing the 70S stalled ribosome and the 30S of the collided ribosome, as well as the full disome.
Extended Data Fig. 6:

Analysis of the E. coli disome structure and comparison of different disome structures. a, The architecture of the E. coli disome is not compatible with bS1 remaining bound to the stalled ribosome. Aligned models of the 30S subunits of the collided (left) and the stalled (middle) ribosomes are shown in surface representation. The position of bS1 as observed in the collided ribosome is shown in purple and the same position of bS1 in the stalled ribosome is indicated by a dashed line. The clash between bS1 of the stalled ribosome and the 30S subunit of the collided ribosome that would occur upon disome formation is shown on the right. b, Cartoon representation of the individual interactions as they occur at the E. coli disome interface. c, 2D class averages and cryo-EM structure model of an E. coli trisome. d&e, Comparison of the E. coli (E.c.) and B. subtilis (B.s.) disomes displaying full and cut views. Note the smaller space between stalled and collided ribosomes in the B.s. disome interface as illustrated by comparing the positions of uS2 proteins in the zoomed view in c. f, Surface representation of the structural model of the S. cerevisiae disome. g&h, Surface representation of the E. coli and B. subtilis hibernation disomes.
Extended Data Fig. 7:

Cryo-EM data processing for the B. subtilis disome and E. coli trisome sample. a, Shown are the classification scheme, and the Gold standard Fourier Shell Correlation (GSFSC) curves for the final volumes of the B. subtilis disome containing the 70S stalled ribosome and the 70S of the collided ribosome. b, Shown are the 2D class averages, classification scheme, and the Gold standard Fourier Shell Correlation (GSFSC) of the E. coli trisome.
Extended Data Fig. 8:

Production of collided and non-collided disomes and relative peak areas of monosomes and disomes in the SmrB nuclease assay. a, mRNA construct to create the collided E. coli disomes and trisomes and below the sucrose density gradient after in vitro translation. The ribosome stalling site is indicated by an asterisk. b, mRNA construct to create the non-collided disomes that were used in the nuclease assay and below the sucrose density gradient after in vitro translation. c, Relative monosome and disome peak area calculated from the sucrose gradient profiles of the SmrB nuclease assay, showing the mean and standard deviation of three replicates. d, The relative decrease of the area of the disome peak upon addition of SmrB is shown as the mean and standard deviation of three replicates. (The mean difference of the relative disome peak area of collided ribosomes between control and SmrB reaction was set to 1).
Extended Data Fig. 9:

Cryo-EM data processing for the E. coli disome sample. Shown are the classification scheme, representative micrographs (the scale bar is 500 Å), 2D class averages and the Gold standard Fourier Shell Correlation (GSFSC) curve for the respective 3D reconstructions. The segmented density for SmrB is colored according to local resolution.
Extended Data Fig. 10:
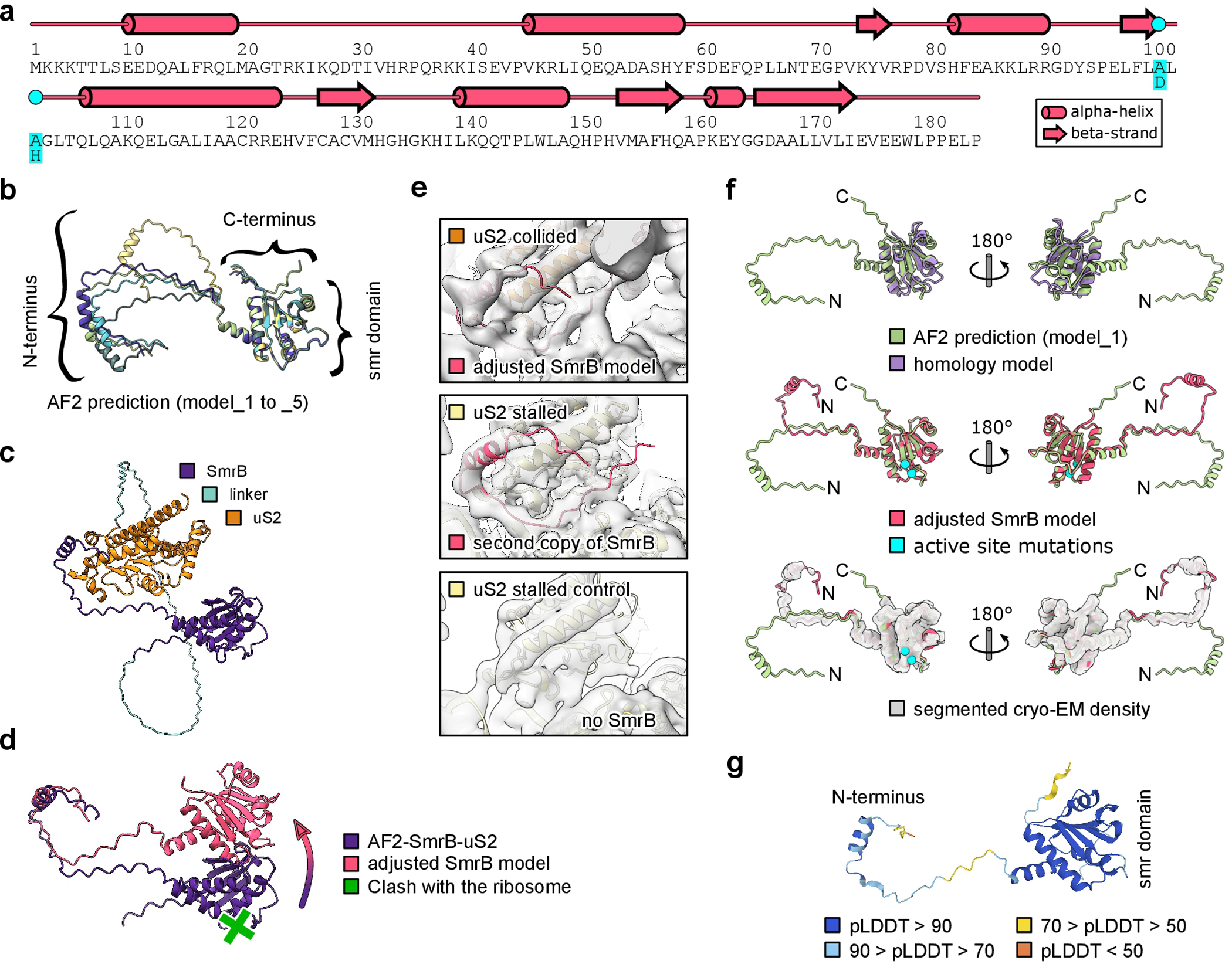
Structural model of SmrB. a, Secondary structure of SmrB. The DLH to ALA mutation is indicated. b, AF2 prediction models 1–5 as predicted through the API from the Söding lab. The SMR domain is predicted with high confidence, while the linker to the N-terminal helix appears flexible. c, AF2 prediction of the interaction between SmrB and uS2. For this prediction uS2 was fused to the C-terminus of SmrB with a glycine serine linker (39 copies of GS). The prediction shows the N-terminal helix of SmrB folded back onto uS2. d, Adjustment of the AF2 predicted model of SmrB-uS2. Without adjustment according to the cryo-EM density (as shown in D) the SMR domain would clash with the ribosome. e, Top: Cryo-EM density and adjusted model of the SmrB. Middle: Cryo-EM density and rigid body docked model of the N-terminus of SmrB from the collided 30S onto the stalled 30S. A second copy of SmrB was found anchored to uS2 of the stalled ribosome. However, there was no density for the SMR domain of the second SmrB, indicating a high degree of flexibility due to the lack of another ribosome in front of the stalled one. Bottom: in the control disome without SmrB, there is no density for the N-terminus of SmrB. f, Comparison of the AF2 prediction, the homology model, and the adjusted model of SmrB. Compared to the AF2 prediction, the homology model is missing the two N-terminal helices and most of the loops are slightly different (top). The AF2 prediction almost perfectly matched the cryo-EM density map and the corresponding adjusted model (middle and bottom). Only the catalytic loop (carrying the active site mutations) had to be slightly adjusted to prevent clashes with the mRNA. The N-terminus was adjusted as discussed above. g. During the preparation of this manuscript the AF2 prediction for SmrB (YfcN) became available at the alphafold database at EMBL-EBI. The deposited model resembles our final adjusted model very well including the position of the N-terminus. The confidence of the prediction (pLDDT) is indicated.
Extended Data Fig. 11:

Testing the importance of structural interactions for SmrB activity. a, Examples of operons containing both uS21 and SMR-domain proteins. b, The distribution of FLAG-tagged full-length SmrB and a construct with only the SMR domain (residues 88–183) was determined by fractionation over sucrose gradient and detection with an anti-FLAG antibody. A non-specific band is marked with *. c, Northern blots using the 3’-probe against the CRP reporters with the short SecM stalling motif in wild-type cells, bL9-deletion strain (ΔrplI), and a strain where mCherry is fused to the C-terminus of bL9 (bL9-mCherry). Ethidium bromide staining of 16S rRNA serves as a loading control. d, Northern blots using the 3’-probe against the CRP reporters with the short SecM stalling motif in wild-type cells, a strain where MBP is fused to the N-terminus of uS21, and a strain where GFP is fused to the C-terminus of uS6.
Extended Data Table 1:
Data collection and refinement statistics.
| E. coli disome-SmrB stalled 70S; validation against focus refined map (EMDB-13956) (PDB 7QGN) | E. coli disome-SmrB collided 70S with SmrB and mRNA; validation against focus refined map (EMDB-13958) (PDB 7QGR) | E. coli disome stalled 70S ribosome; validation against focus refined map (EMDB-13952) (PDB 7QG8) | E. coli disome collided 70S ribosome; validation against focus refined map (EMDB-13955) (PDB 7QGH) | E. coli tri some (EMDB-13964) | B. subtilis disome stalled 70S ribosome (EMDB-13959) (PDB 7QGU) | B. subtilis disome collided 70S ribosome (EMDB-13961) (PDB 7QH4) | |
|---|---|---|---|---|---|---|---|
| Data collection and processing | |||||||
| Magnification | 130,000 | 130,000 | 75,000 | 75,000 | 75.000 | 75,000 | 75.000 |
| Voltage (kV) | 300 | 300 | 300 | 300 | 300 | 300 | 300 |
| Electron exposure (e-/Å2) | 42.4 | 42.4 | 40 | 40 | 40 | 25 | 25 |
| Defocus range (μm) | 0.5–3.5 | 0.5–3.5 | 0.5–4 | 0.5–4 | 0.5–4 | 0.5–4 | 0.5–4 |
| Pixel size (Å) | 1.059 | 1.059 | 1.09 | 1.09 | 1.09 | 1.084 | 1.084 |
| Symmetry imposed | C1 | C1 | C1 | C1 | C1 | C1 | C1 |
| Initial particle images (no.) | 651,789 | 651,789 | 238,082 | 238,082 | 448,847 | 2,761,261 | 2,761,261 |
| Final particle images (no.) | 32,412 | 32,412 | 75,081 | 75,081 | 45,952 | 12,739 | 12,739 |
| Map resolution (Å) | 3.37 | 3.78 | 3.97 | 4.48 | 13.29 | 4.75 | 5.45 |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 |
| Map resolution range (Å) | 2.9–17 | 3.3–18.6 | 3.5–14 | 3.7–15.6 | 4.4–25.7 | 4.8–21.6 | |
| Refinement | |||||||
| Initial model used | 5NWY | 5NWY | 5NWY | 5NWY | 3J9W | 3J9W | |
| (PDB code) | 6WD1 (bL9, L1 stalk) | 6WD1 (bL9, L1 stalk) | 6WD1 (bL9, L1 stalk) | 6WD1 (bL9, L1 stalk) | |||
| 3L0U (tRNA) | 3L0U (tRNA) | 3L0U (tRNA) | 3L0U (tRNA) | ||||
| Model resolution (Å) | 3.4 | 4.3 | 4.9 | 5.0 | 4.5 | 5.1 | |
| FSC threshold | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | 0.143 | |
| Map sharpening B factor (Å2) | −53.6 | −63.0 | −145.2 | −167.3 | −158.1 | −198.7 | |
| Model composition | |||||||
| Non-hydrogen atoms | 147,306 | 149,850 | 147,112 | 147,973 | 136,024 | 136,043 | |
| Protein residues | 5,975 | 6,223 | 5,936 | 6,046 | 5,791 | 5,573 | |
| Nucleotide residues | 4,695 | 4,746 | 4,695 | 4,716 | 4,656 | 4,733 | |
| R.m.s. deviations | |||||||
| Bond lengths (Å) | 0.016 | 0.016 | 0.016 | 0.016 | 0.009 | 0.009 | |
| Bond angles (°) | 1.647 | 1.641 | 1.648 | 1.642 | 1.462 | 1.452 | |
| Validation | |||||||
| MolProbity score | 1.95 | 1.96 | 1.95 | 1.95 | 2.14 | 2.07 | |
| Clashscore | 4.89 | 5.83 | 4.85 | 5.43 | 12.74 | 11.82 | |
| Poor rotamers (%) | 1.61 | 1.38 | 1.61 | 1.41 | 0.53 | 0.19 | |
| Ramachandran plot | |||||||
| Favored (%) | 90.40 | 90.33 | 90.35 | 90.20 | 91.02 | 92.01 | |
| Allowed (%) | 8.60 | 8.66 | 8.63 | 8.79 | 6.64 | 6.06 | |
| Disallowed (%) | 1.01 | 1.01 | 1.01 | 1.01 | 2.34 | 1.93 | |
| Map vs. Model CC | 0.23 | 0.25 | 0.45 | 0.45 | 0.63 | 0.49 |
Supplementary Material
Acknowledgements:
The authors thank Suparna Sanyal for sharing E. coli strains QC101 and QC901, H. Hao at the JHMI Transcriptomics and Deep Sequencing Core for assistance with high-throughput sequencing, B. Cole and T. Boronina at JHMI in the Mass Spectrometry and Proteomics Facility, J. Musial for assistance during protein purifications, T. Mackens-Kiani for helping with the nuclease assay data analysis, C. Ungewickell and S. Rieder for technical assistance, and L. Kater and K. Best for support with the pre-processing pipeline of cryo-EM data. This work was supported by NIH grant GM136960 (ARB), HHMI (RG), the Intramural Research Program of the National Library of Medicine at the NIH (AMB and LA), and by the German Research Council (TRR174) (RB). HK is supported by a DFG fellowship through the Graduate School of Quantitative Bioscience Munich (QBM).
Footnotes
Code availability: Custom python scripts used to analyse the Tn-seq and RACE data are freely available at https://github.com/greenlabjhmi/2021_SmrB.
Competing Interests: The authors declare that there are no competing interests.
Supplementary Information is available for this paper.
Data availability:
Cryo-EM volumes and molecular models have been deposited at the Electron Microscopy Data Bank and Protein Data Bank with the accession codes for the E. coli disome EMDB-13952 & 7QG8 (stalled 70S) and EMDB-13955 & 7QGH (collided 70S); the E. coli trisome EMDB-13964; the B. subtilis disome EMDB-13959 & 7QGU (stalled 70S) and EMDB-13961 & 7QH4 (collided 70S); and the E. coli disome SmrB complex EMDB-13956 & 7QGN (stalled 70S) and EMDB-13958 & 7QGR (collided 70S). Gel source images are provided in Supplementary Figures 1 and 2.
References:
- 1.Müller C, Crowe-McAuliffe C & Wilson DN Ribosome Rescue Pathways in Bacteria. Front. Microbiol. 12, 652980 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Keiler KC, Waller PR & Sauer RT Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science 271, 990–993 (1996). [DOI] [PubMed] [Google Scholar]
- 3.Ivanova N, Pavlov MY, Felden B & Ehrenberg M Ribosome rescue by tmRNA requires truncated mRNAs. J. Mol. Biol. 338, 33–41 (2004). [DOI] [PubMed] [Google Scholar]
- 4.Hayes CS & Sauer RT Cleavage of the A site mRNA codon during ribosome pausing provides a mechanism for translational quality control. Mol. Cell 12, 903–911 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Subramaniam AR, Zid BM & O’Shea EK An integrated approach reveals regulatory controls on bacterial translation elongation. Cell 159, 1200–1211 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janssen BD, Garza-Sánchez F & Hayes CS A-site mRNA cleavage is not required for tmRNA-mediated ssrA-peptide tagging. PLoS One 8, e81319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moore SD & Sauer RT Ribosome rescue: tmRNA tagging activity and capacity in Escherichia coli. Mol. Microbiol. 58, 456–466 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Yan LL & Zaher HS How do cells cope with RNA damage and its consequences? J. Biol. Chem. 294, 15158–15171 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomas EN, Kim KQ, McHugh EP, Marcinkiewicz T & Zaher HS Alkylative damage of mRNA leads to ribosome stalling and rescue by trans translation in bacteria. eLife 9, e61984 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roche ED & Sauer RT SsrA-mediated peptide tagging caused by rare codons and tRNA scarcity. EMBO J. 18, 4579–4589 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sunohara T, Jojima K, Tagami H, Inada T & Aiba H Ribosome stalling during translation elongation induces cleavage of mRNA being translated in Escherichia coli. J. Biol. Chem. 279, 15368–15375 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Hayes CS, Bose B & Sauer RT Proline residues at the C terminus of nascent chains induce SsrA tagging during translation termination. J. Biol. Chem. 277, 33825–33832 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Neubauer C, Gillet R, Kelley AC & Ramakrishnan V Decoding in the absence of a codon by tmRNA and SmpB in the ribosome. Science 335, 1366–1369 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikeuchi K et al. Collided ribosomes form a unique structural interface to induce Hel2-driven quality control pathways. EMBO J. 38, e100276 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Juszkiewicz S et al. ZNF598 Is a Quality Control Sensor of Collided Ribosomes. Mol. Cell 72, 469–481 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsuo Y et al. Ubiquitination of stalled ribosome triggers ribosome-associated quality control. Nat. Commun. 8, 159 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simms CL, Yan LL & Zaher HS Ribosome Collision Is Critical for Quality Control during No-Go Decay. Mol. Cell 68, 361–373 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Langridge GC et al. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Res. 19, 2308–2316 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kannan K et al. The general mode of translation inhibition by macrolide antibiotics. Proc. Natl. Acad. Sci. U.S.A. 111, 15958–15963 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beckert B et al. Structural and mechanistic basis for translation inhibition by macrolide and ketolide antibiotics. Nat. Commun. 12, 4466 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou W et al. PPR-SMR protein SOT1 has RNA endonuclease activity. Proc. Natl. Acad. Sci. U.S.A. 114, E1554–E1563 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu WJ et al. SOT1, a pentatricopeptide repeat protein with a small MutS-related domain, is required for correct processing of plastid 23S-4.5S rRNA precursors in Arabidopsis thaliana. Plant J. 85, 607–621 (2016). [DOI] [PubMed] [Google Scholar]
- 23.D’Orazio KN et al. The endonuclease Cue2 cleaves mRNAs at stalled ribosomes during No Go Decay. eLife 8, e49117 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu S, Melonek J, Boykin LM, Small I & Howell KA PPR-SMRs: ancient proteins with enigmatic functions. RNA Biol. 10, 1501–1510 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glover ML et al. NONU-1 Encodes a Conserved Endonuclease Required for mRNA Translation Surveillance. Cell Rep. 30, 4321–4331 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohammad F, Green R & Buskirk AR A systematically-revised ribosome profiling method for bacteria reveals pauses at single-codon resolution. eLife 8, e42591 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chiba S, Lamsa A & Pogliano K A ribosome-nascent chain sensor of membrane protein biogenesis in Bacillus subtilis. EMBO J. 28, 3461–3475 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishii E et al. Nascent chain-monitored remodeling of the Sec machinery for salinity adaptation of marine bacteria. Proc. Natl. Acad. Sci. U.S.A. 112, E5513–E5522 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su T et al. The force-sensing peptide VemP employs extreme compaction and secondary structure formation to induce ribosomal stalling. eLife 6, e25642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuwirth BS et al. Structures of the bacterial ribosome at 3.5 Å resolution. Science 310, 827–834 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Selmer M, Gao YG, Weixlbaumer A & Ramakrishnan V Ribosome engineering to promote new crystal forms. Acta Crystallogr. D 68, 578–583 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Atkins JF & Bjork GR A Gripping Tale of Ribosomal Frameshifting: Extragenic Suppressors of Frameshift Mutations Spotlight P-Site Realignment. Microbiol. Mol. Biol. R. 73, 178 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Beckert B et al. Structure of the Bacillus subtilis hibernating 100S ribosome reveals the basis for 70S dimerization. EMBO J. 36, 2061–2072 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beckert B et al. Structure of a hibernating 100S ribosome reveals an inactive conformation of the ribosomal protein S1. Nat. Microbiol 3, 1115 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferrin MA & Subramaniam AR Kinetic modeling predicts a stimulatory role for ribosome collisions at elongation stall sites in bacteria. eLife 6, e23629 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simms CL, Yan LL, Qiu JK & Zaher HS Ribosome Collisions Result in +1 Frameshifting in the Absence of No-Go Decay. Cell Rep. 28, 1679–1689 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith AM, Costello MS, Kettring AH, Wingo RJ & Moore SD Ribosome collisions alter frameshifting at translational reprogramming motifs in bacterial mRNAs. Proc. Natl. Acad. Sci. U.S.A. 116, 21769–21779 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chai Q et al. Organization of Ribosomes and Nucleoids in Escherichia coli Cells during Growth and in Quiescence. J. Biol. Chem. 289, 11342–11352 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Datsenko KA & Wanner BL One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crameri A, Whitehorn EA, Tate E & Stemmer WPC Improved green fluorescent protein by molecular evolution using DNA shuffling. Nat. Biotechnol. 14, 315–319 (1996). [DOI] [PubMed] [Google Scholar]
- 42.Jiang HS, Lei R, Ding SW & Zhu SF Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 15, 182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011). [Google Scholar]
- 44.Langmead B, Trapnell C, Pop M & Salzberg SL Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Altschul SF et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eddy SR A new generation of homology search tools based on probabilistic inference. Genome Inform. 23, 205–211 (2009). [PubMed] [Google Scholar]
- 47.Finn RD et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lassmann T, Frings O & Sonnhammer ELL Kalign2: high-performance multiple alignment of protein and nucleotide sequences allowing external features. Nucleic Acids Res. 37, 858–865 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cole C, Barber JD & Barton GJ The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36, W197–W201 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Price MN, Dehal PS & Arkin AP FastTree 2-Approximately Maximum-Likelihood Trees for Large Alignments. PLoS One 5, e9490 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shevchenko A, Wilm M, Vorm O & Mann M Mass spectrometric sequencing of proteins from silver stained polyacrylamide gels. Anal. Chem. 68, 850–858 (1996). [DOI] [PubMed] [Google Scholar]
- 52.Sohmen D et al. Structure of the Bacillus subtilis 70S ribosome reveals the basis for species-specific stalling. Nat. Commun. 6, 6941 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schafer H et al. The alarmones (p)ppGpp are part of the heat shock response of Bacillus subtilis. PLoS Genet. 16, e1008275 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wells JN et al. Structure and function of yeast Lso2 and human CCDC124 bound to hibernating ribosomes. PLoS Biol. 18, e3000780 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rohou A & Grigorieff N CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zivanov J, Nakane T & Scheres SHW Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 7, 253–267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Loveland AB, Demo G & Korostelev AA Cryo-EM of elongating ribosome with EFTu-GTP elucidates tRNA proofreading. Nature 584, 640–645 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Byrne RT, Konevega AL, Rodnina MV & Antson AA The crystal structure of unmodified tRNA(Phe) from Escherichia coli. Nucleic Acids Res. 38, 4154–4162 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Loveland AB & Korostelev AA Structural dynamics of protein S1 on the 70S ribosome visualized by ensemble cryo-EM. Methods 137, 55–66 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mirdita M, Steinegger M & Soding J MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics 35, 2856–2858 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Emsley P & Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 64.Goddard TD et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM volumes and molecular models have been deposited at the Electron Microscopy Data Bank and Protein Data Bank with the accession codes for the E. coli disome EMDB-13952 & 7QG8 (stalled 70S) and EMDB-13955 & 7QGH (collided 70S); the E. coli trisome EMDB-13964; the B. subtilis disome EMDB-13959 & 7QGU (stalled 70S) and EMDB-13961 & 7QH4 (collided 70S); and the E. coli disome SmrB complex EMDB-13956 & 7QGN (stalled 70S) and EMDB-13958 & 7QGR (collided 70S). Gel source images are provided in Supplementary Figures 1 and 2.