

Abstract
Background & Aims:
Sulfoconjugation of small molecules or protein peptides is a key mechanism to ensure biochemical and functional homeostasis in mammals. The PAPS synthase 2 (PAPSS2) is the primary enzyme to synthesize the universal sulfonate donor 3′-phosphoadenosine 5′-phosphosulfate (PAPS). Acetaminophen (APAP) overdose is the leading cause of acute liver failure (ALF), in which oxidative stress is a key pathogenic event, whereas sulfation of APAP contributes to its detoxification. The goal of this study is to determine whether and how PAPSS2 plays a role in APAP-induced ALF.
Methods:
Gene expression was analyzed in APAP-induced ALF in patients and mice. Liver-specific Papss2 knockout mice using Alb-Cre (Papss2ΔHC) or AAV8-TBG-Cre (Papss2iΔHC) were created and subjected to APAP-induced ALF. Primary human and mouse hepatocytes were used for in vitro mechanistic analysis.
Results:
The hepatic expression of PAPSS2 was decreased in APAP-induced ALF in patients and mice. Surprisingly, Papss2ΔHC mice were protected from APAP-induced hepatotoxicity despite having a decreased APAP sulfation, which was accompanied by increased hepatic antioxidative capacity through the activation of the p53-p2-Nrf2 axis. Treatment with a sulfation inhibitor also ameliorated APAP-induced hepatotoxicity. Gene knockdown experiments showed that the hepatoprotective effect of Papss2ΔHC was Nrf2-, p53- and p21-dependent. Mechanistically, we identified p53 as a novel substrate of sulfation. Papss2 ablation led to p53 protein accumulation by preventing p53 sulfation, which disrupts p53-MDM2 interaction and p53 ubiquitination, and increases p53 protein stability.
Conclusions:
We have uncovered a previously unrecognized and p53-mediated role of PAPSS2 in controlling oxidative response. Inhibition of p53 sulfation may be explored for the clinical management of APAP overdose.
Keywords: Oxidative hepatotoxicity, Acetaminophen, PAPSS2, Sulfation, p53
Graphical Abstract
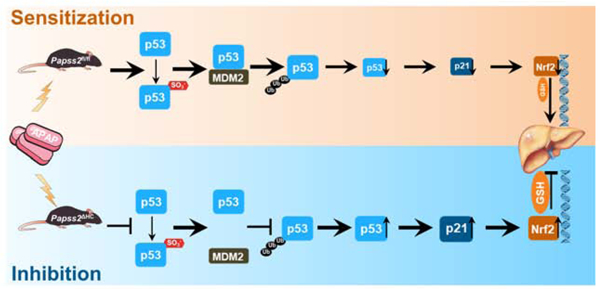
Lay Summary
Inhibition of sulfation prevents oxidative liver injury and acute liver failure by activating the p53-p21-Nrf2 signaling axis.
Introduction
Sulfoconjugation is a widely occurring conjugation reaction. The substrates of sulfoconjugation include small molecules of endogenous or exogenous origins. Sulfation is an important detoxification pathway for many clinical drugs including acetaminophen (APAP), the overdose of which is the leading cause of acute liver failure (ALF) 1. Meanwhile, protein peptides can also be sulfonated by targeting the tyrosine residues, and protein tyrosine sulfation is a common post-translational modification that affects protein-protein interactions and signaling pathways 2, 3. It has been estimated that approximately 1% of the tyrosine residues can be sulfated in a given organism 4.
Sulfoconjugation is catalyzed by families of sulfotransferases 5. Sulfoconjugation reactions depend on the universal sulfate donor substrate 3′-phosphoadenosine-5′-phosphosulfate (PAPS) 6. PAPS is synthesized by PAPS synthase 1 and 2 (PAPSS1/2) from adenosine triphosphate (ATP) and inorganic sulfate (SO42−). PAPSS2 is the major isoform in the liver, where the expression of PAPSS1 is very low 7.
APAP, the active ingredient of Tylenol®, is one of the most often used analgesic and antipyretic medications to treat conditions ranging from cold to fever, muscle ache, toothache, headache, arthritis, and backache 8, 9. APAP overdose is the leading cause of ALF, which accounts for 57% and 39% of the cases recorded in the United Kingdom and United States, respectively 10, 11. In the United States, APAP overdose results in approximately 30,000 hospital presentations each year 12.
APAP is extensively metabolized in the hepatocytes via glucuronidation, sulfation, and cytochrome P 450 2E1 (CYP2E1) oxidation at the recommended therapeutic doses 13, 14. The majority (∼90%) of APAP is rapidly conjugated to nontoxic glucuronidated and sulfated metabolites by uridine 5′-diphospho-glucuronosyltransferases (UGTs) and sulfotransferases (SULTs). Of the remaining APAP, approximately 2% is excreted unchanged via the urine and 5–9% is converted by CYP2E1 to form a reactive noxious quinone metabolite, N-acetyl-p-benzoquinone imine (NAPQI). NAPQI can be rapidly eliminated by binding with the cysteine thiol or glutathione (GSH) to form nontoxic mercapturic (APAP-Mer) and cysteine (APAP-Cys) conjugates 15, 16. In the event of APAP overdose, the sulfate conjugation becomes saturated. When the majority of GSH is depleted, NAPQI begins to accumulate in the hepatocytes, resulting in mitochondrial oxidative stress, generation of reactive oxygen species (ROS), and induction of cell death and liver necrosis 17, 18. In the clinic, N-acetylcysteine (NAC) is an effective antidote to prevent APAP overdose-induced liver injury by replenishing GSH. However, NAC has side effects of anaphylactic reactions and fluid overload, and delayed administration of NAC fails to prevent APAP-induced ALF 19. Thus, novel therapeutic approaches are urgently needed to combat APAP overdose.
In this study, we uncovered a surprising function of hepatic Papss2 in APAP-induced oxidative stress and hepatotoxicity. The hepatocyte-specific Papss2 knockout mice were protected from APAP toxicity through an augmented and p53-p21-Nrf2 mediated anti-oxidative response.
Materials and Methods
Animals and treatment; Human liver sections, patient datasets, and scRNA-seq analyses; Isolation of primary hepatocytes and non-parenchymal liver cells; Histology and immunostaining analyses; Serum and liver biochemical analyses; APAP metabolite analysis; Quantitative real-time PCR; Plasmid construction, cell transfection, and luciferase reporter gene assays; Co-immunoprecipitation, ubiquitination, and western blotting analyses; Chromatin immunoprecipitation assay; Statistics
See Supplementary Materials and Methods. Information on patients, antibodies and primer sequences is provided in Supplementary Tables 1, 2, 3, and 4, respectively.
Results
Hepatic expression of PAPSS2 decreases in APAP-induced hepatotoxicity and acute liver failure in patients and mice
To determine whether PAPSS2 plays a role in APAP-induced hepatotoxicity, we measured the hepatic expression of PAPSS2 in APAP-induced liver injury in patients and mice, as well as in primary human and mouse hepatocytes treated with APAP. Immunohistochemistry showed a markedly decreased expression of PAPSS2 in the liver of APAP overdose patients compared to the normal subjects (Figure 1A). The protein (Figure 1B) and mRNA (Figure 1C) expression of Papss2 was also decreased in the liver of mice treated with a toxic dose of APAP (200 mg/kg) for 6 or 24h. Our bioinformatic analysis of deposited microarray results (GSE13430) 20 derived human primary hepatocytes showed that the mRNA expression of PAPSS2 was decreased by APAP treatment in a dose-dependent manner (Figure 1D). A decreased hepatic expression of PAPSS2 was also observed in a cohort of patients with APAP-induced ALF (GSE74000) 21 (Figure 1E).
Figure 1. Hepatic expression of PAPSS2 decreases in APAP-induced hepatotoxicity and acute liver failure in patients and mice.
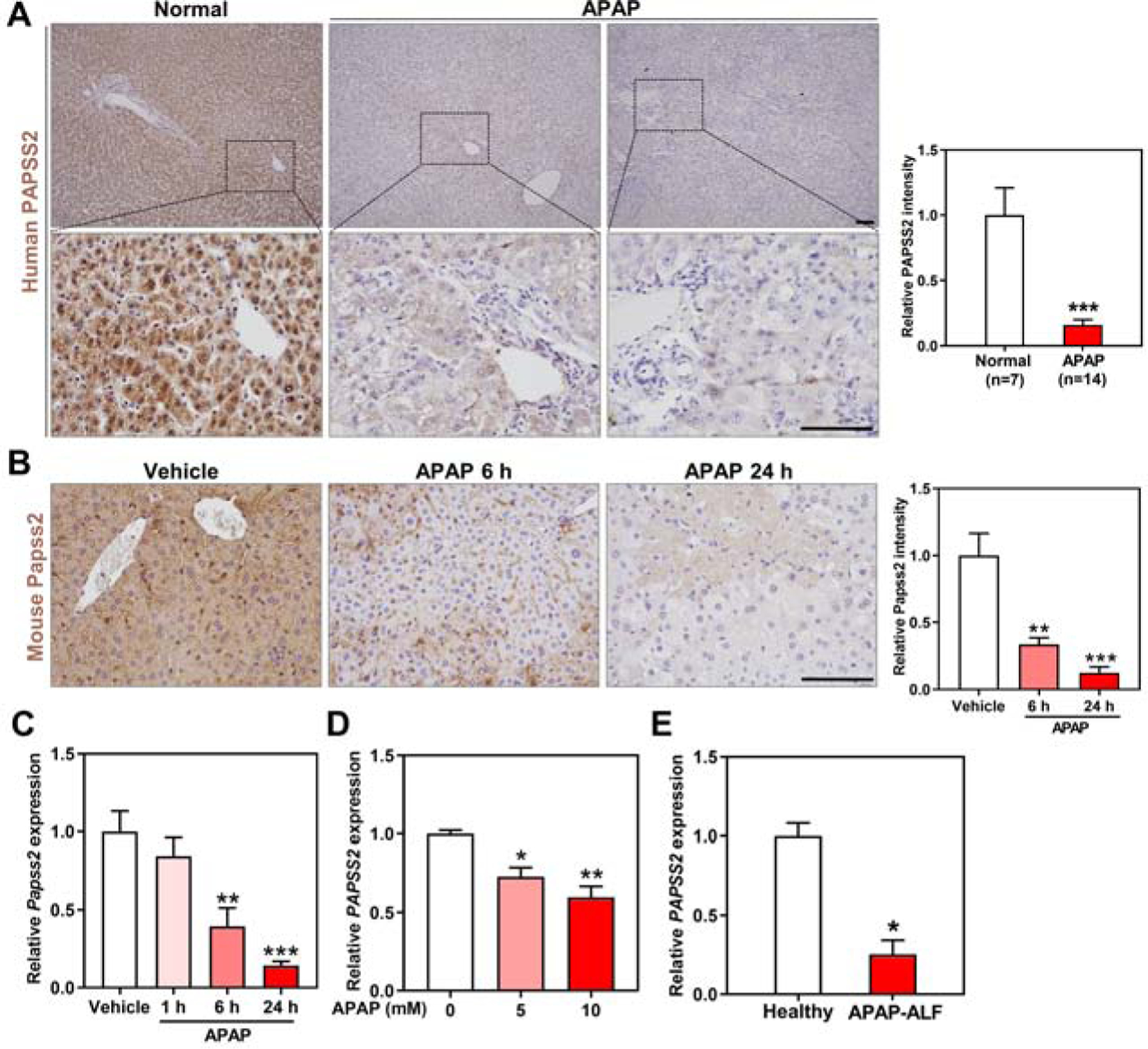
(A) Representative IHC staining of PAPSS2 in human liver sections derived from normal subjects or APAP overdose patients undergone liver transplantations. Shown on the right are the quantifications of relative PAPSS2 signals. Scale bars: 100 μm. (B) Representative IHC staining of Papss2 in livers of WT mice treated with vehicle or APAP (200 mg/kg) for indicated amount of time. Shown on the right are the quantifications of relative Papss2 signals. Scale bars: 100 μm. (C) Relative Papss2 mRNA expression in the livers of vehicle- and APAP-treated mice. (n=4–6). (D) Relative PAPSS2 mRNA expression in human primary hepatocytes treated with different doses of APAP (0, 5, 10 mM) (GSE13430). (E) PAPSS2 mRNA expression in the liver of healthy subjects and APAP-induced ALF patients (GSE74000). *P < .05, **P < .01, ***P < .01, compared to the indicated controls. Data are presented as mean ± SEM.
Hepatocytes are the major cellular source of PAPSS2 in the liver, and creation and characterization of hepatocyte-specific Papss2 knockout mice
Liver is an organ of multiple cell types. To define the cellular source of PAPSS2, we analyzed a publicly available single-cell RNA-seq (scRNA-seq) dataset derived from normal human livers (GSE136103) 22, and found that among hepatic cells, hepatocytes had the highest expression of PAPSS2 (Figure 2A). In mice, the expression of Papss2 in primary hepatocytes (HEP) was also the highest, compared to hepatic stellate cells (HSC), Kupffer cells (KC), and liver sinusoidal endothelial cells (LSEC) isolated from the same mice (Figure 2B).
Figure 2. Hepatocytes are the major cellular source of PAPSS2 in the liver, and creation and characterization of hepatocyte-specific Papss2 knockout mice.
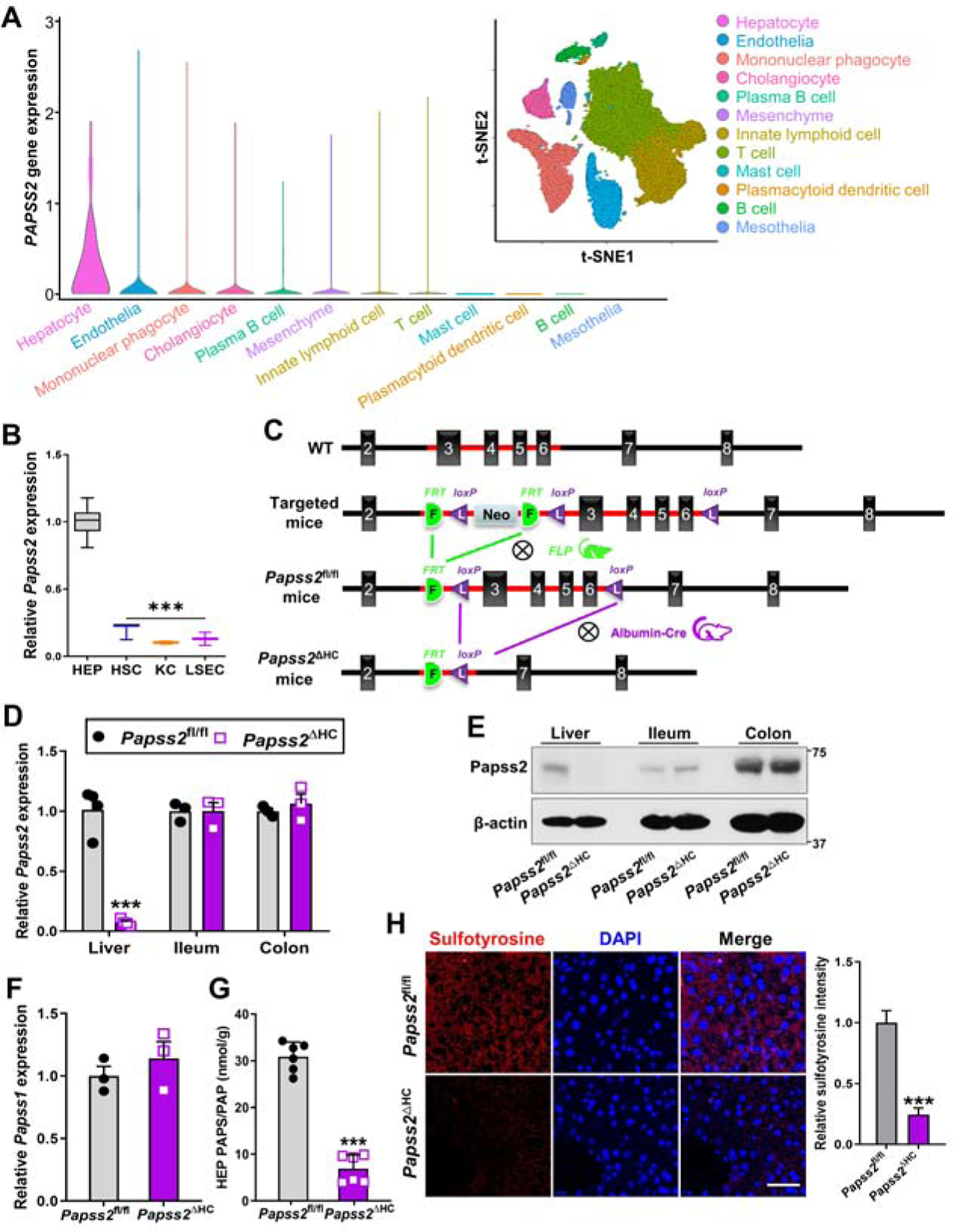
(A) Bioinformatic analyses of scRNA-seq dataset GSE136103 derived from normal human livers. Shown are scaled expression of PAPSS2 in different cell lineages and clustering of cell lineages. Shown on the upper right is cell lineages inferred from expression of marker gene signatures. t-SNE, t-distributed stochastic neighbor embedding. (B) Relative Papss2 mRNA expression in primary mouse hepatocytes (HEP), hepatic stellate cells (HSC), Kupffer cells (KC), and liver sinusoidal endothelial cells (LSEC). (n=7). (C) Strategies to create the Papss2fl/fl and Papss2ΔHC mice. (D to F) The mRNA (D) and protein (E) expression of Papss2 in the liver, ileum and colon, and mRNA expression of Papss1 in the liver (F) of Papss2fl/fl and Papss2ΔHC mice. (n=3–4). (G) Quantification of PAPS and PAP contents in primary hepatocytes. (n=6). (H) Immunofluorescent images of sulfotyrosine (red) in liver sections of intact Papss2fl/fl and Papss2ΔHC mice. Shown on the right are the quantifications of relative sulfotyrosine signals. Scale bar: 50 μm. ***P < .001. Data are presented as violin, box plots, or mean ± SEM.
To investigate the hepatocyte function of Papss2 in vivo, we generated hepatocyte-specific Papss2 knockout (Papss2ΔHC) mice by crossbreeding the floxed Papss2fl/fl mice with the hepatocyte-specific Albumin-Cre transgenic mice 23. The Papss2fl/fl mice were generated by floxing exons 3–6 of the mouse Papss2 gene in embryonic stem cells via homologous recombination, followed by the removal of the Neo cassette by mating with the FLP mice as outlined in Figure 2C. The Papss2fl/fl and Papss2ΔHC mice were genotyped by PCR (Supplementary Figure 1A). The efficiencies of Papss2 knockout at the mRNA (Figure 2D) and protein (Figure 2E) levels in the liver, but not in the ileum and colon, were confirmed by real-time PCR and western blotting, respectively. The liver expression of the Papss1 was not affected by Papss2 ablation (Figure 2F). Consistent with the role of PAPSS2 in generating the sulfate donor, the PAPS/PAP content was markedly reduced in primary hepatocytes isolated from the Papss2ΔHC mice (Figure 2G). Tyrosine sulfation is the hallmark of protein sulfation and as expected, the total sulfotyrosine level in the liver of Papss2ΔHC mice was substantially reduced as shown by immunofluorescence (Figure 2H). The unchallenged Papss2ΔHC mice were healthy and fertile without noticeable gross phenotypes, including gross appearance (Supplementary Figure 1B) and weight of major organs (Supplementary Figure 1C). We also detected a decreased sulfotyrosine in the liver of Papss2ΔHC mice treated with APAP as shown by western blotting (Supplementary Figure 1D).
Hepatocyte-specific knockout of Papss2 protects mice from the toxicity of APAP, paraquat, and thioacetamide
To investigate whether hepatocyte ablation of Papss2 affects APAP-induced liver injury, Papss2ΔHC mice and their Papss2fl/fl littermates were treated with a single dose of 200 mg/kg APAP after 16h fasting, and tissues were harvested 1, 3, 6 and 24h after the APAP administration. At 24h and compared to the expected liver injury in APAP-treated Papss2fl/fl mice, APAP-treated Papss2ΔHC mice exhibited little signs of liver toxicity as shown by histology (Figure 3A). The APAP-induced elevations of serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were also attenuated in Papss2ΔHC mice at 6 and 24h (Figure 3B). There were no appreciable differences in the liver histology (Figure 3A), and ALT and AST (Supplementary Figure 2A) between vehicle treated Papss2fl/fl and Papss2ΔHC mice. TUNEL staining showed that the APAP-induced nuclear DNA fragmentation was markedly decreased in the liver of Papss2ΔHC mice (Figure 3C). Papss2ΔHC mice also exhibited an improved survival rate than Papss2fl/fl mice when treated with a lethal dose (600 mg/kg) of APAP (Figure 3D). In an independent hepatocyte Papss2 knockout model, the knockout was induced by infecting Papss2fl/fl mice with AAV8-TBG-Cre for two weeks (Papss2iΔHC) before challenging with APAP (200 mg/kg) for 24h. Papss2iΔHC mice showed a similar pattern of protection from APAP-induced toxicity as shown by histology (Supplementary Figure 2B) and serum levels of ALT and AST (Supplementary Figure 2C).
Figure 3. Hepatocyte-specific knockout of Papss2 protects mice from the toxicity of APAP, paraquat, and thioacetamide.
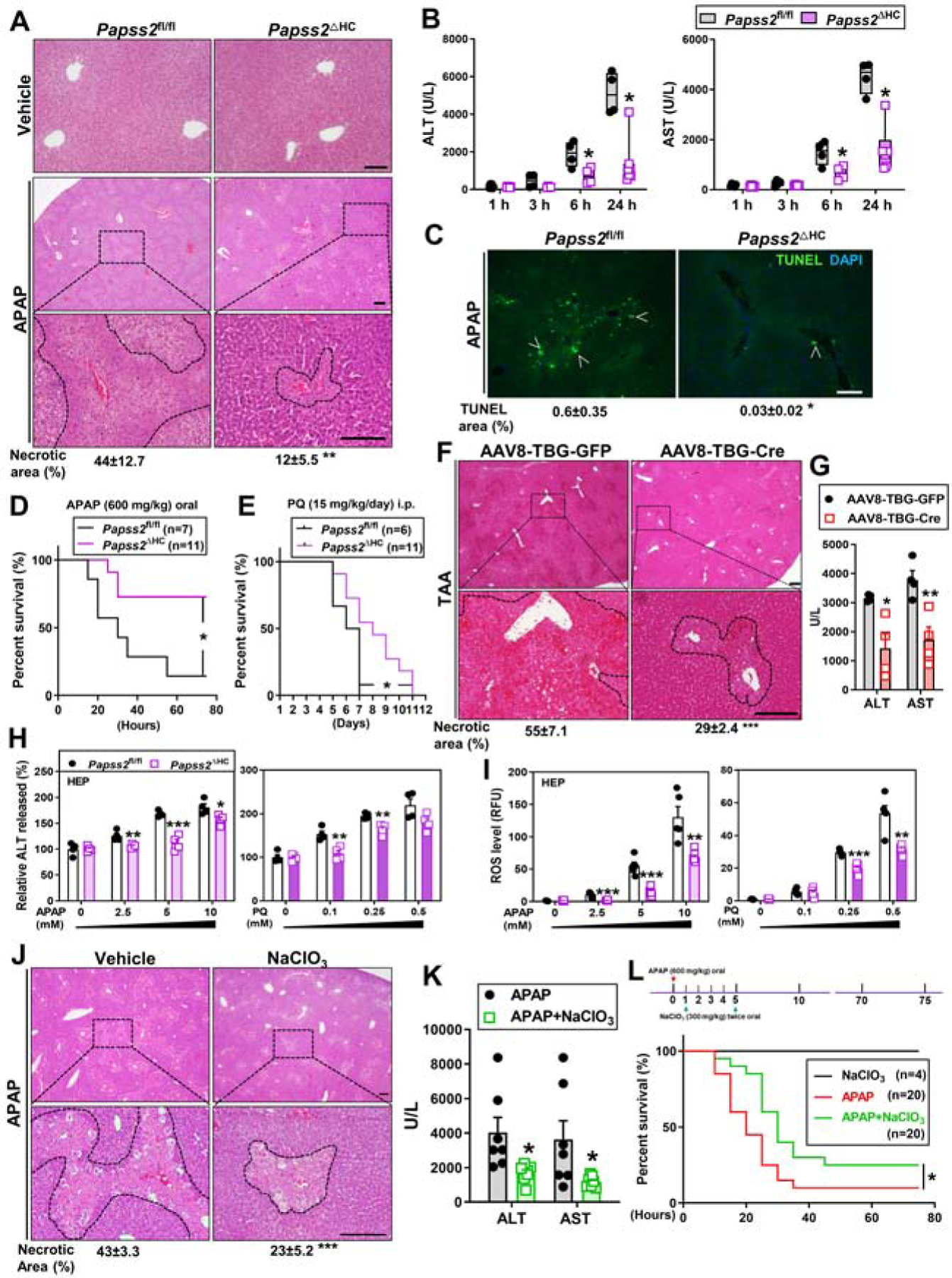
(A-C) Eight-week-old male Papss2fl/fl and Papss2ΔHC mice were treated with vehicle or APAP (200 mg/kg) for 1, 3, 6, or 24h. Shown are representative H&E staining of the liver sections (24h) with the quantifications of the necrotic areas shown below (scale bar: 100 μm. n=3–6) (A), serum levels of ALT and AST (B), representative images of TUNEL staining on the liver sections (24h). Arrowheads indicate positive staining with the quantifications shown below (scale bar: 100 μm.) (C). (D and E) Survival of mice subjected to oral APAP (600 mg/kg) (D) or daily i.p. injections of paraquat (PQ, 15 mg/kg) (E). (F and G) Eight-week-old male Papss2fl/fl mice were infected with AAV8-TBG-GFP or AAV8-TBG-Cre two weeks before being treated with TAA (300 mg/kg) for 24h. (n=4). Shown are representative H&E staining of the liver sections with the quantifications of the necrotic areas shown below (Scale bar: 100 μm) (F), and the serum levels of ALT and AST(G). (H and I) The percentage of ALT release (H) and relative ROS levels (I) in Papss2fl/fl and Papss2ΔHC primary hepatocytes treated with indicated doses of APAP (left) or PQ (right) for 24h. (n=4). (J and K) Eight-week-old WT male mice were treated with vehicle (saline) or NaClO3 (300 mg/kg, gavage twice at 2h before, and 2h after the APAP treatment) and treated with APAP (200 mg/kg) for 24h. (n=7). Shown are representative H&E staining of the liver sections with the quantifications of the necrotic areas shown below (Scale bar: 100 μm) (J), and serum levels of ALT and AST (K). (L) Survival of mice subjected to oral gavage of APAP (600 mg/kg), APAP + NaClO3 (300 mg/kg, gavage twice at 1h and 5h after the APAP gavage), or NaClO3 alone. Data are presented as box plots or mean ± SEM. *P < .05, **P < .01, ***P < .001. The Survival curves in (D, E and L) were analyzed by Log-rank (Mantel-Cox) test.
The Papss2ΔHC mice also showed protection against two other oxidative toxicants, paraquat (PQ) and thioacetamide (TAA). PQ is potent ROS inducer whose overdose leads to lethality in mice 24, whereas ROS induced by TAA results in hepatic damage 25. As shown in Figure 3E, Papss2ΔHC mice showed improved survival upon PQ challenge. Papss2iΔHC mice also exhibited protection against TAA-induced hepatotoxicity as shown by histology (Figure 3F) and serum levels of ALT and AST (Figure 3G). The protective effect of Papss2 ablation was also observed in vitro, as primary hepatocytes isolated from Papss2ΔHC mice showed decreased APAP-or PQ-responsive release of ALT (Figure 3H) and accumulation of ROS (Figure 3I).
Sodium chlorate (NaClO3) is a sulfation inhibitor 26. Treatment with a non-toxic dose of sodium chlorate (300 mg/kg) twice, dosed 2h before and 2h after the APAP (200 mg/kg) treatment, also protected WT mice from APAP-induced hepatotoxicity as shown by histology (Figure 3J) and serum levels of ALT and AST (Figure 3K). Moreover, post-APAP treatment of mice with sodium chlorate twice, 1h and 5h after the APAP treatment, improved the animal survival upon a lethal dose of APAP (600 mg/kg) (Figure 3L).
Hepatocyte-specific knockout of Papss2 enhances antioxidant capacity upon APAP challenge, and Nrf2 is required for the hepatoprotective effect of Papss2 ablation
Both APAP sulfation and oxidative response are involved in APAP hepatotoxicity 27, 28. To understand the mechanism by which Papss2 ablation protected mice from APAP-induced hepatotoxicity, we analyzed APAP metabolites in the liver after APAP treatment. In APAP-treated Papss2ΔHC mice, the content of APAP-sulfate was decreased as expected, whereas the levels of APAP-glucuronide and APAP-GSH were not significantly affected in the Papss2ΔHC mice (Figure 4A). We further analyzed APAP metabolites in the total urine of mice treated with APAP for 24h. The contents of APAP-Mer and APAP-Cys, two metabolites indicative of detoxification of the reactive metabolite NAPQI 15, were increased in Papss2ΔHC mice (Figure 4B). When the antioxidative capacity was evaluated, we found the hepatic total GSH (Total GSH=GSH + 2* glutathione disulfide (GSSG)), GSH content, and ratio of GSH/GSSG in APAP-treated Papss2ΔHC mice were significantly elevated (Figure 4C). A more efficient replenishment GSH was also observed in APAP-treated primary hepatocytes isolated from the Papss2ΔHC mice (Figure 4D). These results suggested that the APAP-treated Papss2ΔHC mice and hepatocytes had increased antioxidative capacity. Indeed, the hepatic mRNA expression of genes encoding enzymes responsible for GSH biosynthesis, including Gclc, Gclm, Gpx1, and their transcriptional activator Nrf2, was increased in the liver of APAP-treated Papss2ΔHC mice at 3h, 6h (Supplementary Figure 3A) and 24h (Figure 4E). The same pattern of increased protein expression of Nrf2, Gclc, and Gclm in APAP-treated Papss2ΔHC mice was confirmed by western blotting (Supplementary Figure 3B and Figure 3F). The increased mRNA (Supplementary Figure 3C) and protein (Supplementary Figure 3D) expression of Nrf2, Gclc, and Gclm was also observed in APAP-treated Papss2iΔHC mice. Consistent with the transcriptional induction of Nrf2 target genes, the recruitment of Nrf2 onto the Gclc and Gclm gene promoters was increased in the liver of APAP-treated Papss2ΔHC mice as shown by chromatin immunoprecipitation (ChIP) assay (Figure 4G). Increased Nrf2 nuclear translocation was demonstrated by immunofluorescence in the liver of APAP-treated Papss2ΔHC mice (Figure 4H), as well as in primary human hepatocytes treated with the sulfation inhibitor sodium chlorate (Figure 4I).
Figure 4. Knockout of Papss2 enhances antioxidant capacity upon APAP challenge, and Nrf2 is required for the hepatoprotective effect of Papss2 ablation.
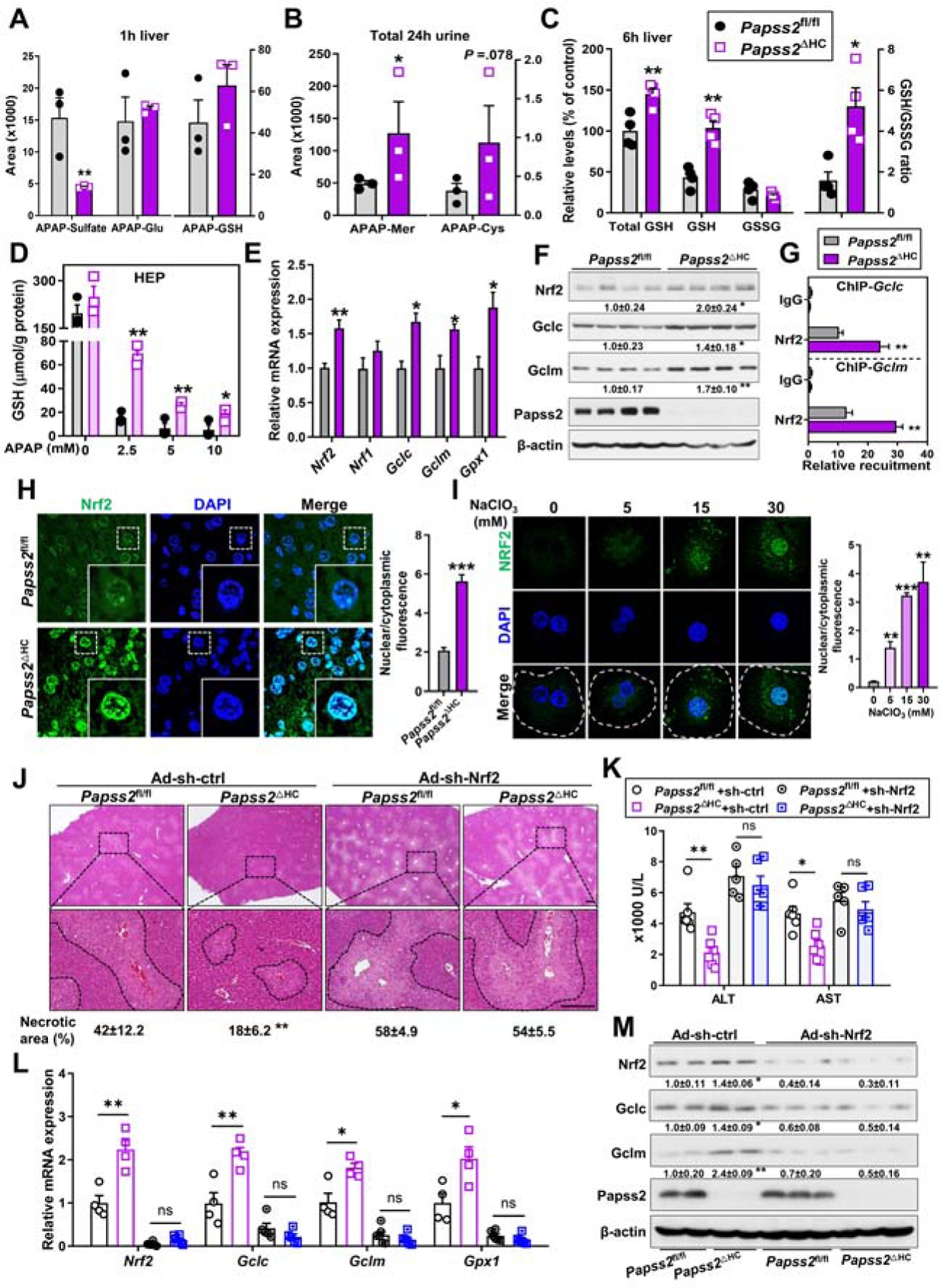
(A) The contents of APAP-sulfate, APAP-glucuronide and APAP-GSH in the livers of Papss2fl/fl and Papss2ΔHC mice treated with APAP (200 mg/kg) for 1h. (n=3). (B) The contents of APAP-Mer and APAP-Cys in the total 24h urine of mice treated with APAP (200 mg/kg). (n=3). (C) The relative hepatic total GSH, GSH, and GSSG levels, and GSH/GSSG ratio in mice treated with APAP (200 mg/kg) for 6h. (n=4). (D) The levels of GSH in primary mouse hepatocytes treated with different doses of APAP (0, 2.5, 5, 10 mM) for 24h. (E to G) Relative mRNA (E) and protein (F) expression of Nrf2 and its target genes, and recruitment of Nrf2 onto the Gclc and Gclm gene promotors as determined by ChIP (G) in the livers of mice treated with APAP for 24h. (n=3–4). The relative values of protein expression in the western blotting are labeled. (H and I) Representative immunofluorescent images of Nrf2 (green) in the liver sections of 6h APAP-treated mice (H) and in 6h APAP (5 mM)-treated primary human hepatocyte in the absence or presence of NaClO3 co-treatment (I). Shown on the right is the quantifications of nuclear to cytoplasmic ratio of Nrf2 fluorescence. Dotted lines indicate the outline of single cells. (J to M) Eight-week-old male Papss2fl/fl and Papss2ΔHC mice were infected with Ad-sh-Nrf2 or Ad-sh-ctrl one week before being treated with APAP (200 mg/kg) for 24h. (n=5–6). Shown are representative H&E staining of the liver sections with the quantifications of the necrotic areas shown below (Scale bar: 100 μm) (J), serum levels of ALT and AST(K), and relative mRNA (L) and protein (M) expression of Nrf2 and its target genes in the livers. The relative values of protein expression are labeled. Data are presented as mean ± SEM. *P < .05, **P < .01, ***P < .001.
Among the phase I and II drug-metabolizing enzymes known to be involved in APAP metabolism, the expression of Cyp2e1, Gstπ, and Gstμ was modestly but significantly increased in the liver of APAP-treated Papss2ΔHC mice (Supplementary Figure 4A). The expression of several nuclear receptors known to play roles in APAP hepatotoxicity, including constitutive androstane receptor (CAR) 29, pregnane X receptor (PXR) 30, 31, retinoid X receptor 32, farnesoid X receptor 33, and liver X receptor α 14, was not affected in Papss2ΔHC mice (Supplementary Figure 4B). Treatment of WT mice with sodium chlorate also induced the hepatic expression of Nrf2 and its target GSH synthesizing enzyme genes in response to APAP (Supplementary Figure 5A), with the expression pattern of phase I and II enzyme genes similar to that of APAP-treated Papss2ΔHC mice (Supplementary Figure 5B).
To determine whether Nrf2 is required for the hepatoprotective effect of Papss2 ablation, Papss2fl/fl and Papss2ΔHC mice were given a tail-vein injection of adenovirus expressing a Nrf2-targeting shRNA (Ad-sh-Nrf2) or the control shRNA (Ad-sh-ctrl) before being treated with a single dose of APAP (200 mg/kg) for 24h. The Ad-sh-ctrl infected Papss2ΔHC mice remained protected from APAP hepatotoxicity. In contrast, infection with Ad-sh-Nrf2 re-sensitized Papss2ΔHC mice to APAP-induced liver injury, as evidenced by increased histological liver necrosis (Figure 4J), and serum levels of ALT and AST (Figure 4K). Knockdown of Nrf2 also decreased the mRNA (Figure 4L) and protein (Figure 4M) expression of Nrf2 and its target genes.
The hepatoprotective effect of Papss2 ablation is p53 and p21 dependent
Interestingly, the hepatoprotective phenotype of APAP-treated Papss2ΔHC mice was accompanied by increased protein expression of the tumor suppressor gene p53 and its primary target gene p21 (Figure 5A). The accumulation of p53 and p21 proteins was also observed in Papss2ΔHC mice treated with vehicle, or APAP for 1, 3, and 6h (Supplementary Figure 6A). At the mRNA level, the expression of p21, but not p53 was induced in APAP-treated Papss2ΔHC mice (Figure 5B). The recruitment of p53 onto the p21 gene promoter was increased in the liver of APAP-treated Papss2ΔHC mice as shown by ChIP assay (Figure 5C). p21 has been reported to enhance Nrf2-mediated antioxidative response through its stabilization of the Nrf2 protein 34, 35. Thus, the Papss2ΔHC-p53-p21-Nrf2 axis provides a plausible mechanism by which Papss2 ablation protects mice from APAP hepatotoxicity.
Figure 5. Papss2 ablation activates the p53-p21-NRF2-antioxidant axis upon APAP challenge.
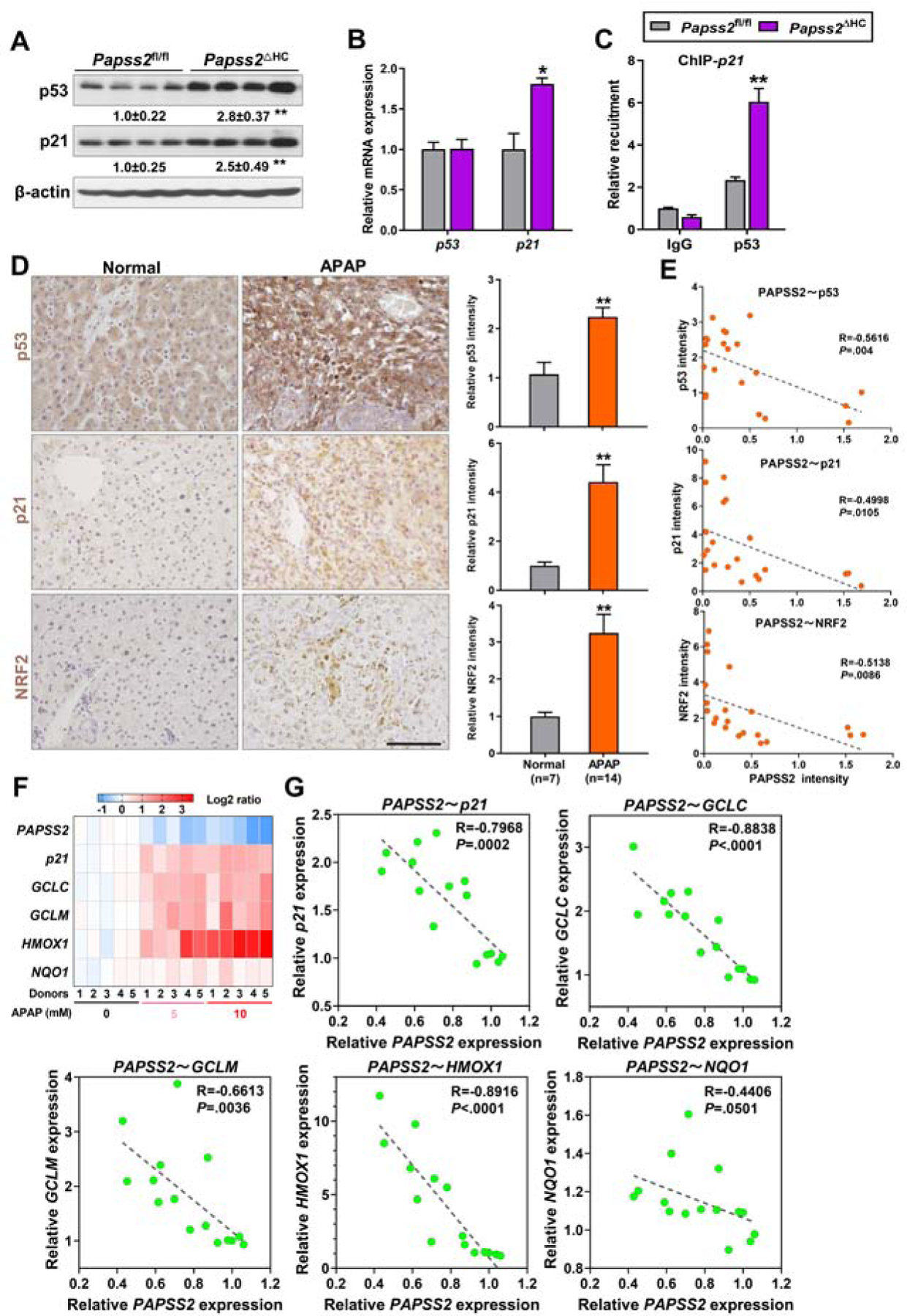
(A to C) Relative protein (A) and mRNA (B) expression of p53 and p21, and recruitment of p53 onto the p21 gene promotor as determined by ChIP (C) in the livers of 24h APAP-treated Papss2fl/fl and Papss2ΔHC mice. (n=3–4). The relative values of protein expression are labeled. (D) Representative IHC staining of p53, p21, and NRF2 in human liver sections of normal subjects or APAP overdose patients undergone liver transplantations. Shown on the right are the quantifications of relative IHC signals. The numbers of cases are labeled. Scale bars: 100 μm. (E) Correlations between the IHC intensity of PAPSS2 and p53, NRF2, and p21 in the same cohort of samples shown in (D). (F and G) Heat map comparing gene signatures (F) and correlations between the expression of PAPSS2 and p21, GCLC, GCLM, HMOX1 and NQO1 (G) in primary human hepatocytes treated with different doses of APAP (0, 5, 10 mM) (GSE13430). Data are presented as the mean ± SEM. *P < .05, **P < .01. The correlations in (E and G) were analyzed by Pearson correlation coefficient analysis.
The increased expression of p53, p21, and NRF2 was also observed in the livers of APAP overdose patients compared to the normal subjects as shown by immunohistochemistry (Figure 5D). In the same cohort of human liver samples, the immunostaining signal of PAPSS2 showed inverse correlations with those of p53, p21, and NRF2 (Figure 5E). Analysis of the GEO dataset GSE13465 showed that the suppression of PAPSS2 in APAP-treated human primary hepatocytes was also accompanied by increased expression of p21 and a panel of NRF2 responsive GSH biosynthesis genes (Figure 5F), whereas the expression of PAPSS2 showed inverse correlations with the expression of p21, GCLC, GCLM, HMOX1, and NQO1 (Figure 5G). In the pharmacological model, increased nuclear accumulation of p53 was observed in APAP-treated human primary hepatocytes co-treated with the sulfation inhibitor sodium chlorate (Supplementary Figure 7). p53 was reported to protect against APAP-induced hepatotoxicity via the acute suppression of JNK activation 36. Indeed, we observed an inhibition of JNK activation in Papss2ΔHC mice treated with APAP for 1, 3, or 6h (Supplementary Figure 3B), and knockdown of p53 restored the JNK activation in Papss2ΔHC mice (Supplementary Figure 6B). These results suggest that the inhibition of JNK activation may have contributed to the protective effect of Papss2 ablation on APAP-induced liver injury. Receptor interacting protein kinase 3 (RIP3) is a necroptosis mediator of APAP hepatotoxicity 37. We found an inhibition RIP3 expression in Papss2ΔHC mice treated with APAP for 6h (Supplementary Figure 3B), which was consistent with the protective phenotype.
To determine whether the expression and accumulation of p53 are required for the hepatoprotective effect of Papss2 ablation, Papss2fl/fl and Papss2ΔHC mice were given adenovirus expressing a p53-targeting shRNA (Ad-sh-p53) or Ad-sh-ctrl before being treated with APAP. Compared to mice infected with Ad-sh-ctrl, infection with Ad-sh-p53 abolished the protective effect of Papss2 ablation, as evidenced by the gross appearance and increased histological liver necrosis (Figure 6A) and increased serum levels of ALT and AST (Figure 6B). Knockdown of p53 also abolished the antioxidative benefits of Papss2 ablation, as evidenced by decreased hepatic total GSH and GSH content and the ratio of GSH/GSSG (Figure 6C), decreased mRNA (Figure 6D) and protein (Figure 6E) expression of p21, Nrf2 and GSH biosynthesis enzyme genes, and inhibition of the nuclear translocation of p53 and Nrf2 (Figure 6F).
Figure 6. The hepatoprotective effect of Papss2 ablation is p53 dependent.
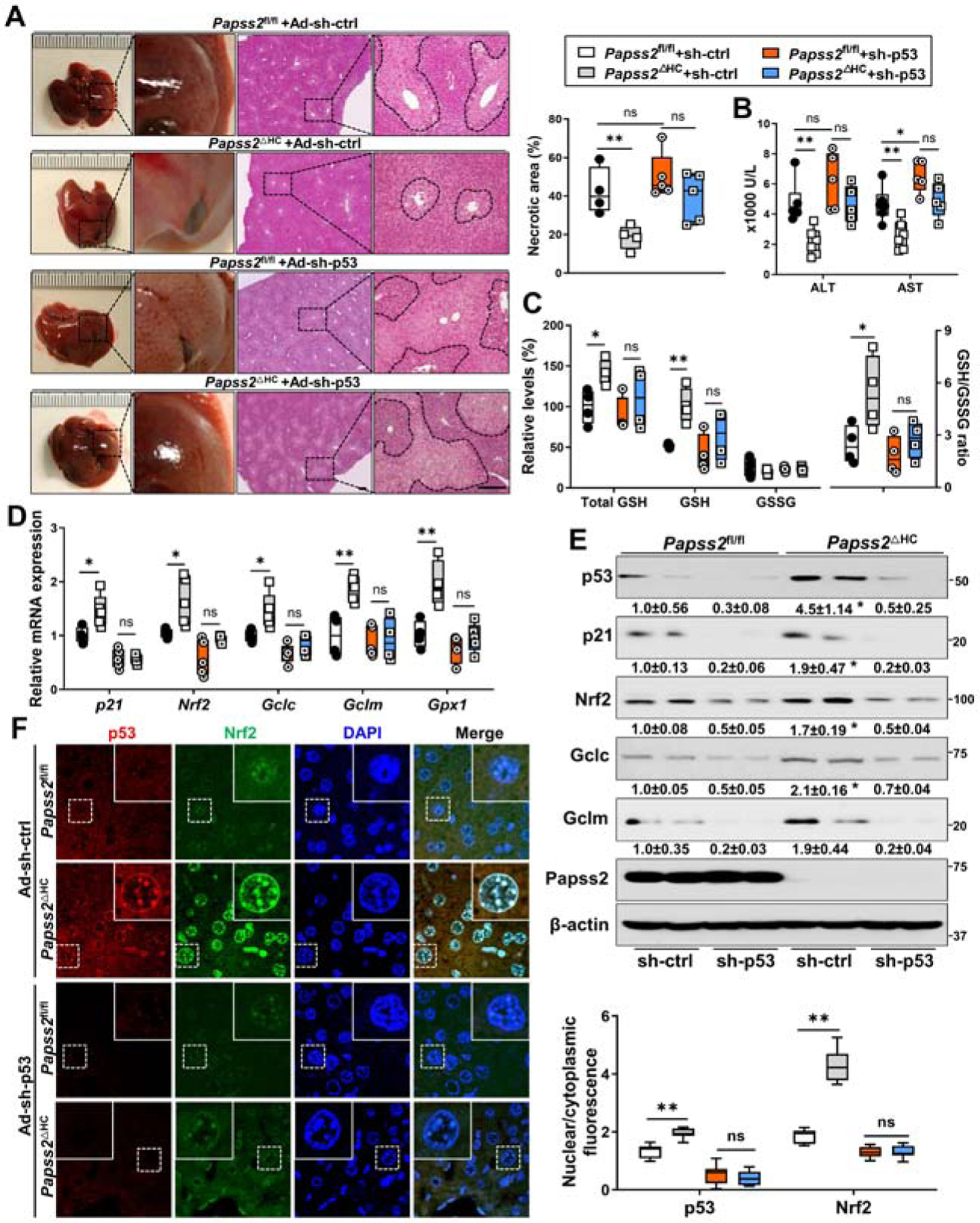
Eight-week-old Papss2fl/fl and Papss2ΔHC mice were infected with Ad-sh-p53 or Ad-sh-ctrl one week before being treated with APAP (200 mg/kg) for 24h. (n=4–6). (A) Representative gross appearance and H&E staining of the liver sections. Shown on the upper right is the quantifications of the necrotic areas. Scale bar: 100 μm. (B) Serum levels of ALT and AST. (C) The relative hepatic total GSH, GSH, and GSSG levels, and GSH/GSSG ratio in mice treated with APAP (200 mg/kg) for 6h. (D and E) Relative mRNA (D) and protein (E) expression of p53, p21, Nrf2 and its target genes in the livers. The relative values of protein expression are labeled. (F) Representative immunofluorescent images of p53 (red) and Nrf2 (green) in liver sections of Papss2fl/fl and Papss2ΔHC mice infected with Ad-sh-p53 or Ad-sh-ctrl one week before being treated with APAP for 6h. Shown on the lower right are the quantifications of the nuclear to cytoplasmic ratio of p53 and Nrf2 fluorescence. Data are presented as box plots or mean ± SEM. *P < .05, **P < .01.
We also showed the hepatoprotective effect of Papss2 ablation was p21 dependent. In this experiment, Papss2fl/fl and Papss2ΔHC mice were given adenovirus expressing a p21-targeting shRNA (Ad-sh-p21) or Ad-sh-ctrl before treatment with APAP. Compared to mice infected with Ad-sh-ctrl, infection with Ad-sh-p21 abolished the protective effect of Papss2 ablation, as evidenced by increased liver necrosis (Supplementary Figure 8A) and increased serum levels of ALT and AST (Supplementary Figure 8B). Knockdown of p21 also decreased the mRNA (Supplementary Figure 8C) and protein (Supplementary Figure 8D) expression of Nrf2 and its target genes.
p53 is a novel sulfation substrate, and Papss2 ablation accumulates p53 by blocking p53 sulfation at Tyr160, leading to the inhibition of p53-MDM2 interaction and p53 ubiquitination
The induction of p53 in Papss2ΔHC mice was at the protein level, but not at the mRNA level, suggesting that the accumulation of p53 was post-transcriptional. Knowing protein tyrosine sulfation is a post-translational modification important for protein interactions and signaling 3, 38, we speculated that p53 might be a novel sulfation substrate and sulfation of p53 may affect the stability of this protein. Indeed, immunoprecipitation followed by western blotting showed that in HEK293T cells, overexpression of PAPSS2 increased the tyrosine sulfation of p53 (Figure 7A), and this effect was abolished by the treatment of sodium chlorate (Figure 7B). In the liver of Papss2ΔHC mice, the tyrosine sulfation of p53 was decreased compared to the Papss2fl/fl controls (Figure 7C). MDM2 is a p53 interacting protein that negatively regulates the transcriptional activity and stability of p53 by ubiquitination 39. Co-IP analyses, using IP antibody of Mdm2 (Figure 7D, left) or p53 (Figure 7D, right) showed the binding between p53 and Mdm2 was decreased in the liver of Papss2ΔHC mice.
Figure 7. p53 is a novel sulfation substrate, and Papss2 ablation accumulates p53 by blocking p53 sulfation at Tyr160, leading to the inhibition of p53-MDM2 interaction and p53 ubiquitination.
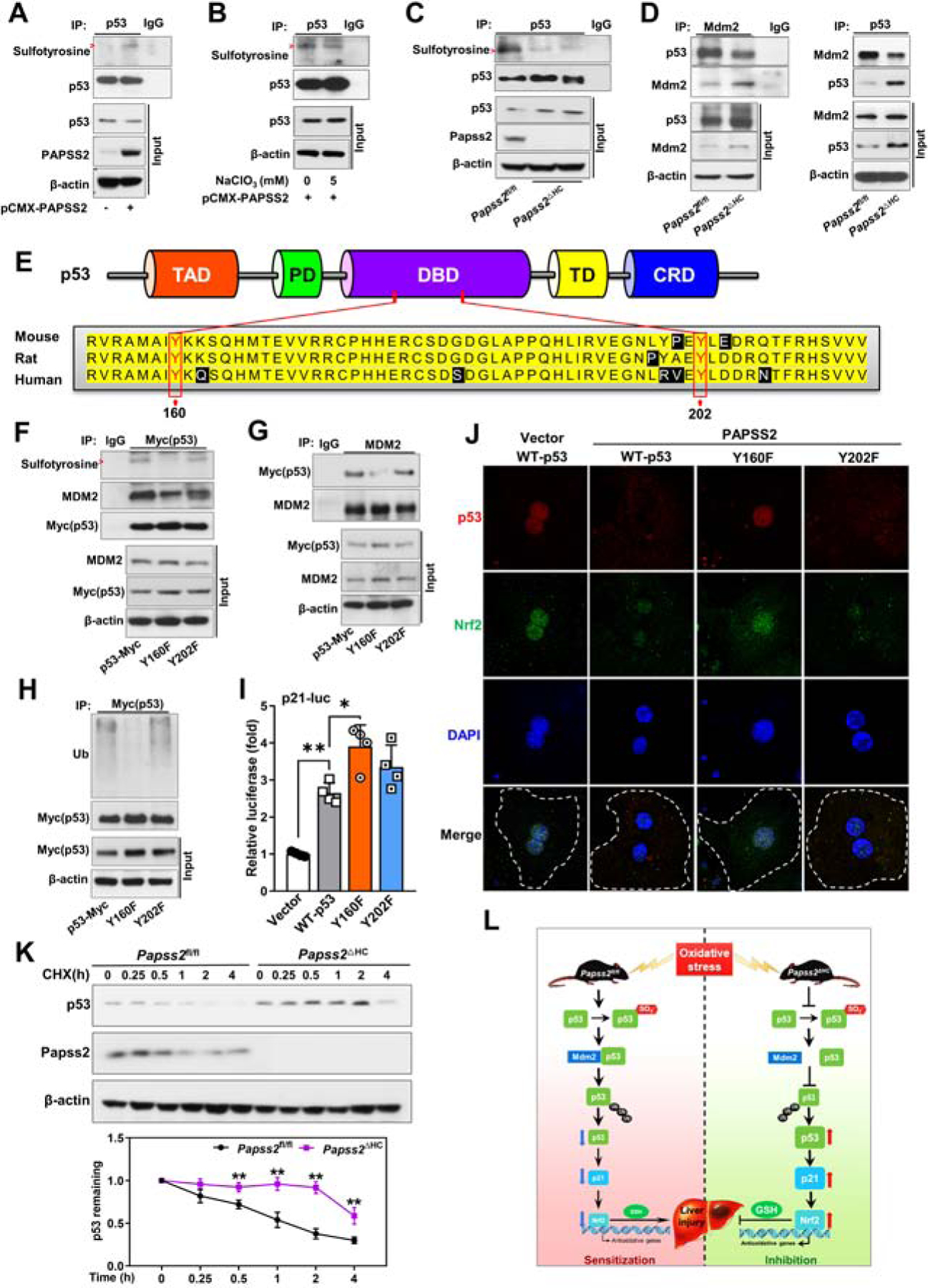
(A and B) Tyrosine sulfation of p53 was assessed by immunoprecipitation (IP) with a p53 antibody, followed by western blotting with an anti-sulfotyrosine antibody in HEK293T cells transfected with pCMX-PAPSS2 without (A) or with the treatment of NaClO3 (5 mM) (B) for 48h. (C) Tyrosine sulfation of p53 was assessed in the livers of Papss2fl/fl and Papss2ΔHC mice. (D) Protein interactions were determined by co-IP with Mdm2 (left) or p53 (right) antibody followed by western blotting. (E) Domain structures of p53 and the alignment of sequences. (F and G) Tyrosine sulfation of p53 and p53-MDM2 interaction in HEK293T cells co-transfected with pCMX-PAPSS2 and Myc-tagged WT p53, or Y160F, or Y202F mutant expression plasmid for 48h. The IP antibodies are anti-Myc (p53) and anti-MDM2 in (F) and (G), respectively. (H) Ubiquitination of Myc-tagged WT p53, or the Y160F or Y202F mutant in HEK293T cells co-transfected with pCMX-PAPSS2 and HA-ubiquitin for 24h and treated with MG132 (10 µM) for 4h. (I) Luciferase reporter assay in HEK293T cells transfected with the p21-luciferase reporter and WT p53, Y160F, or Y202F mutant expression vector. (n=4). (J) Representative immunofluorescent images of p53 (red) and Nrf2 (green) in primary mouse hepatocytes transfected with vector or pCMX-PAPSS2 and Myc-tagged WT p53, or Y160F, or Y202F mutant expression plasmid and treated with APAP (5 mM) for 6h. Dotted lines indicate the outline of single cells. (K) p53 protein stability was examined by western blotting in primary hepatocytes of indicated genotypes and treated with 100 µM cycloheximide (CHX) for indicated durations. (L) Proposed model by which inhibition of Papss2 protects mice from oxidative stress and APAP-induced acute liver failure. Red arrows indicate the sulfotyrosine bands in (A-C and F). Data are presented as mean ± SEM. *P < .05, **P < .01.
By using the SulfoSite prediction tool (http://sulfosite.mbc.nctu.edu.tw) 40, we predicted the tyrosine (Tyr) sulfation sites of the mouse p53 at Tyr160 and Tyr202. The domain structures of p53 and the Tyr160 and Tyr202 sites and the surrounding sequence in different species are outlined in Figure 7E. To confirm the tyrosine sulfation sites of p53, we over expressed Myc-tagged wild-type mouse p53, or its Y160F and Y202F mutants in HEK293T cells. Co-IP analysis showed that the Y160F mutant exhibited a noticeable decrease in sulfotyrosine of p53 compared to the WT and Y202F (Figure 7F). The binding between p53 and MDM2 was also decreased in the Y160F mutant (Figure 7F and 7G), which was accompanied by the decreased ubiquitination of Y160F (Figure 7H). At the functional level, Y160F, but not Y202F exhibited a transcriptional activity higher than the WT p53 in cells co-transfected with the p53-responsive p21 promoter-luciferase reporter gene p21-Luc (Figure 7I). Moreover, overexpression PAPSS2 inhibited WT p53, Y202F, and Nrf2 nuclear translocation in mouse primary hepatocytes treated with APAP, and this effect was blunted in Y160F transfected cells (Figure 7J), and a similar pattern of results was observed in the human hepatoma Huh7 cells (Supplementary Figure 9). These results suggested that Papss2 ablation may have increased the stability of p53 by blocking p53 sulfation at Tyr160, leading to the inhibition of p53-MDM2 interaction and p53 ubiquitination. Indeed, the stability of the endogenous p53 was increased in cycloheximide-treated primary hepatocytes isolated from Papss2ΔHC mice, compared to their Papss2fl/fl counterparts (Figure 7K). Figure 7L summarizes our proposed model by which inhibition of Papss2 protects mice from oxidative stress and APAP-induced acute liver failure.
Discussion
The most intriguing finding of ours is that the liver-specific Papss2ΔHC mice were protected from APAP-induced acute liver failure despite having a decreased APAP sulfation. The role of sulfation in APAP detoxification has been well documented 14,41. Results from the current study suggested that a decreased sulfation of APAP alone was not sufficient to lead to an APAP hypersensitivity. Interestingly, increased APAP sulfation was not sufficient to confer protection either. We recently reported that transgenic overexpression of the sulfotransferase SULT2B1b sensitized mice to APAP-induced liver injury, despite having an increased APAP sulfation. In contrast, Sult2B1b knockout mice showed resistance to APAP toxicity, but the mechanism remains to be defined 41.
In our Papss2ΔHC mouse model, we reason the hepatoprotective effect of Papss2 ablation was due to the inhibition of p53 sulfoconjugation, which led to the stabilization and accumulation of the p53 protein, and subsequent transcriptional activation of p21. p21 enhances antioxidant response through its stabilization of Nrf2 34, 35. Induction of p53 and p21 may enhance cell senescence42, but p53 has been reported to protect against APAP-induced hepatotoxicity in several studies36, 43. Of course, we cannot exclude the possibility that effect of p53 and p21 on hepatocyte senescence may have also contributed to the phenotypic exhibition. By using a series of loss of function models, we showed the hepatoprotective effect of Papss2 ablation was p53, p21, and Nrf2 dependent. Collectively, our results strongly suggested that the Papss2ΔHC-p53-p21-Nrf2-antioxidative axis is the mechanism underlying the protective effect of Papss2 ablation on APAP hepatotoxicity. This interpretation is consistent with the notion that oxidative stress plays a key role in the pathogenesis of APAP-induced liver injury 44, 45. The anti-oxidative phenotype of the hepatocyte-specific Papss2 knockout mice did not appear to be APAP-specific, because these mice were also protected from other two oxidative xenotoxicants PQ and TAA. Among limitations, we used adenovirus to knockdown p53 one week prior to APAP treatment. It will be interesting to know whether a pharmacological inhibition of p53 after APAP treatment can also abolish the protective effect of Papss2 ablation.
Post-translational modification is an efficient and precise mechanism to regulate p53 functions. Many types of p53 modifications have been reported, including phosphorylation, ubiquitination, sumoylation, neddylation, acetylation, methylation, O-GlcNAcylation, ADP-ribosylation, hydroxylation, and β-hydroxybutyrylation 46. To our knowledge, the current study provided the first evidence that p53 can be subjected to sulfoconjugation, and sulfation of p53 led to attenuation of antioxidant capacity and sensitization of mice to oxidative toxicants.
Our results are of high clinical relevance. A decreased expression of PAPSS2 was observed in APAP overdose patient livers and in APAP-treated primary human hepatocytes. These results were further verified by our bioinformatic analysis of the GEO datasets derived from human samples. Moreover, we found that the lower expression of PAPSS2 negatively correlated with the expression of p21 and NRF2 target antioxidant genes in APAP-treated primary human hepatocytes and in APAP overdose patients. We speculate that the decreased expression of PAPSS2 in APAP-induced hepatotoxicity and ALF may have represented a protective response to improve the host redox homeostasis, but that was not sufficient to overcome the overwhelming ALF caused by APAP overdose.
In summary, we have uncovered a novel function of PAPSS2 and p53 sulfoconjugation in antioxidative response and APAP-induced hepatotoxicity. Our results suggest that sulfation of p53 is required for sensitivity to APAP-induced acute liver failure. Inhibition of p53 sulfation is beneficial in preventing APAP toxicity through the up-regulation of the p53-p21-Nrf2 axis of antioxidative response.
Supplementary Material
What You Need to Know.
BACKGROUND AND CONTEXT
The PAPS synthase 2 (PAPSS2) is the key enzyme to synthesize the universal sulfate donor PAPS used for protein sulfation. Acetaminophen overdose is the leading cause of acute liver failure.
NEW FINDINGS
The expression of PAPSS2 is decreased in acetaminophen-induced acute liver failure. Knockout of Papss2 or inhibition of sulfation prevents acetaminophen hepatotoxicity through the up-regulation of the p53-p21-Nrf2 signaling axis.
LIMITATIONS
Future studies are necessary to determine whether and how PAPSS2 sensitizes oxidative hepatotoxicity in humans.
IMPACT
Hepatic expression of PAPSS2 is a biomarker indicative of redox homeostasis and antioxidant defense. Inhibition of sulfation, especially p53 sulfation, is a promising strategy for clinical management of acetaminophen overdose.
Acknowledgments:
We are thankful to Drs. Song Li, Da Yang, and Christian Fernandez and other individuals at the Center for Pharmacogenetics for their insightful comments and technical support. This work was supported in part by National Institutes of Health grant ES030429 (to WX). YX is a visiting student from Sun Yat-sen University supported by a Visiting Student Scholarship from the Government of China’s China Scholarship Council (File no. 201806380128). A portion of the human liver samples were obtained from the Clinical Biospecimen Repository and Processing Core of the Pittsburgh Liver Research Center, which is supported by NIH grant 1P30DK120531.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: None.
References
- 1.Lee WM. Acetaminophen (APAP) hepatotoxicity-Isn’t it time for APAP to go away? J Hepatol 2017;67:1324–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang YS, Wang CC, Chen BH, et al. Tyrosine sulfation as a protein post-translational modification. Molecules 2015;20:2138–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yeoh S, Bayliss R. New tools for evaluating protein tyrosine sulfation and carbohydrate sulfation. Biochem J 2018;475:3035–3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huttner WB. Protein Tyrosine Sulfation. Trends in Biochemical Sciences 1987;12:361–363. [Google Scholar]
- 5.Xie Y, Xie W. The Role of Sulfotransferases in Liver Diseases. Drug Metab Dispos 2020;48:742–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leung AW, Backstrom I, Bally MB. Sulfonation, an underexploited area: from skeletal development to infectious diseases and cancer. Oncotarget 2016;7:55811–55827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu ZH, Otterness DM, Freimuth RR, et al. Human 3′-phosphoadenosine 5′-phosphosulfate synthetase 1 (PAPSS1) and PAPSS2: gene cloning, characterization and chromosomal localization. Biochem Biophys Res Commun 2000;268:437–44. [DOI] [PubMed] [Google Scholar]
- 8.Chen D, Ni HM, Wang L, et al. p53 Up-regulated Modulator of Apoptosis Induction Mediates Acetaminophen-Induced Necrosis and Liver Injury in Mice. Hepatology 2019;69:2164–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Amar PJ, Schiff ER. Acetaminophen safety and hepatotoxicity--where do we go from here? Expert Opin Drug Saf 2007;6:341–55. [DOI] [PubMed] [Google Scholar]
- 10.Stravitz RT, Lee WM. Acute liver failure. Lancet 2019;394:869–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bernal W, Auzinger G, Dhawan A, et al. Acute liver failure. Lancet 2010;376:190–201. [DOI] [PubMed] [Google Scholar]
- 12.Lutkewitte AJ, Schweitzer GG, Kennon-McGill S, et al. Lipin deactivation after acetaminophen overdose causes phosphatidic acid accumulation in liver and plasma in mice and humans and enhances liver regeneration. Food Chem Toxicol 2018;115:273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee SS, Buters JT, Pineau T, et al. Role of CYP2E1 in the hepatotoxicity of acetaminophen. J Biol Chem 1996;271:12063–7. [DOI] [PubMed] [Google Scholar]
- 14.Saini SP, Zhang B, Niu Y, et al. Activation of liver X receptor increases acetaminophen clearance and prevents its toxicity in mice. Hepatology 2011;54:2208–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen C, Krausz KW, Idle JR, et al. Identification of novel toxicity-associated metabolites by metabolomics and mass isotopomer analysis of acetaminophen metabolism in wild-type and Cyp2e1-null mice. J Biol Chem 2008;283:4543–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torres S, Baulies A, Insausti-Urkia N, et al. Endoplasmic Reticulum Stress-Induced Upregulation of STARD1 Promotes Acetaminophen-Induced Acute Liver Failure. Gastroenterology 2019;157:552–568. [DOI] [PubMed] [Google Scholar]
- 17.Yoon E, Babar A, Choudhary M, et al. Acetaminophen-Induced Hepatotoxicity: a Comprehensive Update. J Clin Transl Hepatol 2016;4:131–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ni HM, McGill MR, Chao X, et al. Removal of acetaminophen protein adducts by autophagy protects against acetaminophen-induced liver injury in mice. J Hepatol 2016;65:354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaeschke H, Akakpo JY, Umbaugh DS, et al. Novel Therapeutic Approaches Against Acetaminophen-induced Liver Injury and Acute Liver Failure. Toxicol Sci 2020;174:159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kienhuis AS, van de Poll MC, Wortelboer H, et al. Parallelogram approach using rat-human in vitro and rat in vivo toxicogenomics predicts acetaminophen-induced hepatotoxicity in humans. Toxicol Sci 2009;107:544–52. [DOI] [PubMed] [Google Scholar]
- 21.Rodrigues RM, Govaere O, Roskams T, et al. Gene expression data from acetaminophen-induced toxicity in human hepatic in vitro systems and clinical liver samples. Data Brief 2016;7:1052–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramachandran P, Dobie R, Wilson-Kanamori JR, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019;575:512–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan J, Tung HC, Li S, et al. Aryl Hydrocarbon Receptor Signaling Prevents Activation of Hepatic Stellate Cells and Liver Fibrogenesis in Mice. Gastroenterology 2019;157:793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gong H, Singh SV, Singh SP, et al. Orphan nuclear receptor pregnane X receptor sensitizes oxidative stress responses in transgenic mice and cancerous cells. Mol Endocrinol 2006;20:279–90. [DOI] [PubMed] [Google Scholar]
- 25.Dwivedi DK, Jena GB. Simultaneous Modulation of NLRP3 Inflammasome and Nrf2/ARE Pathway Rescues Thioacetamide-Induced Hepatic Damage in Mice: Role of Oxidative Stress and Inflammation. Inflammation 2021. [DOI] [PubMed] [Google Scholar]
- 26.Safaiyan F, Kolset SO, Prydz K, et al. Selective effects of sodium chlorate treatment on the sulfation of heparan sulfate. J Biol Chem 1999;274:36267–73. [DOI] [PubMed] [Google Scholar]
- 27.Yamamoto A, Liu MY, Kurogi K, et al. Sulphation of acetaminophen by the human cytosolic sulfotransferases: a systematic analysis. J Biochem 2015;158:497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Du K, Ramachandran A, Jaeschke H. Oxidative stress during acetaminophen hepatotoxicity: Sources, pathophysiological role and therapeutic potential. Redox Biol 2016;10:148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J, Huang W, Chua SS, et al. Modulation of acetaminophen-induced hepatotoxicity by the xenobiotic receptor CAR. Science 2002;298:422–4. [DOI] [PubMed] [Google Scholar]
- 30.Guo GL, Moffit JS, Nicol CJ, et al. Enhanced acetaminophen toxicity by activation of the pregnane X receptor. Toxicol Sci 2004;82:374–80. [DOI] [PubMed] [Google Scholar]
- 31.Cheng J, Ma X, Krausz KW, et al. Rifampicin-activated human pregnane X receptor and CYP3A4 induction enhance acetaminophen-induced toxicity. Drug Metab Dispos 2009;37:1611–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dai G, Chou N, He L, et al. Retinoid X receptor alpha Regulates the expression of glutathione s-transferase genes and modulates acetaminophen-glutathione conjugation in mouse liver. Mol Pharmacol 2005;68:1590–6. [DOI] [PubMed] [Google Scholar]
- 33.Lee FY, de Aguiar Vallim TQ, Chong HK, et al. Activation of the farnesoid X receptor provides protection against acetaminophen-induced hepatic toxicity. Mol Endocrinol 2010;24:1626–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen W, Sun Z, Wang XJ, et al. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell 2009;34:663–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toledano MB. The guardian recruits cops: the p53-p21 axis delegates prosurvival duties to the Keap1-Nrf2 stress pathway. Mol Cell 2009;34:637–9. [DOI] [PubMed] [Google Scholar]
- 36.Huo Y, Yin S, Yan M, et al. Protective role of p53 in acetaminophen hepatotoxicity. Free Radic Biol Med 2017;106:111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramachandran A, McGill MR, Xie Y, et al. Receptor interacting protein kinase 3 is a critical early mediator of acetaminophen-induced hepatocyte necrosis in mice. Hepatology 2013;58:2099–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moore KL. Protein tyrosine sulfation: a critical posttranslation modification in plants and animals. Proc Natl Acad Sci U S A 2009;106:14741–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vassilev LT, Vu BT, Graves B, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004;303:844–8. [DOI] [PubMed] [Google Scholar]
- 40.Lee TY, Huang HD, Hung JH, et al. dbPTM: an information repository of protein post-translational modification. Nucleic Acids Res 2006;34:D622–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.An Y, Wang P, Xu P, et al. An Unexpected Role of Cholesterol Sulfotransferase and its Regulation in Sensitizing Mice to Acetaminophen-Induced Liver Injury. Mol Pharmacol 2019;95:597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jung SH, Lee HC, Yu DM, et al. Heparan sulfation is essential for the prevention of cellular senescence. Cell Death Differ 2016;23:417–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou Y, Fan X, Jiao T, et al. SIRT6 as a key event linking P53 and NRF2 counteracts APAP-induced hepatotoxicity through inhibiting oxidative stress and promoting hepatocyte proliferation. Acta Pharm Sin B 2021;11:89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaplowitz N, Than TA, Win S. Acetaminophen Hepatotoxicity: Strong Offense and Weakened Defense. Hepatology 2020;71:1530–1532. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Liu K, Zhang J, et al. c-Jun NH2-Terminal Protein Kinase Phosphorylates the Nrf2-ECH Homology 6 Domain of Nuclear Factor Erythroid 2-Related Factor 2 and Downregulates Cytoprotective Genes in Acetaminophen-Induced Liver Injury in Mice. Hepatology 2020;71:1787–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y, Tavana O, Gu W. p53 modifications: exquisite decorations of the powerful guardian. J Mol Cell Biol 2019;11:564–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.