

Abstract
Background & Aims:
Cirrhosis is a deadly liver disease; fibrosis being a key feature. Due to the lack of genetic animal models exhibiting clinical characteristics, molecular pathogenesis of cirrhosis has been so far poorly characterized, and treatments remain limited.
Methods:
We report the first murine genetic model mimicking human cirrhosis induced by hepatocyte-specific elimination of microspherule 1 (MCRS1), a member of non-specific lethal (NSL) and INO80 chromatin-modifier complexes. Using this genetic tool with other mouse models, cell culture and human samples, combined with quantitative proteomic, single nuclear/cell RNA sequencing and chromatin immunoprecipitation assays, mechanisms of cirrhosis are investigated.
Results:
MCRS1 loss in mouse hepatocytes modulates the expression of bile acid (BA) transporters, with a pronounced downregulation of Na+-taurocholate cotransporting polypeptide (NTCP), concentrating BAs in sinusoids, thereby activating hepatic stellate cells (HSCs) via nuclear farnesoid X receptor (FXR); FXR being predominantly expressed in human and mouse HSCs. Consistently, re-expression of NTCP in mice reduces cirrhosis, and genetic ablation of FXR in HSCs suppresses fibrotic marks in mice and in vitro cell culture. Mechanistically, deletion of a putative SANT domain from MCRS1 evicts histone deacetylase 1 (HDAC1) from its histone H3 anchoring sites, increasing histone acetylation of BA transporter genes, modulating their expression and perturbing BA flow. Accordingly, human cirrhosis displays decreased nuclear MCRS1 and NTCP expression.
Conclusions:
Our data reveal a previously unrecognized function of MCRS1 as a critical histone acetylation regulator, maintaining gene expression and liver homeostasis. MCRS1 loss induces BA transporter acetylation, perturbation of BA flow, and consequently, FXR activation in HSCs. This axis represents a central and universal signaling event in liver cirrhosis, and targeting it would have significant implications for cirrhosis treatment.
Keywords: MCRS1, NTCP, Bile acid transporter, Bile acids, FXR, Fibroblasts, Hepatic stellate cells, Histone acetylation, Liver fibrosis, Cirrhosis
Lay summary
By genetic ablation of MCRS1 in mouse hepatocytes, Garrido et al. generate the first genetic mouse model of liver cirrhosis, recapitulating human features. Garrido et al. demonstrate that the activation of the bile acid/FXR axis in liver fibroblasts is key in liver cirrhosis development.
Introduction
Cirrhosis is a chronic liver disease characterized by disrupted liver architecture and zonation, along with neovascularization, and the formation of fibrotic bands and parenchymal nodules, severely impairing liver function1. Cirrhosis has a high risk of mortality and poor prognosis, with complications such as hepatic encephalopathy, a neuropsychiatric disorder caused by ammonia (NH3) accumulation that eventually leads to coma1. Cellular mechanisms of liver fibrosis imply the transdifferentiation of HSCs into myofibroblasts that synthesize the extracellular matrix (ECM) to form fibrotic bands1. Although research over the last 20 years has increased our understanding of liver fibrosis, effective antifibrotic therapies are still lacking, most likely because mechanisms of HSC activation remain elusive. Murine genetic models mimicking the human disease are needed to study the processes underlying fibrogenesis, identify potential therapeutic targets, and evaluate the effectiveness of antifibrotic therapies.
Cirrhosis is mostly caused by hepatitis B and C viral infection, alcohol abuse or obesity1. Less frequently, cirrhosis can originate from genetic disorders2,3 or autoimmune liver diseases that provoke the disruption of bile ducts causing BA leakage4. This suggests a causal relationship between perturbation of BA flow and cirrhosis development. Primary BAs are synthesized in hepatocytes via cholesterol oxidation, mainly catalyzed by cholesterol 7 alpha-hydroxylase (CYP7A1). Then, they are conjugated with glycine or taurine before excretion to the gallbladder by the bile salt export pump (BSEP)5,6. Other bile components are excreted via multidrug resistance-associated protein 2/3 (human MDR3, mouse MDR2). BAs are eventually secreted into the gut to facilitate digestion of dietary lipids, where secondary BAs are formed by partial dehydroxylation catalyzed by gut bacteria. Primary and secondary BAs are efficiently recycled via the hepatic portal vein and taken up by hepatocytes via NTCP and organic anion transporting polypeptides (human OATP1B1 and OATP1B3, mouse OATP1B2)5,6, closing the loop of the enterohepatic circulation. Excessive BAs in hepatocytes are released into the sinusoids (to eventually reach the bloodstream) thanks to heteromeric organic solute transporter (OSTα-OSTβ) and multidrug resistance-associated proteins 3 and 4 (MRP3, MRP4)5,6. Disruption of BA homeostasis and flow could therefore lead to the development of severe liver diseases, but mechanisms remain unknown.
Histone acetylation controls gene regulation and plays important roles in many biological processes, but it is reportedly increased in human cirrhosis7, although no mechanism has been proposed to explain how it contributes to cirrhosis development. Several chromatin-modifier complexes participate in the tight regulation of this post-translational modification. These include the NSL complex, which contains the histone acetyltransferase males absent on the first (MOF), KAT8 regulatory NSL complex subunits 1 and 3 (KANSL1 and KANSL3), WD repeat-containing protein 5 (WDR5), and MCRS18. MCRS1 is also part of the INO80 complex together with Pontin (TIP49) and Reptin (TIP48), two conserved ATPases8. MCRS1 might thus play a key role in modulating the cellular epigenetic landscape. Here, we show that MCRS1 is a central component of a chromatin-modifying complex, controlling expression of BA transporter genes by regulating histone acetylation in hepatocytes. Loss of MCRS1 in hepatocytes induces histone acetylation of BA transporter genes, which leads to perturbation of BA flow. Accumulation of BAs in liver sinusoids activates HSCs via FXR, promoting liver fibrosis.
Materials and methods
A full description of the materials and methods used, including generation of mouse models, can be found in the supplementary methods and CTAT table.
Results
MCRS1 Loss in Hepatocytes Promotes Cirrhosis
MCRS1 was knocked-out specifically in hepatocytes by crossing Mcrs1 conditional knockout mice with serum albumin (SA)-creERT2 mice (Fig. S1A). CreERT2-mediated recombination was induced by dietary provision of tamoxifen to 5-week-old mice for 3 weeks, yielding Mcrs1(+/+)hep and Mcrs1(Δ/Δ)hep mice that were sacrificed at either 8 or 15 weeks of age (Fig. 1A and Fig. S1B). Although Mcrs1(Δ/Δ)hep mice gained weight more slowly after tamoxifen treatment than control littermates, they recovered body weight after 4 weeks, and liver to body weight ratio of 15-week-old Mcrs1(Δ/Δ)hep mice was identical to that of control mice (Fig. S1C, D). Histopathological analysis of livers from 8-week-old Mcrs1(Δ/Δ)hep mice showed that hepatocyte proliferation rate was unchanged, with cell cycle arrest in G2 phase and elevated levels of p53 and p21 (Fig. S1, E–I). Moreover, hepatic progenitor cells (HPCs) were proliferating (a measure of bile duct reaction), and fibrosis was significantly higher than in littermate controls (Fig. S1J–M). Fibrosis was accompanied by increased apoptosis and angiogenesis, elevated expression of alanine aminotransferase (ALT), aspartate aminotransferase (AST) and alkaline phosphatase (ALP), and increased levels of bilirubin (Fig. S1N–U). Notably, no significant differences were found in hepatic lipid accumulation (Fig. S1V, W). Moreover, T cells and macrophages were increased, but not neutrophils (Fig. S2A–I). Thus, 8-week-old Mcrs1(Δ/Δ)hep mice display fibrotic livers and significant liver injury with loss of hepatocyte proliferative capacity and increased inflammation.
Fig. 1. MCRS1 loss in hepatocytes promotes cirrhosis.
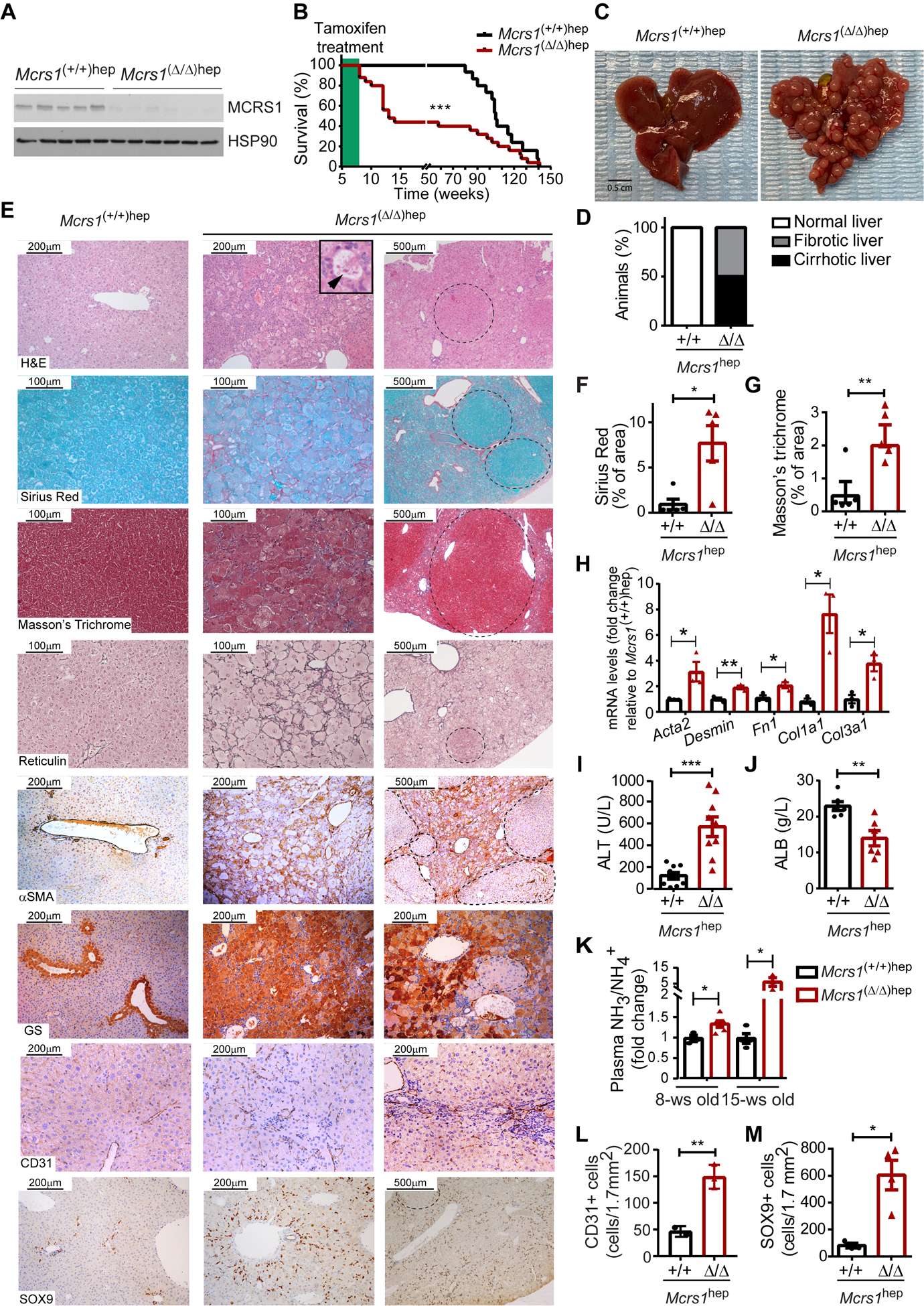
(A) WB from 8-week-old Mcrs1(+/+)hep and Mcrs1(Δ/Δ)hep mouse livers. (B) Survival of Mcrs1(+/+)hep and Mcrs1(Δ/Δ)hep mice (n=15,25). (C) Representative pictures of livers from 15-week-old Mcrs1(+/+)hep and Mcrs1(Δ/Δ)hep mice. (D) Percentage of Mcrs1(+/+)hep and Mcrs1(Δ/Δ)hep mice with cirrhosis at 15 weeks of age (n=15,9). (E) Representative pictures of indicated stainings and IHC of livers from 15-week-old Mcrs1(+/+)hep and Mcrs1(Δ/Δ)hep mice. Arrowhead points a Mallory-Denk body. Dashed lines delimit regenerative nodules. (F) and (G) Quantification of Sirius Red (F) and Masson’s Trichrome (F) stainings described in (E) (n=5). (H) Relative mRNA levels of fibrotic markers of hepatocytes isolated from 15-week-old Mcrs1(+/+)hep and Mcrs1(Δ/Δ)hep mice (n=3). (I) and (J) Blood alanine aminotransferase (ALT) (n=9) and albumin (ALB) (n=6) levels from 15-week-old Mcrs1(+/+)hep and Mcrs1(Δ/Δ)hep mice. (K) Plasma ammonia (NH3/NH4+) levels from 8- and 15-week-old Mcrs1(+/+)hep and Mcrs1(Δ/Δ)hep mice (n=3,6,4,3). (L) and (M) Quantification of CD31 (L) (n=3) and SOX9 (M) (n=3,4) IHC described in (E). Scale bar: 0.5 cm in (C), 200 and 500 μm in H&E, αSMA and SOX9 pictures, 200 μm in GS and CD31 pictures, and 100 and 500 μm in Sirius Red, Masson’s Trichrome and Reticulin pictures. Statistical analysis was performed using Mantel-Cox test in (B), and unpaired two-tailed Student’s t test in (F) to (M). Data are represented as means±SEM. *, p≤0.05; **, p≤0.01; ***, p≤0.001.
Surprisingly, ~50% of Mcrs1(Δ/Δ)hep mice died between 8 and 15 weeks of age (Fig. 1B), and they had a cirrhotic phenotype pathologically characterized by the presence of visible regenerative nodules (RNs) (Fig. 1C, D, and supplementary materials and methods)1. Further characterization of fibrotic marks confirmed that livers from Mcrs1(Δ/Δ)hep mice sacrificed at 15±3 weeks of age had strong fibrosis, with de novo deposition of ECM surrounding RNs (Fig. 1E–H). Mcrs1(Δ/Δ)hep mice also exhibited elevated ALT and reduced albumin levels, respectively, indicating that their livers were damaged and dysfunctional (Fig. 1I, J). Moreover, levels of NH3 were elevated in blood of 8-week-old Mcrs1(Δ/Δ)hep mice and higher still at 15 weeks of age (Fig. 1K). No signs of necrosis, cholestasis or preneoplastic lesions were observed in 15-week-old Mcrs1(Δ/Δ)hep mice.
Cirrhosis in 15-week-old Mcrs1(Δ/Δ)hep mice was additionally characterized by increased angiogenesis and impaired hepatocyte turnover, together with drastic changes in metabolic zonation (Fig. 1E, L and Fig. S3A–C). The arrest of hepatocyte proliferation was accompanied by a pronounced bile duct reaction (Fig. 1E, M). Grading of livers from 15-week-old Mcrs1(Δ/Δ)hep mice according to an adapted Child-Pugh score indicated that Mcrs1(Δ/Δ)hep mice had grade 2 and 3 cirrhosis (Fig. S3D–E and supplementary methods). At this time point, the adaptive immune response was resolved, but macrophages and neutrophils were enhanced, likely because of increased cell death (Fig. S3F–I).
Tracing of hepatocytes using the R26_LSL_eYFP reporter line crossed with the Mcrs1(Δ/Δ)hep mouse confirmed a recombination efficiency of 99,97%±0,03% (Fig. S3J, K). Interestingly, ~50% of RNs were eYFP negative, likely originated from HPCs that differentiated into hepatocytes (Fig. S3L, M)9, and which retained MCRS1 expression (Fig. S3N). Importantly, no desmin positive fibroblasts co-expressed eYFP, and eYFP positive hepatocytes did present neither loss of E-cadherin nor expressed vimentin (Fig. S3O–R), suggesting that hepatocytes do neither transdifferentiate nor undergo EMT. Thus, the Mcrs1(Δ/Δ)hep mouse is apparently the first genetic mouse model recapitulating several clinical features of liver cirrhosis.
Expression of BA transporters is deregulated in cirrhotic livers
To determine how MCRS1 loss in hepatocytes promotes cirrhosis, we performed a quantitative proteomic analysis using isobaric tags for relative and absolute quantitation (iTRAQ) in livers from 8-week-old Mcrs1(Δ/Δ)hep mice (Fig. S4A). Global proteomic profiling identified 4,279 proteins, of which 878 were differentially expressed: 416 and 462 downregulated and upregulated, respectively (Fig. S4B). BA transporters in hepatocytes were heavily represented, with predominant downregulation of NTCP, detected to be the second most differentially downregulated protein in 8-week-old Mcrs1(Δ/Δ)hep mice (Fig. 2A and Fig. S4B). Expression of Slc10a1 (NTCP) and Slco1b2 (OATP1B2), responsible for BA uptake from blood to hepatocytes5,6, was significantly downregulated in hepatocytes isolated from 8-week-old Mcrs1(Δ/Δ)hep mice (Fig. 2B–D), whereas expression of Abcc4 (MRP4), implicated in BA export from hepatocytes to blood, was significantly upregulated (Fig. 2B, C). Notably, Abcb11 (BSEP), a transporter specialized in BA export from hepatocytes to bile canaliculi, was also downregulated (Fig. 2B, C). Consequently, deregulation of BA flow caused accumulation of total BAs, including the primary BA cholic acid, in blood and liver of Mcrs1(Δ/Δ)hep mice (Fig. 2E, F), while reducing cholic acid in their intestines and feces (Fig. 2F and Fig. S4C, D). Notably, unchanged blood and liver cholesterol, as well as Cyp7a1 and Nr0b2 (SHP) mRNA levels, indicated that BA synthesis, metabolism and signaling in hepatocytes are not altered (Fig. S4E–G)6. This was further confirmed by unaffected levels of intestinal Fgf15 (Fig. S4H)5,6.
Fig. 2. Expression of BA transporters is deregulated in cirrhotic livers.
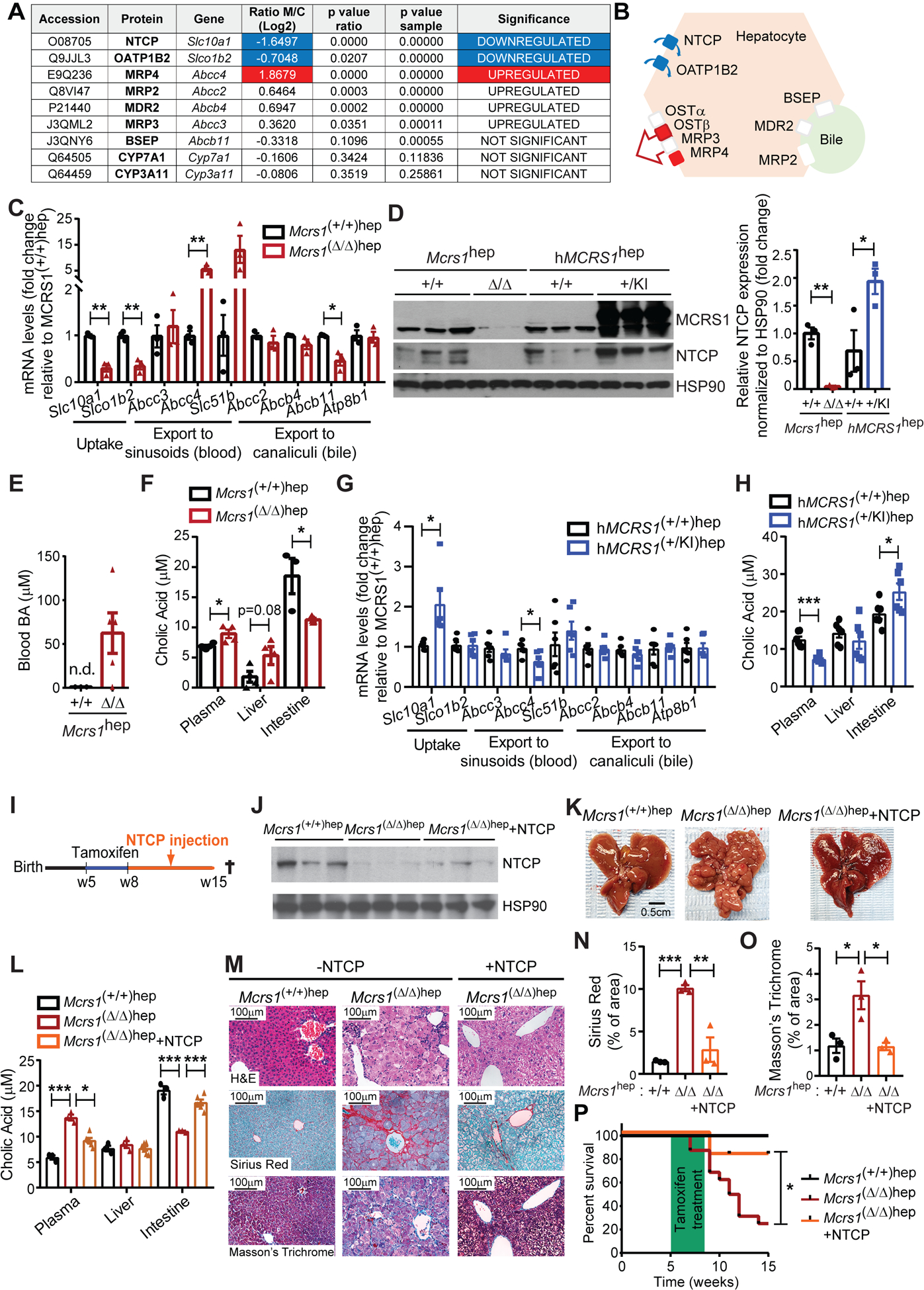
(A) Table representing relative BA-related protein levels detected by iTRAQ in livers from 8-week-old Mcrs1(Δ/Δ)hep mice compared to Mcrs1(+/+)hep mice (n=4). (B) Schematic representation of the location and expression status of BA transporters in the hepatocyte. Red means upregulated and green means downregulated. (C) Relative mRNA levels of BA transporters of hepatocytes isolated from 8-week-old Mcrs1(+/+)hep and Mcrs1(Δ/Δ)hep mice (n=3). (D) WB of isolated hepatocytes from 8-week-old Mcrs1(+/+)hep, Mcrs1(Δ/Δ)hep, hMCRS1(+/+)hep and hMCRS1(+/KI)hep mice. (E) Blood BA levels from 8-week-old Mcrs1(+/+)hep and Mcrs1(Δ/Δ)hep mice (n=3,5). (F) Cholic acid levels in plasma (n=4), liver (n=4) and intestine (n=3,5) from 8-week-old Mcrs1(+/+)hep and Mcrs1(Δ/Δ)hep mice. (G) Relative mRNA levels of BA transporters of hepatocytes isolated from 8-week-old hMCRS1(+/+)hep and hMCRS1(+/KI)hep mice (n=6). (H) Cholic acid levels in plasma, liver and intestine from 8-week-old hMCRS1(+/+)hep and hMCRS1(+/KI)hep mice (n=6). (I) Schematic representation of 5-week-old NTCP-overexpressing Mcrs1hep mice. (J) WB of isolated hepatocytes from the mice described in (I). (K) Representative pictures of livers from the mice described in (I). (L) Cholic acid levels in plasma, liver and intestine from mice described in (I) (n=3,3,6). (M) Representative pictures of indicated stainings of livers from mice described in (I). (N) and (O) Quantification of Sirius Red (N) and Masson’s Trichrome (O) stainings described in (M) (n=3). (P) Survival of mice described in (I) (n=5,16,11). Scale bar: 0.5 cm in (K), and 100 μm in (M). Statistical analysis was performed using unpaired two-tailed Student’s t test in (C), (D) and (F) to (H), two-way ANOVA with Tukey’s multiple comparisons test in (L), (N) and (O), and Mantel-Cox test in (P). n.d. means not detected. Data are represented as means±SEM. *, p≤0.05; **, p≤0.01; ***, p≤0.001.
To corroborate these findings, we generated a Col1a1 knockin mouse expressing human MCRS1 (hMCRS1) via a tetracycline-dependent transactivator controlled by the hepatocyte-specific liver activated protein promoter (Lap) (Fig. S4I). Without doxycycline, hMCRS1 was expressed specifically in hepatocytes (Fig. 2D). These mice were designated “hMCRS1(+/KI)hep mice”; littermate controls are referred to “hMCRS1(+/+)hep mice”. hMCRS1 overexpression in hepatocytes substantially increased NTCP levels, and mirrored the changes observed in the expression pattern of BA transporter genes in 8-week-old Mcrs1(Δ/Δ)hep mice (Fig. 2D, G, H and Fig. S4G). MCRS1 depletion in hepatocytes thus induces BA accumulation in blood by specifically changing the expression pattern of BA transporters, independently of BA synthesis.
Importantly, NTCP was downregulated 1 week after MCRS1 depletion in isolated hepatocytes of asymptomatic 6-week-old Mcrs1(Δ/Δ)hep mice (Fig. S5A–D). While no differences in ALT and BA levels were detected at this time, they increased steadily 2 and 3 weeks after MCRS1 depletion (Fig. S5E, F). However, neither ductular reaction nor inflammation was detected in 1-week-old Mcrs1(Δ/Δ)hep mice (Fig. S5G–K). Therefore, dysregulation of the BA transporters is an early event triggering increased BA levels, prior to proliferation arrest of hepatocytes, inflammation and expansion of HPCs.
To demonstrate the importance of perturbing BA flow in cirrhosis, NTCP was re-expressed in 8-week-old Mcrs1(Δ/Δ)hep mice via hydrodynamic tail vein injection (Fig. 2I, J). NTCP overexpression normalized BA levels, prevented fibrosis, and cirrhosis was not detected in 15-week-old Mcrs1(Δ/Δ)hep mice (Fig. 2K–O). Moreover, ~90% of Mcrs1(Δ/Δ)hep mice overexpressing NTCP survived over 15 weeks of age, while more than 50% of Mcrs1(Δ/Δ)hep mice died before (Fig. 2P). Thus, restoring BA homeostasis by re-expressing NTCP in Mcrs1(Δ/Δ)hep mice was sufficient to reduce fibrosis and extend mouse survival. NTCP downregulation and perturbation of BA flow are hence the causal links between MCRS1 loss and cirrhosis development.
Secondary BAs might not influence the Mcrs1(Δ/Δ)hep mouse phenotype
To exclude the possibility that secondary BAs influence the development of cirrhosis, Mcrs1(Δ/Δ)hep mice were fed a diet containing cholestyramine resin (ChR) (Fig. S6A), a well-known BA sequestrant used in patients with liver diseases5. While ChR-fed Mcrs1(Δ/Δ)hep mice exhibited early body weight loss, their body weight recovered over time (Fig. S6B). Strikingly, although ChR impaired BA recycling and initially reduced blood BA levels in Mcrs1(Δ/Δ)hep mice, BAs accumulated over time in the blood of ChR-fed Mcrs1(Δ/Δ)hep mice to the same extent as in normal diet (ND)-fed Mcrs1(Δ/Δ)hep mice (Fig. S6C, D). Moreover, ChR-fed Mcrs1(Δ/Δ)hep mice had apparently cirrhotic livers with high levels of ALT, and a considerably higher frequency of cirrhosis than their ND-fed counterparts (Fig. S6E–G). However, the adapted Child-Pugh score indicated similar grades of cirrhosis in both ChR- and ND-fed Mcrs1(Δ/Δ)hep mice (Fig. S6H), suggesting that ChR treatment does not aggravate cirrhosis severity. ChR-fed Mcrs1(Δ/Δ)hep mice also exhibited lower survival than ND-fed Mcrs1(Δ/Δ)hep mice (Fig. S6I). Notably, the secondary BA, deoxycholic acid (DCA), was significantly reduced over time in feces of ChR-fed Mcrs1(Δ/Δ)hep mice at 15 and 22 weeks of age when compared to ND-fed mice (Fig. S6J). This indicates an efficient BA chelation in the intestine, and suggests that secondary BAs are unlikely playing a major role in cirrhosis development. Moreover, despite BAs can be chelated and their levels are significantly reduced, the inhibition of BA uptake observed in hepatocytes of Mcrs1(Δ/Δ)hep mice seems to be sufficient to increase blood BAs overtime, triggering fibrosis in the liver. Therefore, ChR cannot prevent blood BA accumulation in Mcrs1(Δ/Δ)hep mice and hence, intestinal chelation of BAs might not be an effective treatment against cirrhosis.
Increased blood BAs exacerbate the Mcrs1(Δ/Δ)hep mouse phenotype
Since reducing BA levels in blood by re-expressing NTCP in Mcrs1(Δ/Δ)hep mice rescues cirrhosis, we next reasoned that increasing their concentration in the liver would exacerbate the cirrhotic phenotype of Mcrs1(Δ/Δ)hep mice. We therefore triggered bile duct injury and bile leakage in livers by genetically suppressing the MDR2 transporter in Mcrs1(Δ/Δ)hep mice, crossing Abcb4 (MDR2) knockout mice (Abcb4(−/−)) with Mcrs1(Δ/Δ)hep mice, generating homozygous Mcrs1(Δ/Δ)hep;Abcb4(−/−) mice (Fig. S7A). MDR2 is a transporter of phosphatidylcholine, which forms micelles around hydrophobic BAs and represent a protective shield to the cells composing the liver bile canaliculi and gallbladder. Loss of MDR2 results into free acidic bile salts in liver bile canaliculi, which disrupt the membranes of bile duct cells, triggering bile leakage. While Abcb4(−/−) mice develop hepatic lesions resembling primary sclerosing cholangitis and spontaneously progress to severe biliary fibrosis, the phenotypic perturbations of Mcrs1(Δ/Δ)hep;Abcb4(−/−) mice were profoundly accelerated, with jaundice reflected by yellowish peritoneum and earlier death (Fig. S7B, C). Moreover, higher total blood BAs, and plasma and liver cholic acid levels were detected (Fig. S7D–F). Direct and total bilirubin were also increased (Fig. S7G, H). This was accompanied by severe fibrosis and a stronger bile duct reaction (Fig. S7I–L).
To confirm these findings, Mcrs1(Δ/Δ)hep mice were fed with a BA-supplemented diet (BD) for 4 weeks after tamoxifen treatment (Fig. S8A). BA supplementation increased plasma and liver cholic acid, ALT, and bilirubin in Mcrs1(+/+)hep mice, which were further enhanced in Mcrs1(Δ/Δ)hep mice (Fig. S8B–G). Expectedly, BA supplementation induced fibrosis in Mcrs1(+/+)hep mice, that was worsened in BA-fed Mcrs1(Δ/Δ)hep mice (Fig. S8H–J). Therefore, high levels of BAs trigger liver fibrosis, and exacerbate the phenotypic consequences of knocking out MCRS1 in mouse livers.
FXR predominantly labels human and mouse HSCs
BAs bind to and activate FXR5,6. Single-nuclei RNA sequencing (snRNA-seq) performed in human livers surprisingly revealed that NR1H4 (FXR) was predominantly expressed in HSCs (Fig. 3A–D, Fig. S9A and Table S2). Notably, expression of other well-known BA receptors was not detected (Fig. S9B–D)5,6. Likewise, single cell (sc) RNA-seq conducted in liver tissue from healthy mouse confirmed that only Nr1h4 was mainly expressed in HSCs (Fig. 3E–H and Fig. S9E–G). Further analysis from publicly available scRNA-seq data sets10 confirmed the expression of Nr1h4 in liver fibroblasts (Fig. 3I, J). Finally, co-IHC using FXR and desmin in Mcrs1(Δ/Δ)hep mice revealed the presence of FXR in 50% of HSCs (Fig. S9H, I). Interestingly, SLC10A1 (NTCP) was detected neither in human nor in mouse HSCs (Fig. S9J, K), confirming previous data11,12, and indicating that other transporters are implicated in BA transport into HSCs. Therefore, activation of FXR in HSCs by BAs could have an unexpected pathophysiological role in liver fibrosis.
Fig. 3. FXR predominantly labels human and mouse HSCs.
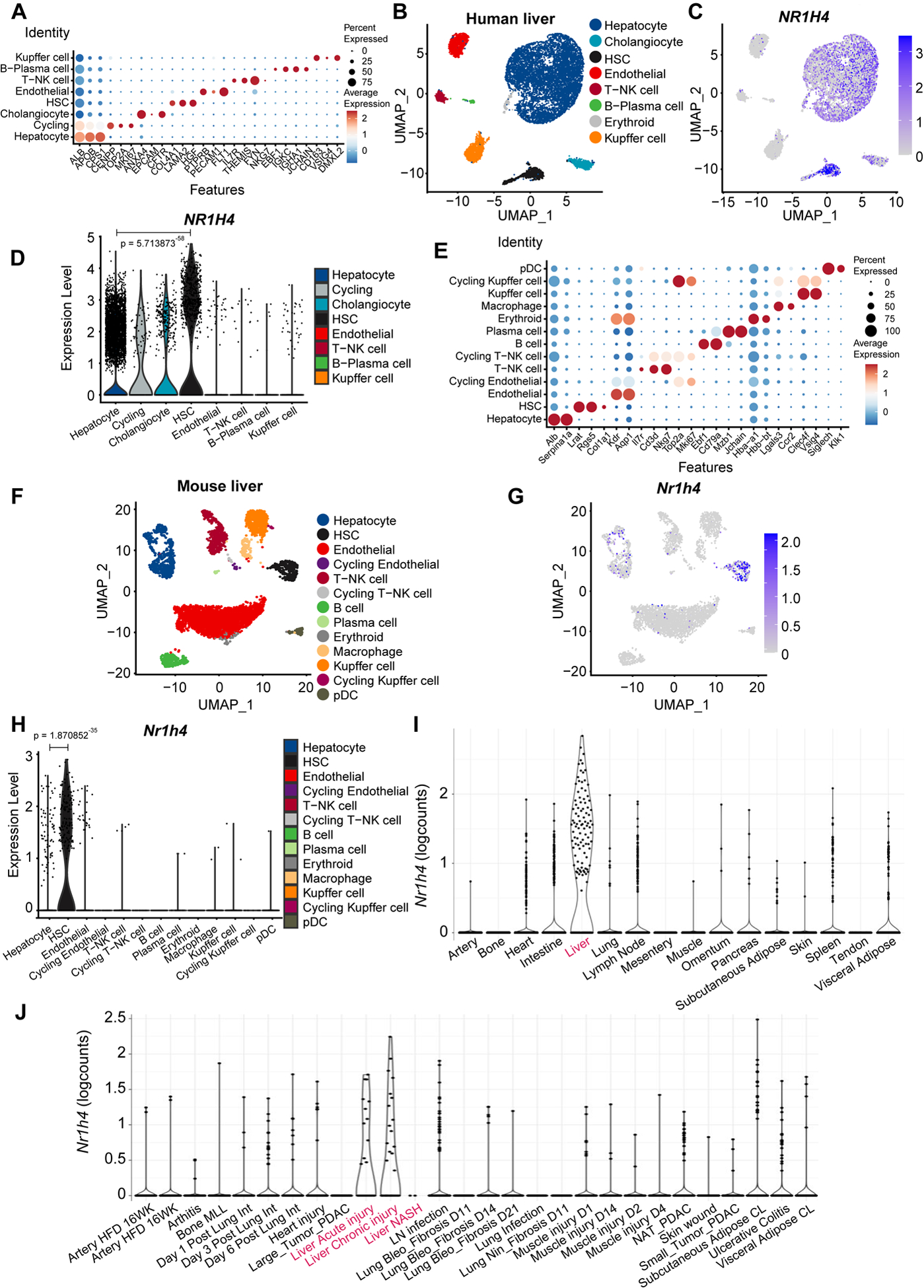
(A) Annotation of cell types and selected marker genes obtained from single nuclei RNA sequencing (snRNA-seq) performed in human livers. Dot size indicates fraction of cells and colour indicates expression level (log-normalized and scaled). (B) Major clusters and respective cell-type assignments in UMAP from the experiment described in (A). (C) RNA expression of NR1H4 in the same embedding as (B). (D) Violin plot representing gene expression levels of NR1H4 from the experiment described in (A). (E) Annotation of cell types and selected marker genes obtained from single cell RNA sequencing (scRNA-seq) performed in liver tissue from healthy mouse. Dot size indicates fraction of cells and colour indicates expression level (log-normalized and scaled). (F) Major clusters and respective cell-type assignments in UMAP from the experiment described in (E). (G) RNA expression of Nr1h4 in the same embedding as (F). (H) Violin plot representing gene expression levels of Nr1h4 from the experiment described in (E). (I) RNA expression of Nr1h4 in fibroblasts from different tissues in steady-state mice. (J) RNA expression of Nr1h4 in fibroblasts from disease mouse models. Statistical analysis was performed using the Wilcoxon rank sum test with continuity correction.
BAs activate FXR in HSCs to promote liver fibrosis
We therefore genetically targeted Nr1h4 in HSCs by crossing the Nr1h4 conditional knockout mouse (Nr1h4 lox) with a transgenic mouse in which cre expression was driven by the lecithin-retinol acyltransferase (Lrat) promoter, reportedly expressed in HSCs (Lrat-cre mouse), and as revealed by scRNA-seq (Fig. 3E), giving Nr1h4(Δ/Δ)HSC mice (Fig. 4A). Specific ablation of Nr1h4 in HSCs was corroborated in isolated HSCs and liver sections (Fig. 4B–E). Notably, loss of FXR was detected neither in hepatocytes nor in immune cells, that apparently did not express FXR, as also confirmed by scRNA-seq (Fig. 3B–D, F-H and Fig. S10A–C). Since crossing Nr1h4(Δ/Δ)HSC and Mcrs1(Δ/Δ)hep mice would lead to deletion of MCRS1 and FXR in both hepatocytes and HSCs, Nr1h4(Δ/Δ)HSC mice were subjected to BDL (Fig. 4F). The liver phenotype of Nr1h4(Δ/Δ)HSC mice with BDL was similar to that of Nr1h4(+/+)HSC mice with BDL, and there were no differences in their: liver to body weight ratios; survival; levels of ALT, blood BAs, total bilirubin; or bile duct reaction, which was elevated in both cases (Fig. S10D–K). This suggests that abolishing FXR signaling in HSCs does not impair liver injury. However, fibrosis was strongly suppressed in Nr1h4(Δ/Δ)HSC mice with BDL (Fig. 4G, H). Interestingly, further analysis showed that bile duct-ligated Nr1h4(+/+)HSC mice had a decrease of NTCP (Slc10a1) expression in isolated hepatocytes (Fig. 4I, J). This confirms that BDL leads to BA accumulation, which feedbacks to hepatocytes, acts on FXR, and suppresses NTCP expression, a well-known target gene negatively regulated by FXR in hepatocytes5. Therefore, BAs activate FXR signaling in HSCs, stimulating HSC transdifferentiation into myofibroblasts to provoke liver fibrosis.
Fig. 4. BAs activate FXR in HSCs to promote liver fibrosis.
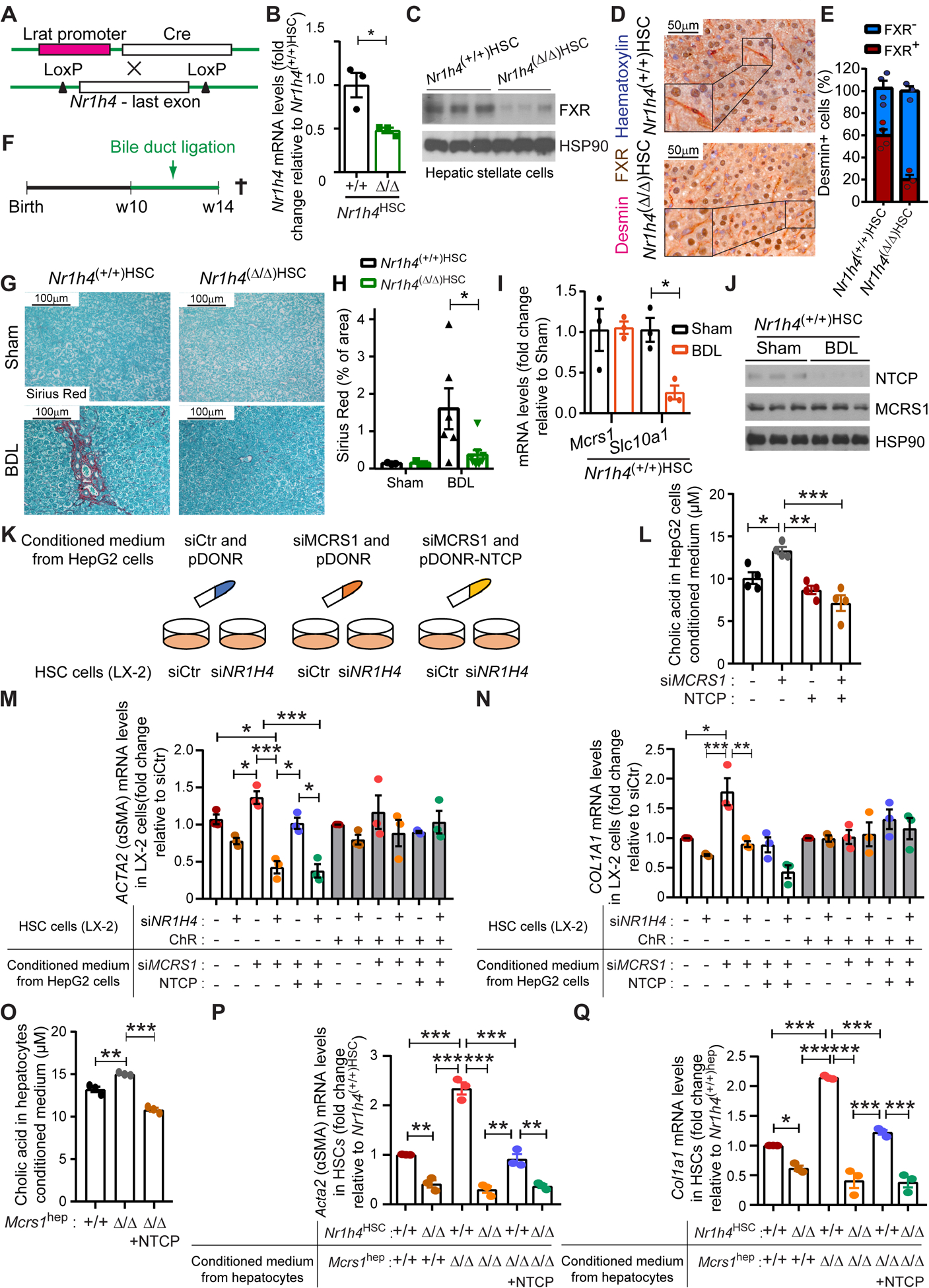
(A) Schematic representation of the Nr1h4HSC mouse model. (B) Relative mRNA levels of Nr1h4 of isolated HSCs from 14-week-old Nr1h4(+/+)HSC and Nr1h4(Δ/Δ)HSC mice (n=3). (C) WB of isolated HSCs from 14-week-old Nr1h4(+/+)HSC and Nr1h4(Δ/Δ)HSC mice. (D) Representative pictures of desmin/FXR co-IHC of livers from 14-week-old Nr1h4(+/+)HSC and Nr1h4(Δ/Δ)HSC mice. (E) Quantification of desmin/FXR co-IHC described in (D) (n=4,3). (F) Schematic representation of the bile duct ligation surgery protocol. (G) Representative pictures of Sirius Red staining of livers from 14-week-old Nr1h4(+/+)HSC and Nr1h4(Δ/Δ)HSC mice sham and bile duct-ligated (BDL). (H) Quantification of Sirius Red staining described in (G) (n=3,3,6,8). (I) Relative mRNA levels of indicated genes of isolated hepatocytes from Nr1h4(+/+)HSC mice sham and BDL (n=3). (J) WB of isolated hepatocytes from Nr1h4(+/+)HSC mice sham and BDL. (K) Schematic representation of the in vitro experiment performed in HepG2 and LX-2 cell lines. (L) Cholic acid levels measured in media from HepG2 cells transfected with siCtr or siMCRS1 and/or NTCP overexpression plasmid as indicated (N=4). (M) Relative mRNA levels of ACTA2 in LX-2 cells transfected with siCtr or siNR1H4 and treated with/without ChR, supplemented with conditioned medium from HepG2 cells transfected with siCtr or siMCRS1 and pDONR-Ctr or pDONR-NTCP as indicated (N=3). (N) Relative mRNA levels of COL1A1 in LX-2 cells transfected with siCtr or siNR1H4 and treated with/without ChR, supplemented with conditioned medium from HepG2 cells transfected with siCtr or siMCRS1 and pDONR-Ctr or pDONR-NTCP as indicated (N=3). (O) Cholic acid levels measured in media from isolated hepatocytes from Mcrs1hep mice and Mcrs1(Δ/Δ)hep mice overexpressing NTCP (N=3). (P) Relative mRNA levels of Acta2 in isolated HSCs from Nr1h4HSC mice supplemented with conditioned medium from isolated hepatocytes from Mcrs1hep mice and Mcrs1(Δ/Δ)hep mice overexpressing NTCP (N=3). (Q) Relative mRNA levels of Col1a1 in isolated HSCs from Nr1h4HSC mice supplemented with conditioned medium from isolated hepatocytes from Mcrs1hep mice and Mcrs1(Δ/Δ)hep mice overexpressing NTCP (N=3). Scale bar: 50 μm in (D) and 100 μm in (G). Statistical analysis was performed using unpaired two-tailed Student’s t test in (B) and (I), and two-way ANOVA with Tukey’s multiple comparisons test in (H), and (L) to (Q). Data are represented as means±SEM. *, p≤0.05; **, p≤0.01; ***, p≤0.001.
To corroborate these findings, FXR was overexpressed in the HSC line LX-2. Overexpression of FXR significantly induced fibrotic marks in LX-2 cells (Fig. S11A, B). Next, MCRS1 was downregulated in the hepatocyte cell line HepG2, and medium conditioned with these cells was used to culture LX-2 cells, in which FXR was depleted via the use of siRNA (Fig. 4K and Fig. S11C, D). MCRS1-depleted HepG2 cells had lower NTCP levels than HepG2 cells treated with control siRNA, and their conditioned medium contained higher levels of primary BAs than control conditioned medium (Fig. 4L and Fig. S11E). Interestingly, FXR-depleted LX-2 cells cultured in BA-enriched conditioned medium from MCRS1-depleted HepG2 cells exhibited lower ACTA2, COL1A1 and COL3A1 expression than siRNA control-treated cells (Fig. 4M, N and Fig. S11F, G). Re-expression of NTCP in MCRS1-depleted HepG2 cells reduced BA levels in their conditioned medium (Fig. 4L and Fig. S11H). When LX-2 cells were grown in conditioned medium from MCRS1-depleted HepG2 cells with overexpression of NTCP, their ACTA2, COL1A1 and COL3A1 expression was significantly reduced in an FXR-dependent manner (Fig. 4M, N and Fig. S11G, H). Interestingly, chelating BAs using ChR in the HepG2 culture medium abolished the expression of fibrotic marks, independently of MCRS1, NTCP or FXR expression (Fig. 4M, N and Fig. S11G, H).
Same data were obtained when isolated HSCs from Nr1h4HSC mice were cultured with conditioned medium of isolated hepatocytes from Mcrs1hep mice (Fig. 4O–Q and Fig. S11I–K). Conditioned medium from isolated hepatocytes from Mcrs1(Δ/Δ)hep mice overexpressing NTCP (Fig. 2I) abrogated fibrotic marks in HSCs from Nr1h4HSC mice (Fig. 4O–Q and Fig. S11K). Taken together, these data indicate that MCRS1 loss in hepatocytes downregulates NTCP, leading to BA efflux, which in turn stimulates HSCs via FXR activation.
MCRS1 overexpression ameliorates liver fibrosis
Since hMCRS1(+/KI)hep mice showed an increase of NTCP levels (Fig. 2D), and mirrored the changes observed in Mcrs1(Δ/Δ)hep mice, we hypothesized that hMCRS1(+/KI)hep mice might be protected against liver fibrosis. Hence, 10-week-old hMCRS1(+/+)hep and hMCRS1(+/KI)hep mice were subjected to BDL (Fig. S12A). Whereas BDL downregulated NTCP in isolated hepatocytes, bile duct-ligated hMCRS1(+/KI)hep mice partially recovered NTCP expression (Fig. 4I, J and Fig. S12B). Notably, BDL did not further increase cholestasis (determined by gallbladder size and number of CK7+ cells and biliary ducts in the lumen) in hMCRS1(+/KI)hep mice when compared to littermate controls (Fig. S12C–F). However, bile duct-ligated hMCRS1(+/KI)hep mice had significantly lower levels of primary BAs in blood, and liver injury and fibrosis were ameliorated when compared to bile duct-ligated hMCRS1(+/+)hep mice (Fig. S12C–N). The same outcome was observed in mice obtained by crossing hMCRS1hep and Abcb4(−/−) mice (Fig. S12O–S). These results indicate that MCRS1 regulates NTCP-mediated BA flow, critical in fibrosis pathogenesis.
MCRS1 regulates histone acetylation of BA transporter genes
Protein sequence analysis revealed a previously unrecognized SANT (standing for switching-defective protein 3 (Swi3), adaptor 2 (Ada2), nuclear receptor co-repressor (N-CoR), transcription factor (TFIIIB)) domain in MCRS1. The SANT domain is found in chromatin remodeling proteins that interact with histones and closely resembles the DNA-binding domain (DBD) of Myb-related proteins (Fig. 5A, B and Fig. S13A). The SANT domain thus has a central role in chromatin remodeling, functioning as a unique histone-interaction module that couples histone binding to deacetyltransferase activity13. To check this, we transfected the HepG2 and Huh7 hepatocyte cell lines with full-length HA-tagged-MCRS1 (HA-MCRS1WT) and mutant HA-tagged-MCRS1 lacking the SANT domain (HA-MCRS1Δ130–192). Protein expression of HA-MCRS1WT and HA-MCRS1Δ130–192 constructs was similar and localized to the nucleus (Fig. S13B, C). Immunoprecipitation assays using HA antibody and blotting with a specific MCRS1 antibody revealed that the SANT domain was required for the interaction between MCRS1 and Histone H3 (HISH3) (Fig. 5C and Fig. S13D). Next, direct HISH3 binding to MCRS1 was tested. Recombinant glutathione S-transferase (GST), GST-tagged-MCRS1WT or GST-tagged-MCRS1Δ130–192 were incubated with in vitro translated (IVT) HA-tagged-HISH3. Pulldown assay revealed that GST-MCRS1WT interacted with HISH3, but this interaction was considerably reduced with GST-MCRS1Δ130–192 (Fig. 5D), indicating that HISH3 binds directly to MCRS1 via SANT domain.
Fig. 5. MCRS1 regulates histone acetylation of BA transporter genes.
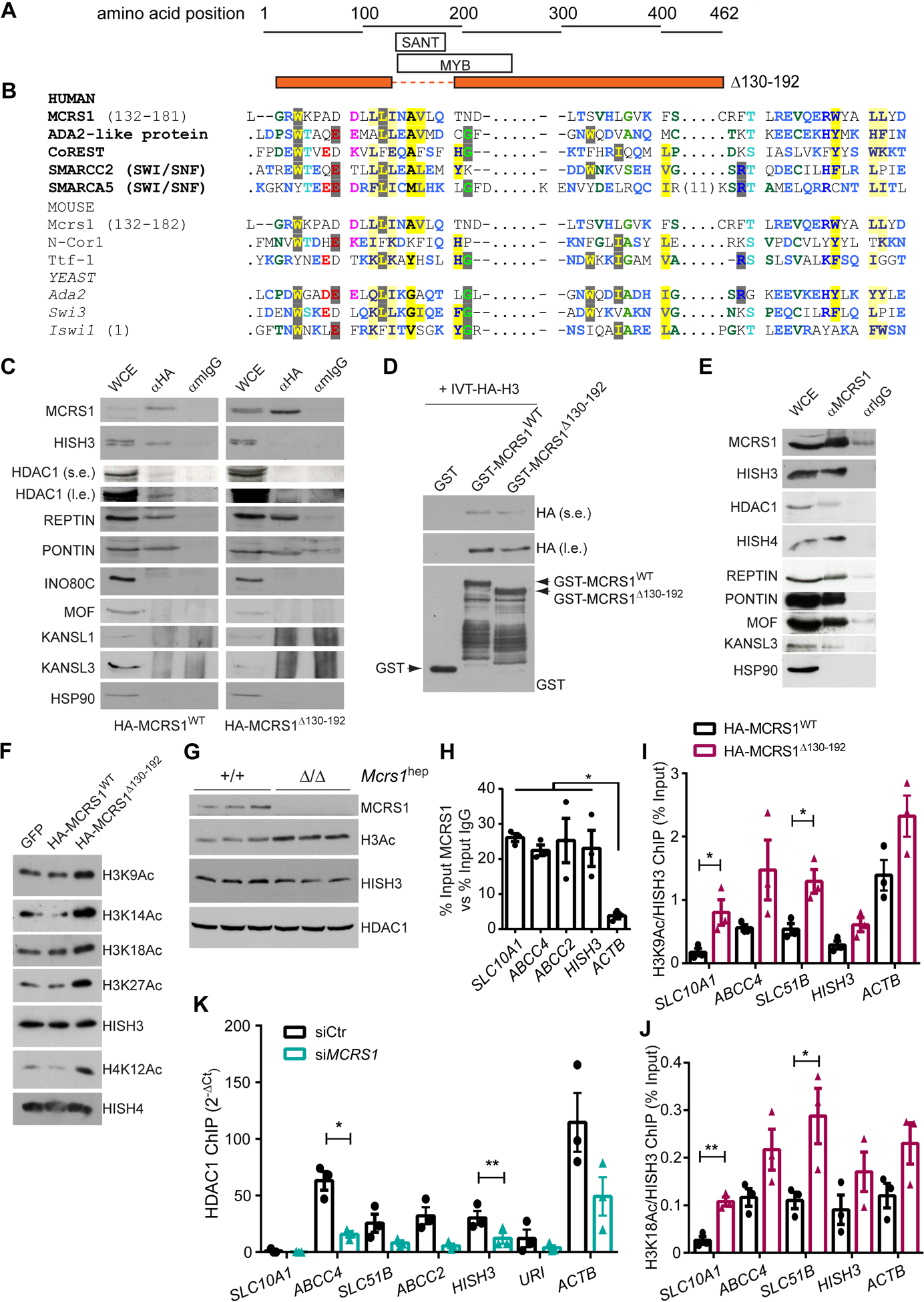
(A) Schematic representation of the protein sequence of MCRS1Δ139–192 mutant. (B) Sequence alignment of human, mouse and yeast MCRS1 protein sequence with proteins containing a SANT domain. (C) IP performed in HepG2 cells overexpressing HA-MCRS1WT and HA-MCRS1Δ130–192 mutant proteins (N=3). s.e. and l.e. mean short exposure and long exposure, respectively. (D) WB performed in preparations of in vitro translated HA-HISH3 incubated with GST-, GST-MCRS1WT and GST-MCRS1Δ130–192. (E) IP performed in HepG2 cells (N=3). (F) WB of indicated proteins of core histones isolated from HepG2 cells overexpressing GFP, HA-MCRS1WT and HA-MCRS1Δ130–192 mutant proteins (N=3). (G) WB performed in isolated hepatocytes from 6-week-old Mcrs1(+/+)hep and Mcrs1(Δ/Δ)hep mice. (H) Chromatin IP (ChIP) using MCRS1 antibody performed in HepG2 cells (N=3). (I) and (J) ChIPs using H3K9Ac (I) and H3K18Ac (J) antibodies performed in HepG2 cells overexpressing HA-MCRS1WT and HA-MCRS1Δ130–192 mutant proteins (N=3). (K) ChIP using HDAC1 antibody performed in HepG2 cells transfected with siCtr or siMCRS1 (N=3). Statistical analysis was performed using two-way ANOVA with Tukey’s multiple comparisons test in (H), and unpaired two-tailed Student’s t test in (I) to (K). Data are represented as means±SEM. *, p≤0.05; **, p≤0.01.
Interestingly, HDAC1 immunoprecipitated with HA-MCRS1WT but not with HA-MCRS1Δ130–192 (Fig. 5C), indicating that MCRS1, HDAC1 and HISH3 form a trimeric complex, in which MCRS1 connects HDAC1 to HISH3 via the SANT domain. Additionally, whereas Pontin and Reptin co-immunoprecipitated with both HA-MCRS1WT and HA-MCRS1Δ130–192, members of neither the NSL complex (MOF, KANSL1 and KANSL3) nor INO80 were detected in the immunoprecipitates of overexpressing HA-tagged MCRS1 constructs (Fig. 5C). However, MOF, KANSL3, Pontin, Reptin, and HISH3 interacted with endogenous MCRS1 (Fig. 5E). Taken together, these data indicate that MCRS1 might act as a histone acetylation regulator by anchoring HDAC1 to HISH3, apparently in an INO80/NSL-independent manner.
In accordance with these findings, histone acetylation (normalized to total HISH3) was substantially increased in core histone extracts from HepG2 cells overexpressing HA-MCRS1Δ130–192 but not in cells overexpressing HA-MCRS1WT (Fig. 5F). Additionally, hepatocytes isolated from Mcrs1(Δ/Δ)hep mice 1 week after MCRS1 deletion, showed significantly increased histone acetylation, without downregulating HDAC1 expression (Fig. 5G). Moreover, an in vitro histone acetylation assay conducted in the presence of HA-MCRS1WT or HA-MCRS1Δ130–192 showed that MCRS1 lacks intrinsic histone deacetylase activity (Fig. S13E). Thus, increased histone acetylation after MCRS1 loss is a very early event, and MCRS1 anchors HDAC1 and HISH3 via the SANT domain. SANT domain deletion, like MCRS1 depletion, seemingly releases HDAC1 from HISH3, increasing histone acetylation.
To determine whether increased histone lysine acetylation affects the expression of BA transporter genes, we performed several chromatin immunoprecipitation (ChIP) assays in HepG2 and Huh7 cells. Interestingly, MCRS1 was enriched in regulatory regions of BA transporter genes (Fig. 5H and Fig. S13F). ChIP experiments using anti-acetylated H3K9 and H3K18 antibodies in HepG2 cells overexpressing either HA-MCRS1WT or the HA-MCRS1Δ130–192 mutant showed that cells overexpressing HA-MCRS1Δ130–192 exhibited increased histone lysine acetylation in regulatory regions of BA transporter genes (Fig. 5I, J). To relate the increase of histone acetylation to the loss of interaction of HDAC1 with the BA transporter genes, we performed ChIP experiments using HDAC1 antibody in HepG2 cells treated with siRNA against MCRS1 (Fig. 5K and Fig. S13G). MCRS1 knockdown reduced HDAC1 binding to BA transporter genes (Fig. 5K). Thus, MCRS1 might be essential for synchronizing lysine histone deacetylation with transcription of BA transporters via its direct binding to HISH3.
Next, we checked whether downregulation of SLC10A1 expression was associated with increased chromatin repressive marks. No differences were detected in H3K9me2, H3K9me3 and H3K27me3 in MCRS1- and control-siRNA-treated cells (Fig. S13G–J). Therefore, downregulation of NTCP upon MCRS1 depletion is likely mediated by gene histone acetylation independently of chromatin repressive marks.
MCRS1 and NTCP expressions correlate in human cirrhosis
To clarify the clinical relevance of our findings, we analyzed MCRS1 expression and NTCP status in two different gene datasets from various liver cirrhotic patients (GSE77627 -14 healthy and 22 cirrhotic samples-, and GSE20140 -307 cirrhotic samples). Analysis of GSE77627 dataset indicated that SLC10A1 expression was significantly downregulated in human cirrhosis when compared to healthy controls (Fig. S14A). Moreover, MCRS1 expression correlated with SLC10A1 and ACTA2 in both datasets (Fig. S14B–E). Although loss of MCRS1 expression was never detected in cirrhotic samples, further analysis of 10 healthy and 50 cirrhotic human livers (Table S3) showed that MCRS1 was mainly nuclear in healthy livers but was diffused throughout the cytoplasm in cirrhotic livers (Fig. S14F–H). Additionally, cytoplasmic MCRS1 correlated significantly with fibrosis marks in these samples (Fig. S14I, J). Therefore, loss of nuclear MCRS1 as well as decreased NTCP levels in human livers might be critical for cirrhosis, and dysregulation of BA flow might represent a central, conserved and universal signaling event in human cirrhotic development.
Discussion
To our knowledge, the Mcrs1(Δ/Δ)hep mouse is the first genetic model of liver cirrhosis. Using this model, we show that histone acetylation-mediated NTCP downregulation in hepatocytes is a primary event triggering liver fibrosis, preceding liver injury and inflammation. Downregulation of NTCP leads to aberrant distribution of the BA pool, and sinusoidal BA accumulation activates FXR in HSCs to promote liver fibrosis.
Human data demonstrates that this mechanism is universally relevant to the variety of ways cirrhosis is induced, and will impact the clinical research to open new avenues for the development of anti-fibrotic treatments and prevention of cirrhosis. Interestingly, obeticholic acid (OA), a semi-synthetic primary BA analogue and potent FXR ligand, was recently approved by the American Food and Drug Administration (FDA) under the name OcalivaR, and is in phase 2/3 clinical studies for cirrhosis14,15. OA supposedly exert beneficial effects by activating FXR in hepatocytes and intestinal epithelial cells16. However, recent clinical data alerted the FDA on adverse effects of OA: its use caused severe fibrosis and fulminant liver damage in some patients. Although more research is needed to understand the effects of primary BAs in other liver cell types to ensure that patients can be safely and effectively treated with OcalivaR, our data indicate that BAs are important in HSC activation and fibrosis, and represent a real paradigm shift with high impact on future therapeutic strategies.
MCRS1 loss or deletion of its SANT domain leads to release of HDAC1 from its anchoring sites on HISH3, preventing histone deacetylation of BA transporter genes. Our data suggest that MCRS1 binds to specific regulatory regions of genes and orchestrates their transcriptional activity, likely in coordination with chromatin remodelers and transcription factors. Transcriptionally active genes are tightly regulated by post-translational chromatin modifications, and in particular by reversible lysine acetylation of histones, which increases accessibility to the transcription machinery. However, enhanced lysine acetylation does not always correlate with increased target gene transcription. Previous work has shown that transcriptional activation is not necessarily associated with increased acetylation17. Indeed, some genes could be downregulated by histone acetylation18 such as individual activators could confer distinct patterns of histone acetylation on target promoters19. Enhanced lysine acetylation of HISH3 and HISH4 lysine residues upon MCRS1 depletion or SANT domain deletion could be implicated in either gene activation or gene repression. Actually, histone acetylation decreases expression of Scl10a1, but increases expression of Abcc4 in Mcrs1(Δ/Δ)hep mice (Fig. 2C). It is hence tempting to speculate that histone acetylation, likely with the cooperation of enhancers and other regulators, might be implicated in the control of gene expression. Despite genome-wide ChIP-sequencing experiments have been conducted in mouse embryonic stem cells20, identifying potential MCRS1 target genes in hepatocytes would give more insights into the role of MCRS1 in gene regulation.
It should also be noted that SANT domain deletion appears to be functionally equivalent to MCRS1 depletion. In agreement with this, a shift of MCRS1 from nucleus to cytoplasm was found in human cirrhosis. This goes in line with recent work indicating that MCRS1 may be localized in different sub-cellular compartments8. Interestingly, MCRS1 regulates mTORC1 activity at the lysosomal surface in response to amino acids21, leading us to speculate that nutritional cues could direct MCRS1 to promoters of specific genes, regulating their transcriptional activity. In this context, it is notable that branched-chain amino acid-based medicine has shown positive results in patients with cirrhosis22.
In sum, MCRS1 is a histone acetylation regulator that anchors HDAC1 to HISH3 via a previously undescribed SANT domain, playing a key role in proper modulation of gene expression and liver health. Loss of MCRS1 in hepatocytes induces liver cirrhosis by increasing histone lysine acetylation of BA transporter genes, accumulating BAs in liver sinusoids to activate FXR on HSCs. Human patient data demonstrate that this mechanism could be general for liver cirrhosis in humans, and targeting histone acetylation in liver cirrhotic patients by inhibiting histone acetyl transferases could be a viable remedial option for this deadly disease.
Supplementary Material
Acknowledgments
We thank all mouse providers. We acknowledge the patients and the BioBank Hospital Ramón y Cajal-IRYCIS (B.0000678), as well the CNIO Biobank, integrated in the Spanish National Biobanks Network and Biomodels Platform of the ISCIII. We are also thankful to the CNIO Mouse Genome Editing Core Unit and Animal Facility for the mouse re-derivation and maintenance, respectively.
Financial support
A.G. is a recipient of Severo Ochoa PhD fellowship. E.K is a recipient of fellowships from the National Research Foundation (NRF) from the Korean Ministry of Education (2020R1A6A3A03037725) and the CNIO-friends, supported by Severo Ochoa32 CNIO funds. This work was funded by grants to N.D. from the European Foundation for the Study of Diabetes (EFSD) award supported by EFSD/JRDF/Lilly programme (EASD 96103), from the Comunidad Autónoma de Madrid (S2017/BMD-3817) and, from the State Research Agency (AEI, 10.13039/501100011033) from the Spanish Ministry of Science and Innovation (projects SAF2016-76598-R, SAF2017-92733-EXP, RTI2018-094834-B-I00 and RED2018-102723-T), cofunded by European Regional Development Fund (ERDF) and from the Asociación Española Contra el Cáncer (AECC) (project PRYGN211184DJOU). This work was developed at the CNIO funded by the Health Institute Carlos III (ISCIII) and the Spanish Ministry of Science and Innovation. G.P.V. was supported by a grant from the State Research Agency from the Spanish Ministry of Science and Innovation (projects PID2019-105173RB42 I00). B.I. was supported by the National Institute of Health, National Cancer Institute (R37CA258829, R21CA263381). R.F.S was funded by NIH grants (1R01DK128955, 5R01DK124104 and 1R01CA262424).
Abbreviations
- ALP
Alkaline phosphatase
- ALT
Alanine aminotransferase
- AST
Aspartate aminotransferase
- BA
Bile acid
- BSEP
Bile salt export pump
- ChIP
Chromatin immunoprecipitation
- DCA
Deoxycholic acid
- ECM
Extracellular matrix
- FDA
Food and Drug Administration
- FXR
Farnesoid X receptor
- GST
Glutathione S-transferase
- HA-MCRS1Δ130–192
HA-tagged-MCRS1 lacking the SANT domain
- HA-MCRS1WT
full-length HA-tagged-MCRS1
- HDAC1
Histone deacetylase 1
- HISH3
histone H3
- HPCs
Hepatic progenitor cells
- HSCs
Hepatic stellate cells
- iTRAQ
isobaric tags for relative and absolute quantitation
- IVT
in vitro translated
- KANSL1 and KANSL3
KAT8 regulatory NSL complex subunits 1 and 3
- MCRS1
Microspherule 1
- MOF
males absent on the first
- MRP2/3
Multidrug resistance-associated protein 2/3
- MRP3, MRP4
Multidrug resistance-associated proteins 3 and 4
- NSL
Non-specific lethal
- NTCP
Na+-taurocholate cotransporting polypeptide
- OA
Obeticholic acid
- OATP
Organic anion transporting polypeptides
- OST
Organic solute transporter
- RNs
Regenerative nodules
- SANT
Switching-defective protein 3 (Swi3), adaptor 2 (Ada2), nuclear receptor co-repressor (N-CoR), transcription factor (TFIIIB)
- SC
Single cell
- snRNA-seq
Single-nuclei RNA sequencing
- TSSs
Transcription start sites
- WB
Western blot
- WDR5
WD repeat-containing protein 5
Footnotes
Conflicts of interest Authors declare no competing financial interests.
Data availability
All data are available in the main text or Methods. Materials are available upon request to N.D. and sharing materials will be subject to standard material transfer agreements. Mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository with the dataset identifier PXD023026 (Username: reviewer_pxd023026@ebi.ac.uk; Password: BqiMEJnA). snRNA- and scRNA-seq data have been deposited to the NCBI with the GEO accession number GSE179548 (Link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE179548; Token: evaliwkctjkbfcl) and GSE158183 GSM4794841, respectively.
References
- [1].Garrido A, Djouder N. Cirrhosis: A Questioned Risk Factor for Hepatocellular Carcinoma. Trends Cancer 2021;7:29–36. [DOI] [PubMed] [Google Scholar]
- [2].Giallourakis CC. Liver complications in patients with congestive heart failure. Gastroenterol Hepatol 2013;9:244–246. [PMC free article] [PubMed] [Google Scholar]
- [3].Karlsen TH, Lammert F, Thompson RJ. Genetics of liver disease: From pathophysiology to clinical practice. J Hepatol 2015;62:S6–S14. [DOI] [PubMed] [Google Scholar]
- [4].Liberal R, Grant CR. Cirrhosis and autoimmune liver disease: Current understanding. World J Hepatol 2016;8:1157–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metab 2013;17:657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shapiro H, Kolodziejczyk AA, Halstuch D, Elinav E. Bile acids in glucose metabolism in health and disease. J Exp Med 2018;215:383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shi Y, Sun H, Bao J, Zhou P, Zhang J, Li L, et al. Activation of inactive hepatocytes through histone acetylation: a mechanism for functional compensation after massive loss of hepatocytes. Am J Pathol 2011;179:1138–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sheikh BN, Guhathakurta S, Akhtar A. The non-specific lethal (NSL) complex at the crossroads of transcriptional control and cellular homeostasis. EMBO Rep 2019;20:e47630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tummala KS, Brandt M, Teijeiro A, Graña O, Schwabe RF, Perna C, et al. Hepatocellular Carcinomas Originate Predominantly from Hepatocytes and Benign Lesions from Hepatic Progenitor Cells. Cell Reports 2017;19:584–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Buechler MB, Pradhan RN, Krishnamurty AT, Cox C, Calviello AK, Wang AW, et al. Cross-tissue organization of the fibroblast lineage. Nature 2021;593:575–579. [DOI] [PubMed] [Google Scholar]
- [11].Kordes C, Sawitza I, Gotze S, Herebian D, Haussinger D. Hepatic stellate cells contribute to progenitor cells and liver regeneration. J Clin Invest 2014;124:5503–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fickert P, Fuchsbichler A, Moustafa T, Wagner M, Zollner G, Halilbasic E, et al. Farnesoid X receptor critically determines the fibrotic response in mice but is expressed to a low extent in human hepatic stellate cells and periductal myofibroblasts. Am J Pathol 2009;175:2392–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Boyer LA, Langer MR, Crowley KA, Tan S, Denu JM, Peterson CL. Essential role for the SANT domain in the functioning of multiple chromatin remodeling enzymes. Mol Cell 2002;10:935–942. [DOI] [PubMed] [Google Scholar]
- [14].Hirschfield GM, Mason A, Luketic V, Lindor K, Gordon SC, Mayo M, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology 2015;148:751–761. [DOI] [PubMed] [Google Scholar]
- [15].Kowdley KV, Luketic V, Chapman R, Hirschfield GM, Poupon R, Schramm C, et al. A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis. Hepatology 2018;67:1890–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Khanna A, Jones DE. Novel strategies and therapeutic options for the management of primary biliary cholangitis. Therap Adv Gastroenterol 2017;10:791–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Deckert J, Struhl K. Histone acetylation at promoters is differentially affected by specific activators and repressors. Mol Cell Biol 2001;21:2726–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Reamon-Buettner SM, Borlak J. A new paradigm in toxicology and teratology: altering gene activity in the absence of DNA sequence variation. Reprod Toxicol 2007;24:20–30. [DOI] [PubMed] [Google Scholar]
- [19].Kaimori JY, Maehara K, Hayashi-Takanaka Y, Harada A, Fukuda M, Yamamoto S, et al. Histone H4 lysine 20 acetylation is associated with gene repression in human cells. Sci Rep 2016;6:24318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chelmicki T, Dundar F, Turley MJ, Khanam T, Aktas T, Ramirez F, et al. MOF-associated complexes ensure stem cell identity and Xist repression. Elife 2014;3:e02024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fawal MA, Brandt M, Djouder N. MCRS1 binds and couples rheb to amino acid-dependent mTORC1 activation. Dev Cell 2015;33:67–82. [DOI] [PubMed] [Google Scholar]
- [22].Tajiri K, Shimizu Y. Branched-chain amino acids in liver diseases. Transl Gastroenterol Hepatol 2018;3:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or Methods. Materials are available upon request to N.D. and sharing materials will be subject to standard material transfer agreements. Mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository with the dataset identifier PXD023026 (Username: reviewer_pxd023026@ebi.ac.uk; Password: BqiMEJnA). snRNA- and scRNA-seq data have been deposited to the NCBI with the GEO accession number GSE179548 (Link: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE179548; Token: evaliwkctjkbfcl) and GSE158183 GSM4794841, respectively.