

Abstract
Fixed dosing of oral targeted therapies is inadequate in the era of precision medicine. Personalized dosing, based on pharmacokinetic (PK) exposure, known as therapeutic drug monitoring (TDM), is rational and supported by increasing evidence.
The purpose of this perspective is to discuss whether randomized studies are needed to confirm the clinical value of precision dosing in oncology.
PK-based dose adjustments are routinely made for many drugs and are recommended by health authorities, e.g. for patients with renal impairment or for drug-drug interaction management strategies. Personalized dosing simply extrapolates this paradigm from selected patient populations to each individual patient with suboptimal exposure, irrespective of the underlying cause.
If it has been demonstrated that exposure is related to a relevant clinical outcome, such as efficacy or toxicity, and that exposure can be optimized by PK-guided dosing, it could be logically assumed that PK-guided dosing would result in better treatment outcomes without the need for randomized confirmatory trials.
We propose a path forward to demonstrate the clinical relevance of individualized dosing of molecularly-targeted anticancer drugs.
Keywords: individualized dosing, pharmacokinetics, precision dosing, personalized medicine, targeted therapies
Introduction
The treatment of cancer is becoming increasingly personalized and attuned to the molecular characteristics of the tumor. The introduction of imatinib and other molecules has widely been hailed as a breakthrough of precision medicine.(1) At the moment, however, these targeted anticancer drugs are still predominantly administered using a one-size-fits-all fixed dose and schedule. Usually, the starting dose is only adjusted in cases of intolerable toxicity. Whilst for some drugs, fixed dosing may be appropriate (e.g. certain monoclonal antibodies(2)), for drugs that exhibit large variability in pharmacokinetic (PK) exposure and have a strong exposure-response or exposure-toxicity relationship, personalized adaptive dosing based on PK measurements, generally known as therapeutic drug monitoring (TDM) or PK-guided dosing, may be superior.(3-6)
Reasons why TDM has not been widely implemented yet could include the lack of acknowledgement by regulatory authorities (i.e. drug label change), the lack of access to appropriate PK tests, concerns about the cost-effectiveness, concerns about reimbursement of PK measurements and treatment costs for higher than approved dosages. Another argument frequently used against the implementation of TDM in routine cancer care is the absence of evidence from randomized controlled trials (RCTs). It could be questioned, though, whether these RCTs are really necessary here before TDM should be applied in daily clinical practice. In this commentary, we discuss whether randomized studies are needed to confirm the clinical value of precision dosing in oncology.
Inadequacy of fixed dosing for targeted anticancer agents
The current model guiding dose finding in oncology is the maximum tolerated dose (MTD). Fixed doses are often determined by selecting the MTD in early clinical trials. This ancient MTD paradigm sets the dose for all patients, based on only a few, sometimes non-representative patients, and does not take into account their exposure.(7,8) These dose-finding studies generally enroll only a small group of patients (with a median sample size of only 26).(9) Importantly, only a very limited number of patients in the highest dose levels (generally including 6–12 patients) will actually receive and determine the recommended dose for future clinical development. This MTD approach would be pharmacologically justifiable if the concentration-effect curves for toxicity and efficacy would be overlapping, as is the case for classical cytotoxic agents, but which is more often not the case for molecularly targeted agents. For these compounds, the MTD usually holds no particular relation to the optimal effective dose, and may, therefore, very well be suboptimal for this class of drugs.(8,10) Although more innovative designs such as the modified toxicity probability interval (mTPI) and other Bayesian adaptive designs are becoming more widely used, the MTD is still the most commonly used model for dose finding. Puzzlingly, once these small dose-finding trials have been completed, the recommended dose is usually not reconsidered or refined in later trials and subsequent clinical use after approval by the respective authorities. For some molecules, though, phase II studies did continue to examine the optimal dose, of which erdafitinib is a recent example.(11) Although there has been an increase in the use of pharmacokinetic-pharmacodynamic (PK-PD) models to guide dose finding, these efforts are still predominantly aimed at identifying a single optimum dose based on population averages, in contrast to an individual level personalized dose.
Many targeted drugs display a solid association between PK exposure and both treatment efficacy and toxicity.(5,6) As PK exposure of anticancer drugs varies greatly between patients receiving the same dose, some patients may be at risk of treatment-related toxicity due to high exposure, while others may experience suboptimal efficacy caused by low exposure. Therefore, using PK exposure to guide dosing decisions in a patient-specific manner (in contrast to using a fixed dose for the whole population) is rational and an important personalized strategy for treatment optimization. Table 1 provides an overview of the extent to which the most frequently prescribed oral targeted anticancer drugs demonstrate exposure-response relationships.
Table 1 –
Exposure-response relationships and feasibility of TDM of some of the most frequently prescribed oral targeted anticancer agents
| Oral targeted anticancer drug |
Exposure- efficacy |
Exposure-toxicity | TDM feasible |
References |
|---|---|---|---|---|
| ALK-inhibitors | ||||
| Alectinib | PFS, tumor size reduction | - | Feasibility study ongoing | (69) |
| Brigatinib | PFS, OS | Diarrhea, CK increase, skin and lung toxicity | Not investigated | FDA & EMA Review |
| Ceritinib | Inconclusive (trend for ORR) | ALT and AST elevation, hyperglycemia | Not investigated | FDA & EMA Review |
| Crizotinib | PFS, ORR | Neutropenia, AST elevation | Feasibility study ongoing | (69) |
| Lorlatinib | - | Hypercholesterolemia, G3/4 events | Not investigated | FDA & EMA Review |
| Anti-hormonal drugs | ||||
| Abiraterone | PFS, PSA response | - | Yes | (20,67,68) |
| Enzalutamide | - | - | Not investigated | (78) |
| Anastrozole | Estradiol suppression | - | Not investigated | (79) |
| Exemestane | - | - | Not investigated | FDA & EMA Review |
| Letrozole | TTP | - | Not investigated | FDA & EMA Review |
| Tamoxifen | Recurrence rate | - | Yes | (22,63) |
| Bcr-Abl inhibitors | ||||
| Dasatinib | MCyR, MR | Pleural effusions, dose adjustments | Not investigated | (80,81) |
| Imatinib | MMR, CCyR, TTP | Neutropenia, rash, diarrhea, arthralgia, edema | Yes | (18,25,72-74,82-84) |
| Nilotinib | TTP, trend for MMR | Bilirubin and liver enzyme elevations | Not investigated | (85,86) |
| BRAF inhibitors | ||||
| Dabrafenib | - | AEs requiring dose reduction | Not investigated | (87) |
| Encorafenib | - | G3/4 events | Not investigated | FDA & EMA Review |
| Vemurafenib | PFS, OS | Rash, QTc prolongation | Feasibility study ongoing | (88-91) |
| CDK 4/6 inhibitors | ||||
| Abemaciclib | PFS, BOR, tumor shrinkage | Neutropenia | Not investigated | FDA & EMA Review |
| Palbociclib | Inconclusive (trend for PFS) | Neutropenia | Feasibility study ongoing | (92) |
| Ribociclib | - | Neutropenia, QTc prolongation | Not investigated | FDA & EMA Review |
| EGFR inhibitors | ||||
| Erlotinib | OS, preclinical efficacy | Skin toxicity | Feasibility study ongoing | (93-96) |
| Gefitinib | OS | Skin toxicity, diarrhea, hepatotoxicity | Feasibility study ongoing | (97,98) |
| Osimertinib | - | Diarrhea, rash | Not investigated | (99) |
| MEK inhibitors | ||||
| Binimetinib | PFS | CK increase, retinopathy | Not investigated | (100) |
| Cobimetinib | - | - | Feasibility study ongoing | FDA & EMA Review |
| Trametinib | PFS | - | Feasibility study ongoing | (101,102) |
| mTOR inhibitor | ||||
| Everolimus | PFS | Stomatitis, lung toxicity | Yes | (103-105) |
| VEGFR inhibitors | ||||
| Axitinib | PFS, OS | Hypertension, diarrhea, fatigue | Feasibility study ongoing | (33,106) |
| Cabozantinib | PFS | HFS, fatigue, diarrhea, hypertension | Feasibility study ongoing | (107,108) |
| Pazopanib | PFS | Hypertension, hand-foot-syndrome | Yes | (19,59,75,76) |
| Sunitinib | TTP, OS | Hypertension, fatigue, anorexia, myelosuppression, HFS, dysgeusia, mucositis | Yes | (21,23,24,62,109-112) |
For all drugs, additional data on exposure-response relationships can be found in the publicly available FDA & EMA Reviews on Clinical Pharmacology.
The feasibility of several compounds in this table is currently being investigated in the Dutch Pharmacology Oncology Group – Therapeutic Drug Monitoring (DPOG-TDM) study.(65,113)
Abbreviations: AE = adverse event, CK = creatine kinase, CCyR = complete cytogenic response, G = grade, HFS = hand foot syndrome, MMR = major molecular response, OS = overall survival, PFS = progression-free survival, TTP = time to tumor progression
As can be appreciated from Table 1, pharmacokinetic exposure to many oral targeted therapies is related to both efficacy and toxicity. However, not for all oral targeted therapies clinically relevant exposure-response relationships were identified (e.g. enzalutamide and osimertinib). This may be explained by a plateau in the exposure-response curve for these drugs, that are dosed at the flat end of this curve. For some molecular targets (i.e. BRAF and EGFR) the therapeutic window appears to be wider than for others (i.e. ALK, MEK and VEGF). Also, newer generation kinase inhibitors may have a more robust formulation resulting in reduced variability.
It is thus essential to study for which oral targeted therapies precision dosing holds promise, and for which it might not be worthwhile. Before solid conclusions can be drawn on the absence of an exposure-response relationship, it should be ensured that the study has sufficient power to demonstrate this. Otherwise, absence of evidence is not evidence of absence, as is the case for several underpowered exposure-response analyses for endoxifen.(12-14)
Progress in implementing precision dosing in oncology
We and others have previously summarized the available data supporting PK-guided dosing of anticancer drugs.(3,5,6,15-17) Moreover, prospective clinical trials have demonstrated the safety and feasibility of PK-guided dosing for several agents.(18-25) When possible, cost-neutral strategies to optimize exposure could be applied, i.e. administration of the drug with food or optimized time of intake.(20,26) In fact, from the patient and treating physician perspectives, PK-guided dosing requires no complicated procedures and consists of simple and convenient interventions. For most compounds, samples could be collected 4, 8 and 12 weeks after start of treatment and every 12 weeks thereafter (except from compounds with intermittent dosing schedules or a long elimination half-life), which could be combined with regular visits to the outpatient clinic and blood sampling for routine safety monitoring. This would thus not be expected to negatively affect quality of life. Alternatively, methods for self-sampling at home could be developed so that results are already available when the patient visits the outpatient clinic. If a dose adjustment is made, the next PK sample could be drawn after 4 weeks for most compounds, so this could again be combined with a regular visit. Although the target exposure is mostly based on a certain trough level (i.e. concentration right before administration of the next dose), it is often sufficient to obtain a single blood sample at a random time point, as trough levels can then be estimated.(27,28) From a financial perspective, these expensive drugs should be supplied for one month at a time, so that the ordered amount is completed by the time a potential dose adjustment would be made. In this way, no medication needs to be thrown away, preventing unnecessary cost implications for the patient or payer.
Several cost-effectiveness analyses have been performed for oral targeted therapies based on retrospective data (i.e. abiraterone, imatinib and tamoxifen), and have demonstrated TDM to be cost effective.(29-31)
Axitinib is an interesting example for which precision dosing is already being performed, with dose titration based on toxicity being included in the drug label.(32) In fact, exposure to axitinib is related to both efficacy and toxicity.(33) Dose titration based on toxicity may result in getting patients within the right exposure range, without measuring PK but by using toxicity as a surrogate for exposure instead. Similarly, toxicity-adjusted dosing was demonstrated to be feasible for sunitinib as well.(34) However, treatment tolerability is only part of what should be aimed for, i.e. we want to maximize efficacy without risking intolerable toxicity by dosing patients within the therapeutic window instead of above of it. An intriguing approach is to use PK-PD modeling to simulate an RCT (instead of actually conducting it) to determine whether PK-guided dosing would be superior to toxicity-adjusted dosing for drugs like axitinib and sunitinib. There are many recent precedents for PK-PD modeling leading to a label change, such as the approvals for less frequent dosing of pembrolizumab and nivolumab.(35,36) Advantages of this approach include that answers on clinically relevant questions can be obtained faster, as conducting the actual clinical trial would take several years, whereas the simulations can be performed within a few months, which would also save considerable amounts of money.
Pharmacogenetically-guided dosing offers an additional strategy for treatment individualization, as the initial dose can be adjusted based on polymorphisms in the genes encoding for metabolizing enzymes and drug efflux transporters. Nevertheless, variability in exposure will remain, as genotype is only one of the many factors affecting exposure. In fact, measured drug concentrations are the translation of all of these factors, allowing for better precision dosing. Ideally, these two approaches should be combined by first selecting the right starting dose based on pharmacogenetics, which could then be further optimized by PK-guided dosing. However, if one of the two approaches should be preferred over the other, PK-guided dosing takes into account all factors that introduce variability, including pharmacogenetics.
Despite the strong rationale for PK-guided dosing in oncology, history suggests that confirmatory RCTs are rarely feasible. Only a very limited number of RCTs of fixed versus personalized dosing have ever been conducted, all of them for classical cytotoxics, underscoring the difficulty and impracticality of conducting RCTs for this specific application.(37-40) Obstacles that must be faced when conducting such RCTs include the large number of patients required, often with rare tumor types, resulting in difficulties in patient accrual, which could be further compounded by competitive studies. Also, the lack of interest by industry and third-party funding makes it challenging to secure sufficient financial support for these types of trials.
Are confirmatory randomized controlled trials needed for implementation?
Although it should be acknowledged that RCTs are currently considered the gold standard for most interventions before implementation in routine clinical practice, RCTs are both impractical and unnecessary when considering TDM. Impractical because large sample sizes would be required, particularly because the majority of patients would not need a PK-guided intervention, which will inevitably increase the costs and burden of the trial. Given the limited scientific rationale for fixed dosing based on the MTD, and the compelling nature of the exposure matching and efficacy arguments (further outlined below), it could be argued that there is no real need to conduct large confirmatory RCTs to investigate individualized dosing for compounds with clearly proven exposure-efficacy relationships.(17)
Interestingly, the paradigm of dose adaptation aiming for a PK exposure is already applied routinely in special patient populations, if not at the individual patient level. Dose recommendations for patients with renal or hepatic impairment, pediatric patients, and to guide drug-drug and drug-food interaction management strategies are based on matching the PK exposure of special populations to that of the reference population at the approved dose.(41-49) This model for dose adjustment based on exposure matching is generally accepted and part of multiple guidelines of regulatory authorities(41-48), and no or very limited follow-up studies on efficacy endpoints are required to support these dosing recommendations (in acknowledgement of their impracticality).
Yet, the logical extrapolation of PK exposure matching to the level of the individual patient by optimizing exposure in patients with very high or very low concentrations, as is done in special populations, is currently considered unconventional in oncology. This is all the more remarkable because interindividual differences in exposure are often much greater than the average impact of organ dysfunction, drug interaction, or food on exposure.(3,50-52)
If it is accepted that drugs should reach a certain target exposure to be effective in special patient populations, it should similarly also be considered beneficial to adapt the dose to target this exposure for any individual patient, which is the core principal of PK-guided dosing.
An additional argument why confirmatory RCTs should not be required in some cases is that retrospective evidence showing that a certain treatment is ineffective in a subgroup of patients is regarded sufficient to formally recommend exclusion of these patients in treatment guidelines and health authority-endorsed drug labels. For example, retrospective analyses for antibodies against the epidermal growth factor receptor (EGFR, i.e. panitumumab and cetuximab) demonstrated that efficacy is limited to KRAS wildtype patients(53,54), resulting in subsequent updates of the drug labels and treatment guidelines and these drugs no longer being prescribed to patients with KRAS-mutated tumors. Analogous to that, EGFR tyrosine kinase inhibitors erlotinib and gefitinib were initially approved for the treatment of all patients with non-small-cell lung cancer (NSCLC), but when retrospective analyses showed that efficacy was confined to patients with activating EGFR mutations(55-57), further development was restricted by targeting these patients. While these two examples concern efficacy, the same logic has been applied regarding toxicity. Recently, the drug label of 5-fluorouracil and capecitabine has been updated to include dose adjustments in patients harboring polymorphisms in the DPYD gene that are associated with an increased risk of severe toxicity.(58) In all three examples, no randomized confirmatory trials have ever been performed apparently and these were not considered essential to persuade the field and to change clinical practice, and could well be considered unethical due to the lack of equipoise.
Indeed, retrospective studies have shown that efficacy of several anticancer drugs is restricted to the subset of patients with a PK exposure above certain efficacy thresholds. For example, progression-free survival (PFS) in patients treated with pazopanib with an exposure below the minimum plasma concentration (Cmin) target of 20.5 mg/L is similar to the placebo arm of the pivotal trial.(59,60) Analogous analyses can be made for several other anticancer drugs, e.g. abiraterone, imatinib and sunitinib.(18,20,21)
Tellingly, TDM has been implemented as routine care based on similar retrospective data and without confirmatory trials having been performed for many other drugs with a narrow therapeutic window (e.g. anti-epileptics, anti-infectives, immunosuppressants and digoxin).
Path to implementation
To advance precision dosing from exploratory studies into standard of care, we propose a development pathway as visualized in Figure 1.(61) First, as part of the learning phase, sufficient information should be obtained to assess whether a compound has a suitable pharmacological profile (i.e. high interindividual variability and relatively lower intra-individual variability). This could be achieved by characterization of the variability in pharmacokinetic exposure within and between patients in the pivotal trials. Also, sound technical support should be in place (i.e. validated bioanalytical methods, convenient blood sampling and logistics). It is important that bioanalytical methods are publicly available so that they can be reproduced by other centers, and that (inter)national cross validation programs are initiated. As samples of most compounds are stable under ambient conditions, regional collaborations can be a viable option in clinical practice. Most available bioanalytical assays measure the total drug concentration (i.e. protein bound plus free drug). As exposure-response analyses are also performed using total drug concentrations, the total drug concentration could be used as a surrogate for the pharmacologically active free drug concentration. It should be kept in mind, though, that under certain circumstances the unbound fraction can change (e.g. organ dysfunction), in which case the total drug concentration may no longer be a representative measure of exposure. Most importantly, well established exposure-therapeutic response and exposure-toxicity relationships should have been demonstrated.(4) These exposure-response analyses could take multiple forms. For some drugs, elaborate model-based analyses are available(62), while only simple analyses (e.g. using exposure quartiles) have been used for others.(63) These more straightforward analyses could be incorporated as pre-specified analyses as an endpoint in pivotal phase 3 trials, as only limited additional sampling is needed (e.g. Cmin), and patients will already need blood sampling for routine safety monitoring. This could be further incentivized by regulatory authorities. In the case of combination therapies, exposure-response relationships would preferably be determined in patients treated with the combination regimen, as it could be imagined that different target exposures would apply for combination therapy compared to monotherapy. Then, based on what has been learned, PK targets and dosing algorithms should be defined. Dosing algorithms should take into account the possibility of cost-neutral interventions (i.e. concomitant intake with food or optimizing the dosing schedule(20,26,64)), the MTD or maximum administered dose in phase I studies, and the available capsule or tablet sizes.
Figure 1 – Proposed development strategy for precision dosing of oral targeted therapies in oncology.
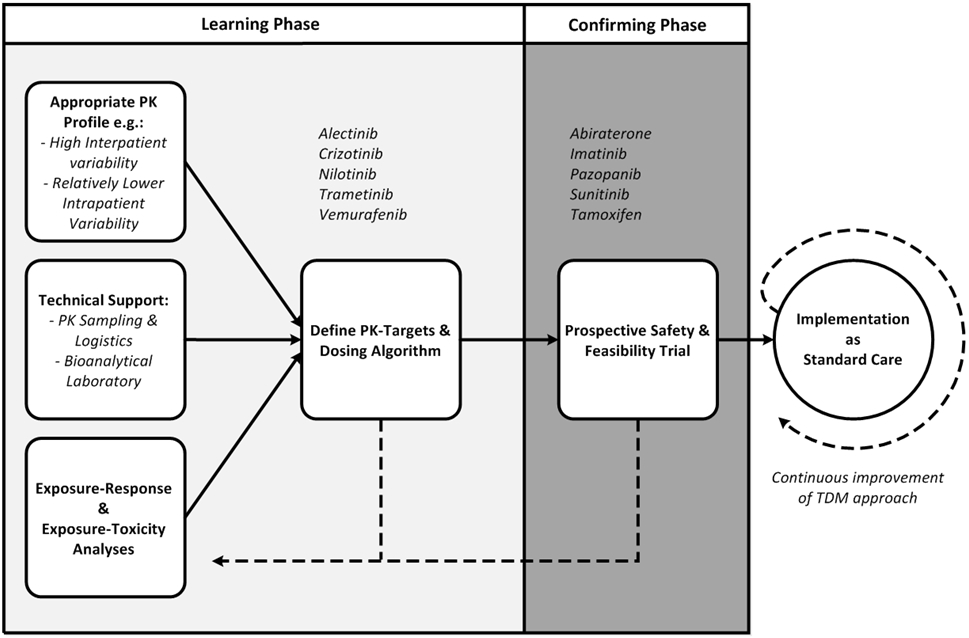
Examples of drugs are given at each stage of development.
PK: pharmacokinetics, TDM = therapeutic drug monitoring
Subsequently, as part of the confirming phase, the safety and feasibility of PK-guided dosing strategies should be demonstrated in clinical trials, preferably in a real-life setting, in which TDM is applied in clinical practice, but in which no control group needs to be used. Several trials in this category have already been performed, e.g. for abiraterone, imatinib, pazopanib, sunitinib and tamoxifen(18-22), and this approach is currently also being applied in a prospective study on TDM by the Dutch Pharmacology Oncology Group.(65) In these studies, individualized dosing may be demonstrated to be logistically feasible, well tolerated and to increase the proportion of patients within the target exposure range (i.e. superiority of TDM compared to fixed dosing). The standard for TDM-guided dosing that needs to be applied in these studies is superiority to fixed dosing with regard to the proportion of patients reaching the target exposure, either compared to a control group or to a historical fixed dosing cohort with available exposure data. As explained in more detail in the sections above, demonstrating superior efficacy might not be feasible (i.e. large sample size) and unnecessary (i.e. efficacy at the target exposure has already been demonstrated to be superior).
At this point, it has been demonstrated that exposure is related to a relevant clinical outcome including efficacy or toxicity, and that exposure can be improved by PK-guided dosing. Hence, it could be logically assumed that individualized dosing would result in better treatment outcomes, at the same level of evidence as Food and Drug Administration and European Medicines Agency endorsed recommendations for dose adjustments in special patient populations. Thus, sufficient evidence for individualized dosing has now been obtained to apply PK-guided dosing in clinical care.
Clinical outcomes and PK data of these patients should then be collected in clinical practice and should be used to further optimize the TDM approach, in order to ensure that with more patients the TDM strategy keeps improving, comparable to a Bayesian algorithm. To facilitate the implementation of individualized dosing, efforts should be made to remove practical barriers as much as possible. This could be done by ensuring that a sound infrastructure is in place for sample collection, shipment, measurement, interpretation and reporting of the results, with a short turn-around time. As PK-guided dosing recommendations should ideally also be included in the drug label, it is important to work together with the regulatory authorities from an early stage in this pathway.
Currently, there appears to be a missing link between the performed feasibility studies and the widespread implementation of PK-guided dosing.(66) Many factors play a role here. First, bioanalytical assays to measure drug concentrations might not be available or a solid infrastructure might be lacking. Second, knowledge about PK-guided dosing and skills to calculate or estimate exposure may be insufficient due to the absence of education and training. Close collaboration between medical oncologists, clinical pharmacologists and pharmacists is essential here. Third, costs might be a concern. Obviously, dose increases will result in higher treatment costs. However, the costs of drug measurements itself is negligible compared to the total treatment costs. For example, in The Netherlands, measurement of one PK sample costs 90 USD (i.e. 540 USD per year), whereas treatment costs of many oral targeted therapies exceed 120 USD per day (i.e. 43800 USD per year). Cost-effectiveness analyses need to be performed to demonstrate the additional value of PK-guided dosing. These are already performed for abiraterone, imatinib and tamoxifen, for all of which PK-guided dosing was cost-effective.(29-31) To limit the financial burden of these expensive treatments, it is essential to perform cost-neutral interventions (i.e. concomitant intake with food or optimization of the dosing schedule) when possible. The above mentioned factors should be addressed to stimulate the implementation of PK-guided dosing in clinical practice. Furthermore, acknowledgement by regulatory authorities (i.e. change in drug label) would support implementation in routine clinical practice, as inclusion in the label would likely produce an incentive for companies to develop commercially available PK tests as a companion diagnostic.
Examples of precision dosing
Abiraterone
According to the label, abiraterone acetate should be administered at a fixed dose of 1000 mg once daily under modified fasting conditions. However, it has been demonstrated that patients with a Cmin ≥ 8.4 ng/mL have a significantly better PFS (i.e. 12.2 vs. 7.4 months).(67) This has later been confirmed in an independent patient cohort.(68) In clinical practice, 35-42% of patients do not reach this efficacy threshold and might thus benefit from PK-guided dosing.(67,68) As it was known that concomitant intake with food resulted in a relevant increase in abiraterone exposure, the DPOG-TDM study investigated whether PK-guided dosing using a food intervention as a first step in case of low exposure was feasible in clinical practice. In this study, 20 out of 32 patients had an abiraterone Cmin < 8.4 ng/mL at a certain timepoint during treatment. These patients were recommended to take abiraterone acetate concomitant with a light meal or a snack, which resulted in an increase in Cmin from 6.9 ng/mL to 27 ng/mL without additional toxicities. This intervention led to adequate exposure in the majority of patients (i.e. 87.5%).(20)
Alectinib
For the ALK-inhibitor alectinib, PFS was significantly longer in patients with Cmin ≥ 435 ng/mL compared to patients with an exposure below this threshold.(69) At the approved dose of 450 mg twice daily, 37% of patients is underexposed and treatment outcomes for this subgroup may be improved by PK-guided dosing. An RCT is planned comparing fixed dosing vs. TDM-guided dosing of alectinib (the Adapt Alec Trial), in which a total of 220 patients needs to be enrolled, which is almost comparable to the phase 3 trial (i.e. ALEX study, in which 303 patients were included) and accrual is planned to take four years.(70,71)
Imatinib
Exposure to imatinib has been related to efficacy for both chronic myeloid leukemia and gastro-intestinal stromal tumors with Cmin thresholds of 1000 ng/mL and 1100 ng/mL, respectively.(72-74) An RCT comparing fixed dosing and TDM-guided dosing failed to demonstrate the additional value of TDM-guided dosing because treating physicians did not implement the recommended dose increases in patients with low exposure.(18) However, several studies have since then shown that PK-guided dosing of imatinib is feasible in clinical practice.(23,25)
Pazopanib
For pazopanib, trough levels ≥ 20 mg/L have been linked to prolonged PFS in different treatment settings.(59,75,76) In clinical practice, 16-30% of patients do not reach this threshold.(59,76) Furthermore, PK-guided dosing of pazopanib was demonstrated to be feasible.(19) Apart from dose increases, cost-neutral interventions can be applied as first steps in case of low exposure (i.e. splitting intake moments from 800 mg once daily to 400 mg twice daily and concomitant intake with food).(26,64,77)
Conclusion
Fixed dosing strategies are suboptimal in the era of precision medicine. PK-guided dosing is a promising tool to lead to more rational, personalized and optimal treatment with targeted anticancer drugs. With the available data, evidence for TDM is already quite robust. Together with continuous optimization of the TDM approach, it is justified to implement individualized dosing based on evidence deduced from exposure-response analyses and feasibility trials, without the need for a large confirmatory RCT. We propose a development pathway to demonstrate the clinical relevance of precision dosing in oncology.
Translational Relevance.
The introduction of oral targeted therapies in oncology has widely been praised as a triumph of precision medicine. While we increasingly select the right drug based on molecular characteristics of the tumor, these drugs are still administered using a one-size-fits-all fixed dosing approach. However, most oral targeted therapies exhibit a high interindividual variability in exposure, and for many of them, drug concentrations are related to both efficacy and toxicity. As a result, a substantial subset of patients is treated outside the therapeutic window. Therefore, rational personalized treatment would not only include selecting the right drug, but also selecting the right dose. Therapeutic drug monitoring, which is adjusting the dose based on measured drug concentrations, is a promising tool to achieve this.
Financial support
No funding was received for this work. Jan H. Beumer was supported by NCI grants UM1CA186690 and P30CA47904.
Footnotes
Conflict of interest
Remy B. Verheijen reports employment at AstraZeneca and Johnson & Johnson and share ownership of AstraZeneca, Johnson & Johnson and Aduro Biotech. Jos H. Beijnen is a part-time employee, shareholder and patent holder of Modra Pharmaceuticals (a spin-out company developing oral taxane formulations, not related to this work). All other authors declare no potential conflicts of interest.
References
- 1.Longo DL. Imatinib changed everything. N Engl J Med. 2017;376:982–3. [DOI] [PubMed] [Google Scholar]
- 2.Hendrikx JJMA, Haanen JBAG, Voest EE, Schellens JHM, Huitema ADR, Beijnen JH. Fixed Dosing of Monoclonal Antibodies in Oncology. Oncologist. 2017;22:1212–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu H, Steeghs N, Nijenhuis C, Schellens J, Beijnen J, Huitema A. Practical guidelines for therapeutic drug monitoring of anticancer tyrosine kinase inhibitors: Focus on the pharmacokinetic targets. Clin Pharmacokinet. 2014;53:305–25. [DOI] [PubMed] [Google Scholar]
- 4.Groenland SL, Mathijssen RHJ, Beijnen JH, Huitema ADR, Steeghs N. Individualized dosing of oral targeted therapies in oncology is crucial in the era of precision medicine. Eur J Clin Pharmacol. 2019;75:1309–18. [DOI] [PubMed] [Google Scholar]
- 5.Verheijen RB, Yu H, Schellens JHM, Beijnen JH, Steeghs N, Huitema ADR. Practical Recommendations for Therapeutic Drug Monitoring of Kinase Inhibitors in Oncology. Clin Pharmacol Ther. 2017;102:765–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Groenland SL, Van Nuland M, Verheijen RB, Schellens JHM, Beijnen JH, Huitema ADR, et al. Therapeutic Drug Monitoring of Oral Anti- Hormonal Drugs in Oncology. Clin Pharmacokinet. 2019;58:299–308. [DOI] [PubMed] [Google Scholar]
- 7.Takimoto CH. Maximum tolerated dose: Clinical endpoint for a bygone era? Target Oncol. 2009;4:143–7. [DOI] [PubMed] [Google Scholar]
- 8.Mathijssen RHJ, Sparreboom A, Verweij J. Determining the optimal dose in the development of anticancer agents. Nat Rev Clin Oncol. 2014;11:272–81. [DOI] [PubMed] [Google Scholar]
- 9.Van Brummelen EMJ, Huitema ADR, Werkhoven E, Beijnen JH, Schellens JHM. The performance of model-based versus rule-based phase I clinical trials in oncology. J Pharmacokinet Pharmacodyn. 2016;43:235–42. [DOI] [PubMed] [Google Scholar]
- 10.Bullock JM, Rahman A, Liu Q. Lessons Learned: Dose Selection of Small Molecule – Targeted Oncology Drugs. Clin Cancer Res. 2016;22:2630–9. [DOI] [PubMed] [Google Scholar]
- 11.Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N Engl J Med. 2019;381:338–48. [DOI] [PubMed] [Google Scholar]
- 12.Sanchez-Spitman A, Dezentje V, Swen J, Moes DJAR, Bohringer S, Batman E, et al. Tamoxifen Pharmacogenetics and Metabolism: Results From the Prospective CYPTAM Study. J Clin Oncol. 2019;37:636–646. [DOI] [PubMed] [Google Scholar]
- 13.Braal CL, Beijnen JH, Koolen SLW, Oomen-de Hoop E, Steeghs N, Jager A, et al. Relevance of Endoxifen Concentrations: Absence of Evidence Is Not Evidence of Absence. J Clin Oncol. 2019;37:1980–1. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez-Spitman AB, Moes DJAR, Swen JJ, Dezentjé VO, Lambrechts D, Neven P, et al. Exposure–response analysis of endoxifen serum concentrations in early-breast cancer. Cancer Chemother Pharmacol. 2020;85:1141–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Wit D, Guchelaar HJ, Den Hartigh J, Gelderblom H, Van Erp NP. Individualized dosing of tyrosine kinase inhibitors: Are we there yet? Drug Discov Today. 2015;20:18–36. [DOI] [PubMed] [Google Scholar]
- 16.Widmer N, Bardin C, Chatelut E, Paci A, Beijnen J, Levêque D, et al. Review of therapeutic drug monitoring of anticancer drugs part two - Targeted therapies. Eur J Cancer. 2014;50:2020–36. [DOI] [PubMed] [Google Scholar]
- 17.Beumer JH, Chu E, Allegra C, Tanigawara Y, Milano G, Diasio R, et al. Therapeutic Drug Monitoring in Oncology: International Association of Therapeutic Drug Monitoring and Clinical Toxicology Recommendations for 5-Fluorouracil Therapy. Clin Pharmacol Ther. 2019;105:598–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gotta V, Widmer N, Decosterd LA, Chalandon Y, Heim D, Gregor M, et al. Clinical usefulness of therapeutic concentration monitoring for imatinib dosage individualization: Results from a randomized controlled trial. Cancer Chemother Pharmacol. 2014;74:1307–19. [DOI] [PubMed] [Google Scholar]
- 19.Verheijen RB, Bins S, Mathijssen RHJ, Lolkema MP, van Doorn L, Schellens JHM, et al. Individualized Pazopanib Dosing: A Prospective Feasibility Study in Cancer Patients. Clin Cancer Res. 2016;22:5738–46. [DOI] [PubMed] [Google Scholar]
- 20.Groenland SL, Van Nuland M, Bergman AM, De Feijter JM, Dezentje VO, Rosing H, et al. Concomitant intake of abiraterone acetate and food to increase pharmacokinetic exposure: real life data from a therapeutic drug monitoring programme. Eur J Cancer. 2020;130:32–8. [DOI] [PubMed] [Google Scholar]
- 21.Lankheet N, Kloth J, Gadellaa-van Hooijdonk C, Cirkel G, Mathijssen R, Lolkema M, et al. Pharmacokinetically guided sunitinib dosing: a feasibility study in patients with advanced solid tumours. Br J Cancer. 2014;110:2441–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fox P, Balleine RL, Lee C, Gao B, Balakrishnar B, Menzies AM, et al. Dose Escalation of Tamoxifen in Patients with Low Endoxifen Level: Evidence for Therapeutic Drug Monitoring - The TADE Study. Clin Cancer Res. 2016;22:3164–71. [DOI] [PubMed] [Google Scholar]
- 23.Lankheet N, Desar I, Mulder S, Burger D, Kweekel D, Van Herpen CML, et al. Optimizing the dose in cancer patients treated with imatinib, sunitinib and pazopanib. Br J Clin Pharmacol. 2017;2195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Westerdijk K, Krens SD, Van der Graaf WTA, Mulder SF, Van Herpen CML, Smilde T, et al. The relationship between sunitinib exposure and both efficacy and toxicity in real-world patients with renal cell carcinoma and gastrointestinal stromal tumour. Br J Clin Pharmacol. 2021;87:326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.IJzerman NS, Groenland SL, Koenen AM, Kerst M, van der Graaf WTA, Rosing H, et al. Therapeutic drug monitoring of imatinib in patients with gastrointestinal stromal tumours – Results from daily clinical practice. Eur J Cancer. 2020;136:140–8. [DOI] [PubMed] [Google Scholar]
- 26.Groenland SL, Van Eerden RAG, Verheijen RB, De Vries N, Thijssen B, Rosing H, et al. Cost-Neutral Optimization of Pazopanib Exposure by Splitting Intake Moments: A Prospective Pharmacokinetic Study in Cancer Patients. Clin Pharmacokinet. 2020;59:941–8. [DOI] [PubMed] [Google Scholar]
- 27.Janssen JM, Dorlo TPC, Beijnen JH, Huitema ADR. Evaluation of extrapolation methods to predict trough concentrations to guide therapeutic drug monitoring of oral anticancer drugs. Ther Drug Monit. 2020;42:532–539. [DOI] [PubMed] [Google Scholar]
- 28.Van Eerden RAG, De Hoop EO, Noordam A, Mathijssen RHJ, Koolen SLW. Feasibility of Extrapolating Randomly Taken Plasma Samples to Trough Levels for Therapeutic Drug Monitoring Purposes of Small Molecule Kinase Inhibitors. Pharmaceuticals. 2021;14:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ten Ham RMT, Van Nuland M, Vreman RA, De Graaf LG, Rosing H, Bergman AM, et al. Cost-Effectiveness Assessment of Monitoring Abiraterone Levels in Metastatic Castration-Resistant Prostate Cancer Patients. Value Heal. 2021;24:121–8. [DOI] [PubMed] [Google Scholar]
- 30.Zuidema S, Desar IME, Van Erp NP, Kievit W. Optimizing the dose in patients treated with imatinib as first line treatment for gastrointestinal stromal tumours: a cost-effectiveness study. Br J Clin Pharmacol. 2019;85:1994–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Nuland M, Vreman RA, Ten Ham RMT, De Vries Schultink AHM, Rosing H, Schellens JHM, et al. Cost-effectiveness of monitoring endoxifen levels in breast cancer patients adjuvantly treated with tamoxifen. Breast Cancer Res Treat. 2018;172:143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Summary of Product Characteristics Axitinib. Available from: https://www.ema.europa.eu/en/documents/product-information/inlyta-epar-product-information_en.pdf
- 33.Rini BI, Garrett M, Poland B, Dutcher JP, Rixe O, Wilding G, et al. Axitinib in Metastatic Renal Cell Carcinoma : Results of a Pharmacokinetic and Pharmacodynamic Analysis. J Clin Pharmacol. 2013;53:491–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sabanathan D, Zhang A, Fox P, Coulter S, Gebski V, Balakrishnar B, et al. Dose individualization of sunitinib in metastatic renal cell cancer: toxicity-adjusted dose or therapeutic drug monitoring. Cancer Chemother Pharmacol. 2017;80:385–93. [DOI] [PubMed] [Google Scholar]
- 35.Long GV, Tykodi SS, Schneider JG, Garbe C, Gravis G, Rashford M, et al. Assessment of nivolumab exposure and clinical safety of 480 mg every 4 weeks flat-dosing schedule in patients with cancer. Ann Oncol. 2018;29:2208–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lala M, Li TR, De Alwis DP, Sinha V, Mayawala K, Yamamoto N, et al. A six-weekly dosing schedule for pembrolizumab in patients with cancer based on evaluation using modelling and simulation. Eur J Cancer. 2020;131:68–75. [DOI] [PubMed] [Google Scholar]
- 37.Joerger M, Von Pawel J, Kraff S, Fisher J, Eberhardt W, Gauler T, et al. Open-label, randomized study of individualized, pharmacokinetically (PK)-guided dosing of paclitaxel combined with carboplatin or cisplatin in patients with advanced non-small-cell lung cancer (NSCLC). Ann Oncol. 2016;27:1895–902. [DOI] [PubMed] [Google Scholar]
- 38.Evans W, Relling M, Rodman J, Crom W, Boyett J, Pui C. Conventional compared with individualized chemotherapy for childhood acute lymphoblastic leukemia. N Engl J Med. 1998;338:499–505. [DOI] [PubMed] [Google Scholar]
- 39.Gamelin E, Delva R, Jacob J, Merrouche Y, Raoul JL, Pezet D, et al. Individual Fluorouracil Dose Adjustment Based on Pharmacokinetic Follow-Up Compared With Conventional Dosage: Results of a Multicenter Randomized Trial of Patients With Metastatic Colorectal Cancer. J Clin Oncol. 2008;26:2099–105. [DOI] [PubMed] [Google Scholar]
- 40.Fety R, Rolland F, Barberi-Heyob M, Hardouin A, Campion L, Conroy T, et al. Clinical Impact of Pharmacokinetically-guided Dose Adaptation of 5-Fluorouracil: Results from a Multicentric Randomized Trial in Patients with Locally Advanced Head and Neck Carcinomas. Clin Cancer Res. 1998;4:2039–45. [PubMed] [Google Scholar]
- 41.Committee for Medicinal Products for Human Use. European Medicines Agency. Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with decreased renal function. 2016; Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-evaluation-pharmacokinetics-medicinal-products-patients-decreased-renal-function_en.pdf
- 42.Food and Drug Administration. Center for Drug Evaluation and Research. Guidance for industry: Pharmacokinetics in patients with impaired renal function - Study design, data analysis, and impact on dosing and labeling (1998). Available from: https://www.fda.gov/media/78573/download
- 43.European Medicines Agency. Committee for Medicinal Products for Human Use. Guideline on the Evaluation of the Pharmacokinetics of Medicinal Products in Patients With Impaired Hepatic Function. 2005; Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-evaluation-pharmacokinetics-medicinal-products-patients-impaired-hepatic-function_en.pdf
- 44.Food and Drug Administration. Center for Drug Evaluation and Research. Guidance for Industry - Pharmacokinetics in Patients with Impaired Hepatic Function: Study Design, Data Analysis, and Impact on Dosing and Labeling. 2003; Available from: https://www.fda.gov/media/71311/download
- 45.Committee for Medicinal Products for Human Use. European Medicines Agency. Guideline on the role of pharmacokinetics in the development of medicinal products in the paediatric population. 2006; Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003066.pdf
- 46.Food and Drug Administration. Center for Drug Evaluation and Research. Draft Guidance for Industry - Pediatric Study Plans: Content of and Process for Submitting Initial Pediatric Study Plans and Amended Pediatric Study Plans. 2016; Available from: https://www.fda.gov/media/86340/download
- 47.Committee for Medicinal Products for Human Use. European Medicines Agency. Guideline on the investigation of drug interactions. 2012; Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf
- 48.Food and Drug Administration. Center for Drug Evaluation and Research. Clinical Drug Interaction Studies — Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. 2020; Available from: https://www.fda.gov/media/134581/download
- 49.Veerman GDM, Hussaarts KGAM, Jansman FGA, Koolen SWL, van Leeuwen RWF, Mathijssen RHJ. Clinical implications of food–drug interactions with small-molecule kinase inhibitors. Lancet Oncol. 2020;21:e265–79. [DOI] [PubMed] [Google Scholar]
- 50.Herbrink M, Nuijen B, Schellens JHM, Beijnen JH. Variability in bioavailability of small molecular tyrosine kinase inhibitors. Cancer Treat Rev. 2015;41:412–22. [DOI] [PubMed] [Google Scholar]
- 51.Hurwitz HI, Dowlati A, Saini S, Savage S, Suttle AB, Gibson DM, et al. Phase I trial of pazopanib in patients with advanced cancer. Clin Cancer Res. 2009;15:4220–7. [DOI] [PubMed] [Google Scholar]
- 52.Attard G, Reid AHM, Yap TA, Raynaud F, Dowsett M, Settatree S, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–71. [DOI] [PubMed] [Google Scholar]
- 53.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–34. [DOI] [PubMed] [Google Scholar]
- 54.Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–5. [DOI] [PubMed] [Google Scholar]
- 55.Lynch TL, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non-Small-Cell Lung Cancer to Gefitinib. N Engl J Med. 2004;350:2129–39. [DOI] [PubMed] [Google Scholar]
- 56.Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR Mutations in Lung Cancer: Correlation with Clinical Response to Gefitinib Therapy. Science (80- ). 2004;304:1497–500. [DOI] [PubMed] [Google Scholar]
- 57.Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Henricks LM, Lunenburg CATC, de Man FM, Meulendijks D, Frederix GWJ, Kienhuis E, et al. DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. Lancet Oncol. 2018;19:1459–67. [DOI] [PubMed] [Google Scholar]
- 59.Suttle AB, Ball HA, Molimard M, Hutson TE, Carpenter C, Rajagopalan D, et al. Relationships between pazopanib exposure and clinical safety and efficacy in patients with advanced renal cell carcinoma. Br J Cancer. 2014;111:1909–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, et al. Pazopanib in Locally Advanced or Metastatic Renal Cell Carcinoma: Results of a Randomized Phase III Trial. J Clin Oncol. 2010;28:1061–8. [DOI] [PubMed] [Google Scholar]
- 61.Sheiner LB. Learning versus confirming in clinical drug development. Clin Pharmacol Ther. 1997;61:275–91. [DOI] [PubMed] [Google Scholar]
- 62.Houk BE, Bello CL, Poland B, Rosen LS, Demetri GD, Motzer RJ. Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: results of a pharmacokinetic/pharmacodynamic meta-analysis. Cancer Chemother Pharmacol. 2009;66:357–71. [DOI] [PubMed] [Google Scholar]
- 63.Madlensky L, Natarajan L, Tchu S, Pu M, Mortimer J, Flatt SW, et al. Tamoxifen Metabolite Concentrations, CYP2D6 Genotype, and Breast Cancer Outcomes. Clin Pharmacol Ther. 2011;89:718–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lubberman FJE, Gelderblom H, Hamberg P, Vervenne WL, Mulder SF, Jansman FGA, et al. The effect of using pazopanib with food vs fasted on pharmacokinetics, patient safety and preference (DIET study). Clin Pharmacol Ther. 2019;106:1076–82. [DOI] [PubMed] [Google Scholar]
- 65.Groenland SL, Van Eerden RAG, Verheijen RB, Koolen SLW, Moes DJAR, Desar IME, et al. Therapeutic drug monitoring of oral anticancer drugs: the DPOG-TDM protocol for a prospective study. Ther Drug Monit. 2019;41:561–7. [DOI] [PubMed] [Google Scholar]
- 66.Menz BD, Stocker SL, Verougstraete N, Kocic D, Galettis P, Stove CP, et al. Barriers and opportunities for the clinical implementation of therapeutic drug monitoring in oncology. Br J Clin Pharmacol. 2020;87:227–236. [DOI] [PubMed] [Google Scholar]
- 67.Carton E, Noe G, Huillard O, Golmard L, Giroux J, Cessot A, et al. Relation between plasma trough concentration of abiraterone and prostate-specific antigen response in metastatic castration-resistant prostate cancer patients. Eur J Cancer. 2017;72:54–61. [DOI] [PubMed] [Google Scholar]
- 68.Van Nuland M, Groenland SL, Bergman AM, Steeghs N, Rosing H, Venekamp N, et al. Exposure-response analyses of abiraterone and its metabolites in real-world patients with metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2020;23:244–251. [DOI] [PubMed] [Google Scholar]
- 69.Groenland SL, Geel DR, Janssen JM, de Vries N, Rosing H, Beijnen JH, et al. Exposure-response analyses of anaplastic lymphoma kinase inhibitors crizotinib and alectinib in non-small-cell lung cancer patients. Clin Pharmacol Ther. 2021;109:394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Netherlands Trial Register. Standard dose alectinib versus Therapeutic Drug Monitoring guided alectinib dosing. Available from: https://www.trialregister.nl/trial/9441
- 71.Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim DW, et al. Alectinib versus Crizotinib in Untreated ALK-positive Non–Small-Cell Lung Cancer. N Engl J Med. 2017;377:829–38. [DOI] [PubMed] [Google Scholar]
- 72.Larson RA, Druker BJ, Guilhot F, O’Brien SG, Riviere GJ, Krahnke T, et al. Imatinib pharmacokinetics and its correlation with response and safety in chronic-phase chronic myeloid leukemia: A subanalysis of the IRIS study. Blood. 2008;111:4022–8. [DOI] [PubMed] [Google Scholar]
- 73.Picard S, Titier K, Etienne G, Teilhet E, Ducint D, Lassalle R, et al. Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood. 2007;109:3496–9. [DOI] [PubMed] [Google Scholar]
- 74.Demetri GD, Wang Y, Wehrle E, Racine A, Nikolova Z, Blanke CD, et al. Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J Clin Oncol. 2009;27:3141–7. [DOI] [PubMed] [Google Scholar]
- 75.Sternberg CN, Donskov F, Haas NB, Doehn C, Russo P, Elmeliegy M, et al. Pazopanib Exposure Relationship with Clinical Efficacy and Safety in the Adjuvant Treatment of Advanced Renal Cell Carcinoma. Clin Cancer Res. 2018;24:3005–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Verheijen RB, Swart LE, Beijnen JH, Schellens JHM, Huitema ADR, Steeghs N. Exposure-survival analyses of pazopanib in renal cell carcinoma and soft tissue sarcoma patients: opportunities for dose optimization. Cancer Chemother Pharmacol. 2017;80:1171–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Groenland SL, Van Eerden RAG, Koolen SLW, Moes DJAR, Imholz A, Wilgenhof S, et al. Therapeutic drug monitoring (TDM) of pazopanib - using cost-neutral PK-guided interventions to optimize exposure. J Clin Oncol. 2020;38 no. 15_suppl: 3598. [Google Scholar]
- 78.Van Nuland M, Bergman AM, Rosing H, De Vries N, Huitema ADR, Beijnen JH. Exposure-Response Assessment of Enzalutamide and Its Major Metabolites in a Real-World Cohort of Patients with Metastatic Castration-Resistant Prostate Cancer. Pharmacotherapy. 2019;39:1137–45. [DOI] [PubMed] [Google Scholar]
- 79.Ingle JN, Kalari KR, Buzdar AU, Robson ME, Goetz MP, Desta Z, et al. Estrogens and their precursors in postmenopausal women with early breast cancer receiving anastrozole. Steroids. 2015;99:32–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang X, Roy A, Hochhaus A, Kantarjian HM, Chen T-T, Shah NP. Differential effects of dosing regimen on the safety and efficacy of dasatinib: retrospective exposure-response analysis of a Phase III study. Clin Pharmacol Adv Appl. 2013;5:85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ishida Y, Murai K, Yamaguchi K, Miyagishima T, Shindo M, Ogawa K, et al. Pharmacokinetics and pharmacodynamics of dasatinib in the chronic phase of newly diagnosed chronic myeloid leukemia. Eur J Clin Pharmacol. 2016;72:185–93. [DOI] [PubMed] [Google Scholar]
- 82.Guilhot F, Hughes TP, Cortes J, Druker BJ, Baccarani M, Gathmann I, et al. Plasma exposure of imatinib and its correlation with clinical response in the Tyrosine Kinase Inhibitor Optimization and Selectivity trial. Haematologica. 2012;97:731–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Widmer N, Decosterd LA, Csajka C, Montemurro M, Haouala A, Leyvraz S, et al. Imatinib plasma levels: correlation with clinical benefit in GIST patients. Br J Cancer. 2010;102:1198–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Delbaldo C, Chatelut E, Ré M, Deroussent A, Séronie-Vivien S, Jambu A, et al. Pharmacokinetic-pharmacodynamic relationships of imatinib and its main metabolite in patients with advanced gastrointestinal stromal tumors. Clin Cancer Res. 2006;12:6073–8. [DOI] [PubMed] [Google Scholar]
- 85.Giles FJ, Yin OQP, Sallas WM, Le Coutre PD, Woodman RC, Ottmann OG, et al. Nilotinib population pharmacokinetics and exposure-response analysis in patients with imatinib-resistant or -intolerant chronic myeloid leukemia. Eur J Clin Pharmacol. 2013;69:813–23. [DOI] [PubMed] [Google Scholar]
- 86.Larson RA, Yin OQP, Hochhaus A, Saglio G, Clark RE, Nakamae H, et al. Population pharmacokinetic and exposure-response analysis of nilotinib in patients with newly diagnosed Ph+ chronic myeloid leukemia in chronic phase. Eur J Clin Pharmacol. 2012;68:723–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rousset M, Dutriaux C, Bosco-Lévy P, Prey S, Pham-Ledard A, Dousset L, et al. Trough dabrafenib plasma concentrations can predict occurrence of adverse events requiring dose reduction in metastatic melanoma. Clin Chim Acta. 2017;472:26–9. [DOI] [PubMed] [Google Scholar]
- 88.Goldwirt L, Chami I, Feugeas J-P, Pages C, Brunet-Possenti F, Allayous C, et al. Reply to “Plasma vemurafenib concentrations in advanced BRAFV600mut melanoma patients: impact on tumour response and tolerance” by Funck-Brentano et al. Ann Oncol. 2016;27:363–4. [DOI] [PubMed] [Google Scholar]
- 89.Funck-Brentano E, Alvarez JC, Longvert C, Abe E, Beauchet A, Funck-Brentano C, et al. Plasma vemurafenib concentrations in advanced BRAFV600mut melanoma patients: Impact on tumour response and tolerance. Ann Oncol. 2015;26:1470–5. [DOI] [PubMed] [Google Scholar]
- 90.Kramkimel N, Thomas-Schoemann A, Sakji L, Golmard JL, Noe G, Regnier-Rosencher E, et al. Vemurafenib pharmacokinetics and its correlation with efficacy and safety in outpatients with advanced BRAF-mutated melanoma. Target Oncol. 2016;11:59–69. [DOI] [PubMed] [Google Scholar]
- 91.Kichenadasse G, Hughes JH, Miners JO, Mangoni AA, Rowland A, Hopkins AM, et al. Relationship between vemurafenib plasma concentrations and survival outcomes in patients with advanced melanoma. Cancer Chemother Pharmacol. 2020;85:615–20. [DOI] [PubMed] [Google Scholar]
- 92.Groenland SL, Martínez-Chávez A, van Dongen MGJ, Beijnen JH, Schinkel AH, Huitema ADR, et al. Clinical Pharmacokinetics and Pharmacodynamics of the Cyclin-Dependent Kinase 4 and 6 Inhibitors Palbociclib, Ribociclib, and Abemaciclib. Clin Pharmacokinet. 2020;59:1501–20. [DOI] [PubMed] [Google Scholar]
- 93.Soulieres D, Senzer NN, Vokes EE, Hidalgo M, Agarvala SS, Siu LL. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J Clin Oncol. 2004;22:77–85. [DOI] [PubMed] [Google Scholar]
- 94.Hidalgo BM, Siu LL, Nemunaitis J, Rizzo J, Hammond L a, Takimoto C, et al. Phase I and Pharmacologic Study of OSI-774, an Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor, in Patients With Advanced Solid Malignancies. J Clin Oncol. 2001;19:3267–79. [DOI] [PubMed] [Google Scholar]
- 95.Tiseo M, Andreoli R, Gelsomino F, Mozzoni P, Azzoni C, Bartolotti M, et al. Correlation between erlotinib pharmacokinetics, cutaneous toxicity and clinical outcomes in patients with advanced non-small cell lung cancer (NSCLC). Lung Cancer. 2014;83:265–271. [DOI] [PubMed] [Google Scholar]
- 96.Lu J-F, Eppler SM, Wolf J, Hamilton M, Rakhit A, Bruno R, et al. Clinical pharmacokinetics of erlotinib in patients with solid tumors and exposure-safety relationship in patients with non-small cell lung cancer. Clin Pharmacol Ther. 2006;80:136–45. [DOI] [PubMed] [Google Scholar]
- 97.Zhao Y-Y, Li S, Zhang Y, Zhao H-Y, Liao H, Guo Y, et al. The relationship between drug exposure and clinical outcomes of non-small cell lung cancer patients treated with gefitinib. Med Oncol. 2011;28:697–702. [DOI] [PubMed] [Google Scholar]
- 98.Kobayashi H, Sato K, Niioka T, Miura H, Ito H, Miura M. Relationship among gefitinib exposure, polymorphisms of its metabolizing enzymes and transporters, and side effects in Japanese patients with non-small-cell lung cancer. Clin Lung Cancer. 2015;16:274–81. [DOI] [PubMed] [Google Scholar]
- 99.Brown K, Comisar C, Witjes H, Maringwa J, De Greef R, Vishwanathan K, et al. Population pharmacokinetics and exposure-response of osimertinib in patients with non-small cell lung cancer. Br J Clin Pharmacol. 2017;83:1216–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wollenberg L, Marchand M, Merdjan H, Litwiler K. Development of a Population Pharmacokinetic Model for Binimetinib with Subsequent Exposure-Response Analyses in NRAS Mutant Melanoma. Am Conf Pharmacometrics. 2018;T-051. [Google Scholar]
- 101.Ouellet D, Kassir N, Chiu J, Mouksassi MS, Leonowens C, Cox D, et al. Population pharmacokinetics and exposure-response of trametinib, a MEK inhibitor, in patients with BRAF V600 mutation-positive melanoma. Cancer Chemother Pharmacol. 2016;77:807–17. [DOI] [PubMed] [Google Scholar]
- 102.Groenland SL, Janssen JM, Nijenhuis C, de Vries N, Rosing H, Wilgenhof S, et al. 567P Exposure-response analyses of dabrafenib and trametinib in melanoma patients. Ann Oncol. 2020;31:S486–7. [DOI] [PubMed] [Google Scholar]
- 103.Ravaud A, Urva SR, Grosch K, Cheung WK, Anak O, Sellami DB. Relationship between everolimus exposure and safety and efficacy: Meta-analysis of clinical trials in oncology. Eur J Cancer. 2014;50:486–95. [DOI] [PubMed] [Google Scholar]
- 104.Thiery-Vuillemin A, Mouillet G, Nguyen Tan Hon T, Montcuquet P, Maurina T, Almotlak H, et al. Impact of everolimus blood concentration on its anti-cancer activity in patients with metastatic renal cell carcinoma. Cancer Chemother Pharmacol. 2014;73:999–1007. [DOI] [PubMed] [Google Scholar]
- 105.Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–11. [DOI] [PubMed] [Google Scholar]
- 106.Tsuchiya N, Igarashi R, Suzuki-Honma N, Fujiyama N, Narita S, Inoue T. Association of pharmacokinetics of axitinib with treatment outcome and adverse events in advanced renal cell carcinoma patients. J Clin Oncol. 2015;33:suppl 7; abstract 506. [Google Scholar]
- 107.Lacy S, Nielsen J, Yang B, Miles D, Nguyen L, Hutmacher M. Population exposure – response analysis of cabozantinib efficacy and safety endpoints in patients with renal cell carcinoma. Cancer Chemother Pharmacol. 2018;81:1061–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nguyen L, Chapel S, Tran BD, Lacy S. Cabozantinib exposure–response analyses of efficacy and safety in patients with advanced hepatocellular carcinoma. J Pharmacokinet Pharmacodyn. 2019;46:577–89. [DOI] [PubMed] [Google Scholar]
- 109.Faivre S, Delbaldo C, Vera K, Robert C, Lozahic S, Lassau N, et al. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol. 2006;24:25–35. [DOI] [PubMed] [Google Scholar]
- 110.Noda S, Otsuji T, Baba M, Yoshida T, Kageyama S, Okamoto K, et al. Assessment of Sunitinib-Induced Toxicities and Clinical Outcomes Based on Therapeutic Drug Monitoring of Sunitinib for Patients with Renal Cell Carcinoma. Clin Genitourin Cancer. 2015;13:350–8. [DOI] [PubMed] [Google Scholar]
- 111.Teo YL, Chue XP, Chau NM, Tan MH, Kanesvaran R, Wee HL, et al. Association of drug exposure with toxicity and clinical response in metastatic renal cell carcinoma patients receiving an attenuated dosing regimen of sunitinib. Target Oncol. 2015;10:429–37. [DOI] [PubMed] [Google Scholar]
- 112.Takasaki S, Kawasaki Y, Kikuchi M, Tanaka M, Suzuka M, Noda A, et al. Relationships between sunitinib plasma concentration and clinical outcomes in Japanese patients with metastatic renal cell carcinoma. Int J Clin Oncol. 2018;23:936–43. [DOI] [PubMed] [Google Scholar]
- 113.Groenland SL, Van Eerden RAG, Koolen SLW, Moes DJAR, Desar IME, Touw DJ, et al. Therapeutic drug monitoring of oral anticancer drugs - preliminary results of a prospective study. Ann Oncol. 2019;30:v159–93. [Google Scholar]