


Abstract
Mobile element insertions (MEIs) are a major class of structural variants (SVs) and have been linked to many human genetic disorders, including hemophilia, neurofibromatosis, and various cancers. However, human MEI resources from large-scale genome sequencing are still lacking compared to those for SNPs and SVs. Here, we report a comprehensive map of 36 699 non-reference MEIs constructed from 5675 genomes, comprising 2998 Chinese samples (∼26.2×, NyuWa) and 2677 samples from the 1000 Genomes Project (∼7.4×, 1KGP). We discovered that LINE-1 insertions were highly enriched in centromere regions, implying the role of chromosome context in retroelement insertion. After functional annotation, we estimated that MEIs are responsible for about 9.3% of all protein-truncating events per genome. Finally, we built a companion database named HMEID for public use. This resource represents the latest and largest genomewide study on MEIs and will have broad utility for exploration of human MEI findings.
INTRODUCTION
Transposable elements (TEs), also known as transposons or mobile elements, comprise a significant portion in mammalian genomes (1–3), approximately half of the human genome (4). Most TEs are transposition incompetent due to accumulated interior mutations and truncation or various host repression mechanisms (5). In humans, Alu, long interspersed nuclear element 1 (L1), and SINE-VNTR-Alu (SVA) elements (and possibly HERV-K elements) are families of TEs which are still active and capable of creating new insertions (6–8), termed mobile element insertions (MEIs). The transposition events have the potential to disrupt normal gene function and alter transcript expression or splicing at the sites of integration, contributing to disease (9). For example, over 120 TE-mediated insertions have been associated with various human genetic diseases, including hemophilia, Dent disease, neurofibromatosis and cancers (10). Apart from the impact through insertion events, intrinsic sequence properties of TEs endow some MEIs with functional effects on the host (9), making MEIs differ qualitatively from typical forms of SVs like copy number variants (CNVs). Another important question related to MEIs is the integration site preference, which are usually non-random and influenced by various factors such as DNA sequences and chromatin context (11).
However, despite these important functions, integrated resources for polymorphic TEs in human genomes are still lacking (12), which could offer a large pool of MEIs to explore TE diversity and serve as bedrock for phenotype-variant association studies. With the advances of whole-genome sequencing (WGS) and improvements in algorithms for MEI detection, MEIs are now increasingly characterized at population scale (13–21). To date, the two largest population studies of SV, including 14 891 genomes (18) and 17 795 genomes (19) respectively, did not perform explicit analysis for MEIs. A comprehensive analysis of variation in mobile elements was conducted by the 1KGP, including >20 000 polymorphic MEIs from 2504 genomes (14,16). Recently, Watkins et al. investigated the global population genetics of MEIs, drawn from 296 genomes across 142 populations (17), extending the findings based on the 1KGP dataset (22). However, these MEI genetic resources are mainly from European ancestry cohorts. Even in the gnomAD SV cohort, only 1304 samples from East Asia (18). As the Han Chinese population is the largest ethnic group in East Asia and in the world (23), the lack of Chinese cohort genomic study on MEIs is a critical part of the missing diversity.
In this study, we employed WGS of 5675 members from newly sequenced Chinese samples and the 1KGP to construct a resource for non-reference MEIs. Although the 1KGP dataset has already been investigated for MEIs (14,16), we included it here to increase population diversity and build a comprehensive MEI map. The NyuWa dataset has been used to study the spectrum of small variants and build a reference panel (24), and the MEIs have not been explored yet. Combining the two cohorts enabled us to systematically analyze the genomic distribution, mutational patterns, and functional impacts of MEIs. From these analyses, we found that L1 MEIs were highly enriched in centromere regions, and we determined that MEIs represent about 9.3% of all protein-truncating events per individual, emphasizing the importance of detecting MEI routinely in WGS studies. We have built a companion database named HMEID (available at http://bigdata.ibp.ac.cn/HMEID/) for polymorphic MEIs, which can be explored for new insights into MEI biology.
MATERIALS AND METHODS
Experimental design
The data in this study were from two sources: low-coverage (∼7.4×) WGS samples from the 1KGP (25) and high-coverage (∼26.2×) WGS samples from the NyuWa dataset (24). For the 1KGP dataset, CRAM-format files of 2691 individuals were downloaded from http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/, which were aligned to the human genome building GRCh38 (26). The CRAM files were then converted to BAMs using SAMtools v1.9 (27). The NyuWa dataset contained 2999 individuals including diabetes and control samples collected from different provinces in China (24), and this cohort was sequenced using the Illumina platform. The processing from raw FASTQs to BAMs was according to the GATK Best Practices Workflows germline short variant discovery pipeline (28), as described in (24). The median depth of the NyuWa samples after genome alignment (GRCh38 human genome build) and removal of PCR duplicates was about 26.2×.
Generation of MEI call set
MELT v2.1.5 (16) was run with default parameters using ‘SPLIT’ mode to identify non-reference MEIs, which detects a wide range of non-reference Alu, L1, SVA and HERV-K insertions. To get the BAM coverage for MELT analysis, we used goleft v0.1.8 (https://github.com/brentp/goleft) ‘covstats’ function to estimate the genomic coverage for each sample. After the initial generation of a unified VCF file by MELT ‘MakeVCF’ function, variants that did not pass the following criteria were filtered to get a high-quality MEI call set: (i) not in low complexity regions; (ii) be genotyped in >25.0% of individuals; (iii) split reads >2; (iv) MELT ASSESS score >3 (at least one-side TSD evidence) and (v) VCF FILTER column be PASS. 2998 of 2999 samples in NyuWa and 2677 of 2691 samples in 1KGP were successfully analyzed, with the final call set consisting of 36 699 MEIs from 5675 genomes. Subfamily characterization for Alu MEIs and L1 MEIs was done using MELT’s CALU and LINEU modules, respectively.
The allele frequency of MEIs was calculated as the ratio of allele count and allele number. The allele count was the number of alternate alleles in the genotype field across all the samples, and the allele number was the total number of alleles in called genotypes. The calculation was done by using BCFtools v1.3.1 (29).
PCR validation of MEIs
Randomly selected, allele frequency adjusted MEI sites detected by MELT were included in PCR validation in the NyuWa samples (Supplementary Table S4). For primer design, the insertion breakpoints were extended by 600 bp on either side to retrieve human reference sequences. Then the Alu elements in the flanking sequences of MEIs were masked to Ns using RepeatMasker (30). Considering the imprecise breakpoint calling, a safety margin of 50 nt up- and down-stream of the insertion locus was granted for each candidate site, as described before (14). Five primer pairs for each site were selected using primer3-py (https://github.com/libnano/primer3-py), which uses Primer3 internally (31). We then used BLAT (32) to align each primer to the reference genome and filtered out primers showing more than one match in the human genome. There were 196 candidate MEIs with at least one primer pair available. For the analysis of L1, SVA and HERV-K sites, previously designed internal primers were also used. All PCR primers were ordered from Sangon Biotech (Shanghai) Co. Ltd. The PCR primer sequences were available in Supplementary Table S4.
The way we performed PCR validation was largely consistent with previous study (16). Candidate Alu loci were amplified using external primers (i.e., two primers flanking the MEI). Candidate L1, SVA and HERV-K sites were amplified with internal primers. In cases of ambiguous or no amplification of the candidate element in the predicted individual, a temperature gradient PCR was performed to optimize the annealing temperature of the reaction. PCR amplification was performed using TOYOBO KOD DNA Polymerase (TOYOBO catalog #: KMM-201). All experiments included: (i) a genomic DNA sample (gDNA) that was expected to have the MEI, (ii) a gDNA sample that was expected to lack MEIs based on the MELT calls and (iii) one PCR reaction that lacked gDNA. PCR reaction conditions were as follows: 3 min at 98°C followed by 30 cycles of 10 s at 98°C, 5 s at 55°C and 10 s at 68°C with a final elongation for 2 min at 68°C. Each PCR reaction contains 15–50 ng of template DNA and 0.3 μM oligonucleotide primer. A test was considered positive only if a PCR product of the expected size was observed in the individual that was predicted by MELT to have the insertion.
After initial testing, L1, SVA and HERV-K sites that were negative were assessed again using long-range PCR approach with two external primers to rule out the possibility of internal sequence changes preventing the binding of the ‘internal’ primer. Long-range PCR using TOYOBO KOD DNA Polymerase (TOYOBO catalog #: KMM-201) was performed for each site with the following reaction conditions: 3 min at 98°C followed by 30 cycles of 10s at 98°C, 5 s at 60°C and 60 s at 68°C with a final elongation for 2 min at 68°C. For long-range PCR reaction, the concentration of primer is 0.15 μM. Sites and primers were reported in Supplementary Table S4.
Detection of L1 3′ transduction and 5′ inversion
Following the generation of a high-quality MEI call set, MELT v2.1.5 was used to detect L1 3' transduction. We followed the instructions of MELT 3' transduction identification pipeline and extracted the METRANS and MESOURCE fields in the resulting VCF manually. The population frequency was calculated with the AC/AN (for offspring MEI set, we used the sum of AC and AN) and normalized across different populations.
The MELT VCF provided the position of a 5' inversion site (from the 3' end) through the ‘ISTP’ field. We subtracted it from the full length of L1 (6019 bp) to obtain the coordinates of the inversion site from the 5' end. Since no inversion events were detected in the first ∼600 bp, we removed the full-length L1 elements from the comparison set while comparing the inversion coordinate and the length of L1. Sites were distributed into 100 bins across the full length of L1. We compared the distribution of sequence length and inversion site position among these bins and calculated the Pearson correlation value.
Analysis of Hardy–Weinberg equilibrium
To evaluate the genotype distributions of each MEI under the null expectations set by the Hardy–Weinberg equilibrium (HWE), we tabulated genotype distributions of autosomal MEIs per dataset and performed exact tests by ‘HWExactStats’ function in R package HardyWeinberg v1.6.3 (33). While disequilibrium may indicate disease association or population stratification, it may be the result of confusion of heterozygotes and homozygotes. We thus used the HWE test for gross quality-check of genotyping accuracy (Supplementary Figure S2), as described in (18).
Comparison with the 1KGP and gnomAD MEI call set
To compare the MEIs generated by the 1KGP (16), we downloaded the GRCh38 version call set from the dbVar database (accession number: nstd144) (34). Then non-reference MEIs were extracted and compared with the MEIs identified in this study, using ‘window’ function from BEDtools v2.26.0 (35). When a site was in ±500 bp of another site, it was considered as a hit.
For comparison with MEIs from gnomAD (18), we downloaded the GRCh38 version call set from the dbVar database (accession number: nstd166) and extracted non-reference MEI sites. We then excluded sites without TE type information and compared them with our call set as described above.
Testing MELT for different genome build and joint calling
To test MELT’s performance on different genome builds, we randomly generated 100 samples from the 1KGP dataset, and we got the alignment files for both GRCh37 and GRCh38 version for these samples. After which we ran MELT v2.1.5 on the two datasets and filtered sites as mentioned above. Finally, we compared the results using the function ‘intersect’ from BEDtools v2.26.0 (35).
To test MELT’s performance with respect to sample size (joint calling), we randomly generated 100 samples from the NyuWa dataset and combined them with 100 random samples from the 1KGP above. We identified MEIs using the same pipeline as before on these 200 samples. After which we compared the call set with the MEIs detected from the 100 samples from the 1KGP with BEDtools ‘intersect’.
Functional annotation
Variant Effect Predictor v99.2 (VEP) (36) with Ensembl database version 99 (37) was used to annotate MEIs, with parameters ‘- -pick - -canonical - -distance 1000 500’. MEIs were also intersected with enhancers from GeneHancer database (38) using BEDtools v2.26.0 ‘intersect’ function (35). Only one functional consequence was kept for each MEI, and enhancers were given higher priority when a MEI was also found in non-coding genes and intergenic regions.
Mapping MEIs to the GWAS signals was done as described in a previous study (39). GWAS SNPs and their related traits were obtained from GWAS Catalog v1.0.2 (40). We first defined the LD block region for each GWAS SNP by its proxy SNPs (r2 > 0.8). The LD between all the SNPs was calculated using the SNP call set generated by 1KGP phase III (25), with plink v2.00a1LM (41). If there were no LD SNPs found on either side of the GWAS SNP, we would use the median length of all predicted LD regions as the block length, centered on the target SNP. Then BEDtools v2.26.0 ‘intersect’ function (35) was employed to identify MEIs falling into these LD block regions. The complete set of these MEIs can be found in Supplementary Table S10.
To qualify the enrichment of MEIs across different genomic features (Figure 4B), we permuted 1,000 times for each MEI type with the same number as the real calls using GAT v1.3.4 (42). Each permutation set was annotated with VEP and BEDtools using the same rules as above. After counting the MEIs in each genomic feature, log2 fold changes and empirical P-values were computed. We repeated 3 times of the permutation procedure to verify the results.
Figure 4.

MEI functional properties. (A) Predicted functional consequences for each type of MEI: (left) cumulative proportion, and (right) cumulative number. Coding Intron, introns of protein-coding genes; UTR5, 5′ UTR; UTR3, 3′ UTR; Non-coding Exon, exons of non-coding genes; Non-coding Intron, introns of non-coding genes; Flank5, 5′ flanking regions of genes; Flank3, 3′ flanking regions of genes. (B) Log2 fold enrichment of the MEI call set compared against the MEIs permutated. The permutation test was repeated 1000 times, and empirical P-values were commutated together with the enrichment values. The enrichment values were scaled row-wise. ns, not significant (P-value > 0.05). (C) Box plots of counts of predicted PTVs by MEI: (left) all the MEIs identified in this study, and (right) rare MEIs (allele frequency < 1%) in this study. (D) Proportions of singleton MEIs in CDS and coding introns for Alu, L1 and SVA. Error bars indicate 95% CIs based on population proportion. P-values were computed using chi-squared test. (E) Proportions of high pLI genes (pLI > 0.9) for genes with MEIs in the CDS and genes with MEIs intron regions. Error bars represent 95% CIs based on population proportion. P-values were computed using chi-squared test. (F) Box plots of LOEUF scores of genes with MEIs in the CDS and genes with MEIs in their introns. Wilcoxon rank sum test was used to compute p-values. Figure D-F uses the same legend beneath. ns, P ≥ 0.05; *P < 0.05; **P < 0.01.
Chromosome-level analyses of MEI density
To check the distribution of MEIs throughout the genome, we used the method described by Collins et al. (18) and we repeat it here for clarity. Focusing on 22 autosomes, each chromosome was segmented into consecutive 100 kb bins and bins overlapped with centromeres were removed. For each MEI type (Alu, L1, SVA and HERV-K), the number of variants in each bin was recorded to get a matrix of MEI counts per 100 kb bins per autosome. The number of variants was the number of unique, non-overlapping insertions we detected. To smooth the MEI counts for each MEI type, an 11-bin (∼1Mb) rolling mean per chromosome was computed. Each bin was then assigned to a percentile based on the position of that bin on its respective chromosome arm relative to the centromere. Specifically, a value of 0 corresponded to the centromere, and a value of −1 and 1 corresponded to the p-arm telomere and q-arm telomere, respectively. Finally, to compute “meta-chromosome”, the normalized bin positions (i.e. −1 to 1) were cut into 500 uniform intervals, and values across all autosomes based on the normalized interval position were averaged. Then the “meta-chromosome” density was normalized by its mean value to get the “fold-enrichment” values shown in Figure 2. For the comparison of chromosome contexts (Figure 2), normalized positions within the outermost 5% of each chromosome arm were considered as “telomeric”, the innermost 5% as ‘centromeric’ and the other 90% of each arm as “interstitial”. The smoothed density of MEIs was first normalized by its mean value to get ‘fold-enrichment’ scores and then whether the ‘fold-enrichment’ values in given chromosome context were greater or smaller than 1 was tested by t-test. The P-values were adjusted using the Bonferroni method.
Figure 2.

Chromosome-level Distribution of MEI Density. (A) Smoothed enrichment of different types of MEIs ascertained in this study. The values were calculated per 100 kb window across the average of all autosomes and normalized by the length of chromosome arms (as ‘meta-chromosome’). (B) Enrichment of MEIs by class and chromosomal context. The dots are the mean values and point ranges represent 95% confidence intervals (CIs). P-values were computed using a two-sided t-test and adjusted using the Bonferroni method. *P ≤ 0.05. C, centromeric; I, interstitial; T, telomeric. The way to compute the chromosomal enrichment and to represent data was from the gnomAD SV paper (18). (C) Smoothed enrichment of L1s with singletons excluded ascertained in this study. (D) Smoothed enrichment of non-reference L1s from the euL1db database (55).
Mutation rates
Before estimating mutation rate, we exclude the MEIs that failed in the HWE test (adjusted P < 0.05) and MEIs in sex chromosomes. MEIs in low complexity regions (43) and in reference TE sequences were also filtered, due to the inability of MELT in these regions (44). The final masked genome size was about 1 920 571 898 bp. Watterson’s Theta (45) was then used to estimate the genome mutation rate of each MEI type:
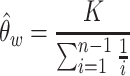 |
where K is the number of MEI sites observed per MEI type in a given population, and n is the total number of chromosomes assessed. Then mutation rates were estimated as:
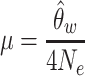 |
with an effective population size (i.e. Ne) of 10 000, consistent with previous studies (14,18,44). The above calculation was performed separately in the NyuWa dataset and the 1KGP dataset (Supplementary Table S6).
SNP heterozygosity and MEI diversity
As described in a previous study (46), SNP heterozygosity was computed as the ratio of heterozygous SNPs over the length of the genome, and the mean value was used when multiple samples were considered. MEI diversity was defined as the average number of MEI differences between individuals in a population. For the NyuWa dataset (24), high-quality SNP calls generated by the GATK v3.7 cohort pipeline (28,47) were used. For 1KGP samples, SNP calls on the human genome build GRCh38 were downloaded from http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/data_collections/1000_genomes_project/release/20190312_biallelic_SNV_and_INDEL/. Number of heterozygous SNPs was computed by VCFtools v0.1.15 (48) and MEI diversity by ‘gtcheck’ function in BCFtools v1.3.1 (29).
Database construction
We collected the MEIs captured in this study and organized them into a MySQL database HMEID. The website was built by Bootstrap and Django. For each population, we calculated allele frequency of each MEI. We drew population plots for each site using the percentages of AF. Site details as well as calling qualities were also extracted from the original VCF and presented on the website. All the data can be browsed in the database and downloaded from the ‘Download’ page. We also provided the website source code on GitHub (https://github.com/oldteng/HMEID).
Statistical analysis
All statistical analyses in this study were briefly described in the main text and performed using R v3.6.2 (http://CRAN.R-project.org/).
RESULTS
A comprehensive map of non-reference human MEIs
To generate a comprehensive map of MEIs from human genomes, we jointly analyzed two WGS datasets using MELT (16), the low-coverage 1KGP dataset consisting of 2677 individuals sequenced to ∼7.4× coverage (14) and the high-coverage NyuWa dataset including 2998 Chinese samples sequenced to ∼26.2× coverage (Supplementary Table S1) (24). After site quality filtering, a total of 36 699 non-reference MEIs were kept, including 26 553 Alus, 7353 L1s, 2667 SVAs and 126 HERV-Ks (Table 1; Supplementary Table S2). Of the 126 non-reference HERV-Ks, 18 sites were already reported before (8,49) and 112 sites had estimated lengths over 8000 bp (Supplementary Table S3). By manually inspecting the IGV screenshots of the read alignment in flanking regions of the HERV-K sites, the evidence of breakpoints for 71 and 23 sites were classified as ‘positive’ and ‘possible’ respectively (Supplementary Table S3; Supplemental Data), accounting for 74.6% of all the HERV-K MEIs detected in this study. Using Hardy-Weinberg equilibrium (HWE) metrics as a rough proxy of genotyping accuracy, we found that about 87% autosomal MEI sites did not violate the HWE, and when restricted to the NyuWa dataset, almost all MEIs (97%) on autosomes had high genotyping accuracy (Supplementary Figure S1). Most Alu and L1 MEIs were well-supported by target site duplications (TSDs) (Supplementary Figure S2A), a hallmark feature of new retrotransposition events. The distributions of TSD lengths were also consistent with previous reports (Supplementary Figure S2B) (50,51). For comparison, there were 55.6% (9756/17 543), 33.0% (1358/4118) and 32.4% (344/1062) MEIs with TSDs in previous 1KGP call set for Alu, L1 and SVA, respectively (16). To validate the sites detected by MELT, 190 sites (66 Alus, 49 L1s, 62 SVAs and 13 HERV-Ks) were tested by PCR in the NyuWa samples. The false discovery rates (FDRs) reported for Alu, L1, SVA and HERV-K sites were 4.55% (3/66), 4.08% (2/49), 6.45% (4/62) and 23.08% (4/13), respectively (Supplementary Figure S2C; Supplementary Table S4). We also compared our MEI call set with the validating data from two previous 1KGP studies, containing 90 sites (16) and 179 sites (14) genotyped by PCR, respectively. The overall detection sensitivity was about 70%, and few false positive calls were found in our call set, notably (Supplementary Table S5). This was reasonable since rather stringent filtering criteria were applied after the initial detection of MEIs and these 1KGP samples were low coverage.
Table 1.
MEI discovery in this study
| Mean sites per doner | Standard deviation | ||||
|---|---|---|---|---|---|
| Total sites | NyuWa | 1KGP | NyuWa | 1KGP | |
| Alu | 26 553 | 1035 | 884 | 25.3 | 153 |
| LINE-1 | 7353 | 145 | 119 | 8.35 | 19.3 |
| SVA | 2667 | 44.4 | 28.8 | 4.83 | 9.9 |
| HERV-K | 126 | 11 | 8.23 | 1.86 | 2.12 |
| Total | 36 699 | 1236 | 1040 | 30 | 178 |
On average, we detected 1236 MEIs with each genome in the NyuWa dataset and 1040 MEIs in the 1KGP dataset (Table 1). The number of MEIs per genome in the NyuWa dataset was consistent with a recent study with comparable sequencing depth (19), which reported a mean of 1199 MEIs per individual. The higher number of MEIs per genome found in the NyuWa dataset than that of the 1KGP dataset was expected, as increased sequencing depth provides more power for MEI detection (Supplementary Figure S2D). The smaller correlation between the MEI number and sequencing coverage in the NyuWa dataset than that of the 1KGP dataset reflected that the MEI detection sensitivity by MELT was close to saturation in ∼30× genomic coverage, consistent with the previous evaluation by the authors of MELT (16). The distribution of MEI numbers per individual, MEI allele frequencies and length estimates largely fit the findings of previous studies (Figure 1; Supplementary Figure S2) (16,44). About 70.7% MEIs are very rare (allele frequency < 0.1%), with over 30% singletons of all four MEI types (Figure 1C; Supplementary Figure S2E). Compared to the 1KGP, more MEIs in the NyuWa dataset were found in the bins with low allele frequency (e.g. allele frequency < 0.1%) (Supplementary Figure S2F). Since a large proportion of MEIs were individual-specific, we next sought to evaluate MEI discovery by increasing sample size. Through randomly down-sampling to different sizes with 100-sample intervals, we estimated the total MEI variants and the increase of variants at different sample sizes. As expected, we found that the number of all four MEI types continued to rise with the increasing sample size, but the growth rate decreased (Supplementary Figure S2H). When restricting this analysis in the NyuWa dataset, the curves of Alu, L1 and SVA MEIs were still steep, indicating high MEI diversity in Han Chinese population (Supplementary Figure S2I).
Figure 1.

The MEI call set. (A) Histograms of the number of MEIs identified per genome in the NyuWa dataset. (B) Histograms of the number of MEIs identified per genome in the 1KGP dataset. (C) Distribution of allele frequency of MEIs of four types: Alu, L1, SVA and HERV-K. ‘Total’ combined the four types of MEIs. The allele frequencies were cut into five bins: 0 ≤ AF < 0.0001, 0. 0001 ≤ AF < 0. 001, 0. 001 ≤ AF < 0. 01, 0. 01 ≤ AF < 0. 1 and 0. 1 ≤ AF < 1 and the proportion of MEIs in each AF bin was calculated. (D) Distribution of insert size estimated by MELT. The x-axis coordinates of peaks were annotated.
Looking at the subfamilies of MEIs, we found that the distributions of active Alu and L1 MEIs were in line with previous observations in humans (13,16,46,52), e.g. AluYa5 and AluYb8 were found to be the most abundant two Alu subfamilies (Supplementary Figure S3), indicating their high retrotransposition activity in modern humans.
Compared to the previous MEI findings of 1KGP samples (16), the total number of non-reference MEIs we detected has increased 61.5%, with 51.4% and 78.6% increase for Alu and L1 insertions, respectively (Supplementary Figure S4A). In addition, large proportions of MEI calls detected by previous study were repeatedly identified in this study, and the allele frequency for overlapping sites also showed high consistency (Supplementary Figure S4B; Pearson’s correlation coefficient = 0.95). In each class of MEI, we found a novel rate from 58% to 83%, and the dominant EAS (East Asian super population)-specific novel MEIs were only detected in the NyuWa dataset (Supplementary Figure S5). Expectedly, most of the novel sites were EAS-specific and enriched in the NyuWa cohort (Supplementary Figure S6), suggesting our cohort provided a huge resource of Chinese-specific (and EAS-specific) MEIs.
Nonetheless, we noticed that many MEIs identified by Gardner et al. (16) were missed in our call set. Considering that stricter filtering criteria was used here than that of Gardner et al., we also applied the same filtering threshold as Gardner et al. (16), to compare with the previous call set. Indeed, only 12.6% of sites from Gardner et al. were not repeatedly captured by us (Supplementary Figure S7). For these sites that were still missing in our call set, we conjectured that it may be due to differences of software version, reference genome build, the way the BAM files were generated, etc. To test this, we performed three runs using three random sample sets: (i) 100 samples from the 1KGP with reads mapping to the GRCh37 genome build; (ii) 100 samples from the 1KGP with reads mapping to the GRCh38 genome build; (iii) 100 samples from the 1KGP and 100 samples from the NyuWa, with reads mapping to the GRCh38 genome build. We found that more MEIs could be detected using the GRCh38 genome build and/or by combining more samples (Supplementary Table S6). This is also in line with the model used by MELT (16), combining the 1KGP dataset with the high-coverage NyuWa dataset would improve MEI detection sensitivity as well as accuracy, with finer resolution of MEI break points. Although the stringent filtering employed by us could possibly lower the detection sensitivity of MEIs, we think it improved the overall accuracy, as described in previous studies (17,44). Collectively, our MEI call set represents a high-quality map of non-reference MEIs for humans.
Enrichment of non-reference L1 insertions in centromeres
It has long been noted that L1s are preferentially found in AT-rich regions but Alus show the opposite trend (4), likely due to different types of selective forces on L1s and Alus. As expected, we also observed this tendency for MEIs (Supplementary Figure S8A). In addition, the GC content of flanking DNA for Alus and L1s was lower than background, while SVAs and HERV-Ks preferred DNA sequences with much higher GC content. We next compared the GC composition of rare MEIs (allele frequency < 1%) and common MEIs (allele frequency ≥ 1%) due to the reported bias shift in GC bias for older and younger short interspersed nuclear elements (SINEs) (1,46,53,54). Significant difference was only observed for HERV-K: rare HERV-K insertions occurred in much higher density in GC-rich regions (Supplementary Figure S8B). For Alus and SVAs, we did not observe marked bias.
We next sought to investigate the distribution of MEIs throughout the genome, like previously Collins et al. done for common SVs (18). Interestingly, L1s were predominantly enriched at centromeric/pericentromeric regions, whereas SVAs and HERV-Ks were enriched at telomeres (Figure 2A and B; Supplementary Figure S9). As insertions found in multiple individuals are more likely to be authentic than singletons, we performed the same analysis on L1 insertions without singleton, and we could still detect the enrichment in centromeres (Figure 2C). In addition, we got similar results when restricting on MEIs with TSD length ≥5 bp (Supplementary Figure S10), representing sites of higher confidence. Importantly, this finding was well-supported by non-reference L1s from euL1db (Figure 2D), which curated human polymorphic L1s from 32 different studies (55). For comparison, similar analysis was applied to TEs in the reference genome (Supplementary Figure S11), but no such patterns for L1s were found. Even in the recent telomere-to-telomere assembly of the human X chromosome, only a single L1 insertion was detected at the centromere region (56). To validate the accuracy of L1 MEIs in centromeric/pericentromeric regions defined by the enrichment calculation method (368 such sites), we used IGV to manually inspect the reads alignment in 500 bp flanking regions of each site. 82.6% (303/368) of these sites had clear breakpoints (‘positive’), and the evidence of breakpoints for 10.0% (37/368) sites was classified as “possible” (Supplementary Table S7; Supplemental Data). In addition, there were 69% (254/368) L1 insertions with ASSESS score of 5 around centromeric/pericentromeric regions, the best accuracy score assigned by MELT. Considering the reduced detection power of short-read WGS in repetitive regions, the enrichment of L1 insertions at centromeric regions could still be underestimated. The enrichment of non-reference L1 insertions at centromeric DNA could be partly attributed to lower GC content, as centromeres contain massive AT-rich alpha satellites (57). Also, active TEs have been found in neocentromere regions, and may contribute to centromere ontogenesis (58–60). The reasons for the dramatic enrichment of L1s in centromere regions are intriguing and further studies are needed in the future.
Strong correlation between MEI diversity and SNP heterozygosity
Since mutations are ultimate sources of genetic innovation and significant causes of human birth defects and diseases, knowledge of mutation rate is a general population genetics question (61,62). Here, we employed the commonly-used Waterson’s estimator (45) of Θ to estimate the mutation rate of each MEI type and found that mutation rates varied markedly by MEI class (Supplementary Table S8). Since MEI detection and genotyping power is profoundly influenced by sample coverage (16), we conducted the analysis separately for the NyuWa and the 1KGP datasets. The resulting calculation provided very close estimates of between 1.609 × 10−11 (NyuWa) and 1.464 × 10−11 (1KGP) de novo MEIs per bp per generation (μ), or roughly one new MEIs genome-wide for every 16-17 live births, which is largely concordant with prior reports (14,44,62).
The availability of SNP genotyping (both the NyuWa and the 1KGP dataset) for the same samples gave us an opportunity to investigate the correlation between MEI diversity and SNP heterozygosity for each population. SNP heterozygosity was computed as the ratio of heterozygous SNPs across the individual’s genome (63) and was compared to the average MEI differences between samples in a given population (64). The diversity for all types of MEIs showed a strong correlation with SNP heterozygosity (R2: 0.64–0.95), with African populations showing the highest MEI diversity and SNP heterozygosity (Figure 3)—consistent with previous study (13).
Figure 3.

Correlation between SNP heterozygosity and MEI diversity. SNP heterozygosities and diversity of (A) Alu MEIs, (B) L1 MEIs, (C) SVA MEIs and (D) HERV-K MEIs were compared in different populations. SNP heterozygosity was computed as the ratio of heterozygous SNPs across the individual’s genome and MEI diversity was computed as the average allele difference in each population. Points were colored by super populations. AFR, African super population; AMR, American super population; EAS, East Asian super population; EUR, European super population; SAS, South Asian super population.
MEI functional properties
Via the local impacts by transposition events or more global post-insertion influence (58), MEIs can disrupt normal gene functions and be disease-causing (9,10). In principle, MEIs in coding regions can result in predicted loss-of-function (pLoF) by altering open-reading frames. To assess the functional impact of MEIs, we annotated the MEI calls using Variant Effect Predictor (VEP) and BEDtools (see Materials and Methods). The vast majority (82.7%) of detected MEIs were in intergenic and intronic regions, while only ∼2.7% MEIs impacted the coding sequences (CDS) (Figure 4A). Varying enrichment levels on different genomic features were observed for different MEI types (Figure 4B), largely consistent with the previous report (17). For example, L1, SVA and HERV-K MEIs were significantly depleted in CDS and non-coding gene exons; SVA and HERV-K sites were enriched in intergenic and non-coding introns. Focusing on protein-truncating variants (PTVs), defined as MEIs in coding regions of protein-coding genes, each genome contained a mean of 24.8 MEIs (12.6 Alu, 7.4 L1, 1.3 SVA and 2.4 HERV-K) directly disrupting CDS, including 1.1 rare pLoF MEIs (allele frequency < 1%) (Figure 4C; Supplementary Table S9). By comparison, Karczewski et al. estimated 98.9 pLoF small variants (SNVs and InDels) per genome (65), and Collins et al. observed 144.3 pLOF SVs per genome (18). We thus estimated that MEIs account for about 9.3% (24.8/268) of all PTVs, among small variants and large SVs in each human genome.
Examining the degree to which evolutionary forces act on coding MEI loci is important to understand the relationship between MEI variation and coding genes. Here we used three different metrics to investigate selective constraints: (i) the proportion of singleton variants (variants observed in only one individual), an established proxy for selection strengths (66); (ii) the proportion of MEIs in genes with high probability of loss-of-function intolerance (pLI) (66); (iii) the loss-of-function observed/expected upper bound fraction (LOEUF) of MEI-containing coding genes, where higher LOEUF scores suggest a relatively higher tolerance to inactivation for a given gene (65). HERV-K MEI was not included in this analysis due to the relatively small number found in coding genes. Higher singleton proportions for Alu and L1 MEIs were found in CDS than that of introns (Figure 4D; χ2P < 0.05), while we did not find a statistically significant bias for SVA MEIs, though there were 166 and 949 SVA insertions found in CDS and coding introns, respectively. Likewise, lower proportions of Alu/L1 MEIs detected in genes with a high pLI score (>0.9) were found in CDS than that of intronic regions (Figure 4E; χ2P < 0.05). Observations from the perspective of enclosing genes fit these results: higher LOEUF score were found for genes with Alu/L1 MEIs (Figure 4F, Wilcoxon P < 0.05). Our results sustained and expanded previous findings on human exome data (44), in which Gardner et al. reported that exonic MEIs were under purifying selection.
Although researchers have long noted that most of reference LTR elements and L1s in gene introns are in the antisense orientation with respect to the host genes (1,53), possibly due to ill effects on transcript processing of sense-oriented elements (67,68), there are no established conclusions about the orientation tendency of other types of non-reference MEIs and the number of sites in previous studies were limited (17,44,46). Our large collection of MEIs found in genes allowed us to closely examine the strand bias of different MEIs. Although the bias for Alu, L1 MEIs and SVA MEIs to be in antisense orientation when found within genes was observed (46), we did not find a statistically significant bias for L1 insertions (Supplementary Figure S12A). Conversely, Alus were found to have strong strand bias when being inserted into protein-coding genes, non-coding genes, protein-coding introns and non-coding introns (Supplementary Figure S12; χ2P < 0.05). For SVA MEIs, protein-coding genes, protein-coding exons, and protein-coding introns were regions where insertion orientation biases were detected (Supplementary Figure S12; χ2P < 0.05). Considering that Alu and SVA elements are non-autonomous TEs that are trans-mobilized by the L1 retrotransposition machinery(69,70), there may be some post-insertion selection forces on Alu/SVA elements which influence these patterns (11). The genes themselves which had MEIs in sense or antisense strand in introns did not show clear differences in terms of selective constraints, by comparing the LOEUF scores of these two kinds of genes (Supplementary Figure S12F). In addition, no significant orientation tendency against the neighboring genes were detected when MEIs were in gene upstream regions (Supplementary Figure S12I).
Alu MEIs have been found to be enriched in regions of the genome associated with human disease risk, suggesting their potential effects on common diseases (9,39). To identify MEIs potentially associated with human trait or disease, we mapped MEIs to regions in linkage disequilibrium (LD) with trait- or disease-associated loci identified by genome-wide association study (GWAS) (P < 10−8) (40). We found that 6457 (about 17.6%) of the MEIs (17.5% for Alu, 15.3% for L1, 24.4% for SVA and 16.6% for HERV-K) were in these regions that tagged by at least one GWAS SNP (Supplementary Table S10), with allele frequency of 738 MEIs over 1%, suggesting the remarkable potential for MEIs to contribute to disease and the utility of our MEI set in future phenotype-variant association studies.
We also evaluated the use of our resources in trait-association genetic studies and medical applications to exemplify the value of our MEI resources. We examined the allele frequencies of MEIs in pigmentation genes (71) and clinically relevant genes (72) across different populations. Uneven allele frequency distribution was found for MEIs in these regions (Supplementary Figure S13; Supplementary Table S11). Skin pigmentation regulator SASH1 has two common Alu insertions in introns, one of which was only observed in Asian populations. Multiple MEIs were detected in CDS regions of disease-associated genes, such as COL3A1, ADGRV1, TMC1, PSPH, RYR3, SCN5A and MYH2. It is generally accepted that there are racial differences in pigmentation and disease susceptibility, and these results implied that MEIs may be potential contributors to color phenotypes and disease risk across populations.
L1 3′ transduction and 5′ inversion
Some L1 elements can bring a 3' readthrough transcript to the offspring insert site, which is called 3' transduction (73). These L1 elements are usually near a strong Poly(A) sequence. Transcription of these L1 elements is not terminated by the original weak poly(A) of the L1 element but by the stronger poly(A) sequence downstream. With the flanking sequences downstream L1 elements, we extracted the correspondence between L1s in different genomic positions. In total, 446 offspring MEIs derived from 57 source MEIs were identified in our samples. These MEI relationships are both interchromosomal and intrachromosomal (Figure 5A, Supplementary Table S12A). Compared with the L1 transduction source sites identified by the 1KGP study (16), we found that most of the sites overlapped with the 1KGP donor sites (Figure 5B). Among these sites, two of the three most active source sites (chr6:13190802, chr1:118858380) were also found in this study, while the site L1RE3 (chr2: 155671336) is in a low complexity region and was filtered in the site filtering. Most of the sources transduced <20 offspring whereas site chrX:11713279 has 186 offspring (41% of all offspring detected). Source and offspring MEIs were distributed into families and population frequency was calculated (Figure 5C and D). Most transduction classes were EAS specific, within which both the source and offspring elements of class chr9:104505307 were detected only in EAS population. Comparing frequencies among the subpopulations of EAS, we noticed that 14 transduction classes were only detected in Chinese people. Inside these classes, only five classes appear in samples of Northern Han Chinese (CHB, CHN) and four classes only appear in Southern Han Chinese (CHS, CHS.1KGP) (Supplementary Tables S12B and S12C).
Figure 5.

L1 3′ Transduction and 5′ Inversion. (A) 3′ transduction source-offspring relations across the whole genome. The heatmap denotes the offspring number. The top 5 sources with the highest numbers of offspring were marked outside the circle. (B) The Venn plot of 3′ transduction sources found by our study and the 1KGP study (16). (C) Source (bottom) and offspring (top) element frequencies in super populations. AFR, African super population; AMR, American super population; EAS, East Asian super population; EUR, European super population; SAS, South Asian super population. (D) Source (bottom) and offspring (top) element frequencies in Asian subpopulations. CDX, Chinese Dai in Xishuangbanna; CHB, Han Chinese in Beijing; CHN, Northern Han Chinese, China; CHS, Southern Han Chinese; CHS.1KGP, Southern Han Chinese from the 1KGP; JPT, Japanese in Tokyo; KHV, Kinh in Ho Chi Minh City. (E) L1 length distribution within our call set. The length was estimated by MELT. (F) 5′ inversion position distribution among all inverted sites. (G) Correlation plot between the distributions shown in (E) and (F). The full length L1 element was excluded from this comparison.
5′ end of the L1 sequence can be inverted during insertion (74). We extracted the 5′ inversion information from the MELT result, and 1606 L1 insertions were detected with a 5′ inversion end. The nearest distance from the 5′ inversion site to the 3′ end of the L1 insertion is 602 bp, which is consistent with the 1KGP study (590 bp) (16). It seems that the inversion does not occur in the first ∼600 bp from the 3′ end, which may indicate that the inversion process requires at least ∼600 bp DNA sequence. The distribution of the 5′ inversion positions highly correlated with the distribution of L1 MEI lengths (R2 = 0.696; Figure 5E–G), which was also found in the 1KGP cohort (16). This trend somehow suggested a dependence of inversion calling on the total L1 length distribution because the inversion can only be observed when the insertion is larger than the breakpoint. We next calculated the percentage of 5′ end inverted MEIs within each 3′ transduction offspring class. The inversion rate across different classes varied and did not correlate with the class size (Supplementary Table S13). For the biggest class which was derived from chrX:11713279, only 25.3% of the offspring had 5′ inversion while a class which only includes 15 offspring had a 40% inversion rate.
A database for polymorphic MEIs
Currently, resources for polymorphic TE findings in human genomes are in high demand (12). There were only two dedicated databases for polymorphic human MEIs: dbRIP (75) and euL1db (55). However, the former had not been updated since 2012 and the latter was only for human specific L1 insertions. To fill this gap, we have designed a companion database called HMEID to archive MEIs identified in this study, and to comprehensively catalog the variants on allele frequencies in the NyuWa dataset and the 1KGP dataset. In addition, variant quality metrics and functional annotations are also presented. Compared to dbRIP, HMEID contained more MEIs; the number of L1 insertions in HMEID was comparable with that of euL1db (Figures 6A and B). Though not explicitly analyzed and discussed in the gnomAD-SV paper, MEIs from 14,891 genomes were also available in gnomAD, including 1,304 samples from East Asia (18). While more non-reference MEIs were in gnomAD than that in HMEID, there were 24 505 MEIs specific to the HMEID, accounting for 66.8% of all MEIs we detected (Figure 6C). For these 24 505 sites, there were still 6122 MEIs remaining when excluding sites identified in the previous 1KGP study (16). When focusing on MEIs from EAS samples of gnomAD, there were 31 709 sites (86.4%) specific to HMEID (Supplementary Figure S14A). Also, the allele frequency for overlapping MEIs from the two databases also showed high correlation (Supplementary Figure S14B). Importantly, HMEID contained MEIs detected from large samples of Han Chinese, which is the largest ethnic group in the world. We anticipate that this resource would facilitate the exploration of TE polymorphisms and benefit future research on TEs as well as human genetics.
Figure 6.
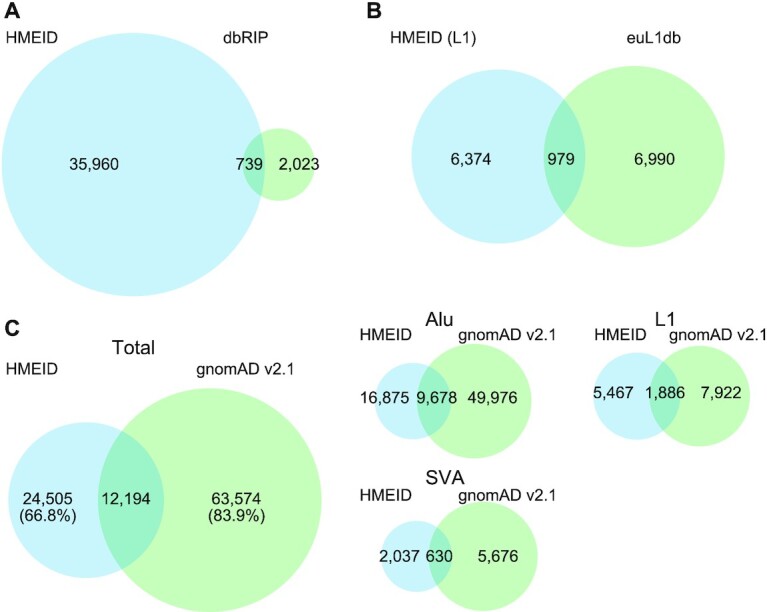
Comparing HMEID with other MEI Databases. (A) Comparison the MEI set in the HMEID with that of from the dbRIP database (75). (B) Comparison of the L1 MEIs in the HMEID with non-reference L1s from the euL1db database (55). (C) Comparison of the L1 MEIs in the HMEID with that of from the gnomAD database (18).
DISCUSSION
Resources for MEIs, an endogenous and ongoing source of genetic variation, are still lacking compared to those for SNVs and SVs. Here, we leveraged 5675 genomes from the NyuWa (24) and 1KGP (25) datasets to create a comprehensive map of non-reference MEIs. After describing the frequency spectrum of variants, we focused on the insertion site preference and functional impacts of MEIs.
We identified 36 699 non-reference MEIs for four types of TEs and determined that individuals harbor a mean of over 1000 non-reference MEIs, mostly contributed by Alu insertions. In line with previous reports (13,16,44), most MEIs were rare and individual-specific, which was also observed for SNVs (25) and SVs (18). With the newly sequenced 2998 genomes from China, this study established a large-scale MEI resource for the genetics of Chinese as well as East Asians. Compared to the previous study conducted by the 1KGP (16), the number of MEIs detected by us has increased about 55%, representing what is to our knowledge one of the most comprehensive sets of human non-reference MEIs and the largest non-reference MEIs for Chinese populations. As expected, most of the novel MEIs detected in this study were EAS-specific and enriched in the NyuWa dataset. There remained 24 505 novel MEIs in our study compared to the MEIs in gnomAD SV study, one of the largest cohorts for SV (18). Considering that the current genetic and genomic resources for MEI were mainly from European ancestry cohorts, our work would benefit future MEI studies by providing a large and high-quality WGS resource for Chinese populations. Analysis of the MEI call set would improve our understanding of how MEIs affect complex traits and disease susceptibility and allow us to map out strategies for efforts focused on populations in East Asia.
We found that non-reference MEIs have non-random distributions along chromosomes, implicating the role of chromosome context in TE insertion. Of note, we found that non-reference L1 MEIs were drastically enriched in centromere regions, which was also supported by independent data from the euL1db (55). This was not likely to be artefactual due to false alignment in repetitive regions of short reads, as MELT did not consider reads aligned to many locations (16) and we have filtered MEIs in low complexity regions flagged by MELT. Unexpectedly, we did not observe the same enrichment in centromeric regions of L1s from the human reference genome. We speculated that the reason for this was because the reference genome was from ‘one’ person, and the enrichment could only be detected at population scale. Also, the phenomenon that L1s accumulate in centromeric/pericentromeric regions was not long enough in human history to make it enriched in each genome.
The genomic distribution of TEs is a result of insertion site preference and post-insertion selection on the host (11). On the one hand, human centromeres are full of AT-rich alpha satellites (57), which could confer insertion preference for L1s, since the target specificity of L1 insertion machinery is TTTT/AA (3,76). Certain centromeric histones and other centromeric proteins may also serve as preferred targets for TEs, as suggested by a study in maize (77). Additionally, studies on HIV integration into the host genome implied that proximity to the nuclear periphery of centromere may facilitate TE targeting (78,79). On the other hand, incorporation of L1s may facilitate the recurring evolutionary novelty of centromeres (58). In support of this, Chueh et al. reported that RNA transcripts from a full-length L1 are the essential structural and functional components in the regulation of a human neocentromere (80). Evidences were also found in the tammar wallaby (Macropus eugenii), where dramatic enrichment of L1s and endogenous retroviruses was found in a latent centromere site (81), and Equus caballus, where evolutionary new centromeres are located in LINE- and AT-rich regions (82). In addition to centromere ontogenesis, a LINE-like element (G2/jockey3) contributes directly to the organization and function of centromeres of D. melanogaster (83). This is also likely true for the non-reference SVA, for which we found an enrichment in telomeres, as TEs were found to be essential in maintaining the telomere length homeostasis in insects (84). However, another plausible explanation for both the enrichment of non-reference L1 MEIs in centromere and non-reference SVA MEIs in telomere is that these regions contain few protein-coding genes, limiting insertional mutagenesis by TEs (11). The reasons for this phenomenon are fascinating, and our study post an important question about the relationship between TEs and centromeres.
Knowing the functional impact of MEIs is fundamental to our understanding of the impact of MEI with respect to human disease or trait and evolution (12). We have estimated that MEIs accounted for about 9.3% of all protein-truncating variants per genome, among small variants (65) and SVs (18). Our estimation was much higher than that determined by whole exome sequencing data (44), possibly due to the limitation of exome baits used before (44). We found that a significant portion of polymorphic MEIs mapping to loci implicated in trait/disease association by GWAS, as increasingly recognized by recent studies (39,85). While previous GWAS have mainly focused on small variants (86), future association studies should consider and evaluate the effects of MEIs on common diseases. We anticipate that the HMEID will serve as a basis for such studies.
Our study is limited in that only one tool was used to identify MEIs. Though the overall performance of MELT outperformed existing MEI discovery tools (16,87) and it has been successfully used in several large-scale studies (16,17,44,62,88,89), but the detection power could be compromised by modest sequencing depth and incompetence in complex genomic regions of short-read WGS etc. In addition, the overall genotyping accuracy by MELT v2 was 87.95% for non-reference Alus (not excluding MEIs in low complexity regions), when compared with PCR generated genotypes (90). As such, we have tried to ensure the site quality by strict filtering, thus resulting in compromised detection sensitivity. In the future, we would consider combining different MEI identification and genotyping tools to improve the detection quality, which has been proved useful in previous reports (12,62,91,92). Also, long-read WGS is promising in detecting MEIs, especially for genomic regions refractory to approaches using short-read sequencing technologies (93–96). Another limitation of our MEI dataset is that reference MEIs (MEIs detected as deletions) have not been included yet, for which the detection is underway, and the results will be integrated into the HMEID for public use.
DATA AVAILABILITY
Complete MEI call set and other related information such as allele frequency and functional annotation are available in the companion database HMEID (available at http://bigdata.ibp.ac.cn/HMEID/). The source code of the HMEID could be accessed at https://github.com/oldteng/HMEID.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Eugene J. Gardner for helping us in using MELT. We thank Jing Wang for valuable comments in the data analysis and critical review of the manuscript. We thank Tingrui Song for assisting the use of high-performance computing platforms. We thank the people for generously contributing samples and sequencing data to the NyuWa dataset and the 1KGP dataset. Data analysis and computing resources were supported by the Center for Big Data Research in Health (http://bigdata.ibp.ac.cn), Institute of Biophysics, Chinese Academy of Sciences.
Author contributions: T.X. and S.M.H. conceptualized and supervised the project. Y.W.N., X.Y.T., Y.R.S., Y.Y.L., Y.H.T. and Q.K. conducted data analysis. H.H.Z. performed all PCR validations. X.Y.T. built the database. Y.W.N., X.Y.T., H.H.Z., H.X.L., P.Z. and S.M.H. drafted the manuscript, and all the primary authors reviewed, edited, and approved the manuscript.
Contributor Information
Yiwei Niu, Key Laboratory of RNA Biology, Center for Big Data Research in Health, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China; College of Life Sciences, University of Chinese Academy of Sciences, Beijing 100049, China.
Xueyi Teng, Key Laboratory of RNA Biology, Center for Big Data Research in Health, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Honghong Zhou, Key Laboratory of RNA Biology, Center for Big Data Research in Health, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China.
Yirong Shi, Key Laboratory of RNA Biology, Center for Big Data Research in Health, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Yanyan Li, Key Laboratory of RNA Biology, Center for Big Data Research in Health, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China; College of Life Sciences, University of Chinese Academy of Sciences, Beijing 100049, China.
Yiheng Tang, Key Laboratory of RNA Biology, Center for Big Data Research in Health, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China; University of Chinese Academy of Sciences, Beijing 100049, China.
Peng Zhang, Key Laboratory of RNA Biology, Center for Big Data Research in Health, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China.
Huaxia Luo, Key Laboratory of RNA Biology, Center for Big Data Research in Health, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China.
Quan Kang, Key Laboratory of RNA Biology, Center for Big Data Research in Health, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China.
Tao Xu, College of Life Sciences, University of Chinese Academy of Sciences, Beijing 100049, China; National Laboratory of Biomacromolecules, CAS Center for Excellence in Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China.
Shunmin He, Key Laboratory of RNA Biology, Center for Big Data Research in Health, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China; College of Life Sciences, University of Chinese Academy of Sciences, Beijing 100049, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Strategic Priority Research Program of Chinese Academy of Sciences [XDB38040300, XDA12030100]; National Natural Science Foundation of China [91940306, 31871294, 31970647, 81902519]; National Key R&D Program of China [2017YFC0907503, 2016YFC0901002, 2016YFC0901702]; 13th Five-year Informatization Plan of Chinese Academy of Sciences [XXH13505-05]; Special Investigation on Science and Technology Basic Resources, Ministry of Science and Technology, China [2019FY100102]; National Genomics Data Center, China. Funding for open access charge: Strategic Priority Research Program of Chinese Academy of Sciences [XDB38040300, XDA12030100]; National Natural Science Foundation of China [91940306, 31871294, 31970647, 81902519]; National Key R&D Program of China [2017YFC0907503, 2016YFC0901002, 2016YFC0901702]; 13th Fiveyear Informatization Plan of Chinese Academy of Sciences [XXH13505-05]; Special Investigation on Science and Technology Basic Resources, Ministry of Science and Technology, China [2019FY100102].
Conflict of interest statement. None declared.
REFERENCES
- 1. Smit A.F. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr. Opin. Genet. Dev. 1999; 9:657–663. [DOI] [PubMed] [Google Scholar]
- 2. Deininger P.L., Moran J.V., Batzer M.A., Kazazian H.H. Jr. Mobile elements and mammalian genome evolution. Curr. Opin. Genet. Dev. 2003; 13:651–658. [DOI] [PubMed] [Google Scholar]
- 3. Cordaux R., Batzer M.A.. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009; 10:691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lander E.S., Linton L.M., Birren B., Nusbaum C., Zody M.C., Baldwin J., Devon K., Dewar K., Doyle M., FitzHugh W.et al.. Initial sequencing and analysis of the human genome. Nature. 2001; 409:860–921. [DOI] [PubMed] [Google Scholar]
- 5. Goodier J.L. Restricting retrotransposons: a review. Mobile DNA. 2016; 7:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mills R.E., Bennett E.A., Iskow R.C., Devine S.E.. Which transposable elements are active in the human genome. Trends Genet. 2007; 23:183–191. [DOI] [PubMed] [Google Scholar]
- 7. Huang C. R.L., Burns K.H., Boeke J.D.. Active transposition in genomes. Ann. rev. Genet. 2012; 46:651–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wildschutte J.H., Williams Z.H., Montesion M., Subramanian R.P., Kidd J.M., Coffin J.M.. Discovery of unfixed endogenous retrovirus insertions in diverse human populations. Proc. Nat. Acad. Sci. U.S.A. 2016; 113:E2326–E2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Payer L.M., Burns K.H.. Transposable elements in human genetic disease. Nat. Rev. Genet. 2019; 20:760–772. [DOI] [PubMed] [Google Scholar]
- 10. Hancks D.C., Kazazian H.H.. Roles for retrotransposon insertions in human disease. Mobile DNA. 2016; 7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sultana T., Zamborlini A., Cristofari G., Lesage P.. Integration site selection by retroviruses and transposable elements in eukaryotes. Nat. Rev. Genet. 2017; 18:292–308. [DOI] [PubMed] [Google Scholar]
- 12. Goerner-Potvin P., Bourque G.. Computational tools to unmask transposable elements. Nat. Rev. Genet. 2018; 19:688–704. [DOI] [PubMed] [Google Scholar]
- 13. Stewart C., Kural D., Strömberg M.P., Walker J.A., Konkel M.K., Stütz A.M., Urban A.E., Grubert F., Lam H.Y., Lee W.-P.et al.. A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genet. 2011; 7:e1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sudmant P.H., Rausch T., Gardner E.J., Handsaker R.E., Abyzov A., Huddleston J., Zhang Y., Ye K., Jun G., Fritz M.H.-Y.et al.. An integrated map of structural variation in 2,504 human genomes. Nature. 2015; 526:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wildschutte J.H., Baron A., Diroff N.M., Kidd J.M.. Discovery and characterization of Alu repeat sequences via precise local read assembly. Nucleic Acids Res. 2015; 43:10292–10307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gardner E.J., Lam V.K., Harris D.N., Chuang N.T., Scott E.C., Pittard W.S., Mills R.E., Devine S.E.1000 Genomes Project Consortium . The Mobile Element Locator Tool (MELT): population-scale mobile element discovery and biology. Genome Res. 2017; 27:1916–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Watkins W.S., Feusier J.E., Thomas J., Goubert C., Mallick S., Jorde L.B.. The Simons Genome Diversity Project: a global analysis of mobile element diversity. Genome Biol. Evol. 2020; 12:779–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Collins R.L., Brand H., Karczewski K.J., Zhao X., Alföldi J., Francioli L.C., Khera A.V., Lowther C., Gauthier L.D., Wang H., et al.. A structural variation reference for medical and population genetics. Nature. 2020; 581:444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abel H.J., Larson D.E., Regier A.A., Chiang C., Das I., Kanchi K.L., Layer R.M., Neale B.M., Salerno W.J., Reeves C.et al.. Mapping and characterization of structural variation in 17,795 human genomes. Nature. 2020; 583:83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Almarri M.A., Bergström A., Prado-Martinez J., Yang F., Fu B., Dunham A.S., Chen Y., Hurles M.E., Tyler-Smith C., Xue Y.. Population structure, stratification, and introgression of human structural variation. Cell. 2020; 182:189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Byrska-Bishop M., Evani U.S., Zhao X., Basile A.O., Abel H.J., Regier A.A., Corvelo A., Clarke W.E., Musunuri R., Nagulapalli K.et al.. High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. 2021; bioRxiv doi:10 November 2021, preprint: not peer reviewed 10.1101/2021.02.06.430068. [DOI] [PMC free article] [PubMed]
- 22. Rishishwar L., Villa C.E.T., Jordan I.K.. Transposable element polymorphisms recapitulate human evolution. Mobile DNA. 2015; 6:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu S., Yin X., Li S., Jin W., Lou H., Yang L., Gong X., Wang H., Shen Y., Pan X.et al.. Genomic dissection of population substructure of Han Chinese and its implication in association studies. Am. J. Hum. Genet. 2009; 85:762–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang P., Luo H., Li Y., Wang Y., Wang J., Zheng Y., Niu Y., Shi Y., Zhou H., Song T.et al.. NyuWa Genome resource: a deep whole-genome sequencing-based variation profile and reference panel for the Chinese population. Cell Rep. 2021; 37:110017. [DOI] [PubMed] [Google Scholar]
- 25. 1000 Genomes Project Consortium A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lowy-Gallego E., Fairley S., Zheng-Bradley H., Clarke L., Flicek P.. Variant calling on the GRCh38 assembly with the data from phase three of the 1000 Genomes. 2018; https://f1000research.com/posters/7-1445. [DOI] [PMC free article] [PubMed]
- 27. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Poplin R., Ruano-Rubio V., DePristo M.A., Fennell T.J., Carneiro M.O., Van der Auwera G.A., Kling D.E., Gauthier L.D., Levy-Moonshine A., Roazen D.et al.. Scaling accurate genetic variant discovery to tens of thousands of samples. 2018; bioRxiv doi:24 July 2018, preprint: not peer reviewedhttps://doi.org/10.1101/201178.
- 29. Danecek P., McCarthy S.A.. BCFtools/csq: haplotype-aware variant consequences. Bioinformatics. 2017; 33:2037–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kapitonov V.V., Jurka J.. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat. Rev. Genet. 2008; 9:411–412. [DOI] [PubMed] [Google Scholar]
- 31. Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B.C., Remm M., Rozen S.G.. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012; 40:e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kent W.J. BLAT—the BLAST-like alignment tool. Genome Res. 2002; 12:656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Graffelman J. Exploring diallelic genetic markers: the hardy weinberg package. J. Stat. Softw. 2015; 64:1–23. [Google Scholar]
- 34. Lappalainen I., Lopez J., Skipper L., Hefferon T., Spalding J.D., Garner J., Chen C., Maguire M., Corbett M., Zhou G.et al.. DbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res. 2012; 41:D936–D941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R., Thormann A., Flicek P., Cunningham F.. The ensembl variant effect predictor. Genome Biol. 2016; 17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Girón C.G.et al.. Ensembl 2018. Nucleic Acids Res. 2018; 46:D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fishilevich S., Nudel R., Rappaport N., Hadar R., Plaschkes I., Iny Stein T., Rosen N., Kohn A., Twik M., Safran M.et al.. GeneHancer: genome-wide integration of enhancers and target genes in GeneCards. Database. 2017; 2017:bax028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Payer L.M., Steranka J.P., Yang W.R., Kryatova M., Medabalimi S., Ardeljan D., Liu C., Boeke J.D., Avramopoulos D., Burns K.H.. Structural variants caused by Alu insertions are associated with risks for many human diseases. Proc. Nat. Acad. Sci. U.S.A. 2017; 114:E3984–E3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Buniello A., MacArthur J.A.L., Cerezo M., Harris L.W., Hayhurst J., Malangone C., McMahon A., Morales J., Mountjoy E., Sollis E.et al.. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019; 47:D1005–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chang C.C., Chow C.C., Tellier L.C., Vattikuti S., Purcell S.M., Lee J.J.. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015; 4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Heger A., Webber C., Goodson M., Ponting C.P., Lunter G.. GAT: a simulation framework for testing the association of genomic intervals. Bioinformatics. 2013; 29:2046–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics. 2014; 30:2843–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gardner E.J., Prigmore E., Gallone G., Danecek P., Samocha K.E., Handsaker J., Gerety S.S., Ironfield H., Short P.J., Sifrim A.et al.. Contribution of retrotransposition to developmental disorders. Nat. Commun. 2019; 10:4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Watterson G. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 1975; 7:256–276. [DOI] [PubMed] [Google Scholar]
- 46. Hormozdiari F., Konkel M.K., Prado-Martinez J., Chiatante G., Herraez I.H., Walker J.A., Nelson B., Alkan C., Sudmant P.H., Huddleston J.et al.. Rates and patterns of great ape retrotransposition. Proc. Nat. Acad. Sci. U.S.A. 2013; 110:13457–13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., Del Angel G., Rivas M.A., Hanna M.et al.. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011; 43:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T.et al.. The variant call format and VCFtools. Bioinformatics. 2011; 27:2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li W., Lin L., Malhotra R., Yang L., Acharya R., Poss M.. A computational framework to assess genome-wide distribution of polymorphic human endogenous retrovirus-K In human populations. PLoS Comput. Biol. 2019; 15:e1006564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kojima K.K. Different integration site structures between L1 protein-mediated retrotransposition in cis and retrotransposition in trans. Mobile DNA. 2010; 1:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kahyo T., Yamada H., Tao H., Kurabe N., Sugimura H.. Insertionally polymorphic sites of human endogenous retrovirus-K (HML-2) with long target site duplications. BMC Genomics. 2017; 18:487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bennett E.A., Keller H., Mills R.E., Schmidt S., Moran J.V., Weichenrieder O., Devine S.E.. Active Alu retrotransposons in the human genome. Genome Res. 2008; 18:1875–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Medstrand P., Van De Lagemaat L.N., Mager D.L.. Retroelement distributions in the human genome: variations associated with age and proximity to genes. Genome Res. 2002; 12:1483–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mikkelsen T., Hillier L., Eichler E., Zody M., Jaffe D., Yang S.-P., Enard W., Hellmann I., Lindblad-Toh K., Altheide T.et al.. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005; 437:69–87. [DOI] [PubMed] [Google Scholar]
- 55. Mir A.A., Philippe C., Cristofari G.. euL1db: the European database of L1HS retrotransposon insertions in humans. Nucleic Acids Res. 2015; 43:D43–D47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Miga K.H., Koren S., Rhie A., Vollger M.R., Gershman A., Bzikadze A., Brooks S., Howe E., Porubsky D., Logsdon G.A.et al.. Telomere-to-telomere assembly of a complete human X chromosome. Nature. 2020; 585:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Manuelidis L., Wu J.C.. Homology between human and simian repeated DNA. Nature. 1978; 276:92–94. [DOI] [PubMed] [Google Scholar]
- 58. Klein S.J., O’Neill R.J.. Transposable elements: genome innovation, chromosome diversity, and centromere conflict. Chromosome Res. 2018; 26:5–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Contreras-Galindo R., Kaplan M.H., He S., Contreras-Galindo A.C., Gonzalez-Hernandez M.J., Kappes F., Dube D., Chan S.M., Robinson D., Meng F.et al.. HIV infection reveals widespread expansion of novel centromeric human endogenous retroviruses. Genome Res. 2013; 23:1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zahn J., Kaplan M.H., Fischer S., Dai M., Meng F., Saha A.K., Cervantes P., Chan S.M., Dube D., Omenn G.S.et al.. Expansion of a novel endogenous retrovirus throughout the pericentromeres of modern humans. Genome Biol. 2015; 16:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kumar S., Subramanian S.. Mutation rates in mammalian genomes. Proc. Nat. Acad. Sci. U.S.A. 2002; 99:803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Feusier J., Watkins W.S., Thomas J., Farrell A., Witherspoon D.J., Baird L., Ha H., Xing J., Jorde L.B.. Pedigree-based estimation of human mobile element retrotransposition rates. Genome Res. 2019; 29:1567–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Prado-Martinez J., Sudmant P.H., Kidd J.M., Li H., Kelley J.L., Lorente-Galdos B., Veeramah K.R., Woerner A.E., O’Connor T.D., Santpere G.et al.. Great ape genetic diversity and population history. Nature. 2013; 499:471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hedges D.J., Callinan P.A., Cordaux R., Xing J., Barnes E., Batzer M.A.. Differential Alu mobilization and polymorphism among the human and chimpanzee lineages. Genome Res. 2004; 14:1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Karczewski K.J., Francioli L.C., Tiao G., Cummings B.B., Alföldi J., Wang Q., Collins R.L., Laricchia K.M., Ganna A., Birnbaum D.P.et al.. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020; 581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B., et al.. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016; 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. van de Lagemaat L.N., Medstrand P., Mager D.L.. Multiple effects govern endogenous retrovirus survival patterns in human gene introns. Genome Biol. 2006; 7:R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang Y., Romanish M.T., Mager D.L.. Distributions of transposable elements reveal hazardous zones in mammalian introns. PLoS Comput. Biol. 2011; 7:e1002046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dewannieux M., Esnault C., Heidmann T.. LINE-mediated retrotransposition of marked Alu sequences. Nat. Genet. 2003; 35:41–48. [DOI] [PubMed] [Google Scholar]
- 70. Raiz J., Damert A., Chira S., Held U., Klawitter S., Hamdorf M., Löwer J., Strätling W.H., Löwer R., Schumann G.G.. The non-autonomous retrotransposon SVA is trans-mobilized by the human LINE-1 protein machinery. Nucleic Acids Res. 2012; 40:1666–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Baxter L.L., Watkins-Chow D.E., Pavan W.J., Loftus S.K.. A curated gene list for expanding the horizons of pigmentation biology. Pigm. Cell Melanoma. R. 2019; 32:348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rehm H.L., Berg J.S., Brooks L.D., Bustamante C.D., Evans J.P., Landrum M.J., Ledbetter D.H., Maglott D.R., Martin C.L., Nussbaum R.L.et al.. ClinGen—the clinical genome resource. N. Engl. J. Med. 2015; 372:2235–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Goodier J.L., Ostertag E.M., Kazazian H.H. Jr. Transduction of 3’-flanking sequences is common in L1 retrotransposition. Hum. Mol. Genet. 2000; 9:653–657. [DOI] [PubMed] [Google Scholar]
- 74. Ostertag E.M., Kazazian H.H.. Twin priming: a proposed mechanism for the creation of inversions in L1 retrotransposition. Genome Res. 2001; 11:2059–2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang J., Song L., Grover D., Azrak S., Batzer M.A., Liang P.. dbRIP: a highly integrated database of retrotransposon insertion polymorphisms in humans. Human Mutat. 2006; 27:323–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Feng Q., Moran J.V., Kazazian H.H. Jr, Boeke J.D.. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 1996; 87:905–916. [DOI] [PubMed] [Google Scholar]
- 77. Schneider K.L., Xie Z., Wolfgruber T.K., Presting G.G.. Inbreeding drives maize centromere evolution. Proc. Nat. Acad. Sci. U.S.A. 2016; 113:E987–E996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lelek M., Casartelli N., Pellin D., Rizzi E., Souque P., Severgnini M., Di Serio C., Fricke T., Diaz-Griffero F., Zimmer C.et al.. Chromatin organization at the nuclear pore favours HIV replication. Nat. Commun. 2015; 6:6483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Marini B., Kertesz-Farkas A., Ali H., Lucic B., Lisek K., Manganaro L., Pongor S., Luzzati R., Recchia A., Mavilio F.et al.. Nuclear architecture dictates HIV-1 integration site selection. Nature. 2015; 521:227–231. [DOI] [PubMed] [Google Scholar]
- 80. Chueh A.C., Northrop E.L., Brettingham-Moore K.H., Choo K.A., Wong L.H.. LINE retrotransposon RNA is an essential structural and functional epigenetic component of a core neocentromeric chromatin. PLoS Genet. 2009; 5:e1000354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Longo M.S., Carone D.M., Green E.D., O’Neill M.J., O’Neill R.J.. Distinct retroelement classes define evolutionary breakpoints demarcating sites of evolutionary novelty. BMC Genomics. 2009; 10:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nergadze S.G., Piras F.M., Gamba R., Corbo M., Cerutti F., McCarter J.G., Cappelletti E., Gozzo F., Harman R.M., Antczak D.F.et al.. Birth, evolution, and transmission of satellite-free mammalian centromeric domains. Genome Res. 2018; 28:789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Chang C.-H., Chavan A., Palladino J., Wei X., Martins N.M., Santinello B., Chen C.-C., Erceg J., Beliveau B.J., Wu C.-T.et al.. Islands of retroelements are major components of Drosophila centromeres. PLoS Biol. 2019; 17:e3000241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Pardue M.-L., DeBaryshe P.. Retrotransposons that maintain chromosome ends. Proc. Nat. Acad. Sci. U.S.A. 2011; 108:20317–20324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wang L., Norris E.T., Jordan I.. Human retrotransposon insertion polymorphisms are associated with health and disease via gene regulatory phenotypes. Front. Microbiol. 2017; 8:1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Visscher P.M., Wray N.R., Zhang Q., Sklar P., McCarthy M.I., Brown M.A., Yang J.. 10 years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 2017; 101:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Vendrell-Mir P., Barteri F., Merenciano M., González J., Casacuberta J.M., Castanera R.. A benchmark of transposon insertion detection tools using real data. Mobile DNA. 2019; 10:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Werling D.M., Brand H., An J.-Y., Stone M.R., Zhu L., Glessner J.T., Collins R.L., Dong S., Layer R.M., Markenscoff-Papadimitriou E.et al.. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat. Genet. 2018; 50:727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Torene R.I., Galens K., Liu S., Arvai K., Borroto C., Scuffins J., Zhang Z., Friedman B., Sroka H., Heeley J.et al.. Mobile element insertion detection in 89,874 clinical exomes. Genet. Med. 2020; 22:974–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Goubert C., Thomas J., Payer L.M., Kidd J.M., Feusier J., Watkins W.S., Burns K.H., Jorde L.B., Feschotte C.. TypeTE: a tool to genotype mobile element insertions from whole genome resequencing data. Nucleic Acids Res. 2020; 48:e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ewing A.D. Transposable element detection from whole genome sequence data. Mobile DNA. 2015; 6:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Rishishwar L., Mariño-Ramírez L., Jordan I.K.. Benchmarking computational tools for polymorphic transposable element detection. Brief. Bioinformatics. 2017; 18:908–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Audano P.A., Sulovari A., Graves-Lindsay T.A., Cantsilieris S., Sorensen M., Welch A.E., Dougherty M.L., Nelson B.J., Shah A., Dutcher S.K.et al.. Characterizing the major structural variant alleles of the human genome. Cell. 2019; 176:663–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chaisson M.J., Sanders A.D., Zhao X., Malhotra A., Porubsky D., Rausch T., Gardner E.J., Rodriguez O.L., Guo L., Collins R.L.et al.. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun. 2019; 10:1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Zhou W., Emery S.B., Flasch D.A., Wang Y., Kwan K.Y., Kidd J.M., Moran J.V., Mills R.E.. Identification and characterization of occult human-specific LINE-1 insertions using long-read sequencing technology. Nucleic Acids Res. 2020; 48:1146–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Ebert P., Audano P.A., Zhu Q., Rodriguez-Martin B., Porubsky D., Bonder M.J., Sulovari A., Ebler J., Zhou W., Mari R.S.et al.. Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science. 2021; 372:eabf7117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Complete MEI call set and other related information such as allele frequency and functional annotation are available in the companion database HMEID (available at http://bigdata.ibp.ac.cn/HMEID/). The source code of the HMEID could be accessed at https://github.com/oldteng/HMEID.