

Abstract
Our understanding of epithelial-to-mesenchymal transition (EMT) has slowly evolved from a simple two state, binary model to a multi-step, dynamic continuum of epithelial-to-mesenchymal plasticity, with metastable intermediate transition states that may drive cancer metastasis. Head and neck cancer is no exception, and in this review, we use head and neck as a case study for how partial-EMT (p-EMT) cell states may play an important role in cancer progression. In particular, we summarize recent in vitro and in vivo studies that uncover these intermediate transition states, which exhibit both epithelial and mesenchymal properties and appear to have distinct advantages in migration, survival in the bloodstream, and seeding and propagation within secondary metastatic sites. We then summarize the common and distinct regulators of p-EMT as well as methodologies for identifying this unique cellular subpopulation, with a specific emphasis on the role of cutting-edge technologies, such as single cell approaches. Finally, we propose strategies to target p-EMT cells, highlighting potential opportunities for therapeutic intervention to specifically target the process of metastasis. Thus, although significant challenges remain, including numerous gaps in current knowledge, a deeper understanding of EMT plasticity and a genuine identification of EMT as spectrum rather than a switch will be critical for improving patient diagnosis and treatment across oncology.
INTRODUCTION
Metastasis is a complex, multistep process whereby primary cancer cells disseminate to regional or distant secondary sites, resulting in substantial patient morbidity and death across numerous solid malignancies (1). Although the biological basis of metastasis has been extensively investigated, the mechanisms that trigger metastasis remain poorly understood. Among carcinomas -- those tumors derived from the epithelium -- one compelling mechanism that has garnered interest is epithelial-to-mesenchymal transition (EMT) (2). In this conceptual framework of metastasis, malignant cells absolve their epithelial identity and adopt a mesenchymal expression state, thereby becoming more motile as they are imbued with the capacity to remodel the extracellular matrix (ECM) and invade local tissues, ultimately entering and exiting the circulation to seed distant colonies. Early studies of EMT and cancer suggested EMT was akin to a switch, with cells ostensibly in an epithelial or mesenchymal state and mesenchymal cells representing the infiltrative subpopulation of a tumor (3). By contrast, intermediary states adopted by cells as they underwent EMT were considered to be incidental, inconsequential, and entirely transient.
However, it is now widely agreed that EMT is a complex and dynamic cell biologic process, with cells existing in a number of meta-stable, intermediary states between the epithelial and mesenchymal poles (so-called “hybrid-EMT” or “partial-EMT” states) (4). These partial-EMT (p-EMT) cells may invade collectively via oligocellular clusters, maintaining some cell-to-cell adhesive properties while escaping from the stromal scaffold and remodeling the ECM (5). Indeed, p-EMT cells are now believed to play essential roles in local tissue invasion (6), collective migration (7), circulating tumor cells (8), and, ultimately, both locoregional (9) and distant metastases (10). Importantly, these aggressive biological features have clinical implications including treatment resistance (10,11) and effects on overall survival (9,12). The p-EMT cell state is driven both by intrinsic genetic, epigenetic, and post-translational alterations among primary malignant cells as well as paracrine signaling via supportive stromal cells in the tumor microenvironment (TME) (4,13,14). Clearly, the identification of p-EMT represents an important aspect of EMT biology that continues to mature within oncology, yet much remains unknown (15–17).
In this review, we discuss the importance of p-EMT in tumor progression and metastasis and the evolving landscape regarding EMT plasticity, utilizing head and neck squamous cell carcinoma (HNSCC) as a case study for this discussion. Identification of a hybrid p-EMT state and its distinction from completely transformed mesenchymal or epithelial cells represents the first and most important step in improving our understanding of these new cell states. We then describe markers that help identify p-EMT cells, while also exploring the important drivers and regulators of p-EMT and potential strategies to target this expression state (Figure 1). Finally, we discuss the challenges that remain in studying p-EMT and the opportunities for new therapeutics targeting this essential program.
Figure 1:

Understanding partial EMT cells in cancer progression.
Classical EMT: From Embryology to Oncology
EMT was first described within the context of developmental biology based on a series of experiments by Greenburg and Hay in the 1980s (18). They noted that epithelial cells from embryonic and adult anterior lenses that were subsequently cultured in three-dimensional collagen matrices lost cell-to-cell attachments, migrated as individual cells, and appeared morphologically similar to mesenchymal cells. Subsequent work demonstrated EMT to be vital for tissue remodeling events, most notably mesoderm and neural crest development (19,20). In addition, mesenchymal cells were demonstrated to undergo the reverse process, mesenchymal-to-epithelial transition (MET) and contribute to the formation of epithelial organs (3).
Subsequently, many investigators suspected a similar EMT-like program may drive invasion and metastasis in carcinomas. This hypothesis was supported by the experimental observation that expression or suppression of the epithelial adherens junction protein E-cadherin can impair or confer invasiveness, respectively (21–23). In this model of EMT, epithelial cells at the invasive front of a primary tumor undergo a loss of epithelial markers such as tight junction proteins (claudins and occludins), adherens junction proteins (E-cadherin, alpha and beta catenin), and cytokeratins. Cells simultaneously acquire a mesenchymal phenotype with the concomitant expression of Vimentin, N-cadherin, Fibronectin, α-SMA, FSP1, Integrin α5/β1, and Desmin (2), allowing increased directional invasion through the basement membrane and ECM followed by intravasation into the circulation and locoregional or distant dissemination (24,25).
In head and neck squamous cell carcinoma (HNSCC), an EMT cell state has been associated with cancer aggressiveness and poor prognosis (26,27). Jung et al. classified three independent cohorts of HNSCC patients into epithelial or mesenchymal subgroups according to an EMT gene signature comprised of 82 genes, with worse survival observed among patients in the mesenchymal subgroup (26). Similarly, high expression of the mesenchymal marker Vimentin and correspondingly low levels of E-cadherin have been associated with increased rates of metastasis (28). From a cell biological perspective, a mesenchymal, spindle shaped morphology and low E-cadherin expression has also been observed in an invasive HNSCC cell line derived from a lymph node metastasis (29). Indeed, spindle cell SCC of the head and neck, which is a pathologic variant, has shown increased lymph node metastasis with a decrease in epithelial markers responsible for adhesion, like E-cadherin, and components of desmosomes, such as desmogleins and desmocollins (30). Recent global expression profiling studies have further extended on these findings, identifying a distinct subgroup of more mesenchymal tumors among HNSCC patients that is associated with more aggressive biology (31). Pooled meta-analyses of well-known EMT transcription factors (TFs) in HNSCC, for example, show a strong correlation between the expression of EMT TFs and poor overall survival (31). The overexpression of TFs TWIST1, SNAI1, SNAI2 and ZEB1, in particular, show a significant association with poor overall survival among HNSCC patients (27).
While several of these studies demonstrate that the mesenchymal phenotype is associated with increased cancer metastasis (28,29), analysis of human primary tumors and their matched metastatic lesions in many carcinomas has surprisingly shown similar, or even augmented, epithelial characteristics in secondary, metastatic tumors compared to primary tumors (32). Indeed, the inability to observe a mesenchymal phenotype in metastatic lesions fueled interest in MET, whereby metastatic mesenchymal cells might embrace an epithelial program which enables post-seeding tumor growth at secondary sites (33,34). Interestingly, pre-clinical models in which a mesenchymal state was stably induced prevented the formation of metastasis (35,36). While these observations were used to question the role of EMT in metastasis, in the broader model of a dynamic continuum of epithelial and mesenchymal phenotypes, such a finding is wholly consistent with this process: Cells stuck at one axis of the spectrum cannot undergo the required changes to support cancer phenotypes such as metastasis that require transitions in cell state.
The Emerging Understanding of EMT as a Continuous Spectrum in Tumor Development and Progression
The idea of EMT and its reverse process, MET, functioning as dynamic switches in cell state has further evolved into considering epithelial and mesenchymal states as poles along a broader, continuous spectrum. While such a continuum had long been suspected, in vivo evidence of intermediary cell states has only recently been reported, largely as the result of technical innovations in cell sorting by surface markers, the advent of single cell sequencing, and clever experimental design with in vivo models (13,14,37,38). Early EMT investigations were performed using cancer cell lines in vitro or through bulk assessment of primary human tumor samples, and consequently, were unable to evaluate the plasticity and functional states of EMT states in vivo (14).
The combined use of genetically engineered mice and lineage tracing has allowed for the study of dynamic p-EMT transition states. Rhim and colleagues used Pdx1CRE/KRasG12D/P53cKO/Rosa-YFP or Pdx1CRE/KRasG12D/Ink4a+/−/Rosa-YFP mice to tag and track pancreatic epithelial cells in an in vivo model of pancreatic cancer (39). This strategy allowed them to evaluate cell states of malignant cells at multiple time points between tumor development, invasion, circulation, and distant seeding. While they found that the majority of circulating pancreatic cells had mesenchymal features and expressed stem cell-associated markers, a smaller group of circulating pancreatic cells (18%) exhibited co-expression of E-cadherin and Zeb1, classical epithelial and mesenchymal markers, respectively. Similarly, Ruscetti et al. utilized an autochthonous murine model of prostate cancer (CPKV) to describe malignant cell states associated with metastasis (40). Using a Vimentin-GFP reporter, they defined three classes of malignant cells: epithelial (EpCAM+/Vim-GFP−), EMT (EpCAM+/Vim-GFP+), and mesenchymal-like (EpCAM−/Vim-GFP+). While both the EMT and mesenchymal-like cells displayed increased stemness and invasiveness relative to epithelial cells, only the EMT cells (i.e. cells with epithelial and mesenchymal features) were able to reliably establish macrometastatic colonies, presumably due to their partially retained epithelial identity and apparent plasticity. Thus, this study provided robust in vivo evidence of a metastatic advantage for p-EMT cells.
It remained unclear, however, whether these intermediary EMT cells represented a temporary transition state along the EMT continuum or whether they existed as a distinct and stable entity. To address the nature of these intermediary states, Pastushenko and colleagues screened a large panel of cell surface markers in skin and mammary cancer models (13). They found that loss of EpCAM was universally linked to gain of Vimentin expression, suggesting conversion to a mesenchymal state. However, strikingly a fraction of Vimentin+ cells also expressed Keratin 14 (KRT14), and they were able to consistently identify six distinct cell signatures of the EMT continuum across skin, mammary, and esophageal cancer models. Again, these hybrid cells were found to be more invasive and better able to seed metastatic colonies, similar to prior studies (39,40). In these distant colonies, they found that all EpCAM− negative cells were able to revert back to EpCAM expression, suggesting that even the most phenotypically mesenchymal of cells can undergo MET in the context of the lung microenvironment. In addition, they found that these hybrid-type cells were able to manipulate the surrounding stroma, with more mesenchymal cells preferentially co-localizing with inflammatory and endothelial cells, thereby conferring increased invasive and metastatic potential. In a subsequent investigation, Kröger et al. found that not only was this hybrid p-EMT an independent and stable phase, but it was also essential for tumorigenesis in models of breast cancer (41). These p-EMT cells were regulated by expression of Snail proteins and associated with expression of adult stem cell programs, namely canonical Wnt signaling.
Based on this body of prior work, it appears that EMT states can be represented in a one-dimensional three-well landscape (with the mesenchymal marker along the x-axis) (Figure 2). Cells that undergo EMT must sequentially overcome the activation energy required to reach the next stable or metastable state, represented as one of three wells. In this model, each well represents a steady state during EMT: 1) Fully epithelial, 2) a partial epithelial/mesenchymal state, and 3) a complete mesenchymal state. While EMT can have multiple transition states with the same energy barriers, certain cancer cells might meta-stabilize as specific intermediate cells with the help (or under the direction) of distinct regulators. This p-EMT intermediate cell state – or cell states – may therefore be more stable than the other transition states with a higher energy barrier to the full mesenchymal phenotype.
Figure 2:

Energy model of EMT transition states.
While these well-designed studies convincingly established the existence of p-EMT intermediate states in several in vivo cancer models, establishing p-EMT in primary human carcinomas has been largely limited by available bulk genomic sequencing and immunohistochemical modalities. The advent of single cell genomics afforded an unprecedented level of resolution in dissecting tumor heterogeneity and the cellular components of the TME (15). To explore tumor heterogeneity in HNSCC, our group recently utilized single cell RNA-sequencing (scRNA-seq) to analyze 18 primary, treatment-naïve tumors and five matched lymph node metastases (6,16). We identified a p-EMT program, which displayed features reminiscent of classical EMT yet also appeared distinct: EMT markers Vimentin and Integrin α5 were identified and Transforming Growth Factor β Induced (TGFBI) was one of the top scoring genes among these cells, suggesting potential regulation by the classical EMT inducer TGFβ. Other hallmark EMT markers, however, were absent and p-EMT cells retained expression of epithelial markers, such as various cytokeratins. For example, among the classical EMT TFs only Snail2 was detected, which peaks relatively early in the EMT process (42). Interestingly, p-EMT cells were exquisitely localized at the leading edge of tumors and had increased invasive potential in vitro, suggesting a potential role for the p-EMT program in invasion and metastasis in HNSCC. To explore this possibility further, we utilized our scRNA-seq data to deconvolve bulk expression data from The Cancer Genome Atlas into inferred malignant profiles (43). In tumors with enrichment of p-EMT among malignant cells, there was a strong association with nodal metastasis, tumor grade, and adverse pathological features, including extracapsular extension (ECS) and lymphovascular invasion. Indeed, the p-EMT program was found to be a stronger predictor of nodal metastasis than a classical EMT signature. While prior studies have associated EMT and increased matrix metalloproteinase-12 expression with evidence of ECS on histology, a specific mechanistic role of the p-EMT cell state in ECS has not been defined (44,45).
Subsequent studies have further validated our initial observations. Using some of the top markers (PDPN, LAMB3, LAMC2) of the p-EMT program, we computed a p-EMT score based on immunohistochemical staining of these markers on a tissue microarray from 99 oral cavity squamous cell carcinoma patients. p-EMT was associated with higher tumor grade and nodal metastasis as well as other adverse pathologic features, representing an orthogonal validation of our initial findings. Importantly, we found that p-EMT had an effect on overall survival, with worse survival among p-EMThigh patients (9). Another group recently focused on 15 representative p-EMT genes and 10 variable p-EMT genes from our signature and re-analyzed the TCGA dataset (27). Genes including SERPINE1, TGFBI, ITGA5, CDH13, P4HA2 and LAMC2 were found to have prognostic value in these analyses, with p-EMT related genes demonstrating increase expression in HNSCC primary tumor samples compared to normal tissue. Gene ontogeny enrichment analysis revealed that these p-EMT genes correlated with processes of cell substrate adhesion and angiogenesis, both of which are related to metastasis. Importantly, TGFBI, which was identified by our group as one of the top scoring p-EMT related genes, was also among the list of genes with significant prognostic value. A more recent study investigated the relationship between p-EMT specifically in circulating tumor cells (CTCs) from HNSCC patients: Hybrid cells co-expressing the epithelial marker Keratin 19 and mesenchymal marker Vimentin were found to be the dominant CTC subpopulation among recurrent and metastatic HNSCC patients (46). Together, these findings suggest that p-EMT expression has both biologic and clinical significance, with tremendous potential to serve as a biomarker and/or therapeutic target in HNSCC.
Outside of HNSCC, additional investigations have leveraged multi-omics strategies to characterize intermediate EMT states in primary human malignancies. Examining cutaneous SCC, Ji et al. used scRNA-seq, spatial transcriptomics, and multiplexed ion beam imaging (MIBI) to identify a population of “tumor specific keratinocytes” (TSKs) that exhibited both epithelial differentiation, yet had high expression of EMT markers including VIM and ITGA5 (47). Similar to p-EMT cells in HNSCC, these TSK cells localized to the leading edge and co-localized with cancer-associated fibroblasts (CAFs) and endothelial cells, highlighting the presence of a fibrovascular niche and suggesting invasive potential.
In breast cancer patients, a p-EMT phenotype was identified in a subset of patients that correlated with expression of the TF NRF2 (48). NRF2 prevented completion of the EMT cascade, stabilizing cells in a p-EMT state. Accordingly, this p-EMT group was associated with a high NRF2 score and poor patient survival. Similarly, in a recent study of urothelial carcinomas, a morphological approach based on sequential immunohistochemistry on the same tissue section combined with slide digitization and image processing was adopted to detect and quantify cancer cells with a p-EMT phenotype (49). These analyses demonstrated a strong correlation between p-EMT at the time of diagnosis and eventual poor prognosis. Likewise, in patients with pancreatic ductal adenocarcinoma, immunohistochemical analysis was completed for EMT markers and tumor budding. Tumor budding is the phenomenon of local dissemination of a single tumor cell or cluster of cells from the invasive front into the surrounding tissue. Tumor budding has been shown to have prognostic value in colorectal adenocarcinoma and several other cancers (50–52). Detailed intra- and inter-tumoral analysis of EMT and tumor budding markers revealed induction of p-EMT at the tumor-stromal interface and in tumor buds (53). Thus, p-EMT appears to be represented in invasive cells across a number of epithelial malignancies, highlighting its importance in oncology as a bona fide intermediate state with relevance to human biology.
The Role of Partial-EMT in Collective Migration, Circulating Tumor Cells, and Distant Metastases
In contrast to individual mesenchymal cells invading through the basement membrane, the intra-tumoral localization of p-EMT cells at the leading edge of a tumor suggests that p-EMT cells may invade through a collective migration model (6,16,47). Epithelial adhesion molecules expressed by p-EMT cells may facilitate cell-cell adhesion, while mesenchymal properties allow cell-ECM interactions that enable migration. In Drosophila intestinal tumors, p-EMT cells were shown to invade the basal lamina of the midgut epithelium and exhibit collective migration with subsequent polyclonal seeding in metastatic sites, an effect seen most clearly with high levels of Sna expression (Drosophila homolog of Snail) (54). The Sna overexpressed cells demonstrated a characteristic p-EMT phenotype with loss of epithelial traits, such as cell shape and polarity, and a gain of mesenchymal characteristics, most notably formation of protrusive membranes and the ability to invade basal lamina (54). Additionally, in vitro p-EMT tumor spheres retain cell-cell contacts and invade as a collective group compared to “complete” EMT (i.e. tumor spheres with transcriptional repression of Ecad) (55).
The role of p-EMT in intravasation, clustering of CTCs, and dissemination appears to represent an orthogonal, yet important, function of p-EMT in oncology. CTCs are often detected as clusters of 2-50 cells, sometimes termed “circulating tumor microemboli” (56,57). These CTC clusters are more robust and demonstrate increased metastatic potential compared to singular CTCs due to increased resistance to apoptosis and stress and they have a higher probability of being trapped in narrow blood vessels, likely facilitating extravasation (56,58–60). Interestingly, CTC clusters have been found to contribute to 50% of total metastases despite constituting only 3% of total CTC events (56). Hybrid cells co-expressing epithelial and mesenchymal markers have been detected among CTC clusters in the bloodstream of breast, lung, colon and prostate cancer patients (61–64). CTC clusters containing both epithelial and mesenchymal markers are resistant to anoikis (65), able to adapt to a foreign microenvironments to form macrometastatic colonies (7), and demonstrate increased chemotherapeutic drug resistance (11,66). Thus, p-EMT may augment the metastatic potential of CTC clusters (Figure 3).
Figure 3:
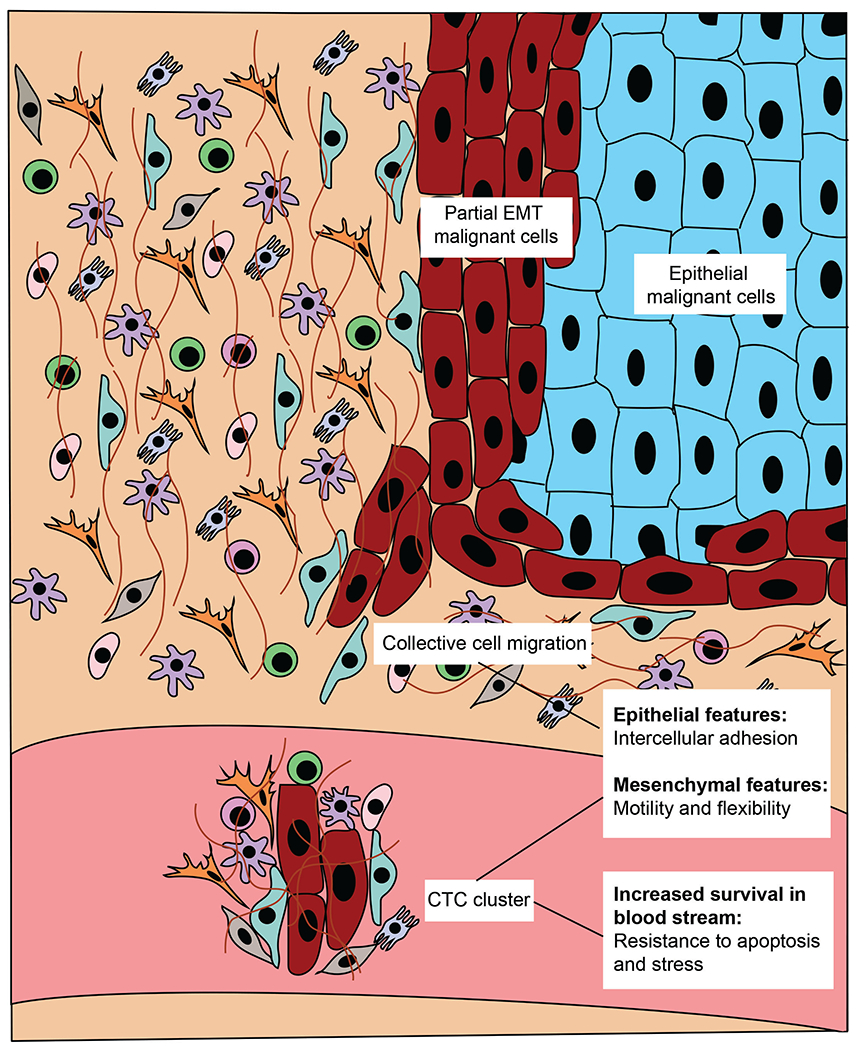
Partial EMT cells at the leading edge of the tumor are prone to collective migration and CTC cluster formation with increased metastatic rate.
However, many questions remain. For one, metastases histologically recapitulate findings of the primary lesion across cancer types (1). While MET is a broadly accepted phenomenon, the cell-intrinsic pathways and TME cues that drive this shift in cell state within a novel, secondary microenvironment remain poorly understood. Furthermore, different primary cancers have proclivities toward certain metastatic sites (e.g. bone in prostate cancer, regional lymphatics in HNSCC, etc.). While p-EMT has been associated with increased invasiveness and metastatic potential across a number of carcinomas, including HNSCC, prostate, skin, lung, and breast (11,13,66,67), it is not well understood what drives this stereotyped behavior. Future studies must define subtypes of p-EMT cells and describe epigenetic, transcriptional, and post-translational alterations that prime CTC clusters for successful seeding of a given metastatic location.
Partial-EMT promotes malignant cell stemness
Besides having heightened invasive capacity to circulate to distant metastatic sites, p-EMT cells may also be more likely to gain stemness properties (40,63,68,69). Cancer stem cells are a subpopulation of cancer cells that are similar to their physiological counterparts, but have the capacity to self-renew and differentiate, giving rise to heterogeneous, malignant subclones (70). Upon metastatic seeding to secondary sites, these cells may retain their ability to engage in tumor initiation, and they may also be more resistant to cell death and cancer therapeutics (71). Indeed, induction of classical EMT has been associated with the acquisition of stem cell properties in several carcinomas (72). It was first shown in immortalized human mammary epithelial cells (HMEC), where ectopic expression of the EMT TFs Snail/Twist1 triggered alterations in cell surface markers resembling a stem phenotype (CD44high/CD24low) along with enhanced ability to form mammospheres and tumors when transplanted into mice (73). These studies provided a preliminary explanation for how metastatic cells might maintain self-renewal capacity: EMT appeared to confer both invasiveness and stemness.
However, the stemness of fully mesenchymal EMT cells has been challenged by research showing that classical EMT prevents the tumor initiating capacity of cells in secondary locations (35,74,75). Subsequent studies have attempted to resolve this controversy by investigating stemness among cells with a more intermediate p-EMT phenotype. Indeed in breast cancer, hybrid EMT cells displayed several stem cell properties including increased plasticity, enhanced self-renewal, increased mammosphere formation and production of ALDH+ progenitors (76). Another orthogonal study provided corollary evidence of a p-EMT cell state with increased stemness including self-renewal and tumor-initiating capacity in ovarian cancer (68). However, direct in vivo evidence of p-EMT features being associated with cancer stemness has remained sparse. Detection of a p-EMT phenotype was observed for the first time in vivo in an autochthonous murine model of prostate cancer, in which isolated cells expressing both epithelial and mesenchymal markers showed enhanced stemness, plasticity, and tumor-initiating capacity (40). Interestingly, a recent study suggests that p-EMT and stemness may also be uniquely relevant among CTCs. In particular, Papadaki et al. found that among metastatic breast cancer patients, CSC+/p-EMT+ CTCs correlated with lung metastasis and poor survival, with an increase in these CTCs upon chemotherapeutic interference (10). Thus, there is likely to be complex interplay between the influence of p-EMT on stemness, collective migration, and CTCs, which will be an important area for further research.
Common and Distinct Regulators of Partial-EMT
There are a multitude of genes that converge to regulate EMT-related states, including epigenetic modulators (77), EMT TFs (6), and microRNAs (78). While these regulators are all critical to EMT initiation and plasticity, EMT TFs comprise the most well-defined group in the published literature. The canonical EMT TFs can be divided into three gene families: Snail, Zeb, and Twist (79). Evidence indicates that the expression of these EMT TFs is both context and tissue dependent (4). Historically, prior work has emphasized the role of Snail1/2, Zeb1/2, and Twist1/2 in EMT within HNSCC cell lines and tissue samples (79–81). However, many of these studies were published prior to single cell approaches and the identification of p-EMT among malignant cells. The premise that all six EMT TFs are all required for HNSCC metastasis has been challenged by our subsequent work (6). Among the 18 tumor samples we analyzed, none of the canonical EMT TFs were expressed at high levels except for Snail2 (also known as Slug) (6). Moreover, one of the genes most upregulated among p-EMT cells was TGFBI, consistent with studies showing that expression of Snail family members can be stimulated by TGFβ during EMT (82–85). Using immunohistochemical staining, we found that p-EMT cells at the leading edge were in close proximity to CAFs. Indeed, ligand-receptor analysis suggested that TGFβ may be secreted by CAFs and induce p-EMT among malignant cells, an observation which was then validated in vitro. Together, these data suggest that signals from CAFs in the stroma may play a role in inducing p-EMT at the leading edge of HNSCC tumors, potentially promoting invasive properties of this subpopulation by stimulating Snail2 expression (6).
Beyond EMT TFs, preliminary data suggests that p-EMT may also be regulated by long noncoding RNAs, such as MYOSLID, which has an effect on invasion and migration (86). Other non-cell intrinsic, tumor microenvironment factors that might influence the tumor p-EMT state, include hypoxia. Under oxygen-deprived hypoxic conditions, hypoxia inducible factors proteins (HIF1α and HIF1β) bind to hypoxia responsive element (HRE) on target gene promoters and serve as the major mediators of hypoxia regulated gene expression alterations (87). HIF1α has been shown to induce the p-EMT phenotype in pancreatic cancer cells when grown under hypoxic condition (88). HIF1α has also been shown to mediate several factors which have relevance in p-EMT, including coactivation with TGFβ (89,90), and induction of EMT-TFs like SNAIL (91), SLUG (92), ZEB1/2 (93,94). As stated before, our study has shown that paracrine interactions between cancer associated fibroblasts (CAFs) and malignant cells at the leading edge in HNSCC tumors may induce the p-EMT program (6). Evidence of activation of CAFs by HIF1α suggests another possible route for hypoxia mediated p-EMT induction.
These observations create meaningful opportunities to explore mechanisms of p-EMT regulation besides transcriptional regulators. Regardless, specific signaling pathways that contribute to the induction and maintenance of p-EMT in HNSCC and across oncology certainly warrant further study. These future studies must critically address how p-EMT may vary between in vitro and in vivo studies, while also clarifying the extent to which p-EMT versus classical EMT is present in a cancer and how each may (or may not) contribute to tumorigenesis and metastasis. Undoubtedly, greater reliance on in vivo models of cancer and patient-derived xenografts will help to address these challenges and more rigorously understand the regulation of p-EMT.
Techniques for Identifying Partial-EMT Markers
Given the poor clinical outcomes associated with p-EMT expressing tumors, the identification of reliable biomarkers to detect the p-EMT phenotype in patients has the potential to improve prognostication, guide primary or adjuvant treatment planning, and inform longitudinal surveillance. Due to the inherent plasticity of p-EMT cells, developing a broadly applicable and valid approach has proven challenging. However, a strategy that allows for the identification of simultaneously expressed epithelial and mesenchymal markers represents a logical strategy to identify p-EMT cell states from patient samples. Here, we discuss several emerging and theoretical strategies for translating p-EMT biomarkers into clinical application (Table 1).
Table 1:
Techniques and markers for identification of p-EMT.
| Technique | Rationale | Supporting Evidence | Limitations | References |
|---|---|---|---|---|
| Histologic Methods | ||||
| Novel IHC Markers | scRNA-seq has captured a wide swath of surface markers specific to p-EMT cells. Use of multiple markers through traditional IHC methods may capture patients along the spectrum of p-EMT phenotypes. | p-EMT specific surface markers identified with scRNA-seq (PDPN, LAMB3, LAMC2) predictive of survival outcomes and adverse pathologic features in cohort of oral cavity HNSCC patients. | Specific markers only validated in HNSCC. May not be relevant to p-EMT programs in other primary cancer sites. | Puram et al., 2017, Parikh et al., 2019 |
| Spatial transcriptomics and artificial intelligence | ST can capture expression signatures in situ overlain on traditional H&E sections. AI methods like computer vision may connect spatial expression matrices with clinically routine histologic images. | TSK cells that express epithelial and mesenchymal markers localize to leading edge on ST analysis in study of cutaneous SCC. | Adoption of AI techniques will require higher resolution of ST and large number of ST samples. | Ji et al., 2020 |
| Computational Methods | ||||
| Scoring with p-EMT gene sets | Machine learning methods have potential to take high dimension expression data from annotated cell lines as input and output optimized scoring tool based on small number of markers. | Computed scoring system able to accurately predict epithelial, mesenchymal or hybrid EMT phenotype in annotated cell lines. | EMT scoring model trained on cell lines included in NCI-60. Survival predictions across cancer tissue of origin inconsistent and cannot be generalised. | George et al., 2017 |
| Digital Deconvolution | Single-cell signature scoring of bulk RNA-seq data can leverage power of scRNA-seq in scalable manner without cost/expertise required for scRNA-seq. | Imputation of cell-type proportions in bulk RNA-seq samples from HNSCC TCGA through deconvolution with scRNA-seq cell signatures. | Requires available, confidently annotated single-cell reference dataset. | Qi et al., 2021 |
| Serum-Based Methods | ||||
| Peripheral blood sampling | A p-EMT cell-specific secretome may release factors detectable on peripheral blood samples. | Secreted factors Fibronectin 1 (FN1), Collagen Type II (COL2A1), and Native Fibrinogen Gamma Chain (FGG) were upregulated in p-EMT HCC cells. | Secreted factors Fibronectin 1 (FN1), Collagen Type II (COL2A1), and Native Fibrinogen Gamma Chain (FGG) were upregulated in p-EMT HCC cells. | Karaosmanoğlu et al., 2018 |
Abbreviations: AI = artificial intelligence, EMT = epithelial-mesenchymal transition, HCC = hepatocellular carcinoma, HNSCC = head and neck squamous cell carcinoma, IHC = immunohistochemistry, H&E = hematoxylin and eosin, p-EMT = partial epithelial-mesenchymal transition, scRNA-seq = single-cell RNA sequencing, ST = spatial transcriptomics, TCGA = The Cancer Genome Atlas, TSK = tumor specific keratinocytes
Early studies of EMT in human tumor samples focused on a small number of markers, typically E-cadherin and Vimentin, to define an epithelial or mesenchymal phenotype, respectively (3). As described, scRNA-seq now allows for the expression profiling of hybrid EMT cells in primary human tumor specimens, thereby expanding this limited set of markers to potentially dozens of genes that comprise a p-EMT “meta-signature” that may be partially or completely recapitulated across tumors of a given type, or even across cancer types (15). These meta-signatures provide multiple potential biomarkers, which can be used to generate a quantifiable score when probed in aggregate.
Such scoring can be derived from clinically feasible assays, such as traditional histologic techniques. For example, in the p-EMT cells identified in our scRNA-seq analysis of HNSCC tumors, we identified several p-EMT specific cell surface markers, including Podoplanin (PDPN), Transforming Growth Factor Receptor 1 (TGFβR1), Laminin Subunit Gamma 2 (LAMC2), and Laminin Subunit Beta 3 (LAMB3) (6). We recently validated the use of PDPN, LAMB3, and LAMC2 as predictive of prognosis in an independent cohort of HNSCC patients via traditional immunohistochemistry (IHC) techniques (9), and other groups have found associations between these specific p-EMT markers with poor prognosis (95). However, much additional work is needed to either validate these particular genes as representative of a p-EMT state across other primary epithelial malignancies or define similar sets of sensitive and specific p-EMT surface markers to characterize tumors on a site-by-site basis.
Given the high dimensional nature of p-EMT gene expression data, machine learning approaches represent another attractive option to quantify p-EMT status of clinical samples. To this end, George et al. developed a machine learning model trained on gene expression profiles from cell lines in the NCI-60 cohort – which are annotated as epithelial, mesenchymal, or hybrid – and then subsequently used their scoring model to predict survival in existing clinical datasets (5). Interestingly, while they found that a higher hybrid EMT score was predictive of worse disease-free survival and overall survival in lung cancer, this was not true for breast cancer and ovarian cancer patients, suggesting either that the prognostic implications of p-EMT states may be heterogeneous across tissue types or that the authors’ model insufficiently captures p-EMT in human subjects across cancer types.
Another computational approach to score the p-EMT phenotype in clinical samples is deconvolution of bulk expression data with annotated single-cell gene expression matrices (also known as “digital cytometry”) (96). Such methods can leverage single-cell level data without the associated costs and technical expertise required of single-cell experiments. However, even bulk RNA-sequencing is not routinely performed in clinical practice and the clinical adoption of deconvolution methods will depend on reductions in cost, labor, and time required of RNA sequencing.
Other potential methods may translate multi-omics characterization of p-EMT cell states into routinely used clinical assays. Spatial transcriptomic (ST) methods can simultaneously show a p-EMT expression signature that localizes to the histologic leading tumor edge (47). Increased resolution of ST platforms along with advances in and integration with computer vision techniques may allow for p-EMT predictions based solely on histologic sections. Additionally, further characterization of the secretome of p-EMT cells may one day allow for p-EMT classification based on peripheral blood assays (97). Clearly, the spectrum of EMT plasticity has implications for patient outcomes, though defining these intermediary p-EMT states and reliable markers that define them are important steps that will necessarily precede the clinical application of p-EMT markers to guide treatment or predict outcomes.
Targeting Partial-EMT and its Therapeutic Potential
Classical EMT has been shown to be induced by different exogenous stimuli which induce the stepwise transition from epithelial to mesenchymal phenotype through paracrine and autocrine signaling (98). These includes growth factors like TGFβ (98), EGF (99), FGF (100), PDGF (101), HGF (102), IGF (103), interleukins like interleukin-6, and BMP (104). Downstream to these exogenous ligands, numerous signaling pathways are involved, which include Wnt, Notch, Hippo, JAK-STAT, AP-1, NF-κB and PI3K/AKT (105–107). While inhibitors are available against several of these factors owing to their involvement in many other common oncogenic pathways (108–111), we will focus our discussion on TGFβ inhibitors considering their specific relevance in p-EMT (6,112). At the outset, it is worth noting that TGFβI has been shown by our group to be among the top scoring p-EMT genes in HNSCC (6). In line with this finding, TGFβ induced a p-EMT program in HNSCC cell lines and triggered invasion, which was reversed upon treatment with TGFβ/SMAD inhibitors. More broadly, inhibitors have been designed against various components of TGFβ signaling – TGFβ ligands, receptors, and their downstream signaling effectors – including small-molecule inhibitors, antisense oligonucleotides, monoclonal blocking antibodies, and receptor tyrosine kinase inhibitors (Tables 2 and 3). The oncogenic functions of TGFβ have long been established in different cancers with TGFβ expression being associated with cancer aggressiveness, metastasis and poor survival (113,114). However, the relation between p-EMT and TGFβ is relatively new (38). The secretory nature of TGFβ enables an easy detection of TGFβ expression through measuring TGFβ blood concentration, suggesting its potential as a clinically relevant biomarker (115). TGFβ inhibitors can be a proposed therapeutic intervention in cancer patients exhibiting p-EMT features with a high TGFβ expression.
Table 2:
Inhibitors of TGFβ signaling in various cancers that are in clinical trial.
| Target molecule | Drug Name | Clinical trial phase | Cancer Type | Result |
|---|---|---|---|---|
| TGF-β1, 2 ligands | Fresolimum ab (GC-1008) (Human monoclonal-antibody) | Phase I NCT00356460 | Advanced malignant melanoma or renal cell carcinoma (metastatic or non-resectable with at least one previous therapy) | Well tolerated. Preliminary antitumor activity needing further studies. |
| Phase I, II NCT02581787 | Non-small cell lung carcinoma (Stage IA, IB, non-operable, high surgical risk or patient refuses surgery) | With stereotactic ablative radiotherapy. Result not yet posted. | ||
| Phase II NCT01401062 | Metastatic breast cancer (persistent or recurrent with at least one failed therapy (endocrine or chemotherapy) | With local radiation therapy. Well tolerated. Showed higher systemic immune response and OS. | ||
| Phase II NCT0112293 | Relapsed malignant pleural mesothelioma (1-2 previous systemic therapies, at least one therapy with pemetrexed) | Result not yet posted. | ||
| ALK1 (type 1 subclass of TGF-β receptor) | PF-03446962 (Human monoclonal-antibody) | Phase II NCT01486368 | Advanced malignant pleural mesothelioma (disease progression after treatment with one line of platinum-based doublet chemotherapy) | Well tolerated but failed to show efficacy. |
| Phase I NCT01911273 | Advanced hepatocellular carcinoma (disease progression or intolerance after treatment with VEGFR-TKIs) | Well tolerated with modest single agent antitumor activity, support further evaluation. | ||
| Phase I NCT00557856 | Advanced solid tumors (treatment refractory or no available treatment) | Well tolerated. Single agent antitumor activity. | ||
| TGFβRI | Galunisertib (LY-2157299) (Small molecule inhibitor) | Phase II NCT01582269 | Recurrent intracranial glioblastoma (Grade IV) | With lomustine. Combination failed to show improved OS. |
| Phase I, II NCT03470350 | Metastatic colorectal cancer (chemotherapy resistant activated TGF-β signature like) | With capecitabine. Result not yet verified. | ||
| Phase I NCT02154646 | Advanced or metastatic pancreatic cancer (tumors not manageable by resection) | With gemcitabine. Well tolerated. Combination showed efficacy. Further investigation needed. | ||
| Phase I NCT02734160 | Recurrent metastatic pancreatic adenocarcinoma (disease progression, refractory or intolerant to <2 systemic regimens) | With durvalumab. Result not yet posted. | ||
| Phase I NCT02240433 | Unresectable hepatocellular carcinoma | With Sorafenib. Well tolerated, Promising antitumor activity | ||
| Phase I NCT02906397 | Advanced hepatocellular carcinoma (inoperable, not eligible, failed or discontinued sorafenib therapy) | With stereotactic body radiotherapy. Result not yet posted |
Abbreviations: OS= Overall survival
Table 3:
Inhibitors of TGFβ signaling tested in various preclinical cancer models.
| Target molecule | Drug Name | Preclinical Model | Cancer Type | Result |
|---|---|---|---|---|
| TGFβRI | EW-7203 (Small molecule inhibitor) Chul-Yong et al., 2011 | 4T1 orthotopic-grafted mice | Mammary cancer | Inhibited lung metastasis. |
| TGFβRI | EW-7197, IN-1130, EW-7195 (Small molecule inhibitor) Ji-Yeon et al., 2014, Chul-Yong 2014, Chul-Yong et al., 2011 | (MMTV)/c-Neu mouse mammary tumor virus mice and 4T1 orthotopic-grafted mice | Breast cancer | Inhibited lung metastasis. |
A number of factors – including GRHL2, OVOL1/2, ΔNP63α, NRF2(14) and NFATc (116) – have been shown to stabilize the p-EMT state in cell models, effectively augmenting the mean residence time cells exist between epithelial and mesenchymal states (116). Oxygen availability, or lack thereof, may also drive plastic EMT cells from one pole to the other. As discussed in our previous section on p-EMT regulators, hypoxia seems to be an important cell-extrinsic, microenvironmental factor that plays an important role in induction of p-EMT. Thus pharmacological targeting of hypoxia might provide another strategy to destabilize the hybrid E/M state in tumors.
Unfortunately, most of these current inhibitors of EMT plasticity are relatively imprecise, either inhibiting EMT or MET, but not both. Therefore, they pose a potential risk: Inhibiting only EMT might promote MET, resulting in increased colonization at secondary sites, while only targeting MET might increase metastatic proliferation (117). This unidirectional approach also poses the risk of incomplete inhibition of the transition process and arrest of cells in a more metastatic, but hybrid or intermediate p-EMT state (118). Accordingly, there is growing interest in targeting both forward and reverse regulatory networks of EMT. In theory, targeting tumor-specific EMT-inducing stimuli will limit the induction of p-EMT phenotype in the primary tumor and limit their invasion and extravasation in the blood stream, while inhibition of the MET inducing factors in the secondary site might curtail the seeding and propagating capacity of the metastatic cells (119). These strategies might be most effective if they are adopted at different time points in the patients depending on tumor stage. Ultimately, however, many gaps in our collective understanding of the biology underlying EMT plasticity remain and precisely targeting this pathway in patients remains a distant goal.
Challenges and Future Directions in EMT Plasticity
EMT and MET has been long studied in the context of invasion, metastasis, and cancer progression across oncology. However, identification of a metastable p-EMT state exhibiting concurrent epithelial and mesenchymal phenotypes represents a relatively new and emerging principle. This hybrid cell state that resembles an intermediate step in the dynamic process of EMT represents a missing link in our understanding of this important cellular process – with reasonable confidence, we can now say that this process is a spectrum rather than a switch. While the impact of a fully mesenchymal state on cancer phenotypes such as metastasis, treatment resistance, and recurrence remain controversial (120), expanding evidence has confirmed the presence of p-EMT cells and identified a clinically relevant association with metastasis, chemo-resistance, cancer stem cell characteristics and poor patient outcomes (5,14). While the vast majority of research, including our own, has emphasized an interest in p-EMT and metastasis, an improved understanding of p-EMT and its relationship with these latter characteristics of stemness and treatment resistance represent an important line of further investigation.
Research into identification of p-EMT in human tumors has rapidly progressed with the introduction of single cell technologies (Table 4). Single cell profiling of cells in HNSCC, skin cancer models, as well as lung cancer and high grade serous ovarian cancer have all uncovered the presence of intermediate, p-EMT cell states (6,13,47,121,122). While these single cell techniques are a powerful tool to profile large numbers of individual cells in a relatively unbiased manner, the impact of sample processing and tissue dissociation on expression profiles is poorly understood (123). These limitations become even more critical for p-EMT cells that exist in a metastable, intermediate state primed for further cell state transitions. Certainly, further validation and downstream analysis of hypotheses based on single cell data will be critical, and an essential component of such work will be the confirmation of findings in large bulk datasets through the use of deconvolution and other computational algorithms (124).
Table 4:
Single cell technologies used to study partial EMT in cancer progression.
| Tumor type and samples | Method | Partial EMT (p-EMT) characterization | Major finding | References |
|---|---|---|---|---|
| Single cell RNA sequencing | ||||
| Using single cell RNA sequencing (scRNA-seq) data to analyze bulk RNA sequencing (bulk RNA-seq) data. | ||||
| Head and neck squamous cell carcinoma 18 treatment naive patients and 5 matched LN metastasis. |
Expression programs identified from scRNA-seq data of patient tumors used to de-convolute bulk expression data. | p-EMT cells expressed EMT TF SNAIL2 but lacked other EMT TFs ZEB1/2, TWIST1/2 and SNAIL1, localized to leading edge of tumor and are highly metastatic. | Explores HNSCC heterogeneity with the identification of cell type specific expression programs and infers a strategy to extract information from bulk expression data. | Puram et al., 2017 |
| HMLE breast cancer cell lines | scRNA-seq of cell lines used to generate breast cancer prognosis method, scPrognosis, validated in bulk breast cancer RNA sequencing data sets. | Most of the identified breast cancer signature genes peak at hybrid E/M stage. | Signature genes detected, link EMT with clinical outcomes of breast cancer. | Xiaomei et al., 2020 |
| Single cell RNA sequencing on time course experiments | ||||
| 4 different cancer cell lines lung, prostate, breast and ovarian cancer |
Multiplexed scRNA-seq (MULTI-seq) of 12 distinct EMT time-course experiments of cancer cells treated with different EMT inducers. | EMT transition was not a linear process but involved combinations of discrete transcriptional events indicating hybrid intermediate states. | Provides a thorough comparison of context dependent variabilities in the EMT program. | Cook et al., 2020 |
| Single cell DNA methylation | ||||
| Progressive breast cancer Matched single and clustered CTCs from 4 patients and 3 mouse-xenografts. |
Combination of single-cell resolution DNA methylation and RNA expression analysis with a drug screen with 2,486 FDA-approved compounds. | Binding sites for stemness and proliferation associated transcription factors were hypomethylated compared to the single CTCs. | Demonstrate a connection between phenotypic features such as CTC clustering and DNA methylome landscape alterations. | Gkountela et al., 2019 |
| Single cell mass cytometry | ||||
| Non-small cell lung carcinoma (adenocarcinoma) 3 NSCLC adenocarcinoma cell lines and 5 fresh NSCLC adenocarcinoma patient samples. |
Single cell mass cytometry time-course experiment on NSCLC cells undergoing EMT and MET was done to construct EMT-MET PHENOSTAMP for evaluating EMT and MET states of clinical samples. | Was able to identify heterogeneity within p-EMT states (co-expressed E-cadherin and Vimentin) p-EMT 1, 2, 3 with p-EMT 2 and 3 having a subgroup of Twist+ cells. | This integrated approach provides in vitro insights on EMT–MET biology and establishes a framework to translate in vitro observations to clinical samples. | Karacosta et al., 2019 |
| High grade serous ovarian cancer Single cells from 17 newly diagnosed patient tumors. |
Multiparametric single-cell mass cytometry, CyTOF. | Seven cell clusters co-expressed epithelial marker E-cadherin and mesenchymal marker Vimentin with protein deregulations in stem cell, cell cycle and metastasis. | CyTOF enabled detailed characterization of subtly differing cell populations. | Gonzalez et al., 2018 |
Abbreviations: LN = lymph node, sc-RNA-seq = single-cell RNA sequencing, p-EMT = partial epithelial-mesenchymal transition, EMT = epithelial-mesenchymal transition, TF = transcription factors, HNSCC = head and neck squamous cell carcinoma, HMLE = immortalized human mammary epithelial cells, CTC = circulating tumor cell, FDA = food and drug administration, NSCLC = non-small cell lung carcinoma, MET = mesenchymal to epithelial transition.
Computational and mathematical modeling is being increasingly utilized to understand the complex regulatory network of EMT plasticity and how it affects tumor progression (125–127). These models help bridge gaps in existing experimental data and developing testable hypotheses. Different models have helped predict the steps involved in EMT, including mechanisms underlying the transition from one cell state to another and the relative stability of different cell states. Given the complexity of the EMT plasticity, a more in-depth understanding of the molecular players of importance and the interactions among them may expedite the development of models that can trace these changes during EMT and MET, potentially highlighting novel avenues for targeted therapy (127,128).
In parallel, we must improve our understanding of the role of EMT plasticity in tumor progression through more sensitive lineage tracing techniques and animal models. While in vitro cell lines and patient tumor samples provide us with important insights into EMT, the dynamic nature of EMT plasticity and the effect of its cross-talk with the tumor microenvironment cannot be adequately captured by in vitro studies or perturbed in human samples. In HNSCC, for example, while patient scRNA-seq data suggested CAFs may release ligands that interact with p-EMT cells at the leading edge, such ligands were all lost in vitro upon culture of patient-derived CAFs isolated from primary tumors (6). We believe that patient-derived xenografts grown as adherent cultures or organoids may represent the “sweet spot” where human patient biology is recapitulated, yet the tissue/cells may still be expanded, grown, perturbed, and then studied.
Lastly, to target a metastable, dynamic p-EMT state, detailed analysis of the bidirectional regulatory networks that direct cells towards or away from this hybrid state is essential. Indeed, as noted, bidirectional inhibition that targets both EMT and MET might be an effective strategy to destabilize transient hybrid states, while avoiding an escape route to a different resistant state. While there has been significant progress, we anticipate the next few decades will represent a major acceleration in our knowledge of EMT and its entire spectrum of states. A deeper understanding of this fundamental biology is likely to present new opportunities for stratification of patients into low and high-risk cohorts, while opening up entirely new avenues in the treatment of epithelial tumors.
Figure 4:
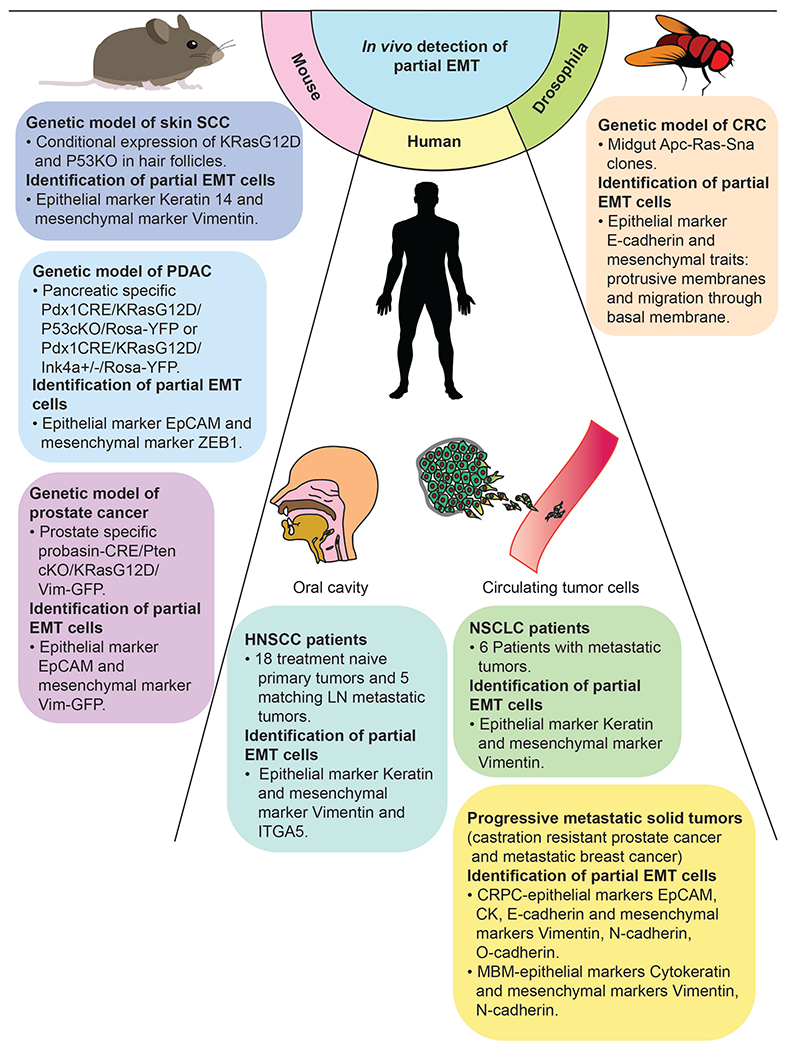
Evidence of in vivo detection of partial EMT cells co-expressing epithelial and mesenchymal markers.
Acknowledgements:
This work was graciously supported by an NIH/NCI K08CA237732 as well as the Cancer Research Foundation: P20-0563, Doris Duke Foundation, American Cancer Society, V Foundation: V2019-005, and the Washington University Dean’s Scholars Program (S.V.P.). This work was also supported by the National Institute of Deafness and Other Communication Disorders (T32DC000022) (T.F.B.).
Footnotes
Competing Interests: The authors have no competing interests to declare.
References
- 1.Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell. 2017. Feb;168(4):670–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang J, Weinberg RA. Epithelial-Mesenchymal Transition: At the Crossroads of Development and Tumor Metastasis. Developmental Cell. 2008. Jun 10;14(6):818–29. [DOI] [PubMed] [Google Scholar]
- 3.Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nature Reviews Cancer. 2002. Jun;2(6):442–54. [DOI] [PubMed] [Google Scholar]
- 4.Yang J, Antin P, Berx G, Blanpain C, Brabletz T, Bronner M, et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nature Reviews Molecular Cell Biology. 2020;21(6):341–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.George JT, Jolly MK, Xu S, Somarelli JA, Levine H. Survival Outcomes in Cancer Patients Predicted by a Partial EMT Gene Expression Scoring Metric. Cancer Research. 2017. Nov;77(22):6415-LP–6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell. 2017. Dec;171(7):1611–1624.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jolly MK, Mani SA, Levine H. Hybrid epithelial/mesenchymal phenotype(s): The “fittest” for metastasis? Biochim Biophys Acta Rev Cancer. 2018;1870(2):151–7. [DOI] [PubMed] [Google Scholar]
- 8.Polioudaki H, Agelaki S, Chiotaki R, Politaki E, Mavroudis D, Matikas A, et al. Variable expression levels of keratin and vimentin reveal differential EMT status of circulating tumor cells and correlation with clinical characteristics and outcome of patients with metastatic breast cancer. BMC Cancer. 2015. Dec;15(1):399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parikh AS, Puram SV, Faquin WC, Richmon JD, Emerick KS, Deschler DG, et al. Immunohistochemical quantification of partial-EMT in oral cavity squamous cell carcinoma primary tumors is associated with nodal metastasis. Oral Oncology. 2019. Dec;99:104458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Papadaki MA, Stoupis G, Theodoropoulos PA, Mavroudis D, Georgoulias V, Agelaki S. Circulating Tumor Cells with Stemness and Epithelial-to-Mesenchymal Transition Features Are Chemoresistant and Predictive of Poor Outcome in Metastatic Breast Cancer. Mol Cancer Ther. 2019. Feb 1;18(2):437–47. [DOI] [PubMed] [Google Scholar]
- 11.Fustaino V, Presutti D, Colombo T, Cardinali B, Papoff G, Brandi R, et al. Characterization of epithelial-mesenchymal transition intermediate/hybrid phenotypes associated to resistance to EGFR inhibitors in non-small cell lung cancer cell lines. Oncotarget. 2017. Nov 28;8(61):103340–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saitoh M Involvement of partial EMT in cancer progression. J Biochem. 2018. Oct 1;164(4):257–64. [DOI] [PubMed] [Google Scholar]
- 13.Pastushenko I, Brisebarre A, Sifrim A, Fioramonti M, Revenco T, Boumahdi S, et al. Identification of the tumour transition states occurring during EMT. Nature. 2018;556(7702):463–8. [DOI] [PubMed] [Google Scholar]
- 14.Pastushenko I, Blanpain C. EMT Transition States during Tumor Progression and Metastasis. Trends in Cell Biology. 2019. Mar;29(3):212–26. [DOI] [PubMed] [Google Scholar]
- 15.Qi Z, Barrett T, Parikh AS, Tirosh I, Puram SV. Single-cell sequencing and its applications in head and neck cancer. Oral Oncology. 2019. Dec;99:104441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puram SV, Parikh AS, Tirosh I. Single cell RNA-seq highlights a role for a partial EMT in head and neck cancer. Mol Cell Oncol. 2018;5(3):e1448244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stepan KO, Li MM, Kang SY, Puram SV. Molecular margins in head and neck cancer: Current techniques and future directions. Oral Oncology. 2020. Nov;110:104893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Greenburg G, Hay ED. Epithelia suspended in collagen gels can lose polarity and express characteristics of migrating mesenchymal cells. J Cell Biol. 1982. Oct;95(1):333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Viebahn C. Epithelio-mesenchymal transformation during formation of the mesoderm in the mammalian embryo. Acta Anat (Basel). 1995;154(1):79–97. [DOI] [PubMed] [Google Scholar]
- 20.Tucker GC, Duband JL, Dufour S, Thiery JP. Cell-adhesion and substrate-adhesion molecules: their instructive roles in neural crest cell migration. Development. 1988;103Suppl:81–94. [DOI] [PubMed] [Google Scholar]
- 21.Frixen UH, Behrens J, Sachs M, Eberle G, Voss B, Warda A, et al. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol. 1991. Apr;113(1):173–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perl AK, Wilgenbus P, Dahl U, Semb H, Christofori G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature. 1998. Mar 12;392(6672):190–3. [DOI] [PubMed] [Google Scholar]
- 23.Tomson AM, Scholma J, Meijer B, Koning JG, de Jong KM, van der Werf M. Adhesion properties, intermediate filaments and malignant behaviour of head and neck squamous cell carcinoma cells in vitro. Clin Exp Metastasis. 1996. Nov;14(6):501–11. [DOI] [PubMed] [Google Scholar]
- 24.Birchmeier C, Birchmeier W, Brand-Saberi B. Epithelial-mesenchymal transitions in cancer progression. Acta Anat (Basel). 1996;156(3):217–26. [DOI] [PubMed] [Google Scholar]
- 25.Langley RR, Fidler IJ. The seed and soil hypothesis revisited - the role of tumor-stroma interactions in metastasis to different organs. Int J Cancer. 2011. Jun 1;128(11):2527–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung AR, Jung C-H, Noh JK, Lee YC, Eun Y-G. Epithelial-mesenchymal transition gene signature is associated with prognosis and tumor microenvironment in head and neck squamous cell carcinoma. Scientific Reports. 2020. Feb 27;10(1):3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wan Y, Liu H, Zhang M, Huang Z, Zhou H, Zhu Y, et al. Prognostic value of epithelial-mesenchymal transition-inducing transcription factors in head and neck squamous cell carcinoma: A meta-analysis. Head & Neck. 2020. May 1;42(5):1067–76. [DOI] [PubMed] [Google Scholar]
- 28.Nijkamp MM, Span PN, Hoogsteen IJ, van der Kogel AJ, Kaanders JHAM, Bussink J. Expression of E-cadherin and vimentin correlates with metastasis formation in head and neck squamous cell carcinoma patients. Radiother Oncol. 2011. Jun;99(3):344–8. [DOI] [PubMed] [Google Scholar]
- 29.Nguyen PT, Kudo Y, Yoshida M, Kamata N, Ogawa I, Takata T. N-cadherin expression is involved in malignant behavior of head and neck cancer in relation to epithelial-mesenchymal transition. Histol Histopathol. 2011;26(2):147–56. [DOI] [PubMed] [Google Scholar]
- 30.Zidar N, Boštjančič E, Gale N, Kojc N, Poljak M, Glavač D, et al. Down-regulation of microRNAs of the miR-200 family and miR-205, and an altered expression of classic and desmosomal cadherins in spindle cell carcinoma of the head and neck--hallmark of epithelial-mesenchymal transition. Hum Pathol. 2011. Apr;42(4):482–8. [DOI] [PubMed] [Google Scholar]
- 31.De Cecco L, Nicolau M, Giannoccaro M, Daidone MG, Bossi P, Locati L, et al. Head and neck cancer subtypes with biological and clinical relevance: Meta-analysis of gene-expression data. Oncotarget. 2015. Apr 20;6(11):9627–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chui MH. Insights into cancer metastasis from a clinicopathologic perspective: Epithelial-Mesenchymal Transition is not a necessary step. Int J Cancer. 2013. Apr 1;132(7):1487–95. [DOI] [PubMed] [Google Scholar]
- 33.Craene BD, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nature Reviews Cancer. 2013. Feb;13(2):97–110. [DOI] [PubMed] [Google Scholar]
- 34.Brabletz T To differentiate or not — routes towards metastasis. Nature Reviews Cancer. 2012. Jun;12(6):425–36. [DOI] [PubMed] [Google Scholar]
- 35.Ocaña OH, Córcoles R, Fabra Á, Moreno-Bueno G, Acloque H, Vega S, et al. Metastatic Colonization Requires the Repression of the Epithelial-Mesenchymal Transition Inducer Prrx1. Cancer Cell. 2012. Dec 11;22(6):709–24. [DOI] [PubMed] [Google Scholar]
- 36.Giampieri S, Manning C, Hooper S, Jones L, Hill CS, Sahai E. Localised and reversible TGFβ signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol. 2009. Nov;11(11):1287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Panchy N, Azeredo-Tseng C, Luo M, Randall N, Hong T. Integrative Transcriptomic Analysis Reveals a Multiphasic Epithelial–Mesenchymal Spectrum in Cancer and Non-tumorigenic Cells. Front Oncol. 2020. Jan 22;9:1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J, Tian X-J, Zhang H, Teng Y, Li R, Bai F, et al. TGF-β-induced epithelial-to-mesenchymal transition proceeds through stepwise activation of multiple feedback loops. Sci Signal. 2014. Sep 30;7(345):ra91. [DOI] [PubMed] [Google Scholar]
- 39.Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and Dissemination Precede Pancreatic Tumor Formation. Cell. 2012. Jan 20;148(1):349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruscetti M, Quach B, Dadashian EL, Mulholland DJ, Wu H. Tracking and Functional Characterization of Epithelial-Mesenchymal Transition and Mesenchymal Tumor Cells during Prostate Cancer Metastasis. Cancer Res. 2015. Jul 1;75(13):2749–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kröger C, Afeyan A, Mraz J, Eaton EN, Reinhardt F, Khodor YL, et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. PNAS. 2019. Apr 9;116(15):7353–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dijk D van, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell. 2018. Jul 26;174(3):716–729.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.The Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015. Jan;517(7536):576–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim JM, Kim HJ, Koo BS, Rha KS, Yoon Y-H. Expression of matrix metalloproteinase-12 is correlated with extracapsular spread of tumor from nodes with metastasis in head and neck squamous cell carcinoma. Eur Arch Otorhinolaryngol. 2013. Mar 1;270(3):1137–42. [DOI] [PubMed] [Google Scholar]
- 45.Lee W-Y, Shin D-Y, Kim HJ, Ko Y-H, Kim S, Jeong H-S. Prognostic Significance of Epithelial-Mesenchymal Transition of Extracapsular Spread Tumors in Lymph Node Metastases of Head and Neck Cancer. Ann Surg Oncol. 2014. Jun 1;21(6):1904–11. [DOI] [PubMed] [Google Scholar]
- 46.Tada H, Takahashi H, Ida S, Nagata Y, Chikamatsu K. Epithelial-Mesenchymal Transition Status of Circulating Tumor Cells Is Associated With Tumor Relapse in Head and Neck Squamous Cell Carcinoma. Anticancer Res. 2020. Jun;40(6):3559–64. [DOI] [PubMed] [Google Scholar]
- 47.Ji AL, Rubin AJ, Thrane K, Jiang S, Reynolds DL, Meyers RM, et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell. 2020. Jul;182(2):497–514.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bocci F, Tripathi SC, Vilchez Mercedes SA, George JT, Casabar JP, Wong PK, et al. NRF2 activates a partial epithelial-mesenchymal transition and is maximally present in a hybrid epithelial/mesenchymal phenotype. Integrative Biology. 2019. Jun 1;11(6):251–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Godin L, Balsat C, Van Eycke Y-R, Allard J, Royer C, Remmelink M, et al. A Novel Approach for Quantifying Cancer Cells Showing Hybrid Epithelial/Mesenchymal States in Large Series of Tissue Samples: Towards a New Prognostic Marker. Cancers (Basel). 2020. Apr 8;12(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cho S-J, Kakar S. Tumor Budding in Colorectal Carcinoma: Translating a Morphologic Score Into Clinically Meaningful Results. Archives of Pathology & Laboratory Medicine. 2018. Aug 1;142(8):952–7. [DOI] [PubMed] [Google Scholar]
- 51.Liang F, Cao W, Wang Y, Li L, Zhang G, Wang Z. The prognostic value of tumor budding in invasive breast cancer. Pathology - Research and Practice. 2013. May 1;209(5):269–75. [DOI] [PubMed] [Google Scholar]
- 52.Şirin AH, Sökmen S, Ünlü SM, Ellidokuz H, Sarioğlu S. The prognostic value of tumor budding in patients who had surgery for rectal cancer with and without neoadjuvant therapy. Tech Coloproctol. 2019. Apr;23(4):333–42. [DOI] [PubMed] [Google Scholar]
- 53.Kohler I, Bronsert P, Timme S, Werner M, Brabletz T, Hopt UT, et al. Detailed analysis of epithelial-mesenchymal transition and tumor budding identifies predictors of long-term survival in pancreatic ductal adenocarcinoma. Journal of Gastroenterology and Hepatology. 2015;30(S1):78–84. [DOI] [PubMed] [Google Scholar]
- 54.Campbell K, Rossi F, Adams J, Pitsidianaki I, Barriga FM, Garcia-Gerique L, et al. Collective cell migration and metastases induced by an epithelial-to-mesenchymal transition in Drosophila intestinal tumors. Nature Communications. 2019. May 24;10(1):2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aiello NM, Maddipati R, Norgard RJ, Balli D, Li J, Yuan S, et al. EMT subtype influences epithelial plasticity and mode of cell migration. Dev Cell. 2018. Jun 18;45(6):681–695.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, et al. Circulating Tumor Cell Clusters are Oligoclonal Precursors of Breast Cancer Metastasis. Cell. 2014. Aug 28;158(5):1110–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Amintas S, Bedel A, Moreau-Gaudry F, Boutin J, Buscail L, Merlio J-P, et al. Circulating Tumor Cell Clusters: United We Stand Divided We Fall. Int J Mol Sci. 2020. Apr 10;21(7):2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hou J-M, Krebs M, Ward T, Sloane R, Priest L, Hughes A, et al. Circulating Tumor Cells as a Window on Metastasis Biology in Lung Cancer. Am J Pathol. 2011. Mar;178(3):989–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Joosse SA, Gorges TM, Pantel K. Biology, detection, and clinical implications of circulating tumor cells. EMBO Mol Med. 2015. Jan;7(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hou J-M, Krebs MG, Lancashire L, Sloane R, Backen A, Swain RK, et al. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J Clin Oncol. 2012. Feb 10;30(5):525–32. [DOI] [PubMed] [Google Scholar]
- 61.Lecharpentier A, Vielh P, Perez-Moreno P, Planchard D, Soria JC, Farace F. Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br J Cancer. 2011. Oct 25;105(9):1338–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull J, et al. Circulating Tumor Cells from Patients with Advanced Prostate and Breast Cancer Display Both Epithelial and Mesenchymal Markers. Mol Cancer Res. 2011. Aug;9(8):997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu M, Bardia A, Wittner BS, Stott SL, Smas ME, Ting DT, et al. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013. Feb 1;339(6119):580–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu S, Liu S, Liu Z, Huang J, Pu X, Li J, et al. Classification of Circulating Tumor Cells by Epithelial-Mesenchymal Transition Markers. PLoS One. 2015. Apr 24;10(4):e0123976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang RY-J, Wong MK, Tan TZ, Kuay KT, Ng AHC, Chung VY, et al. An EMT spectrum defines an anoikis-resistant and spheroidogenic intermediate mesenchymal state that is sensitive to e-cadherin restoration by a src-kinase inhibitor, saracatinib (AZD0530). Cell Death & Disease. 2013. Nov;4(11):e915–e915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Biddle A, Gammon L, Liang X, Costea DE, Mackenzie IC. Phenotypic Plasticity Determines Cancer Stem Cell Therapeutic Resistance in Oral Squamous Cell Carcinoma. EBioMedicine. 2016. Jan 9;4:138–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jolly MK, Boareto M, Debeb BG, Aceto N, Farach-Carson MC, Woodward WA, et al. Inflammatory breast cancer: a model for investigating cluster-based dissemination. NPJ Breast Cancer. 2017. Jun 6;3:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Strauss R, Li Z-Y, Liu Y, Beyer I, Persson J, Sova P, et al. Analysis of epithelial and mesenchymal markers in ovarian cancer reveals phenotypic heterogeneity and plasticity. PLoS ONE. 2011. Jan 14;6(1):e16186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jolly MK, Huang B, Lu M, Mani SA, Levine H, Ben-Jacob E. Towards elucidating the connection between epithelial-mesenchymal transitions and stemness. J R Soc Interface. 2014. Dec 6;11(101):20140962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Visvader JE, Lindeman GJ. Cancer Stem Cells: Current Status and Evolving Complexities. Cell Stem Cell. 2012. Jun;10(6):717–28. [DOI] [PubMed] [Google Scholar]
- 71.Plaks V, Kong N, Werb Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell. 2015. Mar 5;16(3):225–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scheel C, Weinberg RA. Cancer stem cells and epithelial-mesenchymal transition: concepts and molecular links. Semin Cancer Biol. 2012. Oct;22(5–6):396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mani SA, Guo W, Liao M-J, Eaton ENg, Ayyanan A, Zhou AY, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008. May 16;133(4):704–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal Regulation of Epithelial-Mesenchymal Transition is Essential for Squamous Cell Carcinoma Metastasis. Cancer Cell. 2012. Dec 11;22(6):725–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Celià-Terrassa T, Meca-Cortés O, Mateo F, Martínez de Paz A, Rubio N, Arnal-Estapé A, et al. Epithelial-mesenchymal transition can suppress major attributes of human epithelial tumor-initiating cells. J Clin Invest. 2012. May;122(5):1849–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Grosse-Wilde A, d’Hérouël AF, McIntosh E, Ertaylan G, Skupin A, Kuestner RE, et al. Stemness of the hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival. PLOS ONE. 2015. May 28;10(5):e0126522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Latil M, Nassar D, Beck B, Boumahdi S, Wang L, Brisebarre A, et al. Cell-Type-Specific Chromatin States Differentially Prime Squamous Cell Carcinoma Tumor-Initiating Cells for Epithelial to Mesenchymal Transition. Cell Stem Cell. 2017. Feb;20(2):191–204.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zaravinos A The Regulatory Role of MicroRNAs in EMT and Cancer. Journal of Oncology. 2015;2015:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sánchez-Tilló E, Liu Y, de Barrios O, Siles L, Fanlo L, Cuatrecasas M, et al. EMT-activating transcription factors in cancer: beyond EMT and tumor invasiveness. Cellular and Molecular Life Sciences. 2012;69(20):3429–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dohadwala M, Wang G, Heinrich E, Luo J, Lau O, Shih H, et al. The role of ZEB1 in the inflammation-induced promotion of EMT in HNSCC. Otolaryngology–Head and Neck Surgery. 2010. May;142(5):753–9. [DOI] [PubMed] [Google Scholar]
- 81.Kojc N, Zidar N, Gale N, Poljak M, Fujs Komloš K, Cardesa A, et al. Transcription factors Snail, Slug, Twist, and SIP1 in spindle cell carcinoma of the head and neck. Virchows Archiv. 2009;454(5):549–55. [DOI] [PubMed] [Google Scholar]
- 82.Dhasarathy A, Phadke D, Mav D, Shah RR, Wade PA. The Transcription Factors Snail and Slug Activate the Transforming Growth Factor-Beta Signaling Pathway in Breast Cancer. PLOS ONE. 2011. Oct;6(10):e26514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cai J, Xia L, Li J, Ni S, Song H, Wu X. Tumor-Associated Macrophages Derived TGF-β–Induced Epithelial to Mesenchymal Transition in Colorectal Cancer Cells through Smad2,3-4/Snail Signaling Pathway. Cancer research and treatment: official journal of Korean Cancer Association. 2019. Jan;51(1):252–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Horiguchi K, Shirakihara T, Nakano A, Imamura T, Miyazono K, Saitoh M. Role of Ras Signaling in the Induction of Snail by Transforming Growth Factor-β. Journal of Biological Chemistry. 2009. Jan;284(1):245–53. [DOI] [PubMed] [Google Scholar]
- 85.Medici D, Hay ED, Olsen BR. Snail and Slug Promote Epithelial-Mesenchymal Transition through β-CateninβT-Cell Factor-4-dependent Expression of Transforming Growth Factory-β3. Molecular Biology of the Cell. 2008. Sep;19(11):4875–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xiong H-G, Li H, Xiao Y, Yang Q-C, Yang L-L, Chen L, et al. Long noncoding RNA MYOSLID promotes invasion and metastasis by modulating the partial epithelial-mesenchymal transition program in head and neck squamous cell carcinoma. Journal of Experimental & Clinical Cancer Research. 2019;38(1):278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kaelin WG, Ratcliffe PJ. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Molecular Cell. 2008. May 23;30(4):393–402. [DOI] [PubMed] [Google Scholar]
- 88.Chen S, Chen X, Li W, Shan T, Lin WR, Ma J, et al. Conversion of epithelial-to-mesenchymal transition to mesenchymal-to-epithelial transition is mediated by oxygen concentration in pancreatic cancer cells. Oncol Lett. 2018. May;15(5):7144–52. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 89.Zhang H, Akman HO, Smith ELP, Zhao J, Murphy-Ullrich JE, Batuman OA. Cellular response to hypoxia involves signaling via Smad proteins. Blood. 2003. Mar 15;101(6):2253–60. [DOI] [PubMed] [Google Scholar]
- 90.McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem. 2006. Aug 25;281(34):24171–81. [DOI] [PubMed] [Google Scholar]
- 91.Zhang L, Huang G, Li X, Zhang Y, Jiang Y, Shen J, et al. Hypoxia induces epithelial-mesenchymal transition via activation of SNAI1 by hypoxia-inducible factor-1 [alpha] in hepatocellular carcinoma. BMC Cancer. 2013. Mar 9;13–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Storci G, Sansone P, Mari S, D’Uva G, Tavolari S, Guarnieri T, et al. TNFalpha up-regulates SLUG via the NF-kappaB/HIF1 alpha axis, which imparts breast cancer cells with a stem cell-like phenotype. J Cell Physiol. 2010. Nov;225(3):682–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Deng S, Chen H, Ye Z, Deng S, Zhu S, Zeng Z, et al. Hypoxia-induced LncRNA-BX111 promotes metastasis and progression of pancreatic cancer through regulating ZEB1 transcription. Oncogene. 2018. Nov;37(44):5811–28. [DOI] [PubMed] [Google Scholar]
- 94.Evans AJ, Russell RC, Roche O, Burry TN, Fish JE, Chow VWK, et al. VHL promotes E2 box-dependent E-cadherin transcription by HIF-mediated regulation of SIP1 and snail. Mol Cell Biol. 2007. Jan;27(1):157–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kisoda S, Shao W, Fujiwara N, Mouri Y, Tsunematsu T, Jin S, et al. Prognostic value of partial EMT-related genes in head and neck squamous cell carcinoma by a bioinformatic analysis. Oral Dis. 2020. Apr 11; [DOI] [PubMed] [Google Scholar]
- 96.Qi Z, Liu Y, Mints M, Mullins R, Sample R, Law T, et al. Single-Cell Deconvolution of Head and Neck Squamous Cell Carcinoma. Cancers (Basel) [Internet], 2021. Mar 11;13(6):1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Karaosmanoğlu O, Banerjee S, Sivas H. Identification of biomarkers associated with partial epithelial to mesenchymal transition in the secretome of slug over-expressing hepatocellular carcinoma cells. Cellular Oncology. 2018;41(4):439–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tretbar S, Krausbeck P, Müller A, Friedrich M, Vaxevanis C, Bukur J, et al. TGF-β inducible epithelial-to-mesenchymal transition in renal cell carcinoma. Oncotarget. 2019. Feb 19;10(15):1507–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hugo HJ, Wafai R, Blick T, Thompson EW, Newgreen DF. Staurosporine augments EGF-mediated EMT in PMC42-LA cells through actin depolymerisation, focal contact size reduction and SnaiH induction – A model for cross-modulation. BMC Cancer. 2009. Jul 15;9:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.KURIMOTO R, IWASAWA S, EBATA T, ISHIWATA T, SEKINE I, TADA Y, et al. Drug resistance originating from a TGF-β/FGF-2-driven epithelial-to-mesenchymal transition and its reversion in human lung adenocarcinoma cell lines harboring an EGFR mutation. Int J Oncol. 2016. Mar 4;48(5):1825–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Devarajan E, Song Y-H, Krishnappa S, Alt E. Epithelial–mesenchymal transition in breast cancer lines is mediated through PDGF-D released by tissue-resident stem cells. International Journal of Cancer. 2012;131(5):1023–31. [DOI] [PubMed] [Google Scholar]
- 102.Suárez-Causado A, Caballero-Díaz D, Bertrán E, Roncero C, Addante A, García-Álvaro M, et al. HGF/c-Met signaling promotes liver progenitor cell migration and invasion by an epithelial–mesenchymal transition-independent, phosphatidyl inositol-3 kinase-dependent pathway in an in vitro model. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2015. Oct 1;1853(10, Part A):2453–63. [DOI] [PubMed] [Google Scholar]
- 103.Wang R, Li H, Guo X, Wang Z, Liang S, Dang C. IGF-I Induces Epithelial-to-Mesenchymal Transition via the IGF-IR-Src-MicroRNA-30a-E-Cadherin Pathway in Nasopharyngeal Carcinoma Cells. Oncol Res. 2016;24(4):225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Miao J-W, Liu L-J, Huang J Interleukin-6-induced epithelial-mesenchymal transition through signal transducer and activator of transcription 3 in human cervical carcinoma. Int J Oncol. 2014. Jul;45(1):165–76. [DOI] [PubMed] [Google Scholar]
- 105.Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-Mesenchymal Transitions in Development and Disease. Cell. 2009. Nov 25;139(5):871–90. [DOI] [PubMed] [Google Scholar]
- 106.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial–mesenchymal transitions. Nature Reviews Molecular Cell Biology. 2006;7(2):131–42. [DOI] [PubMed] [Google Scholar]
- 107.Xu W, Yang Z, Lu N. A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh Migr. 2015. Aug 4;9(4):317–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Seshacharyulu P, Ponnusamy MP, Haridas D, Jain M, Ganti AparK, Batra SK. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012. Jan;16(1):15–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Harb J, Lin P-J, Hao J. Recent Development of Wnt Signaling Pathway Inhibitors for Cancer Therapeutics. Curr Oncol Rep. 2019. 04;21(2):12. [DOI] [PubMed] [Google Scholar]
- 110.Espinoza I, Miele L. Notch inhibitors for cancer treatment. Pharmacology & Therapeutics. 2013. Aug 1;139(2):95–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Simpson A, Petnga W, Macaulay VM, Weyer-Czernilofsky U, Bogenrieder T. Insulin-Like Growth Factor (IGF) Pathway Targeting in Cancer: Role of the IGF Axis and Opportunities for Future Combination Studies. Target Oncol. 2017;12(5):571–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009. Feb;19(2):156–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ivanović V, Demajo M, Krtolica K, Krajnović M, Konstantinović M, Baltić V, et al. Elevated plasma TGF-beta1 levels correlate with decreased survival of metastatic breast cancer patients. Clin Chim Acta. 2006. Sep;371 (1–2):191–3. [DOI] [PubMed] [Google Scholar]
- 114.Li J, Shen C, Wang X, Lai Y, Zhou K, Li P, et al. Prognostic value of TGF-β in lung cancer: systematic review and meta-analysis. BMC Cancer. 2019. Jul 15;19(1):691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ciftci R, Tas F, Kilic L, Vatansever S, Karabulut S. Clinical significance of serum transforming growth factor beta 1 (TGFB1) level in breast cancer. JCO. 2014. May 20;32(15_suppl):e11526–e11526. [Google Scholar]
- 116.Subbalakshmi AR, Kundnani D, Biswas K, Ghosh A, Hanash SM, Tripathi SC, et al. NFATc Acts as a Non-Canonical Phenotypic Stability Factor for a Hybrid Epithelial/Mesenchymal Phenotype. Front Oncol. 2020;10:553342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bhatia S, Monkman J, Toh AKL, Nagaraj SH, Thompson EW. Targeting epithelial-mesenchymal plasticity in cancer: clinical and preclinical advances in therapy and monitoring. Biochem J. 2017. 20;474(19):3269–306. [DOI] [PubMed] [Google Scholar]
- 118.Jolly MK, Celià-Terrassa T. Dynamics of Phenotypic Heterogeneity Associated with EMT and Stemness during Cancer Progression. J Clin Med. 2019. 25;8(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bhatia S, Wang P, Toh A, Thompson EW. New Insights Into the Role of Phenotypic Plasticity and EMT in Driving Cancer Progression. Front Mol Biosci. 2020. Apr 23;7:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Williams ED, Gao D, Redfern A, Thompson EW. Controversies around epithelial–mesenchymal plasticity in cancer metastasis. Nature Reviews Cancer. 2019. Dec;19(12):716–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Karacosta LG, Anchang B, Ignatiadis N, Kimmey SC, Benson JA, Shrager JB, et al. Mapping lung cancer epithelial-mesenchymal transition states and trajectories with single-cell resolution. Nature Communications. 2019. Dec 6;10(1):5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gonzalez VD, Samusik N, Chen TJ, Savig ES, Aghaeepour N, Quigley DA, et al. Commonly Occurring Cell Subsets in High-Grade Serous Ovarian Tumors Identified by Single-Cell Mass Cytometry. Cell Rep. 2018. 13;22(7):1875–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kinker GS, Greenwald AC, Tal R, Orlova Z, Cuoco MS, McFarland JM, et al. Pan-cancer single-cell RNA-seq identifies recurring programs of cellular heterogeneity. Nat Genet. 2020. Nov;52(11):1208–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Newman AM, Steen CB, Liu CL, Gentles AJ, Chaudhuri AA, Scherer F, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019. Jul;37(7):773–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sha Y, Wang S, Zhou P, Nie Q. Inference and multiscale model of epithelial-to-mesenchymal transition via single-cell transcriptomic data. Nucleic Acids Res. 2020. Sep 25;48(17):9505–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.He P, Qiu K, Jia Y. Modeling of mesenchymal hybrid epithelial state and phenotypic transitions in EMT and MET processes of cancer cells. Scientific Reports. 2018. Sep 25;8(1):14323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Burger GA, Danen EHJ, Beltman JB. Deciphering Epithelial–Mesenchymal Transition Regulatory Networks in Cancer through Computational Approaches. Front Oncol. 2017. Aug 3;7:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jolly MK, Tripathi SC, Somarelli JA, Hanash SM, Levine H. Epithelial/mesenchymal plasticity: how have quantitative mathematical models helped improve our understanding? Mol Oncol. 2017. Jul;11(7):739–54. [DOI] [PMC free article] [PubMed] [Google Scholar]