

Abstract
Advances in clinical diagnostics and molecular tools have improved our understanding of the genetically heterogeneous causes underlying congenital anomalies of kidney and urinary tract (CAKUT). However, despite a sharp incline of CAKUT reports in the literature within the past two decades, there remains a plateau in the genetic diagnostic yield that is disproportionate with the accelerated ability to generate robust genome-wide data. Explanations for this observation include: (1) diverse inheritance patterns with incomplete penetrance and variable expressivity; (2) rarity of single-gene drivers such that large sample sizes are required to meet the burden of proof; and (3) multi-gene interactions that might produce either intra- (e.g. copy number variants) or inter- (e.g. effects in trans) locus effects. These challenges present an opportunity for the community to implement innovative genetic and molecular avenues to explain the missing heritability and to better elucidate the mechanisms that underscore CAKUT. Here, we review recent multidisciplinary approaches at the intersection of genetics, genomics, in vivo modeling and in vitro systems toward refining a blueprint for overcoming the diagnostic hurdles that are pervasive in urinary tract malformation cohorts. These approaches will not only benefit clinical management by reducing age at molecular diagnosis and prompting early evaluation for comorbid features, but will also serve as a springboard for therapeutic development.
Keywords: CAKUT, genomics, copy number variant, kidney organoid, zebrafish, embryonic development
Overview
Congenital anomalies of the kidney and urinary tract (CAKUT) are a commonly occurring group of birth defects that constitute the primary cause of end-stage renal disease (ESRD) in children. These anomalies occur in ~5 per 1,000 live births and represent 40%−50% of pediatric ESRD cases worldwide 1–6. CAKUT is also associated with increased incidence of chronic kidney disease (CKD), hypertension and cardiovascular disease with advanced age 7. We and others have shown that the risk of CKD increases over time among individuals with CAKUT and most cases show signs of renal impairment after the third decade of life 8–17. Individuals with CKD are 5–10 times more likely to die before reaching ESRD, and ESRD cases have a high cardiovascular mortality 18–22. Thus, understanding the pathobiology of CAKUT is paramount for developing new strategies to preserve kidney function, reduce cardiovascular-related morbidity, and ultimately diminish the burden that this population places on the health care system.
An entry point toward understanding the molecular underpinnings of CAKUT is the identification and functional assessment of genetic lesions contributing to the disorder (reviewed extensively elsewhere23–26). To date, genetic studies on CAKUT have been based predominantly on small cohorts or pedigrees, are often studied using targeted approaches, and have historically been restricted to monogenic inheritance paradigms 24. However, such approaches have been only partially successful toward explaining the genetic architecture hallmarked by CAKUT. Additionally, improved sequencing technologies and DNA array-based techniques have uncovered myriad of molecular insults ranging from single nucleotide variants (SNV) to copy number variants (CNV) encompassing tens to hundreds of genes. In this review, we summarize briefly the clinical and genetic complexity of CAKUT, and highlight recent combinatorial approaches which leverage large-scale genomic studies coupled to functional modeling in vertebrates and in vitro models. Together, these multidisciplinary approaches aim to overcome the limitations of traditional methodology and accelerate our understanding of CAKUT.
Introduction to CAKUT and the current state of the field
Clinical characteristics of CAKUT.
The clinical presentation of CAKUT is complex, from the standpoints of both multi-organ as well as multi-system involvement. More than 100 syndromes have been described in association with renal or urinary tract anomalies 27. Although various forms of CAKUT may appear as part of a systemic condition involving multi-organ manifestations, the majority of cases present nonsyndromic forms confined to the kidney and urinary tract 28. This observation suggests that aspects of genitourinary development are regulated by a repertoire of cell-type specific effectors. CAKUT can affect various parts of the kidney and urinary tract (Figure 1)29–35, including the renal parenchyma (agenesis, dysplasia), ureter (duplication, dilation, obstruction, reflux), bladder (exstrophy, diverticulum), or urethra (posterior urethral valves, PUV), all of which are governed by tight spatio-temporal developmental regulation that is sensitive to the hormonal and metabolic environment 36. Defects in urinary system morphogenesis range from alterations in the number, structure and/or position of the kidneys; obstructive or non-obstructive dilatation of the urinary tract; to dysplastic kidney lesions, including cystic disorders. Moreover, sex-specific differences in kidney development and, subsequently, in the prevalence of kidney disease and ESRD have been noted 9, 37–39. Therefore, it is not surprising that multiple signaling cascades and pathways are required for normal kidney development in a complex yet interactive fashion.
Figure 1. Schematic of the human kidney system depicting proximity and prevalence based distribution of congenital anomalies of kidney and urinary tract.
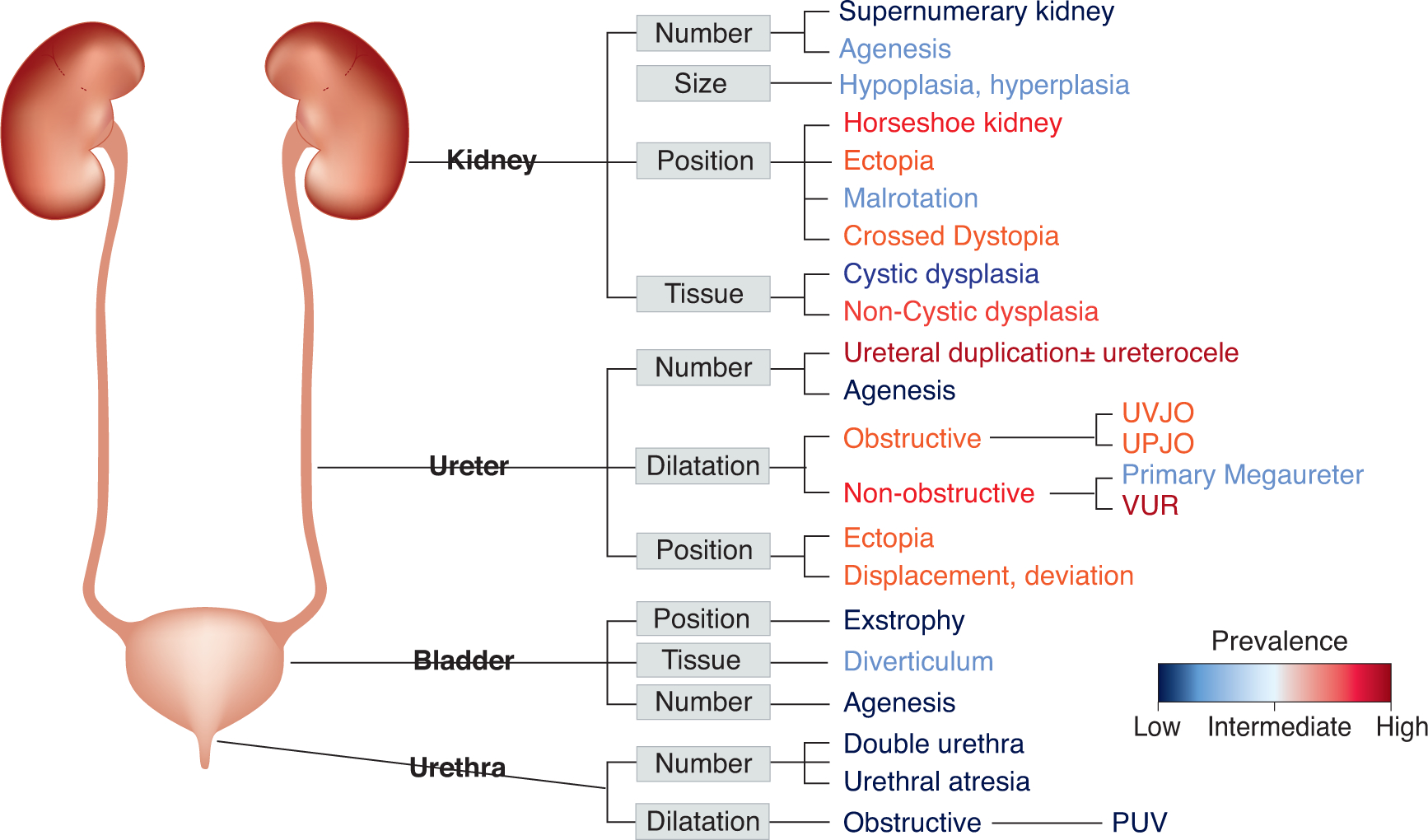
Topological subdivision of the phenotypic complexity of CAKUT delineated at the anatomical structural level. This classification is commonly used for clinical purposes. The coloring scale reflects the estimated prevalence of conditions represented in the figure on the basis of large scale cohort and autopsy study findings 30, 32, 35. The prevalence estimates of conditions range from rare (urethral atresia: 1 in 100,000)31, (bladder exstrophy: 1 in 50,000)34, to common (vesicoureteral reflux and duplicated ureter: 1/100)29, 33.
Variable penetrance and expressivity are reported within and across families who present with the condition. Phenotypic variability has been documented among family members with the same genetic defect, ranging from asymptomatic structural abnormalities to advanced kidney disease 40, 41. Diverse forms of CAKUT have also been observed among members of the same family, emphasizing the phenotypic complexity of the disease 42, 43. Moreover, monozygotic twins with phenotypically discordant CAKUT may serve as a model to study somatic mosaicism, stochastic effects, and epigenetic regulation 44, 45. The clinical heterogeneity of CAKUT sub-phenotypes poses significant challenges for human genetics studies aimed at the identification of genes underlying these conditions. While certain disease phenotypes seem to be enriched for rare variants in specific genes 46, 47, in the majority of cases the correlation between clinical presentation and the underlying genetic causal mutations is limited.
Genetic etiology of CAKUT: Twin, family, and cohort studies uncover novel candidates.
Genetic, environmental, or pharmacologic disruption of distinct stages in kidney and urinary tract development can cause the phenotypic spectrum observed in CAKUT 48 (Figure 1). Although several environmental factors, including angiotensin converting enzyme (ACE)-inhibitors or maternal diabetes and obesity 49–53 have links to CAKUT, a strong genetic predisposition to the disease is supported by the occurrence of familial forms 54–56. While most individuals with CAKUT are sporadic cases, the recurrence risk of CAKUT among relatives (15–20%) in several family-based studies indicates a strong genetic contribution to the pathogenesis of the disease 55, 57–60. One study reported CAKUT incidence as high as 51% among first-degree relatives, although this estimate is likely inflated due the high consanguinity of the cohort studied 59. Further, there are high concordance rates in twin and sibling studies for CAKUT phenotypes; for instance, concordance rates of almost 80% and 35% in monozygotic and dizygotic twins, respectively, are observed in primary vesicoureteral reflux (VUR) 61, 62.
Diverse inheritance patterns and single gene drivers.
Several lines of evidence support the role of single-gene disease drivers in both syndromic and nonsyndromic forms of CAKUT 55. To date, ~50 single-gene causes for isolated CAKUT and >100 syndromic forms of CAKUT caused by mutations in single genes are described in the Online Mendelian Inheritance in Man (OMIM) database (https://www.omim.org) 63, 64. Genes linked to Mendelian CAKUT were initially mapped using single or few, clinically ascertained families containing multiple affected individuals, which facilitated gene identification using positional cloning; however, a major caveat is that large families are rare, and the disease burden might affect the ability to procreate 65–67.
Knock-out mouse models manifesting a phenotypic spectrum that recapitulates human CAKUT have been developed 68–70, supporting the notion that a substantial proportion of both syndromic and non-syndromic CAKUT may be caused by single-gene defects in humans. However, monogenic forms of the disease seem to account for a small portion of cases and accurate heritability estimates in humans remain poorly deduced 53, 71–73. In family-based linkage analysis studies, multigenerational disease segregation suggested multifactorial or dominant inheritance with variable expressivity and reduced penetrance in most kindreds 74–77. Autosomal and X-linked recessive inheritance paradigms have also been implicated in human CAKUT 78–81.
However, the observation that a majority of CAKUT cases occur sporadically, suggests a role for either de novo mutations, recessive inheritance, or complex disease determination 53, 82.
The reduction in cost, rapid turnaround time, and advancements in bioinformatics approaches have fueled whole exome or whole genome sequencing (WES/WGS) efforts toward the discovery of new genes in sporadic and familial CAKUT 83–86. Next-generation sequencing studies have also influenced the revision of risk estimates and prevalence of genes previously thought to contribute to CAKUT incidence 5, 87. For example, in a study from the Netherlands an unexpectedly low incidence of PAX2 and HNF1B mutations was reported, previously thought to explain up to 5–15% of CAKUT cases 64, 82, 88. Nicolaou et al. analyzed 453 CAKUT patients, PAX2 was found to explain 8% and 0.7% of all cases of kidney dysplasia/hypoplasia and overall CAKUT, respectively, while HNF1B was only identified in 3% and 0.2% of multicystic dysplastic kidney (MCDK) and overall CAKUT, respectively 87. It is possible that the decrease from the original estimates of attributable risk of CAKUT genes is due to: (a) the “regression to the mean” phenomenon where, due to the selection of extreme phenotypes and random effect in discovery studies, there is over-estimation of effect size, and the subsequent risk measures are predicted to regress to the underlying population mean over time; (b) the expansion of sequencing efforts to a broad clinical phenotypic gamut of CAKUT, which is likely more complex than once thought; or (c) the estimates might be diluted by mild CAKUT cases that do not have an underlying genetic etiology. Hence, the attempts to establish population prevalence of mutations based on small and selected cohorts must be taken cautiously.
Multi-gene effects in CAKUT.
Case-control studies have unmasked unappreciated pleiotropic effects, genotype-phenotype correlations, and multi-locus interactions that would be challenging to uncover in single pedigrees. Recently, such approaches have been used to investigate the role of rare CNVs and SNVs in sporadic cases of CAKUT with complex genetic etiologies. Initial studies illustrated that up to 10–15% of kidney malformations are attributable to large rare CNVs 89–93. For example, evaluation of a modestly sized renal hypodysplasia (RHD) cohort offered insights into hitherto unknown pleiotropic effects. We detected pathogenic CNVs in 22.5% of individuals with syndromic malformations and 14.5% of individuals with isolated urinary-tract defects 90. Notably, the majority of known pathogenic CNVs detected in this cohort have reported involvement in developmental delay or neurocognitive disease. This observation not only suggests common molecular pathways in renal and neuronal development, but also bears clinical relevance since most kidney malformations are identified in utero, while cognitive deficits become apparent later in childhood. Thus, identification of a causal CNV in a child with CAKUT can also potentially improve the clinical management of neurocognitive defects. These data have now been validated in several additional studies 89, 92, 94–96.
Assembly of large cohorts has offered further resolution of CAKUT phenotype by CNV type. In a study designed to define the CNV landscape of CAKUT 96, we identified loci that were associated previously with a genomic disorder (GD-CNV) in a significantly enriched fraction of CAKUT patients (4% of 2,824 cases vs 0.6% of 21,498 controls). Our analysis showed that six well-known GD-CNVs account for 65% of patients with a GD-CNV, thus identifying major susceptibility loci for CAKUT. When we investigated the distribution of CNVs among different CAKUT phenotypes, we discovered that upper tract conditions (e.g. kidney agenesis or dysplasia) were associated predominantly with large deletions, whereas lower urinary tract phenotypes such as duplex kidney, VUR, or posterior urethral valves were enriched for duplications.
Genome wide association studies (GWAS) have also contributed to the identification of candidate susceptibility loci in association with some CAKUT phenotypes, including VUR97–99, hypospadias 100, and bladder exstrophy 101. Interestingly, the effect sizes detected for common variants in association with bladder exstrophy, hypospadias, and VUR were unusually large and suggest that moderately penetrant common variants may play a significant role in the predisposition to CAKUT 101. Together, the observation of incomplete penetrance, variable expressivity of disease, and possible complex inheritance in CAKUT posit that focusing on simple models of disease determination is inadequate to resolve the genetic underpinnings of disease.
Second-site modification can also influence CAKUT-related phenotype102–104. Humans with autosomal dominant polycystic kidney disease (ADPKD) exemplify this phenomenon, in which a transcriptional network, in addition to multiple causative genes, might explain onset and severity of polycystic kidney disease 105, 106. ADPKD cases with co-occurrence of HNF1B mutations manifest earlier onset of disease than individuals harboring hallmark PKD1 or PKD2 pathogenic variants in isolation106. Concordantly, a role for contributory modifier genes was demonstrated in mouse models of autosomal-recessive polycystic kidney disease (ARPKD)107. In the mouse, Hnf1b has been shown to bind specifically to the Pkhd1 promoter to stimulate transcription, and as a result, a dominant-negative Hnf1b mutant allele results in the inhibition of Pkhd1 promoter activity 108, 109. Thus, transgenic mice expressing the dominant-negative Hnf1b mutation under the control of a kidney-specific promoter develop kidney cysts 110. Collectively, these studies and similar exemplars provide a molecular basis for the observed variability in disease progression in cystic kidney disease 111.
Mouse models of CAKUT have elucidated mechanisms of kidney development.
Understanding CAKUT has been facilitated by the use of in vivo models, in particular, the mouse. Initial studies involving mouse models were limited to forward genetic approaches, however gene targeting (reverse genetics) strategies have both accelerated the validation of CAKUT associated genes, and also informed the role of these genes in kidney development and disease 70. Mouse models have proven to be a useful tool for identifying and investigating signaling pathways such as glial cell-line-derived neurotrophic factor (GDNF)-RET, renin-angiotensin system (RAS), Wnt, and fibroblast growth factor that mediate kidney and urinary tract development 112–116. Moreover, these animal models provide a spatiotemporally accessible context for defining the genomic and transcriptomic space in which susceptibility and modifier genes impact kidney development and disease.
While many single-gene knockout mouse models result in phenotypes that recapitulate human CAKUT, studies have reported discordance in the effect of genetic mutations on kidney phenotypes between the two species. For instance, mutations in angiotensinogen (AGT), renin (REN), angiotensin converting enzyme (ACE), or angiotensin II receptor type 1 (AGTR1) in humans are associated with autosomal recessive kidney tubular dysgenesis 117, 118. In mice however, mutations in RAS genes result in severe hydronephrosis, medullary hypoplasia and in some cases duplicated ureters, phenotypes not observed in humans with the same gene mutations 119, 120. Alternatively, it is possible that the variable genetic background of inbred mouse strains protect against disease severity or expressivity; for example C57BL/6 are resistant to several kidney phenotypes 70, 121.
Complementary approaches to elucidate the molecular pathogenesis of CAKUT
Current challenges to overcome.
Although the community has much to celebrate with regard to understanding the underpinnings of aberrant kidney development, substantial interpretive challenges remain in human genetic studies. (1) In Mendelian paradigms, private mutations in small pedigrees are insufficient to explain causality. (2) The limited size of many cohort-based association studies often prohibits genome-wide significance for candidate CAKUT loci. (3) For CNVs, driver genes or modulating loci within a CNV can only be assigned with genetic approaches through the identification of phenotypically similar cases with smaller overlapping CNVs or point mutations in single genes encompassed by the CNV. (4) Mutations in some loss-of-function intolerant genes are embryonic lethal in mice and thus limits their utility in studying kidney development and pathology. These challenges bring forth the opportunity to employ alternative research models.
An intermediate vertebrate model: the zebrafish.
The zebrafish (Danio rerio) is a robust vertebrate model for studying kidney development and modeling human kidney disease (reviewed elsewhere 122–124). Anatomical simplicity and rapid development of zebrafish provides the opportunity to study organs early during development 125. The zebrafish pronephros is formed by four days post fertilization (dpf) and can be visualized by either whole-mount immunostaining or with transgenic lines that elaborate kidney structures with fluorescent proteins driven by kidney-specific promoters 126. Importantly, zebrafish larvae display cell type similarity and structural resemblance of pronephric components with the mammalian glomerulus and proximal convoluted tubule (Figure 2A–C) 123, 126–128. Lower urinary tract structures are absent in zebrafish, however, surrogates in the distal anatomical regions can be used to model anomalies of the ureters, bladder, or urethra.
Figure 2. In-vivo and in-vitro models to study CAKUT.
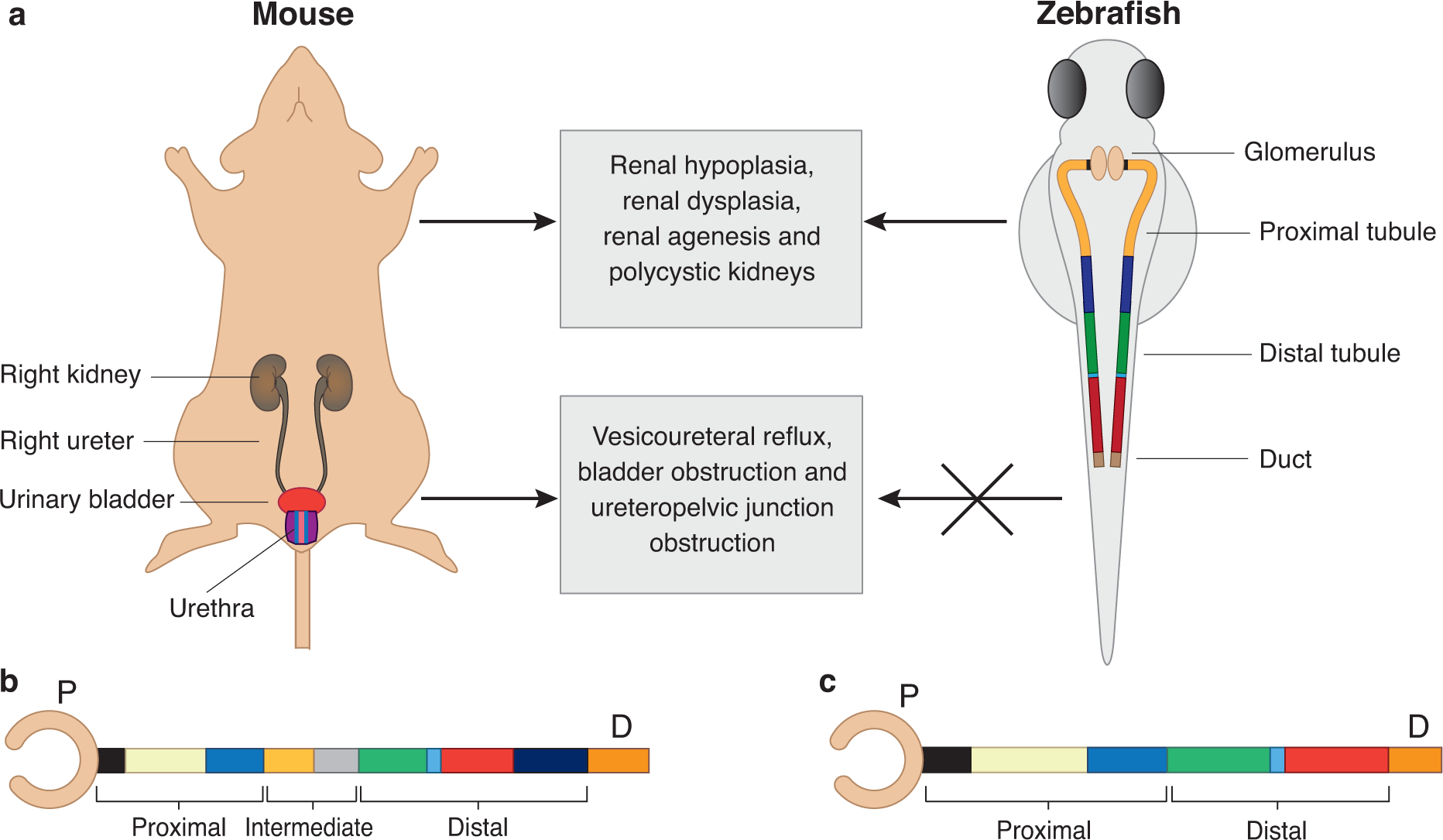
A. Schematics of mouse (left) and zebrafish (right) urinary system. Mammals have two symmetrical kidneys, each of which are connected to the urinary bladder through a collecting ducting (ureter). The mammalian kidney is composed of millions of kidney structural units known as nephrons. The zebrafish urinary system is comprised of two functional pronephric tubules but share structural and functional similarity with mammalian urinary system. The absence of lower urinary tract organs is a limitation of the zebrafish model.
B. Schematic of the mammalian kidney structural unit is represented as a straightened nephron tube. The glomerulus capsule (blood filter) is drawn as a pink cup-like sac structure. The tubule can be subdivided in to three segments including proximal, intermediate and distal tubule. The proximal tubule consists of the neck (black), proximal convoluted tubule (yellow) and proximal straight tubule (blue). The intermediate tubule consists of descending thin limb (light orange) and ascending thin limb (gray). The distal tubule consists of thick ascending limb (green), macula densa (light blue), distal convoluted tubule (red) and connecting tubule (indigo). The last segment of nephron consists of collecting duct (orange).
C. The zebrafish pronephros is drawn as straight tubule. The glomerulus capsule is represented as pink cup-like sac structure. The zebrafish tubule lacks the intermediate segment and contains only the proximal segment (neck, proximal convoluted tubule and proximal straight tubule; black, yellow and blue, respectively) and distal portion (distal early, corpuscle of stannius and distal late tubule; green, light blue and red, respectively). The last segment of nephron is the duct (orange, also known as cloaca).
Physiological relevance and assays of variant pathogenicity.
Zebrafish studies have supported a role for certain genes in both kidney development and disease. Many loci are involved in pathways such as GDNF-RET and Wnt 82. For instance, the transcription factor PAX2 (zebrafish orthologue, pax2a), which is part of the GDNF-RET pathway, has been shown to have an important role in zebrafish pronephros development through the ablation of the orthologous zebrafish locus 127, consistent with PAX2 mutation-bearing humans 129. Additionally, mutations in BMP4 and SIX2 (also GDNF-RET pathway genes) are associated with kidney hypodysplasia; studies carried out in zebrafish provided further evidence of disease association for these BMP4 variants 130.
For decades, the zebrafish molecular toolkit was restricted to labor-intensive forward genetics approaches 127. However, reverse genetics methods including morpholino-based knockdown and CRISPR/Cas9 genome editing accelerated the use of zebrafish as a tractable tool for modeling genetic findings in CAKUT 131–133. Accumulating studies have shown the utility of both stable mutant or transient suppression models for testing disease relevance and the direction of variant effect with in vivo complementation studies 85, 134–140. In a recent example, we showed that CRISPR/Cas9 F0 mosaic mutants or transient knockdown of the candidate gene greb1l in zebrafish results in proximal convolution tubule area reduction, recapitulating human phenotype 85. Additionally, we used this model to determine the pathogenicity of four missense variants identified in cases 85.
Dissection of CNVs.
CNVs are a major contributor to CAKUT 81, 89, 90, 93, 141–143. Recently, zebrafish have been utilized to identify both phenotype driver genes as well as intra-CNV genetic interactions. We reported a significant enrichment of heterozygous deletions of 22q11.2 (a.k.a. DiGeorge syndrome locus) in CAKUT subjects without overt clinical manifestations of DiGeorge syndrome, compared to controls 144. A minimal 370 kb region containing 9 protein-coding genes was exclusive to kidney defects. Seven of nine transcripts had an orthologous locus in the zebrafish genome. For four genes (lztr1, pi4ka, serpind1, and slc7a4), gene suppression models were indistinguishable from controls; however, for crkl, aifm3 and snap29 morphants or F0 mutants, we observed altered kidney convolution and pronephric tubule length. We also tested genetic interaction; pair-wise suppression of subeffective doses of aifm3 with snap29 exacerbated kidney phenotype in zebrafish but loss of crkl in combination with either of the other two genes resulted in no detectable additive or epistatic effects. Together, this study highlights zebrafish as a tractable model for CNV dissection.
Anatomical limitations and surrogate models of CAKUT in zebrafish.
Lower urinary tract structures including the urinary bladder are not present in zebrafish, thus making it difficult to model certain human disorders including VUR, bladder obstruction and ureterovesical junction obstruction. Even so, there are reports in which the zebrafish pronephros terminus (a putative homolog to end of mammalian nephric duct which inserts into the bladder) has been used to model lower urinary tract malformations 139, 145, 146. Recently, WNT3 was associated with severe human congenital urological malformations including bladder exstrophy-epispadias complex; in concordance with the human mutational data, knockdown of wnt3 in zebrafish resulted in cloaca abnormalities including expansion of the cloaca lumen at 3 dpf and 4 dpf 146. In other instances, the gene driver might not be abundantly expressed in the pronephros limiting functional modeling in zebrafish. For example, our recent discovery of high burden of 16p11.2 microdeletions in CAKUT and its attribution to TBX6 gene dosage 96, 143 required direct functional modeling studies in the mouse, since Tbx6 is not readily detectable in the zebrafish pronephros and no reliable readouts were available.
In sum, zebrafish are not perfect anatomical models of CAKUT. However, the functional conservation of certain genes allows for the testing of a genetic hypothesis generated from human data of an imperfectly matched anatomical structure.
Humanized tools: In vitro models of kidney pathology.
In some instances, in vivo vertebrate models are hampered by a lack of genomic conservation, or they cannot provide precise human cellular contexts. Pluripotent Stem Cells (PSCs) are able to differentiate into multiple kidney lineages with self-renewal capabilities when grown under defined conditions 147–150. PSCs constitute a wide variety of cell types including Embryonic Stem Cells (ESC) derived from the mammalian blastocyst stage and somatic cells reprogrammed to an embryonic-like-state (termed Induced Pluripotent Stem Cells; iPSCs) 151, 152. Human kidney tubular cells present in urine can also be used to generate human iPSCs which retain the cell origin epigenetic memory 147, 153.
Together these models can potentially provide a controlled environment to study kidney disease and explore underappreciated pathways involved in kidney development that might be intractable using in vivo models.
Compared to in vivo models, human-derived iPSCs (hiPSCs) may provide increased accuracy for modeling species- and individual-specific aspects of pathophysiology, which are not recapitulated in animal models. This is due to their ability to retain the entire genomic background in which the mutation arose, which is integral for modeling intricate genetic pathways and networks in diseases of multifactorial origin such as CAKUT 147. Genomic backgrounds affect the penetrance and expressivity of disease phenotypes. The use of hiPSC technology preserves such features and allows for the investigation of complex genetic mechanisms, which can be obscured by secondary effects of the genetic background in animal models 154, 155, 156.
Advancement in 3D culture conditions have made it possible to generate mini 3D organ structures termed as “organoids”, multicellular structures that have the capacity to self-organize in-vitro 157. Such methodology has overcome the limitations of 2D cultures, whereby branching of the ureteric bud was unattainable 158. Although recapitulation of the kidney’s structural and cell-type complexity in vitro is challenging, evolving strategies that induce ureteric phenotypes could facilitate modeling of specific CAKUT defects. Human iPSCs have been differentiated into kidney organoids, nephron like structures with characteristics of podocytes, distal and proximal tubules, and loops of Henle (Figure 3) 159–163. For example, induction of hiPSCs into either ureteric tip or nephron progenitors, followed by in vitro recombination, has been shown to generate distal nephron structures that fuse to collecting ducts 161. Additionally, genome editing in hiPSCs can target CAKUT-related genes and subsequently recapitulate human kidney phenotypes. For example, CRISPR/Cas9 disruption of polycystic kidney disease genes PKD1 or PKD2 in hiPSCs did not affect hiPSCs pluripotency or epithelial morphogenesis, and as a promising proof-of-principle, showed tubular expansion by cyst formation in kidney organoids 164, 165. Still, challenges regarding the tractability of kidney organoids in modeling human kidney diseases remain. Furthermore, the structural differences of in vitro tubular organoids from in vivo organ systems have made it difficult to conduct a wide range of functional assays which restrict, at present, the pathobiological hypotheses that can be tested 166, 167. However, efforts to optimize culture conditions are ongoing with the objective of expanding the use of these models 168–170. In the future, complementary studies of 3D tissue architecture with single cell multimodal analysis could help unravel molecular etiology and structural consequences of mutations that predispose to CAKUT.
Figure 3. In vitro models to study CAKUT.
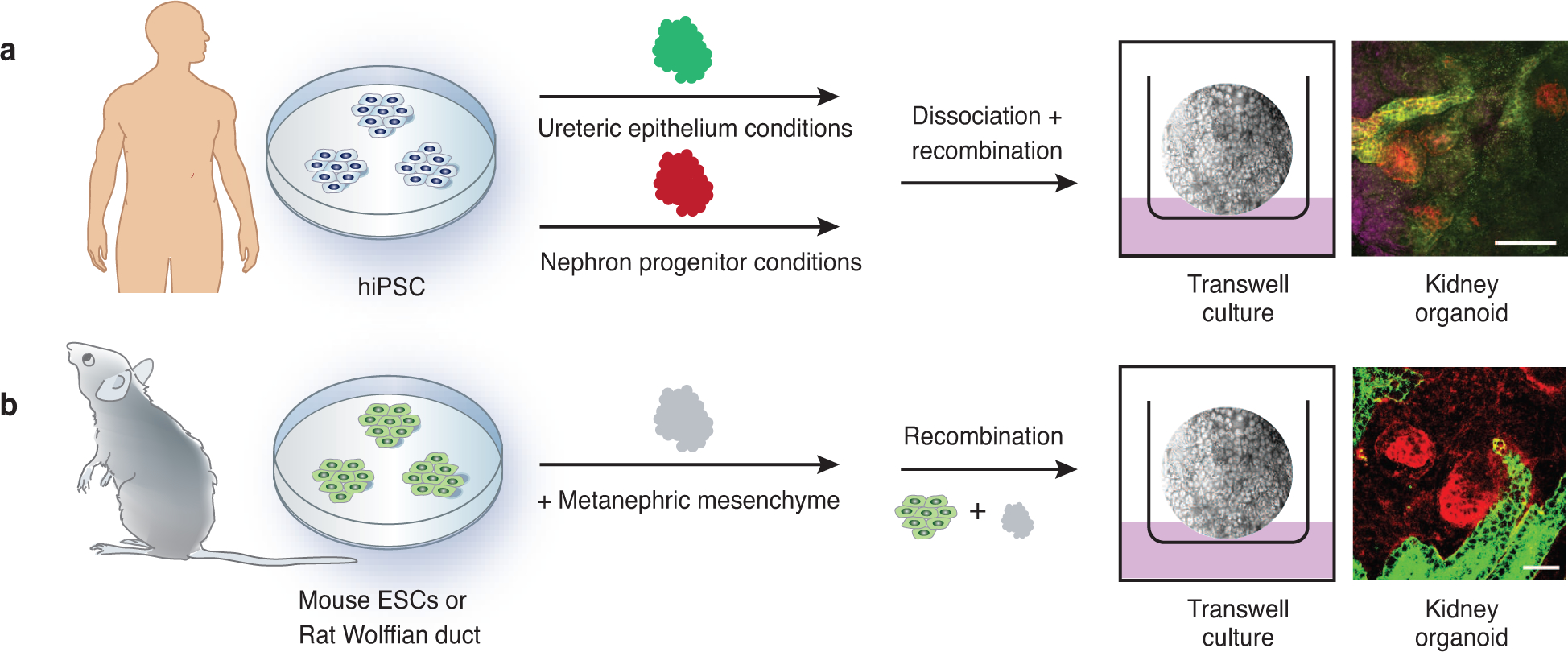
Kidney organoids can be generated from pluripotent stem cells derived from rodents or humans. A. Human fibroblasts or peripheral blood cells can be reprogrammed to generate human induced pluripotent stem cells (hiPSCs) that harbor patient-specific genetic variants. These can be differentiated into nephron/mesodermal progenitors or into ureteric epithelium. Recombination of these lineages in appropriate culture conditions generates cellularly complex 3D organoids. Far right: glomeruli are stained with podocalyxin (purple), proximal tubule epithelial cells are stained with Lotus tetragonolobus lectin (red), and ureteric epithelial cells are stained with E-cadherin (green). Scale bar: 100 μm. B. Similarly, rodent embryonic stem cells (ESCs) or Wolffian duct can be recombined with metanephric mesenchyme (MM) and cultured. Resultant hybrid organoids form segmented nephrons with functional structures. Far right: MM-derived epithelial cells are stained with peanut agglutinin lectin (red) and ureteric epithelial cells are stained with Dolichos biflorus (green). Scale bar: 50 μm; image courtesy of S.K. Nigam 161–163.
Future perspectives
Capturing the “missing heritability” of CAKUT.
For a small proportion of CAKUT cases there is a high-risk actionable genotype underlying the disease. However, for the majority of individuals, genetic risk factors are either elusive, or when present, account for a minimal incremental risk for disease, thus limiting their utility for genetic counseling and management 24, 171. Extending variant identification to systematic inclusion of a wider spectrum of variant types (e.g. protein coding and non-coding variants; single nucleotide variants (SNV) and structural variants (SV)) will address the issue of missing heritability as demonstrated in other complex disease studies 172–176. We anticipate that reduced costs will facilitate a transition from WES to WGS; this will reduce current approaches to a single platform capable of detecting genomic lesions within the coding and non-coding genomic space. Still, complex rearrangements will require more sophisticated methodologies to detect large numbers of breakpoints in a single event (multi-breakpoint SVs) or structurally polymorphic loci with multiple allelic formations (multi-allelic SVs) 177–182. Recently, two platforms that offer single molecule sequencing with long-fragment reads and improved throughput have emerged: (1) Single Molecule Real-Time sequencing (SMRT) from PacBio and, (2) Oxford Nanopore’s MinION, a real time nanopore-based DNA sequencing instrument 183–186. The utility of these technologies has been demonstrated in ways including the reconstruction of long-range haplotypes and the enhanced mapping of complex SV 186–191.
Investigation of non-coding regulatory variants has also contributed to greater understanding of the genetic landscape of several complex genetic diseases 181, 192–196. Regulatory variants play crucial roles in transcriptional regulation by modulating transcriptional factor binding, chromatin states as well as epigenetic modifications 197–202. Combined information from WGS and transcriptomic data from relevant cell types can inform pathogenicity and impact of non-coding variants on mRNA quantity and mRNA splicing 203, 204. One persistent challenge for CAKUT is to obtain the pertinent urinary tract tissue for study since biopsies or surgical resection of kidney and urinary tract are rarely conducted in these patients. Even when a nephrectomy or surgery for correction of a ureteric or urethra defect are performed, the collected specimen is of limited utility due to the temporal window at which it is collected (post-natal rather than at the relevant in utero developmental timepoint). Further, tissue quality is often compromised by secondary changes like fibrosis or infection and inflammation. The use of patient-derived iPSCs in conjunction with genomic analysis will likely overcome these hurdles. Another challenge is to obtain the ideal cell type. Bulk RNA-sequencing is confined to describing the average transcriptional profile across a heterogeneous population of cells, which may obscure signals of interest pertaining to specific cell types. The application of single-cell profiling is anticipated to provide greater understanding of specific kidney cell profiles while revealing novel regulatory mechanisms that underlie cell functions 205–210. These approaches offer specificity by avoiding biologically-relevant variability at the individual cell level and enable reduced cost by averaging across large and diverse cell populations, such as those present in the kidney.
Clinical and translational possibilities for CAKUT
While utilizing rapidly evolving technologies in clinical practice enhances diagnostic capabilities and precision medicine provision, it also creates opportunities for complications that have not been foreseen until now. The perceived benefits present a source for concern, particularly in relation to the paucity of knowledge related to clinical actionability of variants identified, particularly those of unknown significance that have not been reported previously or that are present at low frequencies in the general population and display incomplete penetrance. Evidence supporting clinical actionability for most genetic disorders including kidney pathologies varies 211. The need to devise and implement standardized, evidence-based approaches to accurately characterize the clinical actionability of genomic findings is of particular importance for CAKUT, because almost all patients are now diagnosed in utero while the long-term outcome may remain unpredictable for decades. CAKUT patients are currently at risk of under-detection of severe CKD as well as overtreatment of relatively mild kidney disease, which burdens an individual’s health and also society. The actionability profiles of CAKUT disorders is foreseen to facilitate genetic counseling and improvement of health care decisions. For instance, genetic variants potentially mediate gene functions related to drug absorption, distribution, metabolism, and excretion 212–215. Consequently, genetic associations identified in CAKUT may suggest therapeutic development strategies. However, the direct translation of genomic findings into effective treatments has only been demonstrated for a small subset of genetic variants identified 216, 217. Ultimately, development of multidisciplinary systems capable of integrating and converting genetic data into potential drug candidates may help researchers to uncover novel drug targets to improve kidney function survival of patients.
Acknowledgements
This work was supported by NIH/NIDDK grants R01DK103184, R01DK115574, P20DK116191, R21DK098531, and UL1 TR000040 (to S.S-C.) and R01DK072301 and R01HD042601 (to E.E.D.). K.K. was funded by an International Research Support Initiative Program fellowship from the Higher Education Commission of Pakistan. R.W. is funded by a Consortium grant of the Dutch Kidney Foundation (20OC002). E.E.D. is the Ann Marie and Francis Klocke, MD Research Scholar.
Footnotes
Competing interests
N.K. is a paid consultant and holds founder stock in Rescindo Therapeutics. The other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ardissino G, Dacco V, Testa S, et al. Epidemiology of chronic renal failure in children: data from the ItalKid project. Pediatrics 2003; 111: e382–387. [DOI] [PubMed] [Google Scholar]
- 2.Esbjorner E, Berg U, Hansson S. Epidemiology of chronic renal failure in children: a report from Sweden 1986–1994. Swedish Pediatric Nephrology Association. Pediatr Nephrol 1997; 11: 438–442. [DOI] [PubMed] [Google Scholar]
- 3.Harambat J, van Stralen KJ, Kim JJ, et al. Epidemiology of chronic kidney disease in children. Pediatr Nephrol 2012; 27: 363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White A, Wong W, Sureshkumur P, et al. The burden of kidney disease in indigenous children of Australia and New Zealand, epidemiology, antecedent factors and progression to chronic kidney disease. J Paediatr Child Health 2010; 46: 504–509. [DOI] [PubMed] [Google Scholar]
- 5.Nigam A, Knoers N, Renkema KY. Impact of next generation sequencing on our understanding of CAKUT. Semin Cell Dev Biol 2019; 91: 104–110. [DOI] [PubMed] [Google Scholar]
- 6.Zhou X, Wang Y, Shao B, et al. Molecular diagnostic in fetuses with isolated congenital anomalies of the kidney and urinary tract by whole-exome sequencing. J Clin Lab Anal 2020; 34: e23480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.San Agustin JT, Klena N, Granath K, et al. Genetic link between renal birth defects and congenital heart disease. Nat Commun 2016; 7: 11103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Westland R, Abraham Y, Bokenkamp A, et al. Precision of estimating equations for GFR in children with a solitary functioning kidney: the KIMONO study. Clin J Am Soc Nephrol 2013; 8: 764–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Westland R, Schreuder MF. Gender differences in solitary functioning kidney: do they affect renal outcome? Pediatr Nephrol 2014; 29: 2243–2244. [DOI] [PubMed] [Google Scholar]
- 10.Westland R, Schreuder MF, Bokenkamp A, et al. Renal injury in children with a solitary functioning kidney--the KIMONO study. Nephrol Dial Transplant 2011; 26: 1533–1541. [DOI] [PubMed] [Google Scholar]
- 11.Westland R, Schreuder MF, Ket JC, et al. Unilateral renal agenesis: a systematic review on associated anomalies and renal injury. Nephrol Dial Transplant 2013; 28: 1844–1855. [DOI] [PubMed] [Google Scholar]
- 12.Westland R, Schreuder MF, van der Lof DF, et al. Ambulatory blood pressure monitoring is recommended in the clinical management of children with a solitary functioning kidney. Pediatr Nephrol 2014; 29: 2205–2211. [DOI] [PubMed] [Google Scholar]
- 13.Westland R, Schreuder MF, van Goudoever JB, et al. Clinical implications of the solitary functioning kidney. Clin J Am Soc Nephrol 2014; 9: 978–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Westland R, van Wijk JA, Schreuder MF. The reason why mother nature provided us with two kidneys: the risks of a congenital solitary functioning kidney. Nephrol Dial Transplant 2012; 27: 2603–2604. [DOI] [PubMed] [Google Scholar]
- 15.Sanna-Cherchi S, Ravani P, Corbani V, et al. Renal outcome in patients with congenital anomalies of the kidney and urinary tract. Kidney Int 2009; 76: 528–533. [DOI] [PubMed] [Google Scholar]
- 16.Westland R, Kurvers RA, van Wijk JA, et al. Risk factors for renal injury in children with a solitary functioning kidney. Pediatrics 2013; 131: e478–485. [DOI] [PubMed] [Google Scholar]
- 17.La Scola C, Ammenti A, Puccio G, et al. Congenital Solitary Kidney in Children: Size Matters. J Urol 2016; 196: 1250–1256. [DOI] [PubMed] [Google Scholar]
- 18.Johns T, Jaar BG. U.S. Centers for Disease Control and Prevention launches new chronic kidney disease surveillance system website. BMC Nephrol 2013; 14: 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tesar V [Cardiovascular complications in patients with chronic renal insufficiency and chronic kidney failure]. Vnitr Lek 2003; 49: 383–387. [PubMed] [Google Scholar]
- 20.Wright J, Hutchison A. Cardiovascular disease in patients with chronic kidney disease. Vasc Health Risk Manag 2009; 5: 713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Collins AJ, Li S, Gilbertson DT, et al. Chronic kidney disease and cardiovascular disease in the Medicare population. Kidney Int Suppl 2003: S24–31. [DOI] [PubMed] [Google Scholar]
- 22.Chesnaye NC, Schaefer F, Bonthuis M, et al. Mortality risk disparities in children receiving chronic renal replacement therapy for the treatment of end-stage renal disease across Europe: an ESPN-ERA/EDTA registry analysis. Lancet 2017; 389: 2128–2137. [DOI] [PubMed] [Google Scholar]
- 23.Bodria M, Sanna-Cherchi S. [Genetic Basis of Congenital Anomalies of the Kidney and Urinary Tract]. G Ital Nefrol 2015; 32 Suppl 64. [PubMed] [Google Scholar]
- 24.Sanna-Cherchi S, Westland R, Ghiggeri GM, et al. Genetic basis of human congenital anomalies of the kidney and urinary tract. J Clin Invest 2018; 128: 4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van der Ven AT, Vivante A, Hildebrandt F. Novel Insights into the Pathogenesis of Monogenic Congenital Anomalies of the Kidney and Urinary Tract. J Am Soc Nephrol 2018; 29: 36–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Westland R, Renkema KY, Knoers N. Clinical Integration of Genome Diagnostics for Congenital Anomalies of the Kidney and Urinary Tract. Clin J Am Soc Nephrol 2020; 16: 128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taal MW, Brenner BM, Rector FC. Brenner & Rector’s the kidney, 9th edn. Elsevier/Saunders: Philadelphia, PA, 2012. [Google Scholar]
- 28.Song R, Yosypiv IV. Genetics of congenital anomalies of the kidney and urinary tract. Pediatr Nephrol 2011; 26: 353–364. [DOI] [PubMed] [Google Scholar]
- 29.Arumugam S, Subbiah NK, Mariappan Senthiappan A. Double Ureter: Incidence, Types, and Its Applied Significance-A Cadaveric Study. Cureus 2020; 12: e7760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barakat AJ, Drougas JG. Occurrence of congenital abnormalities of kidney and urinary tract in 13,775 autopsies. Urology 1991; 38: 347–350. [DOI] [PubMed] [Google Scholar]
- 31.Brownlee E, Wragg R, Robb A, et al. Current epidemiology and antenatal presentation of posterior urethral valves: Outcome of BAPS CASS National Audit. J Pediatr Surg 2019; 54: 318–321. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez MM. Congenital Anomalies of the Kidney and the Urinary Tract (CAKUT). Fetal Pediatr Pathol 2014; 33: 293–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sargent MA. What is the normal prevalence of vesicoureteral reflux? Pediatr Radiol 2000; 30: 587–593. [DOI] [PubMed] [Google Scholar]
- 34.Siffel C, Correa A, Amar E, et al. Bladder exstrophy: an epidemiologic study from the International Clearinghouse for Birth Defects Surveillance and Research, and an overview of the literature. Am J Med Genet C Semin Med Genet 2011; 157C: 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshida J, Tsuchiya M, Tatsuma N, et al. Mass screening for early detection of congenital kidney and urinary tract abnormalities in infancy. Pediatr Int 2003; 45: 142–149. [DOI] [PubMed] [Google Scholar]
- 36.dos Santos Junior AC, de Miranda DM, Simoes e Silva AC. Congenital anomalies of the kidney and urinary tract: an embryogenetic review. Birth Defects Res C Embryo Today 2014; 102: 374–381. [DOI] [PubMed] [Google Scholar]
- 37.Iseki K Gender differences in chronic kidney disease. Kidney Int 2008; 74: 415–417. [DOI] [PubMed] [Google Scholar]
- 38.Carrero JJ. Gender differences in chronic kidney disease: underpinnings and therapeutic implications. Kidney Blood Press Res 2010; 33: 383–392. [DOI] [PubMed] [Google Scholar]
- 39.Cobo G, Hecking M, Port FK, et al. Sex and gender differences in chronic kidney disease: progression to end-stage renal disease and haemodialysis. Clin Sci (Lond) 2016; 130: 1147–1163. [DOI] [PubMed] [Google Scholar]
- 40.Schwaderer AL, Bates CM, McHugh KM, et al. Renal anomalies in family members of infants with bilateral renal agenesis/adysplasia. Pediatr Nephrol 2007; 22: 52–56. [DOI] [PubMed] [Google Scholar]
- 41.Kerecuk L, Sajoo A, McGregor L, et al. Autosomal dominant inheritance of non-syndromic renal hypoplasia and dysplasia: dramatic variation in clinical severity in a single kindred. Nephrol Dial Transplant 2007; 22: 259–263. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi M, Kaplan BS, Bellah RD, et al. Infundibulopelvic stenosis, multicystic kidney, and calyectasis in a kindred: clinical observations and genetic analysis. Am J Med Genet 1995; 59: 218–224. [DOI] [PubMed] [Google Scholar]
- 43.Schultza K, Todab LY. Genetic Basis of Ureterocele. Curr Genomics 2016; 17: 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jin M, Zhu S, Hu P, et al. Genomic and epigenomic analyses of monozygotic twins discordant for congenital renal agenesis. Am J Kidney Dis 2014; 64: 119–122. [DOI] [PubMed] [Google Scholar]
- 45.Castillo-Fernandez JE, Spector TD, Bell JT. Epigenetics of discordant monozygotic twins: implications for disease. Genome Med 2014; 6: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakayama M, Nozu K, Goto Y, et al. HNF1B alterations associated with congenital anomalies of the kidney and urinary tract. Pediatr Nephrol 2010; 25: 1073–1079. [DOI] [PubMed] [Google Scholar]
- 47.Madariaga L, Garcia-Castano A, Ariceta G, et al. Variable phenotype in HNF1B mutations: extrarenal manifestations distinguish affected individuals from the population with congenital anomalies of the kidney and urinary tract. Clin Kidney J 2019; 12: 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schedl A Renal abnormalities and their developmental origin. Nat Rev Genet 2007; 8: 791–802. [DOI] [PubMed] [Google Scholar]
- 49.Cooper WO, Hernandez-Diaz S, Arbogast PG, et al. Major congenital malformations after first-trimester exposure to ACE inhibitors. N Engl J Med 2006; 354: 2443–2451. [DOI] [PubMed] [Google Scholar]
- 50.Parikh CR, McCall D, Engelman C, et al. Congenital renal agenesis: case-control analysis of birth characteristics. Am J Kidney Dis 2002; 39: 689–694. [DOI] [PubMed] [Google Scholar]
- 51.Tain YL, Luh H, Lin CY, et al. Incidence and Risks of Congenital Anomalies of Kidney and Urinary Tract in Newborns: A Population-Based Case-Control Study in Taiwan. Medicine (Baltimore) 2016; 95: e2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Macumber I, Schwartz S, Leca N. Maternal obesity is associated with congenital anomalies of the kidney and urinary tract in offspring. Pediatr Nephrol 2017; 32: 635–642. [DOI] [PubMed] [Google Scholar]
- 53.Nicolaou N, Renkema KY, Bongers EM, et al. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat Rev Nephrol 2015; 11: 720–731. [DOI] [PubMed] [Google Scholar]
- 54.Vivante A, Hwang DY, Kohl S, et al. Exome Sequencing Discerns Syndromes in Patients from Consanguineous Families with Congenital Anomalies of the Kidneys and Urinary Tract. J Am Soc Nephrol 2017; 28: 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weber S Novel genetic aspects of congenital anomalies of kidney and urinary tract. Curr Opin Pediatr 2012; 24: 212–218. [DOI] [PubMed] [Google Scholar]
- 56.Hildebrandt F Decade in review--genetics of kidney diseases: Genetic dissection of kidney disorders. Nat Rev Nephrol 2015; 11: 635–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roodhooft AM, Birnholz JC, Holmes LB. Familial nature of congenital absence and severe dysgenesis of both kidneys. N Engl J Med 1984; 310: 1341–1345. [DOI] [PubMed] [Google Scholar]
- 58.Carter CO, Evans K, Pescia G. A family study of renal agenesis. J Med Genet 1979; 16: 176–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bulum B, Ozcakar ZB, Ustuner E, et al. High frequency of kidney and urinary tract anomalies in asymptomatic first-degree relatives of patients with CAKUT. Pediatr Nephrol 2013; 28: 2143–2147. [DOI] [PubMed] [Google Scholar]
- 60.Arora V, Anand K, Chander Verma I. Genetic Testing in Pediatric Kidney Disease. Indian J Pediatr 2020; 87: 706–715. [DOI] [PubMed] [Google Scholar]
- 61.Connolly LP, Treves ST, Connolly SA, et al. Vesicoureteral reflux in children: incidence and severity in siblings. J Urol 1997; 157: 2287–2290. [DOI] [PubMed] [Google Scholar]
- 62.Kaefer M, Curran M, Treves ST, et al. Sibling vesicoureteral reflux in multiple gestation births. Pediatrics 2000; 105: 800–804. [DOI] [PubMed] [Google Scholar]
- 63.Hamosh A, Scott AF, Amberger JS, et al. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res 2005; 33: D514–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vivante A, Kohl S, Hwang DY, et al. Single-gene causes of congenital anomalies of the kidney and urinary tract (CAKUT) in humans. Pediatr Nephrol 2014; 29: 695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hildebrandt F Genetic kidney diseases. Lancet 2010; 375: 1287–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Devuyst O, Knoers NV, Remuzzi G, et al. Rare inherited kidney diseases: challenges, opportunities, and perspectives. Lancet 2014; 383: 1844–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jain S, Chen F. Developmental pathology of congenital kidney and urinary tract anomalies. Clin Kidney J 2019; 12: 382–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sanna-Cherchi S, Sampogna RV, Papeta N, et al. Mutations in DSTYK and dominant urinary tract malformations. N Engl J Med 2013; 369: 621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jain S, Knoten A, Hoshi M, et al. Organotypic specificity of key RET adaptor-docking sites in the pathogenesis of neurocristopathies and renal malformations in mice. J Clin Invest 2010; 120: 778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kuure S, Sariola H. Mouse Models of Congenital Kidney Anomalies. Adv Exp Med Biol 2020; 1236: 109–136. [DOI] [PubMed] [Google Scholar]
- 71.Gallagher AR, Hoffmann S, Brown N, et al. A truncated polycystin-2 protein causes polycystic kidney disease and retinal degeneration in transgenic rats. J Am Soc Nephrol 2006; 17: 2719–2730. [DOI] [PubMed] [Google Scholar]
- 72.Madariaga L, Moriniere V, Jeanpierre C, et al. Severe prenatal renal anomalies associated with mutations in HNF1B or PAX2 genes. Clin J Am Soc Nephrol 2013; 8: 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weber S, Moriniere V, Knuppel T, et al. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: results of the ESCAPE study. J Am Soc Nephrol 2006; 17: 2864–2870. [DOI] [PubMed] [Google Scholar]
- 74.McPherson E, Carey J, Kramer A, et al. Dominantly inherited renal adysplasia. Am J Med Genet 1987; 26: 863–872. [DOI] [PubMed] [Google Scholar]
- 75.Ruf RG, Xu PX, Silvius D, et al. SIX1 mutations cause branchio-oto-renal syndrome by disruption of EYA1-SIX1-DNA complexes. Proc Natl Acad Sci U S A 2004; 101: 8090–8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kaplan BS, Milner LS, Jequier S, et al. Autosomal dominant inheritance of small kidneys. Am J Med Genet 1989; 32: 120–126. [DOI] [PubMed] [Google Scholar]
- 77.Doray B, Gasser B, Reinartz I, et al. Hereditary renal adysplasia in a three generations family. Genet Couns 1999; 10: 251–257. [PubMed] [Google Scholar]
- 78.Saisawat P, Kohl S, Hilger AC, et al. Whole-exome resequencing reveals recessive mutations in TRAP1 in individuals with CAKUT and VACTERL association. Kidney Int 2014; 85: 1310–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Humbert C, Silbermann F, Morar B, et al. Integrin alpha 8 recessive mutations are responsible for bilateral renal agenesis in humans. Am J Hum Genet 2014; 94: 288–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Weng PL, Sanna-Cherchi S, Hensle T, et al. A recessive gene for primary vesicoureteral reflux maps to chromosome 12p11-q13. J Am Soc Nephrol 2009; 20: 1633–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Westland R, Sanna-Cherchi S. Recessive mutations in CAKUT and VACTERL association. Kidney Int 2014; 85: 1253–1255. [DOI] [PubMed] [Google Scholar]
- 82.Capone VP, Morello W, Taroni F, et al. Genetics of Congenital Anomalies of the Kidney and Urinary Tract: The Current State of Play. Int J Mol Sci 2017; 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bekheirnia MR, Bekheirnia N, Bainbridge MN, et al. Whole-exome sequencing in the molecular diagnosis of individuals with congenital anomalies of the kidney and urinary tract and identification of a new causative gene. Genet Med 2017; 19: 412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Heidet L, Moriniere V, Henry C, et al. Targeted Exome Sequencing Identifies PBX1 as Involved in Monogenic Congenital Anomalies of the Kidney and Urinary Tract. J Am Soc Nephrol 2017; 28: 2901–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sanna-Cherchi S, Khan K, Westland R, et al. Exome-wide Association Study Identifies GREB1L Mutations in Congenital Kidney Malformations. Am J Hum Genet 2017; 101: 1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Vivante A, Mann N, Yonath H, et al. A Dominant Mutation in Nuclear Receptor Interacting Protein 1 Causes Urinary Tract Malformations via Dysregulation of Retinoic Acid Signaling. J Am Soc Nephrol 2017; 28: 2364–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Nicolaou N, Pulit SL, Nijman IJ, et al. Prioritization and burden analysis of rare variants in 208 candidate genes suggest they do not play a major role in CAKUT. Kidney Int 2016; 89: 476–486. [DOI] [PubMed] [Google Scholar]
- 88.Caruana G, Bertram JF. Congenital anomalies of the kidney and urinary tract genetics in mice and men. Nephrology (Carlton) 2015; 20: 309–311. [DOI] [PubMed] [Google Scholar]
- 89.Westland R, Verbitsky M, Vukojevic K, et al. Copy number variation analysis identifies novel CAKUT candidate genes in children with a solitary functioning kidney. Kidney Int 2015; 88: 1402–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sanna-Cherchi S, Kiryluk K, Burgess KE, et al. Copy-number disorders are a common cause of congenital kidney malformations. Am J Hum Genet 2012; 91: 987–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Siomou E, Mitsioni AG, Giapros V, et al. Copy-number variation analysis in familial nonsyndromic congenital anomalies of the kidney and urinary tract: Evidence for the causative role of a transposable element-associated genomic rearrangement. Mol Med Rep 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Caruana G, Wong MN, Walker A, et al. Copy-number variation associated with congenital anomalies of the kidney and urinary tract. Pediatr Nephrol 2015; 30: 487–495. [DOI] [PubMed] [Google Scholar]
- 93.Cai M, Lin N, Su L, et al. Copy number variations associated with fetal congenital kidney malformations. Mol Cytogenet 2020; 13: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Faure A, Bouty A, Caruana G, et al. DNA copy number variants: A potentially useful predictor of early onset renal failure in boys with posterior urethral valves. J Pediatr Urol 2016; 12: 227 e221–227. [DOI] [PubMed] [Google Scholar]
- 95.Verbitsky M, Sanna-Cherchi S, Fasel DA, et al. Genomic imbalances in pediatric patients with chronic kidney disease. J Clin Invest 2015; 125: 2171–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Verbitsky M, Westland R, Perez A, et al. The copy number variation landscape of congenital anomalies of the kidney and urinary tract. Nat Genet 2019; 51: 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Darlow JM, Dobson MG, Darlay R, et al. A new genome scan for primary nonsyndromic vesicoureteric reflux emphasizes high genetic heterogeneity and shows linkage and association with various genes already implicated in urinary tract development. Mol Genet Genomic Med 2014; 2: 7–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.van Eerde AM, Duran K, van Riel E, et al. Genes in the ureteric budding pathway: association study on vesico-ureteral reflux patients. PLoS One 2012; 7: e31327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Verbitsky M, Krithivasan P, Batourina E, et al. Copy Number Variant Analysis and Genome-wide Association Study Identify Loci with Large Effect for Vesicoureteral Reflux. J Am Soc Nephrol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Geller F, Feenstra B, Carstensen L, et al. Genome-wide association analyses identify variants in developmental genes associated with hypospadias. Nat Genet 2014; 46: 957–963. [DOI] [PubMed] [Google Scholar]
- 101.Draaken M, Knapp M, Pennimpede T, et al. Genome-wide association study and meta-analysis identify ISL1 as genome-wide significant susceptibility gene for bladder exstrophy. PLoS Genet 2015; 11: e1005024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nie XG, Sun JB, Gordon RE, et al. SIX1 acts synergistically with TBX18 in mediating ureteral smooth muscle formation. Development 2010; 137: 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Marini M, Giacopelli F, Seri M, et al. Interaction of the LMX1B and PAX2 gene products suggests possible molecular basis of differential phenotypes in Nail-Patella syndrome. Eur J Hum Genet 2005; 13: 789–792. [DOI] [PubMed] [Google Scholar]
- 104.Fain PR, McFann KK, Taylor MRG, et al. Modifier genes play a significant role in the phenotypic expression of PKD1. Kidney International 2005; 67: 1256–1267. [DOI] [PubMed] [Google Scholar]
- 105.Gresh L, Fischer E, Reimann A, et al. A transcriptional network in polycystic kidney disease. EMBO J 2004; 23: 1657–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bergmann C, von Bothmer J, Ortiz Bruchle N, et al. Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J Am Soc Nephrol 2011; 22: 2047–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Guay-Woodford LM. Murine models of polycystic kidney disease: molecular and therapeutic insights. Am J Physiol Renal Physiol 2003; 285: F1034–1049. [DOI] [PubMed] [Google Scholar]
- 108.Hiesberger T, Bai Y, Shao XL, et al. Mutation of hepatocyte nuclear factor-1 beta inhibits Pkhd1 gene expression and produces renal cysts in mice. Journal of Clinical Investigation 2004; 113: 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hiesberger T, Shao XL, Sthapit B, et al. Mutations of HNF1 beta inhibit Pkhd1 gene expression and induce cyst formation in transgenic mice. J Am Soc Nephrol 2003; 14: 50a–50a. [Google Scholar]
- 110.Hiesberger T, Bai Y, Shao X, et al. Mutation of hepatocyte nuclear factor-1beta inhibits Pkhd1 gene expression and produces renal cysts in mice. J Clin Invest 2004; 113: 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Persu A, Duyme M, Pirson Y, et al. Comparison between siblings and twins supports a role for modifier genes in ADPKD. Kidney Int 2004; 66: 2132–2136. [DOI] [PubMed] [Google Scholar]
- 112.Vainio S, Lin Y. Coordinating early kidney development: lessons from gene targeting. Nat Rev Genet 2002; 3: 533–543. [DOI] [PubMed] [Google Scholar]
- 113.Torres M, Gomez-Pardo E, Dressler GR, et al. Pax-2 controls multiple steps of urogenital development. Development 1995; 121: 4057–4065. [DOI] [PubMed] [Google Scholar]
- 114.Miyazaki Y, Oshima K, Fogo A, et al. Evidence that bone morphogenetic protein 4 has multiple biological functions during kidney and urinary tract development. Kidney Int 2003; 63: 835–844. [DOI] [PubMed] [Google Scholar]
- 115.Mackie GG, Stephens FD. Duplex kidneys: a correlation of renal dysplasia with position of the ureteral orifice. J Urol 1975; 114: 274–280. [DOI] [PubMed] [Google Scholar]
- 116.Durbec P, Marcos-Gutierrez CV, Kilkenny C, et al. GDNF signalling through the Ret receptor tyrosine kinase. Nature 1996; 381: 789–793. [DOI] [PubMed] [Google Scholar]
- 117.Gribouval O, Gonzales M, Neuhaus T, et al. Mutations in genes in the renin-angiotensin system are associated with autosomal recessive renal tubular dysgenesis. Nat Genet 2005; 37: 964–968. [DOI] [PubMed] [Google Scholar]
- 118.Allanson JE, Hunter AG, Mettler GS, et al. Renal tubular dysgenesis: a not uncommon autosomal recessive syndrome: a review. Am J Med Genet 1992; 43: 811–814. [DOI] [PubMed] [Google Scholar]
- 119.Yosypiv IV. Renin-angiotensin system in ureteric bud branching morphogenesis: implications for kidney disease. Pediatr Nephrol 2014; 29: 609–620. [DOI] [PubMed] [Google Scholar]
- 120.Yosypiv IV. Renin-angiotensin system in ureteric bud branching morphogenesis: insights into the mechanisms. Pediatr Nephrol 2011; 26: 1499–1512. [DOI] [PubMed] [Google Scholar]
- 121.Rabe M, Schaefer F. Non-Transgenic Mouse Models of Kidney Disease. Nephron 2016; 133: 53–61. [DOI] [PubMed] [Google Scholar]
- 122.Elmonem MA, Berlingerio SP, van den Heuvel LP, et al. Genetic Renal Diseases: The Emerging Role of Zebrafish Models. Cells 2018; 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Garrett MR, Korstanje R. Using Genetic and Species Diversity to Tackle Kidney Disease. Trends Genet 2020; 36: 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Outtandy P, Russell C, Kleta R, et al. Zebrafish as a model for kidney function and disease. Pediatr Nephrol 2019; 34: 751–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Davis EE, Frangakis S, Katsanis N. Interpreting human genetic variation with in vivo zebrafish assays. Biochim Biophys Acta 2014; 1842: 1960–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Drummond IA, Davidson AJ. Zebrafish kidney development. Methods in cell biology 2010; 100: 233–260. [DOI] [PubMed] [Google Scholar]
- 127.Drummond IA, Majumdar A, Hentschel H, et al. Early development of the zebrafish pronephros and analysis of mutations affecting pronephric function. Development 1998; 125: 4655–4667. [DOI] [PubMed] [Google Scholar]
- 128.Poureetezadi SJ, Wingert RA. Congenital and Acute Kidney Disease: Translational Research Insights from Zebrafish Chemical Genetics. General medicine (Los Angeles, Calif) 2013; 1: 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sanyanusin P, Schimmenti LA, McNoe LA, et al. Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies and vesicoureteral reflux. Nat Genet 1995; 9: 358–364. [DOI] [PubMed] [Google Scholar]
- 130.Weber S, Taylor JC, Winyard P, et al. SIX2 and BMP4 mutations associate with anomalous kidney development. J Am Soc Nephrol 2008; 19: 891–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hwang WY, Fu Y, Reyon D, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nature biotechnology 2013; 31: 227–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Andrews OE, Cha DJ, Wei C, et al. RNAi-mediated gene silencing in zebrafish triggered by convergent transcription. Scientific reports 2014; 4: 5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nasevicius A, Ekker SC. Effective targeted gene ‘knockdown’ in zebrafish. Nat Genet 2000; 26: 216–220. [DOI] [PubMed] [Google Scholar]
- 134.Khan TN, Khan K, Sadeghpour A, et al. Mutations in NCAPG2 Cause a Severe Neurodevelopmental Syndrome that Expands the Phenotypic Spectrum of Condensinopathies. Am J Hum Genet 2019; 104: 94–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Al-Hamed MH, van Lennep C, Hynes AM, et al. Functional modelling of a novel mutation in BBS5. Cilia 2014; 3: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bolar NA, Golzio C, Zivna M, et al. Heterozygous Loss-of-Function SEC61A1 Mutations Cause Autosomal-Dominant Tubulo-Interstitial and Glomerulocystic Kidney Disease with Anemia. Am J Hum Genet 2016; 99: 174–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dupont MA, Humbert C, Huber C, et al. Human IFT52 mutations uncover a novel role for the protein in microtubule dynamics and centrosome cohesion. Human molecular genetics 2019; 28: 2720–2737. [DOI] [PubMed] [Google Scholar]
- 138.Heon E, Kim G, Qin S, et al. Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21). Human molecular genetics 2016; 25: 2283–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kolvenbach CM, Dworschak GC, Frese S, et al. Rare Variants in BNC2 Are Implicated in Autosomal-Dominant Congenital Lower Urinary-Tract Obstruction. Am J Hum Genet 2019; 104: 994–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lindstrand A, Davis EE, Carvalho CM, et al. Recurrent CNVs and SNVs at the NPHP1 locus contribute pathogenic alleles to Bardet-Biedl syndrome. Am J Hum Genet 2014; 94: 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Materna-Kiryluk A, Kiryluk K, Burgess KE, et al. The emerging role of genomics in the diagnosis and workup of congenital urinary tract defects: a novel deletion syndrome on chromosome 3q13.31–22.1. Pediatr Nephrol 2014; 29: 257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mansilla MA, Sompallae RR, Nishimura CJ, et al. Targeted broad-based genetic testing by next-generation sequencing informs diagnosis and facilitates management in patients with kidney diseases. Nephrol Dial Transplant 2021; 36: 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Yang N, Wu N, Dong S, et al. Human and mouse studies establish TBX6 in Mendelian CAKUT and as a potential driver of kidney defects associated with the 16p11.2 microdeletion syndrome. Kidney Int 2020; 98: 1020–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lopez-Rivera E, Liu YP, Verbitsky M, et al. Genetic Drivers of Kidney Defects in the DiGeorge Syndrome. N Engl J Med 2017; 376: 742–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Slanchev K, Putz M, Schmitt A, et al. Nephrocystin-4 is required for pronephric duct-dependent cloaca formation in zebrafish. Human molecular genetics 2011; 20: 3119–3128. [DOI] [PubMed] [Google Scholar]
- 146.Baranowska Korberg I, Hofmeister W, Markljung E, et al. WNT3 involvement in human bladder exstrophy and cloaca development in zebrafish. Human molecular genetics 2015; 24: 5069–5078. [DOI] [PubMed] [Google Scholar]
- 147.Freedman BS. Modeling Kidney Disease with iPS Cells. Biomark Insights 2015; 10: 153–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006; 126: 663–676. [DOI] [PubMed] [Google Scholar]
- 149.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131: 861–872. [DOI] [PubMed] [Google Scholar]
- 150.Mae SI, Shono A, Shiota F, et al. Monitoring and robust induction of nephrogenic intermediate mesoderm from human pluripotent stem cells. Nat Commun 2013; 4: 1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.de Carvalho Ribeiro P, Oliveira LF, Filho MA, et al. Differentiating Induced Pluripotent Stem Cells into Renal Cells: A New Approach to Treat Kidney Diseases. Stem cells international 2020; 2020: 8894590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sallam M, Palakkan AA, Mills CG, et al. Differentiation of a Contractile, Ureter-Like Tissue, from Embryonic Stem Cell-Derived Ureteric Bud and Ex Fetu Mesenchyme. J Am Soc Nephrol 2020; 31: 2253–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mulder J, Sharmin S, Chow T, et al. Generation of infant- and pediatric-derived urinary induced pluripotent stem cells competent to form kidney organoids. Pediatr Res 2020; 87: 647–655. [DOI] [PubMed] [Google Scholar]
- 154.Shanks N, Greek R, Greek J. Are animal models predictive for humans? Philos Ethics Humanit Med 2009; 4: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Yoshiki A, Moriwaki K. Mouse phenome research: implications of genetic background. ILAR J 2006; 47: 94–102. [DOI] [PubMed] [Google Scholar]
- 156.Shimizu T, Mae SI, Araoka T, et al. A novel ADPKD model using kidney organoids derived from disease-specific human iPSCs. Biochem Biophys Res Commun 2020; 529: 1186–1194. [DOI] [PubMed] [Google Scholar]
- 157.Schutgens F, Verhaar MC, Rookmaaker MB. Pluripotent stem cell-derived kidney organoids: An in vivo-like in vitro technology. European journal of pharmacology 2016; 790: 12–20. [DOI] [PubMed] [Google Scholar]
- 158.Rak-Raszewska A, Hauser PV, Vainio S. Organ In Vitro Culture: What Have We Learned about Early Kidney Development? Stem cells international 2015; 2015: 959807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Takasato M, Er PX, Chiu HS, et al. Kidney organoids from human iPS cells contain multiple lineages and model human nephrogenesis. Nature 2015; 526: 564–568. [DOI] [PubMed] [Google Scholar]
- 160.Morizane R, Lam AQ, Freedman BS, et al. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nature biotechnology 2015; 33: 1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Howden SE, Wilson SB, Groenewegen E, et al. Plasticity of distal nephron epithelia from human kidney organoids enables the induction of ureteric tip and stalk. Cell stem cell 2021; 28: 671–684 e676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rosines E, Sampogna RV, Johkura K, et al. Staged in vitro reconstitution and implantation of engineered rat kidney tissue. Proc Natl Acad Sci U S A 2007; 104: 20938–20943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tan Z, Rak-Raszewska A, Skovorodkin I, et al. Mouse Embryonic Stem Cell-Derived Ureteric Bud Progenitors Induce Nephrogenesis. Cells 2020; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Freedman BS, Brooks CR, Lam AQ, et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat Commun 2015; 6: 8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Kuraoka S, Tanigawa S, Taguchi A, et al. PKD1-Dependent Renal Cystogenesis in Human Induced Pluripotent Stem Cell-Derived Ureteric Bud/Collecting Duct Organoids. J Am Soc Nephrol 2020; 31: 2355–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Little MH. Growing Kidney Tissue from Stem Cells: How Far from “Party Trick” to Medical Application? Cell stem cell 2016; 18: 695–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Schmidt-Ott KM. How to grow a kidney: patient-specific kidney organoids come of age. Nephrol Dial Transplant 2017; 32: 17–23. [DOI] [PubMed] [Google Scholar]
- 168.Li Z, Araoka T, Wu J, et al. 3D Culture Supports Long-Term Expansion of Mouse and Human Nephrogenic Progenitors. Cell stem cell 2016; 19: 516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Yamaguchi S, Morizane R, Homma K, et al. Generation of kidney tubular organoids from human pluripotent stem cells. Scientific reports 2016; 6: 38353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhang D, Du X, Zhang X, et al. In vitro induction and in vivo engraftment of kidney organoids derived from human pluripotent stem cells. Exp Ther Med 2020; 20: 1307–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kohl S, Habbig S, Weber LT, et al. Molecular causes of congenital anomalies of the kidney and urinary tract (CAKUT). Mol Cell Pediatr 2021; 8: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med 2016; 18: 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ziats MN, Rennert OM. The Evolving Diagnostic and Genetic Landscapes of Autism Spectrum Disorder. Front Genet 2016; 7: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sandholm N, Van Zuydam N, Ahlqvist E, et al. The Genetic Landscape of Renal Complications in Type 1 Diabetes. J Am Soc Nephrol 2017; 28: 557–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Genin E Missing heritability of complex diseases: case solved? Hum Genet 2020; 139: 103–113. [DOI] [PubMed] [Google Scholar]
- 176.Sadee W, Hartmann K, Seweryn M, et al. Missing heritability of common diseases and treatments outside the protein-coding exome. Hum Genet 2014; 133: 1199–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Sudmant PH, Kitzman JO, Antonacci F, et al. Diversity of human copy number variation and multicopy genes. Science 2010; 330: 641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Stephens PJ, Greenman CD, Fu B, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011; 144: 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Zhang F, Seeman P, Liu P, et al. Mechanisms for nonrecurrent genomic rearrangements associated with CMT1A or HNPP: rare CNVs as a cause for missing heritability. Am J Hum Genet 2010; 86: 892–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Alkan C, Kidd JM, Marques-Bonet T, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet 2009; 41: 1061–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Collins RL, Brand H, Redin CE, et al. Defining the diverse spectrum of inversions, complex structural variation, and chromothripsis in the morbid human genome. Genome Biol 2017; 18: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zepeda-Mendoza CJ, Morton CC. The Iceberg under Water: Unexplored Complexity of Chromoanagenesis in Congenital Disorders. Am J Hum Genet 2019; 104: 565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ip CLC, Loose M, Tyson JR, et al. MinION Analysis and Reference Consortium: Phase 1 data release and analysis. F1000Res 2015; 4: 1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Jain M, Tyson JR, Loose M, et al. MinION Analysis and Reference Consortium: Phase 2 data release and analysis of R9.0 chemistry. F1000Res 2017; 6: 760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Reid C Company profile: Complete Genomics Inc. Future Oncol 2011; 7: 219–221. [DOI] [PubMed] [Google Scholar]
- 186.Logsdon GA, Vollger MR, Eichler EE. Long-read human genome sequencing and its applications. Nat Rev Genet 2020; 21: 597–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Huang M, Tu J, Lu Z. Recent Advances in Experimental Whole Genome Haplotyping Methods. Int J Mol Sci 2017; 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Merker JD, Wenger AM, Sneddon T, et al. Long-read genome sequencing identifies causal structural variation in a Mendelian disease. Genet Med 2018; 20: 159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Pirola Y, Zaccaria S, Dondi R, et al. HapCol: accurate and memory-efficient haplotype assembly from long reads. Bioinformatics 2016; 32: 1610–1617. [DOI] [PubMed] [Google Scholar]
- 190.Ritz A, Bashir A, Raphael BJ. Structural variation analysis with strobe reads. Bioinformatics 2010; 26: 1291–1298. [DOI] [PubMed] [Google Scholar]
- 191.Cretu Stancu M, van Roosmalen MJ, Renkens I, et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat Commun 2017; 8: 1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Zhang F, Lupski JR. Non-coding genetic variants in human disease. Human molecular genetics 2015; 24: R102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kapoor A, Sekar RB, Hansen NF, et al. An enhancer polymorphism at the cardiomyocyte intercalated disc protein NOS1AP locus is a major regulator of the QT interval. Am J Hum Genet 2014; 94: 854–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Trudu M, Janas S, Lanzani C, et al. Common noncoding UMOD gene variants induce salt-sensitive hypertension and kidney damage by increasing uromodulin expression. Nat Med 2013; 19: 1655–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Rojano E, Seoane P, Ranea JAG, et al. Regulatory variants: from detection to predicting impact. Brief Bioinform 2019; 20: 1639–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Zepeda-Mendoza CJ, Ibn-Salem J, Kammin T, et al. Computational Prediction of Position Effects of Apparently Balanced Human Chromosomal Rearrangements. Am J Hum Genet 2017; 101: 206–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Li MJ, Yan B, Sham PC, et al. Exploring the function of genetic variants in the non-coding genomic regions: approaches for identifying human regulatory variants affecting gene expression. Brief Bioinform 2015; 16: 393–412. [DOI] [PubMed] [Google Scholar]
- 198.Ward LD, Kellis M. Interpreting noncoding genetic variation in complex traits and human disease. Nature biotechnology 2012; 30: 1095–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Boix CA, James BT, Park YP, et al. Regulatory genomic circuitry of human disease loci by integrative epigenomics. Nature 2021; 590: 300–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Moyses-Oliveira M, Yadav R, Erdin S, et al. New gene discoveries highlight functional convergence in autism and related neurodevelopmental disorders. Curr Opin Genet Dev 2020; 65: 195–206. [DOI] [PubMed] [Google Scholar]
- 201.Stolze LK, Conklin AC, Whalen MB, et al. Systems Genetics in Human Endothelial Cells Identifies Non-coding Variants Modifying Enhancers, Expression, and Complex Disease Traits. Am J Hum Genet 2020; 106: 748–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zheng ZH, Sam TW, Zeng Y, et al. Chromatin Regulation in Development: Current Understanding and Approaches. Stem cells international 2021; 2021: 8817581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Cummings BB, Karczewski KJ, Kosmicki JA, et al. Transcript expression-aware annotation improves rare variant interpretation. Nature 2020; 581: 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Mohammadi P, Castel SE, Cummings BB, et al. Genetic regulatory variation in populations informs transcriptome analysis in rare disease. Science 2019; 366: 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Heintze JM. Developmental biology: Single-cell RNA sequencing identifies novel kidney cell types in zebrafish. Nat Rev Nephrol 2017; 13: 663. [DOI] [PubMed] [Google Scholar]
- 206.Wu H, Humphreys BD. The promise of single-cell RNA sequencing for kidney disease investigation. Kidney Int 2017; 92: 1334–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chen L, Chou CL, Knepper MA. Targeted Single-Cell RNA-seq Identifies Minority Cell Types of Kidney Distal Nephron. J Am Soc Nephrol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Hochane M, van den Berg PR, Fan X, et al. Single-cell transcriptomics reveals gene expression dynamics of human fetal kidney development. PLoS Biol 2019; 17: e3000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Schoels M, Zhuang M, Fahrner A, et al. Single-cell mRNA profiling reveals changes in solute carrier expression and suggests a metabolic switch during zebrafish pronephros development. Am J Physiol Renal Physiol 2021. [DOI] [PubMed] [Google Scholar]
- 210.Subramanian A, Sidhom EH, Emani M, et al. Single cell census of human kidney organoids shows reproducibility and diminished off-target cells after transplantation. Nat Commun 2019; 10: 5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Reuter C, Chun N, Pariani M, et al. Understanding variants of uncertain significance in the era of multigene panels: Through the eyes of the patient. J Genet Couns 2019; 28: 878–886. [DOI] [PubMed] [Google Scholar]
- 212.Grossman I ADME pharmacogenetics: current practices and future outlook. Expert Opin Drug Metab Toxicol 2009; 5: 449–462. [DOI] [PubMed] [Google Scholar]
- 213.Santos M, Niemi M, Hiratsuka M, et al. Novel copy-number variations in pharmacogenes contribute to interindividual differences in drug pharmacokinetics. Genet Med 2018; 20: 622–629. [DOI] [PubMed] [Google Scholar]
- 214.Seely JC. A brief review of kidney development, maturation, developmental abnormalities, and drug toxicity: juvenile animal relevancy. J Toxicol Pathol 2017; 30: 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Tennessen JA, Bigham AW, O’Connor TD, et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 2012; 337: 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Muller RU, Haas CS, Sayer JA. Practical approaches to the management of autosomal dominant polycystic kidney disease patients in the era of tolvaptan. Clin Kidney J 2018; 11: 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Temme J, Peters F, Lange K, et al. Incidence of renal failure and nephroprotection by RAAS inhibition in heterozygous carriers of X-chromosomal and autosomal recessive Alport mutations. Kidney Int 2012; 81: 779–783. [DOI] [PubMed] [Google Scholar]