


Abstract
The Sleeping Beauty (SB) transposon system is a popular tool for genome engineering, but random integration into the genome carries a certain genotoxic risk in therapeutic applications. Here we investigate the role of amino acids H187, P247 and K248 in target site selection of the SB transposase. Structural modeling implicates these three amino acids located in positions analogous to amino acids with established functions in target site selection in retroviral integrases and transposases. Saturation mutagenesis of these residues in the SB transposase yielded variants with altered target site selection properties. Transposon integration profiling of several mutants reveals increased specificity of integrations into palindromic AT repeat target sequences in genomic regions characterized by high DNA bendability. The H187V and K248R mutants redirect integrations away from exons, transcriptional regulatory elements and nucleosomal DNA in the human genome, suggesting enhanced safety and thus utility of these SB variants in gene therapy applications.
INTRODUCTION
Most mobile genetic elements, including retroviruses, long terminal repeat (LTR) retrotransposons and DNA-based transposons, share a fundamental step of their life-cycle: genomic integration. Integration of these elements typically involves DNA strand transfer reactions, where 3’-OH groups exposed at the ends of the elements mediate nucleophilic attacks on the two strands of the target DNA (tDNA), thereby directly joining the element to the target without prior tDNA cleavage. This process is executed by dedicated enzymes encoded by the mobile elements, which are called transposases for DNA transposons and integrases (INs) for retroviruses and LTR retrotransposons. There is a wide spectrum of specificity with respect to the genomic sites where integration of mobile genetic elements occurs. Mechanistic studies of retroviral integration have revealed two key factors that determine integration site selection: the retroviral IN protein and its cellular, chromatin-associated binding partners [reviewed in (1–3)]. Due to the interplay of these factors, retroviral/lentiviral integration displays little specificity on the primary DNA sequence level, but shows biased patterns of distribution on the genome level [reviewed in (1–4)].
Transposases (5–7), INs (8) and the RAG1 immunoglobulin gene recombinase (9, 10) all share an evolutionarily related catalytic domain, harboring a triad of negatively charged amino acids, the aspartate-aspartate-glutamate/aspartate (DDE/D) motif. Specific amino acids responsible for target site selection of the bacterial Tn10 transposon and retroviruses have been mapped to the catalytic domain of the transposase or IN, respectively (11–14), suggesting that the DDE/D domain plays a critical role in target site selection.
The crystal structure of the prototype foamy virus (PFV) intasome—an integration-competent retroviral nucleoprotein complex—in its tDNA-bound state (target capture complex, TCC) revealed that the tDNA assumes a severely bent conformation mediated by the IN catalytic domain to accommodate the scissile phosphodiester bonds in the enzyme active sites (Supplementary Figure S1). The major groove of the tDNA is widened such that the pyrimidine (Y)-purine (R) dinucleotide at the center of the integration site is unstacked (15). Notably, YR dinucleotides display lower base-stacking properties than any other dinucleotide steps (16). It was also shown in subsequent structural studies that nucleosomal DNA is lifted from the histone octamer and deformed to a strikingly similar extent by interactions with the PFV intasome, providing a structural basis for retroviral integration into chromatin (17). Furthermore, the crystal structures of the PFV TCC revealed amino acid residues, including A188, in PFV IN that distort tDNA by direct interaction with DNA bases. Consistently, mutant PFV IN proteins carrying amino acid replacements in position 188 displayed defects in strand transfer and altered nucleotide preferences for PFV integration in vitro (15). Subsequent studies of the HIV-1 IN identified a role for its S119 residue analogous to that of A188 in PFV IN (18), and the naturally occurring polymorphism S119G has been shown to retarget viral integration away from gene-dense regions (19). In addition to S119, mutations of N120 of HIV IN have been found to either block integration (20) or to result in changes in integration site preference, suggesting that N120 is also involved in interactions with target DNA (20,21). Finally, amino acid substitutions of S124 in the analogous position of the Rous sarcoma virus (RSV) IN resulted in altered target site preferences (14). Similar to the PFV TCC, the tDNA in the available structures of the RSV intasome in its post-integration conformation (strand transfer complex, STC) revealed a strong bending of ∼90°, which is stabilized by minor groove contacts made by a short helix of IN containing S124 (22) (Supplementary Figure S1). Retroviral INs additionally exhibit a preference for weakly conserved palindromic sequences that center around the integration target site (23–26). Such palindromic DNA sequences represent high DNA bendability with bending likely mediated by interactions between amino acid residues of the INs and the centrally located, flexible YR sequence (15). Taken together, these observations suggest that S119 and N120 in HIV-1 IN, A188 in PFV IN and S124 in RSV IN, all located in analogous positions of the α2 helix of the retroviral integrases (Figure 1A), play a conserved role in interacting with tDNA during viral DNA integration.
Figure 1.

Sequence alignment and structural superposition identify P247 and K248 in the Sleeping Beauty transposase as potential equivalents of amino acids in retroviral integrases responsible for interactions with target DNA. (A) Multiple amino acid sequence alignment of segments of retroviral INs and the SB transposase. The alignment was generated by CLUSTALW and then used to highlight secondary structures of HIV IN (based on PDB ID: 1BIS) with ESPript 3.0 (100). Accordingly, the numbering of alpha helices and beta strands corresponds to HIV IN. The second D and the E residues of the catalytic DDE triads are marked with asterisks. P247 and K248 of SB (arrows) are located three and four amino acids downstream of the second D, respectively, in a region corresponding to the α2 helix of the retroviral INs. (B) Superposition of the SB TCC model with structures of retroviral intasomes. S124 of RSV STC [light blue, PDB ID: 5EJK (22)], S119 of HIV-1 STC [green, PDB ID: 5U1C (54)] and A188 of PFV TCC [dark blue, PDB ID: 3OS1 (15)] all overlay with P247 of the SB transposase (orange). SB’s K248 and HIV IN’s equivalent N120, both situated within the conserved α2 helix, are also displayed. The target DNA is depicted in dark gray.
Cut-and-paste DNA transposons also display a wide spectrum of selectivity with respect to chromosomal integration. Integration of the bacterial Tn10 transposon and of the Tc1 and Tc3 transposons in Caenorhabditis elegans can occur genome-wide, and their target site selection is primarily determined by the transposase itself (11,27). The Mos1 mariner transposase (which has a DDD catalytic triad instead of DDE) preferentially executes transposon integration into TA target dinucleotides. Structural studies of the STC of the Mos1transposase indicated that the tDNA is severely distorted from the B-form conformation with the DNA backbone bent by 147° and the apex of the bend positioned at the TA target dinucleotide (28) (Supplementary Figure S1). Interactions with Mos1 transposase residues, including R186, were shown to stabilize distortions in the tDNA (28). Importantly, R186 was also proposed to be essential for target recognition and its mutations abolish transposon integration (28,29). Notably, R186 does not map to the α2 helix, but to the so-called clamp loop region of the Mos1 transposase (29) situated between the first two aspartates in the DDD domain (Figure 2A). As opposed to retroviruses, recent evidence indicates that Tc1/mariner transposons preferentially integrate at linker regions between nucleosomes (30).
Figure 2.
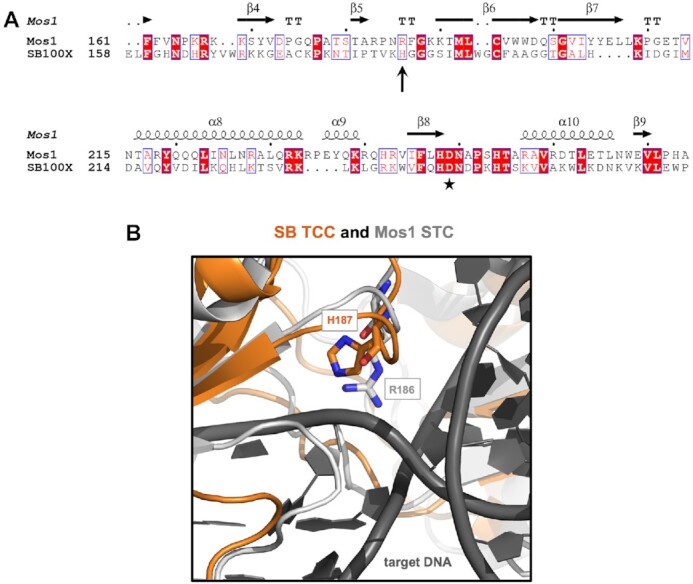
Sequence alignment and structural superposition identifies H187 in the Sleeping Beauty transposase as a potential homologue of R186 in the Mos1 transposase responsible for interactions with target DNA. (A) Amino acid sequence alignment of segments of the SB and Mos1 transposases. The alignment was generated by CLUSTALW and then used to highlight secondary structures of the Mos1 transposase (based on PDB ID: 4U7B) with ESPript 3.0 (100). Accordingly, the numbering of alpha helices and beta strands corresponds to Mos1. The second D residue of the catalytic DDE/D triad is marked by an asterisk. H187 in SB (arrow) aligns with R186 in Mos1. (B) An overlay of the SB TCC model (orange) with the Mos1 STC [gray, PDB ID: 5HOO (28)] shows that H187 in SB aligns with R186 in Mos1. The target DNA is depicted in dark gray.
Sleeping Beauty (SB, a Tc1/mariner superfamily transposon) is a synthetic transposon that was reconstructed based on sequences of transpositionally inactive elements isolated from fish genomes (31). It is the most thoroughly studied vertebrate transposon to date, which supports a wide spectrum of genetic engineering applications, including the generation of transgenic cell lines, induced pluripotent stem cell (iPSC) reprogramming, phenotype-driven insertional mutagenesis screens in the area of cancer biology, germline gene transfer in experimental animals and somatic gene therapy both ex vivo and in vivo [reviewed in (32–41)]. On the genomic scale, SB transposons exhibit a close-to-random integration profile with a slight bias towards integration into genes and their upstream regulatory sequences in cultured mammalian cell lines (42–47). On a local scale, SB preferentially inserts at TA dinucleotides (like Mos1), and shows additional target site preferences based on physical properties of the DNA, including bendability, A-philicity and a symmetrical pattern of hydrogen bonding sites in the major groove of the tDNA (48,49). To date, the structural basis of SB target site selection is unknow, hindering the design of new transposase variants with altered target specificity.
In sum, an emerging theme in integration catalyzed by DDE/D recombinases is a severely bent tDNA structure, unstacking of bases at YR dinucleotides at the center of palindromic target sequences, and stabilization of the structure by direct interaction of bases in the tDNA with amino acid residues in the recombinase proteins (50). In this work, we set out to investigate the molecular basis of target site selection by the SB transposase, using structural modeling and saturation mutagenesis of specific amino acid residues. The results reveal marked changes in target specificity with selected transposase mutants, providing insights into SB’s transposition mechanism and opening new possibilities for genetic engineering. Namely, specific variants feature reduced integration in genic sequences of the human genome, suggesting enhanced safety in gene therapy applications.
MATERIALS AND METHODS
Structural modeling
The pre-integration TCC model was assembled based on the SB paired-end complex (PEC) homology model described in (51). The transposase catalytic domain was replaced with the available crystal structure (PDB ID: 5CR4) (52); amino acids 148-199 (corresponding to the clamp loop and two beta strands flanking this region) that are involved in crystal packing interactions in the catalytic domain structure were maintained from the PEC homology model to retain a DNA-bound conformation of this region. Target DNA was modelled by docking the integration target substrate from the PFV intasome TCC structure (PDB ID: 3OS1) (15). Finally, the assembled model was refined in HADDOCK (53). The SB TCC model was superposed onto the crystal structures of the post-integration Mos1 STC (PDB ID: 5HOO) (28), the RSV intasome STC (PDB ID: 5EJK) (22), the HIV intasome STC (PDB ID: 5U1C) (54) and the PFV intasome TTC (PDB ID: 3OS1) (15). Structural models were superposed and visualized in PyMOL (PyMOL Molecular Graphics System, Version 1.5.0.4, Schrödinger, LLC).
Site-directed mutagenesis of the SB100X transposase
All mutations were generated using the Q5 polymerase (NEB, Ipswich, MA, USA) and the plasmid pCMV(CAT)T7-SB100X (35). 5’-phosphorylated primers for the particular positions were created with ‘NEBaseChanger™’ (http://nebasechanger.neb.com/) and were designed with 5’-ends annealing back-to-back. Primers were synthesized with a 5’-phosphate to enable a downstream intramolecular ligation reaction and were ordered from Eurofins (Eurofins/MWG, Luxembourg). The primer sequences are listed in Supplementary Table S1. PCR cycling conditions were set according to the manufacturer's instructions. The annealing temperatures of the mutagenic primers were calculated with the “NEB Tm calculator™” software (https://www.neb.com/tools-and-resources/interactive-tools/tm-calculator). The PCR products were purified with QIAquick PCR Purification Kit (QIAGEN, Venlo, Holland), eluted in 30 μl elution buffer and digested with 2 μl DpnI (NEB, Ipswich, MA, USA) for 2 h at 37°C, followed by 20 min heat-inactivation at 80°C. The linear, double-stranded PCR products were circularized by ligation with T4 DNA Ligase (NEB, Ipswich, MA, USA) overnight at room temperature. The circularized PCR products were transformed into chemically competent E. coli (Invitrogen/Life Technologies, Carlsbad, CA, USA), grown in Luria-Bertani (LB) medium for 1 h, and selected for chloramphenicol resistance by plating on LB agar plates containing 25 μg/ml chloramphenicol. To confirm the presence of the desired mutations, and the absence of undesired mutations, plasmid DNA from several colonies was purified using the QIAprep spin miniprep kit (QIAGEN, Venlo, Holland) and Sanger sequenced by GATC Biotech (Konstanz, Germany).
Western blotting
One day prior to transfection, 2 × 105 HeLa cells were seeded onto six well plates. 1.5 μl of TransIT-LT1transfection reagent (Mirus Bio LLC, Madison, WI USA) was used to transfect 500 ng of DNA (each transfection reaction was filled up to 500 ng with the plasmid pUC19). 50 ng of transposase plasmid [mutant SB100X transposase or wild-type SB100X (pCMV(CAT)T7-SB100X)] or green fluorescent protein (GFP) expression plasmid (pmaxGFP™; Lonza, Basel, Switzerland) were transfected. 48 h post-transfection, HeLa cells were lysed in RIPA buffer (150 mM NaCl, 1.0% Triton X-100, 1.0% Na-deoxycholate, 0.1% SDS, 25 mM Tris, pH 8.0) supplemented with protease inhibitor cocktail (Complete Mini, Roche, Basel, Switzerland). Protein was extracted using the Bioruptor®Plus (Diagenode, Denville, NJ, USA), 10 cycles, high power, 30 s ON/30 s OFF at 4°C. Total protein was quantified using BCA Protein Assay Kit (Pierce, Rockford, IL). Proteins (10 μg per lane) were loaded onto 10% polyacrylamide gels and subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Gels were transferred to nitrocellulose membrane (Hybond ECL, Amersham Bioscience, Little Chalfont, UK) and immunoblotting was performed according to standard procedures. Proteins were detected with goat monoclonal anti-SB transposase antibody (R&D Systems, Minneapolis, USA) at dilution 1:5000, or mouse monoclonal anti-Vinculin antibody (Abcam, Cambridge, UK) at dilution 1:3000, and chemiluminescence using ECL Prime Western Blotting Detection Kit (Amersham Bioscience). Signals were captured on a film (Hyperfilm ECL High performance chemiluminescence film, Amersham Bioscience, Little Chalfont, UK).
FACS-based excision assay
1.5 × 105 HeLa cells were seeded one day prior to transfection. HeLa cells were transfected with 2 μg of the transposon donor plasmid pCMV(CAT)-GFP//T2Neo (52) and 200 ng of a mutant SB100X transposase, SB100X or a catalytically inactive transposase mutant (E279D, also referred to D3). In the pCMV(CAT)-GFP//T2Neo reporter plasmid a genetically tagged SB transposon disrupts the GFP coding sequence so that cells transfected with this construct do not express GFP. In the presence of SB transposase excision occurs, and in a fraction of the products the GFP coding sequence is restored, thereby leading to green fluorescence that can be quantified, as previously described (55). Six microliters of TransIT-LT1 transfection reagent (Mirus Bio LLC, Madison, WI USA) were used per transfection reaction. Three days post-transfection cells were trypsinized, washed with PBS, fixed in 1% Paraformaldehyde (PFA) in PBS and FACS analyzed with BD LSR II flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). Data were analyzed with FCS Express 4 Flow Cytometry (De Novo Software, Glendale, CA). The % GFP positive cells in cultures transfected with SB100X transposase mutants/pCMV(CAT)-GFP//T2Neo were normalized to that in cells transfected with wild-type SB100X/pCMV(CAT)-GFP//T2Neo.
Transposition assay
For transposition assay in HeLa cells, 2 × 105 cells were seeded onto six-well plates one day prior to transfection. Three μl of TransIT-LT1 transfection reagent (Mirus Bio LLC, Madison, WI, USA) were used to transfect 1 μg of DNA (each transfection reaction was filled up to 1 μg with the plasmid pUC19). To obtain predominantly single-copy transposon insertions (43), 10 ng of transposon donor plasmid (pT2B/puro) were co-transfected with 5 ng of transposase plasmid (mutant transposase, inactive D3 transposase or SB100X). Forty-eight hours after transfection cells were trypsinized and 2.5-5% of the cells were replated to 10-cm plates, and selected for transposon integration using 3 μg/ml puromycin (InvivoGen, San Diago, CA, USA). After 3 weeks of selection, cell colonies were fixed with 10% (vol/vol) formaldehyde in phosphate-buffered saline (PBS), stained with methylene blue in PBS, and counted. For in vitro comparisons of relative transposition efficiencies at least three independent experiments were performed.
Generation of SB insertion libraries
For the analysis of the target site selection properties of the SB100X transposase mutants, 3.5 × 105 HepG2 cells were seeded onto 6-well plates 1 day prior to transfection. Transfections were done with QIAGEN-purified plasmid DNA using TransIT-LT1 transfection reagent according to the manufacturer's protocol. Two μl of TransIT-LT1 transfection reagent were used to transfect 650 ng of DNA, including 500 ng pT2Bpuro, 50 ng helper plasmid expressing the mutant transposases or pCMV(CAT)T7-SB100X and 100 ng of green fluorescent protein (GFP) expression plasmid pmaxGFP (transfection control). 48 h after transfection, cells were trypsinized, diluted to multiple 10 cm dishes containing DMEM supplemented with 3 μg/ml puromycin and selected for growth over a period for 2–3 weeks. At least 10 000 puromycin-resistant HepG2 cell colonies were trypsinized and centrifuged at 1000 rpm for 5 min. The pellet was washed with PBS and genomic DNA was extracted from cells using the Qiagen DNeasy Blood & Tissue Kit according to manufacturer's protocol.
For the generation of SB insertion site libraries, 2 μg DNA was sheared with a Covaris M220 ultra-solicitor device to an average fragment size of 600 bp in Screw-Cap microTUBEs in 50 μl, using the following settings: peak incident power 50 W, duty factor 20%, cycles per burst 200, treatment 28 s. 1.2 μg of the sheared DNA was blunted and 5’-phosphorylated using the NEBNext End Repair Module (NEB), and 3’-A-tailed with NEBNext dA-Tailing Module (NEB) following the recommendations of the manufacturer. The DNA was purified with the Clean and Concentrator Kit (Zymo Research) and eluted in 8 μl 10 mM Tris pH 8.0 (EB) for ligation with 50 pmol of T-linker (see below) with T4 ligase (NEB) in 20 μl volume, at 16 °C, overnight. T-linkers were created by annealing the 100 pmol each of the oligonucleotides Linker_TruSeq_T+ and Linker_TruSeq_T- (Supplementary Table S1) in 10 mM Tris–Cl pH 8, 50 mM NaCl, 0.5 mM EDTA. After heat-inactivation, ligation products enclosing fragments of non-integrated transposon donor plasmid DNA were digested with DpnI (NEB) in 50 μl for 3 h, and the DNA was column-purified and eluted in 20 μl EB. Six μl elute was used for PCR I with 25 pmol of the primers specific for the linker and for the transposon inverted repeat: Linker and T-Bal-Long, respectively, with the conditions: 98 °C 30 s; 10 cycles of 98 °C 10 s, 72 °C 30 s; 15 cycles of 98 °C 10 s, ramp to 62 °C (1 °C/s) 30 s, 72 °C 30 s, 72 °C 5 min. All PCR reactions were performed with NEBNext High-Fidelity 2 × PCR Master Mix (for PCR primer sequences see Supplementary Table S1). The PCR was column purified eluted in 20 μl EB and 10 μl was used for PCR II with primers Nested and LAM-SB-50, with the following program: 98 °C 30 s; 12 cycles of 98 °C 10 s, ramp to 65 °C (1 °C/sec) 30 s, 72 °C 30 s, 72 °C 5 min. One third of the column-purified PCR II was used for PCR III with primers PE-nest-ind-N and SB-20-bc-ill-N (where N is the number of the Illumina TrueSeq indexes) for barcoding the samples using the following PCR program: 98°C 30 s; 12 cycles of 98 °C 10 s, ramp to 64 °C (1 °C/s) 30 s, 72 °C 30 s, 72 °C 5 min. The final PCR products were separated on a 1% agarose gel and the smears of 200–500 bp were isolated and purified.
Sequencing and analyses of the insertion sites
The insertion site libraries were prepared as described earlier (56) and sequenced on Illumina instruments with 150-bp, single-end settings. After adapter and quality trimming (Phred score ≥ 20) with fastp (57) the reads were tested for the transposon sequences downstream of the SB-specific primer and were filtered for the presence of the remaining part of the transposon inverted terminal repeat (ITR) and a minimum length of 28 bases of genomic sequence for mapping with bowtie2 (58) with –sensitive and –end-to-end settings. A mapped locus was considered valid if the mapping quality for the read supporting it was ≥20. Any insertion site needed to be supported by at least 10 independent reads at a TA target site of the human genome (hg38). If multiple insertions were detected within 10 bases the insertion site supported by the largest number of independent reads were consider as the valid one. The coordinates of the insertion sites can be found in Supplementary Datasets 1–7. The sequence logos were plotted with the SeqLogo package of R (https://www.R-project.org/). Gene coordinates of the RefSeq database for the hg38 human genome assembly were downloaded from the UCSC Table Browser (TB) (http://genome.ucsc.edu/cgi-bin/hgTables). MLV and HIV insertion sites have been described previously (59,60). The list of cancer genes was downloaded from http://www.bushmanlab.or/lingks/genelists. Representation of the insertion sites in various genic categories were investigated using the Genomation package (61). A computationally generated random set of 100 000 loci of the human hg38 genome assembly was used as reference for investigations of insertion site representation in various genomic bins. ChIP-Seq peaks of various histone modifications in HepG2 cells were downloaded from the ENCODE project homepage (https://www.encodeproject.org/) using the wgEncodeBroadHistoneHepg2 dataset. Repeat annotations are from Repeat Masker available in the TB. Annotated repeats of the class Simple repeats were defined as TA-rich if their short, repeated sequences contained the AT or TA dinucleotides. Those without AT/TA were designated as not TA-rich. The combined segmentation data of the ChromHMM (62) and Segway (63) softwares for the HepG2 genome were downloaded from the TB. The genomic safe harbor coordinates for the hg38 assembly were created following the earlier defined criteria (64).
For predicting nucleosome occupancy of various DNA sequences, we implemented an algorithm published previously (65). For the nucleosome occupancy prediction on exons, coding exons and TSS the corresponding coordinates were obtained from the RefSeq database available in the TB; the coordinates of Regulatory open and Enhancer are those of the HepG2 combined segmentation data (see above).
Physical properties of the DNA sequences flanking the insertion sites were predicted as follows. The sets of insertion loci sequences of the transposase versions were filtered for matching the corresponding majority-rule consensus motifs revealed by the sequence logos. These were for SB100X: ANNTANNT, for K248R: ATATATAT, for H187V: ATATATAT, for P247R: WNNTANNW. The tables containing various physical parameters of all the possible di-, or tri-nucleotides have been published (66,67). The physical properties were extracted using sliding windows on the insertion site sequences with a step of 1 to obtain the values for all nucleotide steps and the mean values of these for each sequence were calculated.
RESULTS
Structural modeling identifies candidate residues involved in target DNA interactions of the Sleeping Beauty transposase
To shortlist amino acids in the SB transposase potentially involved in shaping its integration site selection, we performed a comparative structural analysis of a structural model of the SB TCC complex and the available crystal structures of retroviral intasomes, including the PFV TCC (PDB ID: 3OS1) (15), the HIV-1 STC (54) and the RSV STC (22) (Figure 1), and the structure of the Mos1 STC (28) (Figure 2). The SB TCC complex model was built based on the catalytic domain crystal structure of the hyperactive SB100X variant using the Mos1 PEC and the PFV TCC as templates (51,52). Despite the low sequence identity between the SB transposase and the retroviral INs or the Mos1 transposase (amino acid identity of 15% with the HIV-1 and PFV INs, 11% with the RSV IN and 20% with the Mos1 transposase over the catalytic domains), their core RNaseH folds superpose very well at the structural level [root mean square deviation (r.m.s.d.) between the SB RNaseH core and the equivalent regions of the HIV-1 IN, RSV IN, PFV IN and Mos1 transposase equals 3.15, 3.20, 2.90 and 1.58 Å, respectively]. Strikingly, A188 of PFV IN, S119 of HIV-1 IN and S124 of RSV IN, all of which are implicated in target DNA recognition, overlay with a single residue, P247 of the SB transposase (Figure 1B). P247 is located three amino acids downstream of the second aspartate of the DDE triad in a transposase segment corresponding to helix α2 of the retroviral INs (Figure 1A), which was shown to be required for retroviral integration (13,14). Of note, a proline residue is also present at this position in numerous retroviral INs (68). Right next to P247 still in the α2 helix, there is K248 that overlays with N120 of HIV IN (Figure 1B), whose mutations have been found to alter the integration site preferences of HIV (20,21). Mutations of K248 of the SB transposase have recently been shown to generate an excision-proficient but integration-deficient phenotype (55,69). This feature phenocopies R186 mutations in Mos1 (28,29), although K248 in SB and R186 in Mos1 are not structurally related (see below). The structural position and documented phenotypes suggest a role for K248 in interaction with tDNA or in strand transfer.
Structural superposition of the SB TCC model with the Mos1 STC structure shows that the SB residue H187 aligns with R186 of Mos1, which is situated between the first and second aspartates of the DDD triad in the clamp loop region (29) (Figure 2). H187 was previously found to localize at the end of the clamp loop delving into the major groove of the tDNA substrate in the SB TCC model (52), which suggested that it might contribute to broadening the groove and/or kinking the tDNA substrate via unstacking bases at the integration site. Notably, H187, P247 and K248 are all predicted to be in close proximity to tDNA in the SB TCC model, supporting the hypothesis that these amino acids play a role in target binding and/or integration.
Saturation mutagenesis of H187, P247 and K248 in the Sleeping Beauty transposase identifies variants with altered transposition efficiencies
In order to assess the relative effects of single amino acid replacements of residues H187, P247 and K248 on transposition, the SB100X transposase was subjected to saturation mutagenesis at these positions by incorporating all possible amino acids by site-directed PCR mutagenesis. All constructs encoding the mutant SB100X transposases showed protein expression levels comparable to that of SB100X by Western blot analysis (Supplementary Figures S2–S4).
Next we evaluated transposition activities of the mutants relative to SB100X by applying a cell-based transposition assay in human cells (31) that was fine-tuned to predominantly obtain a single transposon integration per cell (43). Briefly, a donor plasmid carrying a puromycin (puro) resistance gene-marked transposon was co-transfected with a helper plasmid encoding either SB100X, the inactive E279D transposase (D3) variant or a mutant of the SB100X transposase.
Figure 3A summarizes relative transposition efficiencies of the H187 series of mutated transposases. Overall, most of the mutants retained considerable levels of transposition activity relative to SB100X, indicating a fair degree of flexibility in this particular amino acid position. A group of mutants represented by H187R, H187P, H187V and H187T (P = 0.039) displayed a moderate reduction of transposition efficiency to a level ranging between 60% and 86% relative to SB100X (Figure 3A). In contrast, H187E exhibited a drastically reduced transposition activity of 12% relative to SB100X (P = 0.001), whereas the introduction of an aspartate (H187D) completely abolished transposition. Finally, incorporation of some amino acid residues at position 187, including S, F and Y exhibited mild hyperactive phenotypes relative to SB100X, although these differences were not supported by statistical significance in these datasets. Measuring relative efficiencies of transposon excision catalyzed by the mutant transposases by applying a FACS-based transposon excision assay (55) revealed that all of the SB transposases mutated at H187 were proficient in transposon excision. Most variants showed excision efficiencies close to wild-type levels, only the H187D and H187E mutants displayed a clear reduction in excision activity (42% and 68% excision activity relative to SB100X, respectively), consistent with their strongly diminished transposition activities (Supplementary Figure S5).
Figure 3.

Transposition activities of Sleeping Beauty transposase mutants. Relative transposition efficiencies for H187 (A), P247 (B) and K248 [reproduced from (55)] (C). Plasmids expressing transposase mutants were transiently cotransfected with a transposon-donor plasmid (pT2B/puro) into HeLa cells. Cells were selected for puromycin resistance and stained with methylene blue to identify viable cell colonies. Colony numbers were normalized to the SB100X positive control, whose transposition efficiency was set to 100%. An inactive SB transposase (D3) was included as negative control. Data are represented as mean ± SD, n = 3 biological replicates. Differences in transposition activities are significant as determined by Student's t-test for the indicated mutants. Transpositional differences for all of the other mutants were not supported by statistical significance.
In contrast to the H187 mutations, the vast majority of the P247 mutations was either completely inactive or displayed a severe reduction in both overall transposition (Figure 3B) and excision (Supplementary Figure S5) activities. P247A was the most active mutant with 109% transposition activity relative to SB100X. Of note, the relative transposition activity of P247A exceeded the calculated relative excision activity. This discrepancy between excision- and integration capacities is likely due to the fact that excision and integration rates were determined by two independent assays. The second most active mutant was P247S which retained 73% of transposon integration activity relative to SB100X, followed by P247R (27%, P = 0.017), P247C (7%, P = 0.002) and P247K (3%, P = 0.003) (Figure 3B).
Similar to P247, the amino acid exchanges at position 248 completely abolished transposition in almost all of the 19 mutants (Figure 3C). K248R was the most active mutant with 77% activity relative to SB100X. Only three additional K248 mutants, including K248C (4%, P = 0.003), K248I (4%, P = 0.003) and K248V (9%, P = 0.003), displayed transposition activities at very low levels relative to SB100X. As described recently, some of the K248 mutants displayed a segregation of transposon excision and integration activities (55) (Supplementary Figure S5).
Altered target site preferences of H187, P247 and K248 transposase mutants
Based on the structural modeling and mutagenesis data, the H187, P247 and K248 amino acids are strong candidates for playing a role in interactions with tDNA. Thus, it is reasonable to assume that some mutations in these positions of the SB transposase might lead to a change in the target site selection properties of the affected proteins. To investigate this, we used some of the SB mutants for the generation of transposon insertion site libraries in human HepG2 cells, and compared both the local attributes as well as genome-wide distributions of these insertion sites to those generated by SB100X. For position 187 we selected 6 mutants (H187R, H187E, H187S, H187T, H187V, H187P) with a marked impact on transposition efficiency (Figure 3A), and for positions 247 and 248 we selected all mutants that displayed measurable transposition activity (P247A, P247R, P247C, P247S, P247K and K248C, K248I, K248V, K248R, respectively) (Figure 3B and Figure 3C).
Transposon integration sites were visualized by SeqLogo analysis that not only reports a consensus sequence, but also delivers information on overall sequence conservation and relative frequencies of nucleotides in each position (measured as height of symbols, which are ordered by their relative frequency within the logo). The analysis revealed that the core TA target site dinucleotides are embedded in A/T-rich DNA for all mutants, as noted previously for several SB variants (47,48) (Figure 4). SB100X transposase prefers the 8-bp palindromic AT repeat sequence ATATATAT centered on the actual TA target dinucleotide (48). Because SB almost exclusively integrates into TA, the central TA approaches the maximal value of 2 bits (log2 4) in the logo. The SB100X reference logo also displays a relatively strong overall conservation of the A base in the -3 position and the matching T in the +3 position of the consensus with an overall score of 0.6 bits (Figure 4). These integration preferences appear similar for all of the transposase mutants tested (Figure 4 and Supplementary Figure S6); however, there are notable differences. First, the P247S and P247R mutants display slight deviations in their consensus target sites [AGATATCT for P247S and ATTATATAAT for P247R (Figure 4)]; note that the palindromic nature of the consensus is maintained]. Second, the preference for the alternating AT bases upstream and downstream of the 8-bp consensus becomes more pronounced with the H187P, H187V and K248R mutants, manifesting itself as “shoulders” flanking the 8-bp consensus (Figure 4). Finally, conservation of the A in the -3 position and the T in the +3 position of the consensus is largely increased to ∼1.5 bits with the H187P, H187V and K248R mutants (Figure 4). These findings show that amino acid replacements in positions 187, 247 and 248 of the SB transposase indeed have an impact on the target site selection properties of the respective mutant transposases. The most significant alteration is a more pronounced overall preference for A/T-richness of the tDNA and stronger conservation of palindromic bases within the consensus sequences. These observed changes imply that some of the mutants became more constrained in their target site choice. This is remarkably revealed by analyzing the overall frequencies of transposon insertions into the preferred 8-bp ATATATAT sequence. Although SB100X and the P247S and P247R mutants target this particular sequence only 2-3% of the time, some other mutants display significantly higher frequencies of integration into this motif (18% for H187P, 21% for H187V and 39% for K248R, Figure 4). In sum, specific amino acid substitutions in positions 187, 247 and 248 of the SB transposase result in a phenotype of highly preferential integration into AT-repeats.
Figure 4.

Integration sites of the transposase mutants. The sequence logos show the majority-rule consensus sequences at the genomic insertion loci in a 60-bp window, centered around the target TA dinucleotides. The value 2 (log2 4) on the y axis stands for maximum possible frequency. The pie charts depict the percentages of insertions occurring at the SB-specific ATATATAT consensus motif. The N values represent the numbers of uniquely mappable SB transposon insertions.
The H187V, P247R and K248R transposase mutants generate altered genome-wide distribution of SB insertions
The above data show that some of the tested transposase mutants gained an enhanced preference for integration into AT-rich DNA. Because AT-rich DNA is not distributed uniformly across the human genome, it is reasonable to assume that the preference for integration into such DNA sequences would be manifested at the level of the genome-wide distribution of transposon insertions by these enzymes.
To test this, we determined the relative frequencies of integration into various genomic features, including genic and non-genic regions, cancer genes, exons, introns, 5’- and 3’-UTRs and sequences flanking genes upstream and downstream within 10-kb windows, compared to a computer-generated random data set. We not only compared insertions within these genomic features generated by SB100X and its mutants, but also included MLV gammaretroviral and HIV lentiviral integration sites in the analysis. As established previously (30,60,70), SB transposon insertions show only a minor bias toward transcription units (hereafter referred to as genes) and their flanking regions, in contrast to MLV (59,71,72) and HIV (72) insertions that are markedly enriched in loci flanking transcriptional start sites (TSSs) and within actively transcribed genes, respectively (Figure 5). Out of these three gene vector systems the overall insertion frequencies of SB are the closest to an expected random distribution (Figure 5).
Figure 5.

Representation of insertion sites in genic features and chromatin-defined functional segments. (A) Integration loci of MLV, HIV, SB100X and its mutant derivatives were counted in gene-related segments of the human genome. The numbers represent fold change increase (red) or decrease (blue) in insertion frequencies compared to a theoretical random control (set to 1). The dendogram on the left is based on the mean frequency values of the rows. Statistical significance (Fisher's exact test) measured for the values of the K248R, P247R and H187V mutants versus SB100X is indicated by stars; * P< 0.05, ** P< 0.001, ns: not significant. (B) Representation of insertion sites in functional genomic segments as defined by epigenetic signal patterns. Color code, dendogram and statistical significance as indicated for panel (A).
Next, we selected a small subset of our mutants; namely the P247R, H187V and K248R mutants, and addressed if these mutants generate a recognizably different genome-wide distribution profile than that of SB100X. The overall percentages of insertions into these genomic regions are presented in Supplementary Figure S7. The data presented in Figure 5A reveal a significant depletion in insertion into genes, especially with the H187V and K248R mutants. The most significant differences are within 5’- and 3’-UTRs as well as in exons. The most dramatic change in integration frequency is a 4-fold drop seen with K248R in comparison to SB100X (P < 0.001) in coding exons (Figure 5A). Notably, transposon integration by H187V and K248R within these genomic regions is not only depleted in comparison to SB100X, but also relative to the random dataset.
Beyond driving integration away from exons, a preference of integrating into AT-repeats by our mutants also implies that these insertions might be enriched in repetitive DNA. Indeed, a dramatic enrichment in simple repeats is detected for integrations catalyzed by the H187V and K248R variants; the enrichment in this compartment of the genome is ∼12-fold over the random dataset (P< 0.001) and ∼4-fold over SB100X (P< 0.001) by H187V, and ∼21-fold over the random dataset (P< 0.001) and ∼7-fold over SB100X (P< 0.001) by K248R (Supplementary Figure S8a). Consistent with a preference for integration into ATATATAT, the H187V variant displays a 24-fold and ∼5-fold enrichment in TA-rich simple repeats over random and SB100X-mediated insertions (P< 0.001), respectively, whereas K248R-mediated insertions are ∼43-fold and ∼8-fold enriched in these regions over the random and SB100X datasets (P< 0.001), respectively (Supplementary Figure S8b).
Next, we profiled insertions generated by SB100X and the P247R, H187V and K248R mutants in diverse functional genomic segments. These segments are defined by co-occurring epigenetic signal patterns, which are clustered together computationally to define various functional partitions of the human genome (73). We used 25-state chromatin models of human HepG2 cells. As above, we included MLV and HIV insertions in the analysis. First, in line with previous observations (30,60,70), SB transposon insertions show only a minor bias towards promoters, TSSs, enhancers and transcriptional regulatory regions with an open chromatin structure. This is in contrast to MLV and HIV insertions that display the highest enrichment in promoter regions including TSSs and transcribed regions, respectively (Figure 5B). Second, a strong depletion in enhancers, promoters including TSSs and open regulatory regions is observed for integrations catalyzed by H187V and K248R. The most significant changes are a ∼50-fold, ∼3-fold and ∼5-fold depletion over MLV, SB100X (P< 0.001) and the random dataset, respectively, by H187V in promoters including TSSs, and a ∼14-fold, 4-fold and ∼3-fold depletion over MLV, SB100X (P< 0.001) and the random dataset, respectively, by K248R in enhancers (Figure 5B). Collectively, the data show that the P247R, H187V and K248R transposase mutants direct a significant fraction of transposon integrations away from exons as well as transcriptional regulatory regions including promoters and enhancers.
Enrichment of insertions into genomic safe harbors by the H187V, P247R and K248R transposase mutants
Integration of therapeutic gene constructs into safe sites in the human genome would prevent insertional mutagenesis and associated risks of oncogenesis in gene therapy. Genomic “safe harbors” (GSHs) are regions of the human genome that are able to accommodate the predictable expression of newly integrated DNA without adverse effects on the host cell or organism. Chromosomal sites or regions can be bioinformatically assigned as GSHs if they satisfy the following criteria: (i) no overlap with transcription units, (ii) distance of at least 50 kb from the 5′-end of any gene, (iii) at least 300 kb distance to cancer related genes and (iv) microRNA genes, and (v) regions outside of ultra-conserved elements (UCEs) (64,74). We have previously established that the SB transposon system has a significantly more favorable insertion profile than MLV- and HIV-based viral integration systems with respect to frequencies of insertions into GSHs (30,60,70).
The data presented above establish that some of the identified SB transposase mutants significantly redirect insertions away from exons and transcriptional regulatory regions of genes, thereby inherently implying that a larger fraction of insertions catalyzed by these enzymes lands in GSHs. We analyzed our insertion site datasets with respect to the relative frequencies of integration into GSHs. Figure 6A displays three clusters in the dendogram: the first represented by MLV and HIV, the second by SB100X, P247R and H187V and the third by K248R and random. There is a gradual shift towards a random-like distribution of insertions by the SB100X transposase and its mutants. For example, only 17% of HIV insertions fall outside of genes, whereas this value is 41% for SB100X, 42% for P247R, 44% for H187V, 46% for K248R and 49% for the random dataset (Figure 6A). In line with these observations, our analyses revealed increasing frequencies of insertions into GSHs by simultaneously applying all five GSH criteria. Out of the mutants tested, the K248R transposase variant approached random the closest: overall, 29% of all K248R insertions and 34% of all random insertions fall in a GSH (Figure 6B). Collectively, our analysis reveals a safer, thus favorable transgene insertion profile for the SB system over MLV-, and HIV-based vectors and thus predicts enhanced safety of the P247R, H187V and K248R transposase variants in therapeutic gene transfer in human cells.
Figure 6.
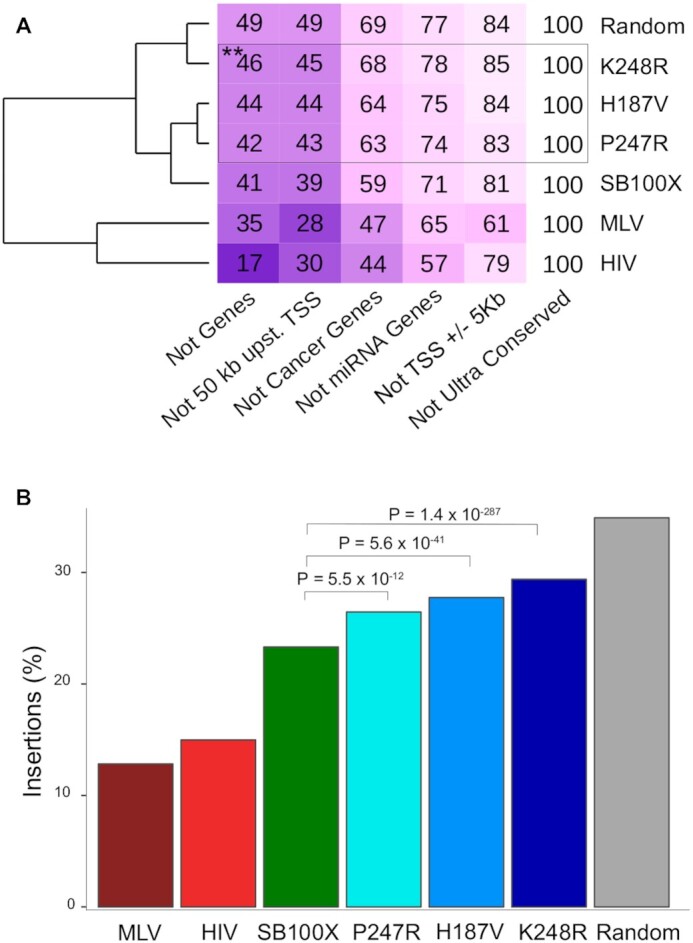
Insertion frequencies in genomic safe harbors. (A) The numbers in the boxes indicate the percentages of the insertions per condition that fall in the in the safe harbor sub-categories listed below the table. Note the overlapping nature of the categories, which explains why the sums of the numbers per condition exceed 100%. Darker boxes correspond to lower values in the boxes, indicating under-representation of insertions in the safe harbor sub-categories. The dendrogram on the left is based on the mean values of the rows; thus, it clusters conditions by their overall similarities of insertion frequencies in genomic safe harbors. Statistical analysis by Fisher's exact test, ** P< 0.001 as compared to SB100X. (B) Overall representation of insertions in genomic safe harbors.
The H187V and K248R transposase variants avoid nucleosomal DNA for integration
High-density integration profiling of the Hermes transposon in yeast revealed a strong association of integration sites with nucleosome-free chromatin (75,76). In addition, recent evidence indicates that Tc1/mariner transposons preferentially integrate at linker regions between nucleosomes (30). We mapped our insertion datasets with respect to nucleosome occupancy as determined by MNase-Seq data (77). As seen previously, transposon insertions mediated by SB100X are underrepresented in nucleosomal DNA (Figure 7). Remarkably, out of the three transposase mutants tested, the H187V and K248R variants displayed a drastic drop in nucleosome occupancy at transposon integration sites, not only as compared to the random control, but also to the SB100X dataset (Figure 7). Intriguingly, we detected an increase in the probability of nucleosome presence in the immediate downstream vicinity of the insertion sites of the P247R and K248R mutants (Figure 7). This finding may imply a preference of these mutants for integrating into DNA entering a downstream nucleosome, thus a preference for distorted, yet not histone-bound DNA for insertion. In sum, the H187V and K248R mutations markedly intensify the phenotype of the SB transposase; namely, avoidance of nucleosomal DNA for transposon integration.
Figure 7.
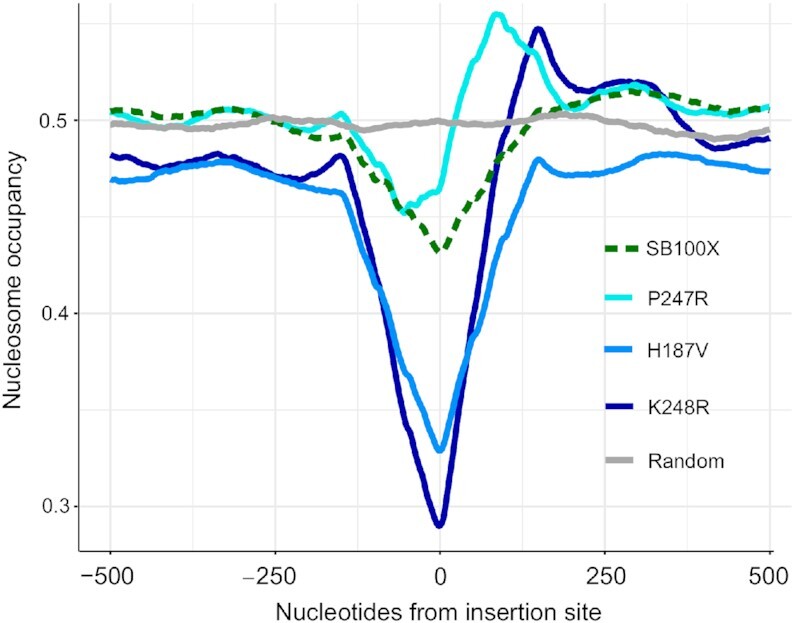
Nucleosome occupancy at transposon insertion sites. Mean calculated probabilities of the nucleotides for being present on a nucleosome are shown on the y-axis. The occupancy prediction was implemented on all the insertion sequences of the corresponding transposase versions. The probabilities are depicted in a window that extends 500 bp upstream and 500 bp downstream from the transposon integration sites. 0 stands for the insertion site.
Molecular features associated with preferential genomic target sites of H187V and K248R transposase mutants
The above data establish that the H187V and K248R transposase mutants detarget exons and transcriptional regulatory regions of genes and avoid nucleosomal DNA for integration. However, these data do not necessarily shed light on causative relationships between these independent observations. For example, exons tend to be associated with nucleosomes (Supplementary Figure S9a); thus, depletion of integrations in exonic sequences by H187V and K248R may be a mere reflection of these transposase variants avoiding nucleosomal DNA. However, transcriptional regulatory regions including enhancers and TSSs are clearly depleted in nucleosomes (Supplementary Figure S9b); yet we observed low integration frequency also in these regions, suggesting that nucleosome occupancy alone cannot explain the integration pattern of H187V and K248R.
Because the genomic segments are defined by chromatin marks, the question arises whether it is the local chromatin structure or the underlying primary DNA sequence that modulates integration frequencies in these segments. Consistent with previous findings (30), the SB100X transposase promotes insertions with an almost random insertion profile in human cells with a slight bias for euchromatin marks (including H3K4me1, H3K27ac, H3K36me3 and H3K29me2) (Figure 8A). We detected a significant (P< 0.001) depletion in chromatin marks at insertions by the H187V and K248R variants as compared to SB100X-mediated insertions (Figure 8A). However, the depletion is not restricted to euchromatin marks. Instead, the H187V and K248R insertion sites are generally depleted in histone modifications, regardless whether they mark transcriptionally active or repressed chromatin (Figure 8A); thus, we consider it unlikely that the H187V and K248R transposase variants would interact with chromatin differently than SB100X.
Figure 8.

Molecular features associated with preferential genomic target sites of the H187V and K248R transposase mutants. (A) Relative co-occurrence of transposon integration sites with histone tail modifications. The numbers represent fold change increase (red) or decrease (blue) in insertion frequencies within ChiP-Seq peaks compared to a theoretical random control (set to 1). The dendogram on the left is based on the mean frequency values of the rows. ** P< 0.001, Fischer's exact test as compared to SB100X. (B) Relative frequencies of TA dinucleotides and ATATATAT octanucleotides in exonic sequences. Genomic mean is included as reference and is set to 1. (C) Relative frequencies of TA dinucleotides and ATATATAT octanucleotides in transcriptional regulatory sequences including enhancers, open regulatory regions and TSSs. Genomic mean is included as reference and set to 1.
The above findings led us to hypothesize that the primary determinant of preferred genomic targets by the H187, P247 and K248 transposase mutants is DNA sequence composition. Exons, in particular coding exons, are relatively poor in TA dinucleotides (Figure 8B), which are the preferred target sites for SB transposition. Having seen a pronounced preference by some of our mutants, especially by H187V and K248R, for the ATATATAT sequence motif implies that detargeting of exon sequences by this mutant is driven by the rare occurrence of this sequence motif in exonic sequences. Indeed, ATATATAT is severely underrepresented in exons, especially so in coding exons (Figure 8B), thereby suggesting that primary DNA sequence composition is the major determinant of driving H187V and K248R insertions away from exonic sequences. Similarly, open regulatory regions, TSSs and enhancers are relatively TA-poor in comparison to the average base composition of the human genome (Figure 8C). As seen for coding exons, the preferred ATATATAT sequence motif of the H187V and K248R mutants is severely underrepresented in these three genomic segments (Figure 8C). The data argue that the major determinant of detargeting exons as well as transcriptional regulatory regions by the H187V and K248R mutants is the reduced availability of the highly preferred ATATATAT sequence in these genomic regions.
Sequence-dependent DNA deformability drives target site selection by the H187V, P247R and K248R transposase mutants
Primary DNA sequence is a major determinant of helix structure, which can shape DNA-protein interactions. Thus, we set out to investigate the differences in the physical properties of the consensus insertion site sequences of the SB100X transposase and its mutants. It has been postulated that DNA-binding proteins can recognize the intrinsic flexibility of the DNA. A crucial determinant of the latter is the energy needed for disrupting the interactions of bases stacking on each other in the helix. The RY or YR base-steps, in particular the AT or TA steps, have the lowest stacking energy (the amount of energy required for disrupting the interaction of the bases stacked on each other in the helix) (78), and are therefore particularly flexible. Similarly, DNA denaturation energy (the energy needed for melting the DNA strands) is lower for A/T-rich DNA. In contrast, stiffness is a measure of resistance against a bending force on the DNA helix, whereas base pair rigidity based on DNase I cleavage frequency is also associated with DNA sequence, in that A/T-rich sequences require less energy for duplex disruption (79,80). We found that the insertion sites preferred by the H187V, P247R and K248R transposase mutants are associated with higher flexibility (lower stacking energy) (Figure 9A), and require less energy for duplex disruption, and are therefore more bendable than the SB100X insertion sites (Figure 9B–D). These results imply a deficiency of these mutant transposases to insert into TA dinucleotides surrounded by rigid (A/T-poor) DNA.
Figure 9.

Physical properties of the DNA sequences around the insertion loci. Shown are average values of individual insertion sites within 60-nucleotide-long windows around the insertion sites. (A) Stacking energy, which is a measure of how much energy is needed for disrupting the interaction of the bases stacked on each other in the helix. This value is the lowest at TA steps (note the negative scale). (B) DNA denaturation energy, which is the energy needed for melting the DNA strands. (C) Bending stiffness, which is a measure of resistance against a bending force on the DNA helix. (D) Rigidity of the DNA segments based on DNase I cutting site frequency between all possible bases. The arrows depict the central TA insertion sequence.
Non-canonical transposon integrations by the H187V transposase variant
A striking feature of the Mos1 STC structure is flipping of the adenines of the TA target sites on both strands into extra-helical positions (28). Adenine-specific interactions with the transposase trap the flipped conformation, which is required for transposon integration at TA dinucleotides. Because SB also integrates almost exclusively to TA dinucleotides, a similar mechanism of base flipping may occur during SB transposition. Nonetheless, dinucleotides other than TA can also serve as target sites for SB transposition at very low frequencies (47,81,82).
We examined non-canonical transposon integration sites used by the H187V transposase variant. Consistent with previous observations for the canonical SB transposase (47,81,82), transposon integrations at non-TA dinucleotides occurred at a frequency of ∼0.5% (712 events out of a total of 130 504 mappable insertions), and the most frequent non-canonical insertion sites were CA and TG (Figure 10A). As discussed above, for base flipping to occur de-stacking of the adjacent bases and DNA backbone deformation need to take place. Consistently, we detected a strong correlation between dinucleotide frequencies at integration sites and the base stacking energy as well as protein-inducible twist [deduced from protein–DNA crystal structures, as measures for sequence–dependent DNA deformability (83)] associated with different base steps (Figure 10A). The highly preferred TA dinucleotide and the CA and TG dinucleotides, whose usage is severely disfavored in SB transposition but nonetheless represent the most frequent non-canonical integration sites, are the most prone to undergo helix deformations when bound by proteins (Figure 10A). This observation offers a physical explanation to target site selection in SB transposition.
Figure 10.

Frequencies and physical properties of non-canonical transposon insertion sites generated by the H187V mutant of the Sleeping Beauty transposase. (A) Relative frequencies of dinucleotides occurring at non-TA sites by the H187V mutant (bars) and the relative base stacking energies (red) and protein-inducible twist (blue) associated with each dinucleotide. (B) SeqLogo analysis of non-canonical insertion sites. (C) DNA flexibility as measured by stacking energy for canonical versus non-canonical insertion sites. Averages flexibilities were calculated from di-nucleotide values applied to all the non-TA insertion sites of the H187V mutant in 60-nucleotide-long windows centered around the insertion (black triangle).
The H187V mutant is indistinguishable from SB100X both its insertion frequency into non-canonical dinucleotides (∼0.5%) and in using the CA and TG dinucleotides as the most preferred non-canonical sites for integration. Nevertheless, SeqLogo analysis of the non-canonical insertion sites upstream and downstream of the target dinucleotides revealed a profound difference as compared to the logo generated for the canonical insertions. Namely, a stronger conservation and more pronounced preference for alternating AT base pairs in the flanks is evident (reaching 0.2–0.3 bits of the “shoulders”) for the non-canonical insertion sites (Figure 10B). In agreement, the non-canonical insertion sites display an even further increased flexibility as compared to canonical insertion sites (Figure 10C), suggesting that H187V can only target non-canonical dinucleotides for integration if they are situated in an A/T-rich, bendable DNA sequence context.
Finally, we assessed the combinatorial phenotype of a H187V/K248R double mutant of the SB transposase. Apparently, the extents to which these particular single mutants target certain DNA sequences and genomic regions are not additive (Supplementary Figure S10). The double mutant displayed a phenotype largely similar to H187V with respect to frequency of targeting the ATATATAT consensus motif (Supplementary Figure S10a), detargeting genic sequences including exons and transcriptional regulatory regions (Supplementary Figure S10b), enrichment in TA-rich repeats (Supplementary Figure S10c), and DNA sequences at non-canonical insertion sites (Supplementary Figure S10d and e)
DISCUSSION
In this work, we establish that amino acid replacements in three positions of the SB transposase lead to profound changes in target site selection in human cells. These findings indicate that H187 of SB is a functional equivalent of R186 of the Mos1 transposase, P247 of SB is a functional equivalent of S119 in HIV-1 IN, A188 in PFV IN and S124 in RSV IN, and K248 of SB is a functional equivalent of N120 in HIV-1 IN. The data imply a shared role of these amino acid residues in interacting with tDNA during mobile element (virus and transposon) integration.
In particular, the H187V and K248R SB transposase variants detarget exons and transcriptional regulatory regions of genes (Figure 5). These transposase mutants display a strong preference for A/T-rich DNA primarily composed of alternating AT (or TA) dinucleotides (Figure 4 and Supplementary Figure S8), in particular for an 8-bp ATATATAT sequence motif (Figure 4). As high as 39% of all integrations catalyzed by the K248R variant occur at such motif (as opposed to 2% by SB100X, Figure 4), indicating a dramatic gain of target site specificity by this mutation. Exons and transcriptional regulatory elements including promoters, TSSs and enhancers are severely depleted in the ATATATAT sequence motif (Figure 8), resulting in disfavored integration into these genomic segments by our mutants.
Primary sequence determines the physical properties of DNA, which in turn has a profound impact on its interactions with proteins. Bendability of tDNA seems to be an important factor in defining integration sites for mobile genetic elements, because it enables proper positioning of the phosphodiester bonds in the transposase active sites. A bendable/flexible structure is likely required for the IN/transposase protein and/or auxiliary host factors to deform the bound DNA into an optimal conformation for strand transfer. Indeed, tDNA bending is a recurring theme in retroviral integration and DNA transposition executed by DDE/D recombinases (50) (Supplementary Figure S1). Structural studies of the TCC comprising four PFV IN molecules, viral DNA ends and tDNA highlighted a severely bent (90°) tDNA conformation, and revealed amino acid residues, including A188, involved in distorting the tDNA by direct interaction with DNA bases. Similarly, the tDNA in the post-integration structure of the RSV intasome STC revealed a strong bending of ∼90°, which is stabilized by DNA contacts made by the α2 helix of IN containing S124 (22). Structures of STCs by DNA transposons, including Mos1 (28) and Mu (84) both revealed a severe tDNA bend (∼140°); in addition, Tn10 transposase (85), P element transposase (86), piggyBac transposase (87) and Transib transposase (88) all favor bent target DNA structures. Importantly, interactions with Mos1 transposase residues, including R186, were shown to stabilize distortions in the tDNA (28). In the absence of high-resolution data for a tDNA-bound SB complex, we used structural modeling to infer that H187, P247 and K248 in the SB transposase execute similar functions; namely, stabilizing a DNA bend by direct interactions with bases of the tDNA. This is supported by both the conserved positions of H187 (with respect to R186 in Mos1 transposase), P247 (with respect to S119 in HIV-1 IN, A188 in PFV IN and S124 in RSV IN) and K248 (with respect to N120 in HIV-1 IN) in the SB TCC model (Figure 1 and Figure 2) as well as a more pronounced requirement for a highly flexible tDNA structure at the integration sites of our mutants as compared to SB100X (Figure 9). Our results are thus most parsimonious with the conclusion that the SB variants that we analyzed here are loss-of-function mutants either defective or strongly attenuated in their abilities to induce or stabilize a bent tDNA structure. This defect must be compensated by enhanced intrinsic bendability of the tDNA for transposon integration to take place, which manifests itself in highly selective integration into AT-repeats that are associated with a bendable structure. This phenotype is further exaggerated when examining non-canonical integration sites generated by H187V. Non-canonical insertions apparently can only take place in DNA with an even higher AT-content associated with higher flexibility than the canonical insertion sites targeted by the same enzyme (Figure 10). These observations are consistent with the proposed role of H187 in broadening the major groove of the tDNA and/or kinking the tDNA substrate via un-stacking bases at the integration site (52). A defect of the enzyme to execute this function properly due to mutations can be, at least to some extent, compensated by selecting and engaging sites associated with a higher level of bendability.
The H187V and K248R mutants also display the salient property of avoiding integration into nucleosomal DNA to a more dramatic extent than seen for the SB100X transposase (Figure 7). The Mos1 STC structure (28) suggests that the severe bending of tDNA by the transpososome that is required for the integration step during transposition can be more easily achieved on flexible linker DNA than on nucleosomal DNA, in the context of which the tight wrapping of the DNA around the nucleosome core represents a constraint to further physical distortion. Thus, we consider it likely that the H187V and K248R mutants markedly disfavor nucleosomes due to their extended dependence on highly flexible DNA sequences for integration. Other transposons, such as the Drosophila P element, show a preference for insertion into the 5′ transcriptional regulatory elements of genes, possibly as a consequence of their preferential integration into nucleosome-free regions (89–91). Moreover, Hermes transposon integrations in yeast were also found to favor nucleosome-free regions, and it has been suggested that these regions are physically more accessible for the transposase (76). However, a preference for nucleosome-free DNA for integration is not a general attribute among mobile genetic elements. Retroviruses have been found to direct integration into outward-facing major grooves on nucleosome-wrapped DNA (17,92–94), and the Ty1 retrotransposon in yeast preferentially integrates into nucleosome-bound DNA near the H2A/H2B interface (95), suggesting that these elements can exploit the nucleosome-induced bending of tDNA for integration. Why not all integrating genetic elements can capitalize on the pre-bent DNA structure as offered by a nucleosome is unclear. Perhaps the almost extreme extent to which the tDNA needs to be bent for integration of certain transposons, including SB, simply cannot be generated and stabilized in a nucleosomal structure due to physical constraints. These transposons will then necessarily select their target sites in regions of the genome, where the required DNA distortions can be accommodated.
Beyond providing important biochemical and biophysical clues for SB transposition, the work presented here also bears practical relevance in the context of genetic engineering, in particular gene therapy. Preferential integrations of gammaretroviral vectors near transcriptional regulatory elements of active genes and the bias of lentiviral vectors towards transcription units bear a finite risk of insertional oncogenesis, which manifested in patients treated with MLV-derived vectors in early gene therapy clinical trials based on gene insertion into hematopoietic stem cells (96–99). The integration profile of the SB variants described here may enable therapeutic cell engineering with a significantly increased safety profile. Directing integration away from the transcriptional regulatory regions and exons of genes to an extent that the frequencies of integration into these genomic regions becomes significantly lower than that produced by fully random distribution (Figure 5) provides a significant improvement in the projected safety of these transposase variants for gene therapy.
DATA AVAILABILITY
All data are available in the main article and in Supplementary Information and Supplementary Datasets.
Supplementary Material
ACKNOWLEDGEMENTS
We thank T. Diem and T. Leyendecker for technical assistance and Y. van de Peer for kindly providing us with conversion tables for DNA physical property predictions.
Author contributions: C.M. generated PCR libraries representing transposon insertions and bioinformatically analyzed the insertion sites and wrote the paper; L.K. generated to SB transposase mutants, tested them for expression and transposition activity and generated PCR libraries representing transposon insertions and wrote the paper; I.Q. performed structural modeling of the SB transposase-target DNA complex and comparison with available structures of retroviral integrase and transposase complexes; A.G. did structural modeling of the SB transposase-target DNA complex and compared this to retroviral integrase and transposase complexes; O.B. supervised structural analyses and wrote the paper; Z.I. conceived the study, supervised the experimental work and wrote the paper.
Contributor Information
Csaba Miskey, Transposition and Genome Engineering, Division of Medical Biotechnology, Paul Ehrlich Institute, Langen 63225, Germany.
Lisa Kesselring, Transposition and Genome Engineering, Division of Medical Biotechnology, Paul Ehrlich Institute, Langen 63225, Germany.
Irma Querques, Structural and Computational Biology Unit, European Molecular Biology Laboratory, Heidelberg 69117, Germany; Department of Biochemistry, University of Zurich, Zurich 8057, Switzerland.
György Abrusán, Institute of Biochemistry, Biological Research Center of the Hungarian Academy of Sciences, Szeged 6726, Hungary.
Orsolya Barabas, Structural and Computational Biology Unit, European Molecular Biology Laboratory, Heidelberg 69117, Germany; Department of Molecular Biology, University of Geneva, Geneva 1211, Switzerland.
Zoltán Ivics, Transposition and Genome Engineering, Division of Medical Biotechnology, Paul Ehrlich Institute, Langen 63225, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
OTKA [PD83571 to G.A.]. Funding for open access charge: Institutional funds.
Conflict of interest statement. Z. Ivics, I. Querques and O. Barabas are co-inventors on several patents relating to SB transposon technology.
REFERENCES
- 1. Lewinski M.K., Yamashita M., Emerman M., Ciuffi A., Marshall H., Crawford G., Collins F., Shinn P., Leipzig J., Hannenhalli S.et al.. Retroviral DNA integration: viral and cellular determinants of target-site selection. PLoS Pathogens. 2006; 2:e60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kvaratskhelia M., Sharma A., Larue R.C., Serrao E., Engelman A.. Molecular mechanisms of retroviral integration site selection. Nucleic Acids Res. 2014; 42:10209–10225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sultana T., Zamborlini A., Cristofari G., Lesage P.. Integration site selection by retroviruses and transposable elements in eukaryotes. Nat. Rev. Genet. 2017; 18:292–308. [DOI] [PubMed] [Google Scholar]
- 4. Mitchell R.S., Beitzel B.F., Schroder A.R., Shinn P., Chen H., Berry C.C., Ecker J.R., Bushman F.D.. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biol. 2004; 2:E234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Luenen H.G., Colloms S.D., Plasterk R.H.. The mechanism of transposition of Tc3 in C. elegans. Cell. 1994; 79:293–301. [DOI] [PubMed] [Google Scholar]
- 6. Mitra R., Fain-Thornton J., Craig N.L.. piggyBac can bypass DNA synthesis during cut and paste transposition. EMBO J. 2008; 27:1097–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Plasterk R.H., Izsvak Z., Ivics Z.. Resident aliens: the Tc1/mariner superfamily of transposable elements. Trends Genet. 1999; 15:326–332. [DOI] [PubMed] [Google Scholar]
- 8. Kulkosky J., Jones K.S., Katz R.A., Mack J.P., Skalka A.M.. Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol. Cell. Biol. 1992; 12:2331–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim D.R., Dai Y., Mundy C.L., Yang W., Oettinger M.A.. Mutations of acidic residues in RAG1 define the active site of the V(D)J recombinase. Genes Dev. 1999; 13:3070–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Landree M.A., Wibbenmeyer J.A., Roth D.B.. Mutational analysis of RAG1 and RAG2 identifies three catalytic amino acids in RAG1 critical for both cleavage steps of V(D)J recombination. Genes Dev. 1999; 13:3059–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Junop M.S., Haniford D.B.. Factors responsible for target site selection in Tn10 transposition: a role for the DDE motif in target DNA capture. EMBO J. 1997; 16:2646–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Katzman M., Sudol M.. Mapping domains of retroviral integrase responsible for viral DNA specificity and target site selection by analysis of chimeras between human immunodeficiency virus type 1 and visna virus integrases. J. Virol. 1995; 69:5687–5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Appa R.S., Shin C.G., Lee P., Chow S.A.. Role of the nonspecific DNA-binding region and alpha helices within the core domain of retroviral integrase in selecting target DNA sites for integration. J. Biol. Chem. 2001; 276:45848–45855. [DOI] [PubMed] [Google Scholar]
- 14. Harper A.L., Sudol M., Katzman M.. An amino acid in the central catalytic domain of three retroviral integrases that affects target site selection in nonviral DNA. J. virol. 2003; 77:3838–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maertens G.N., Hare S., Cherepanov P.. The mechanism of retroviral integration from X-ray structures of its key intermediates. Nature. 2010; 468:326–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson R.C., Stella S., Heiss J.K.. Rice P.A., Correll C.C.. Protein–Nucleic Acid Interactions: Structural Biology. 2008; London: RSC Publishing; 176–220. [Google Scholar]
- 17. Maskell D.P., Renault L., Serrao E., Lesbats P., Matadeen R., Hare S., Lindemann D., Engelman A.N., Costa A., Cherepanov P.. Structural basis for retroviral integration into nucleosomes. Nature. 2015; 523:366–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Serrao E., Krishnan L., Shun M.C., Li X., Cherepanov P., Engelman A., Maertens G.N.. Integrase residues that determine nucleotide preferences at sites of HIV-1 integration: implications for the mechanism of target DNA binding. Nucleic Acids Res. 2014; 42:5164–5176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Demeulemeester J., Vets S., Schrijvers R., Madlala P., De Maeyer M., De Rijck J., Ndung’u T., Debyser Z., Gijsbers R.. HIV-1 integrase variants retarget viral integration and are associated with disease progression in a chronic infection cohort. Cell Host Microbe. 2014; 16:651–662. [DOI] [PubMed] [Google Scholar]
- 20. Lu R., Limon A., Ghory H.Z., Engelman A.. Genetic analyses of DNA-binding mutants in the catalytic core domain of human immunodeficiency virus type 1 integrase. J. Virol. 2005; 79:2493–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Gent D.C., Groeneger A.A., Plasterk R.H.. Mutational analysis of the integrase protein of human immunodeficiency virus type 2. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:9598–9602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yin Z., Shi K., Banerjee S., Pandey K.K., Bera S., Grandgenett D.P., Aihara H.. Crystal structure of the Rous sarcoma virus intasome. Nature. 2016; 530:362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stevens S.W., Griffith J.D.. Sequence analysis of the human DNA flanking sites of human immunodeficiency virus type 1 integration. J. Virol. 1996; 70:6459–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Holman A.G., Coffin J.M.. Symmetrical base preferences surrounding HIV-1, avian sarcoma/leukosis virus, and murine leukemia virus integration sites. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:6103–6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu X., Li Y., Crise B., Burgess S.M., Munroe D.J.. Weak palindromic consensus sequences are a common feature found at the integration target sites of many retroviruses. J. Virol. 2005; 79:5211–5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Berry C., Hannenhalli S., Leipzig J., Bushman F.D.. Selection of target sites for mobile DNA integration in the human genome. PLoS Comput. Biol. 2006; 2:e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van Luenen H.G., Plasterk R.H.. Target site choice of the related transposable elements Tc1 and Tc3 of Caenorhabditis elegans. Nucleic Acids Res. 1994; 22:262–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morris E.R., Grey H., McKenzie G., Jones A.C., Richardson J.M.. A bend, flip and trap mechanism for transposon integration. ELife. 2016; 5:e15537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Richardson J.M., Colloms S.D., Finnegan D.J., Walkinshaw M.D.. Molecular architecture of the Mos1 paired-end complex: the structural basis of DNA transposition in a eukaryote. Cell. 2009; 138:1096–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gogol-Doring A., Ammar I., Gupta S., Bunse M., Miskey C., Chen W., Uckert W., Schulz T.F., Izsvak Z., Ivics Z.. Genome-wide profiling reveals remarkable parallels between insertion site selection properties of the MLV retrovirus and the piggyBac transposon in primary human CD4(+) T cells. Mol. Ther. 2016; 24:592–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ivics Z., Hackett P.B., Plasterk R.H., Izsvak Z.. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997; 91:501–510. [DOI] [PubMed] [Google Scholar]
- 32. Ivics Z., Li M.A., Mates L., Boeke J.D., Nagy A., Bradley A., Izsvak Z.. Transposon-mediated genome manipulation in vertebrates. Nat. Methods. 2009; 6:415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Narayanavari S.A., Chilkunda S.S., Ivics Z., Izsvak Z.. Sleeping Beauty transposition: from biology to applications. Crit. Rev. Biochem. Mol. Biol. 2017; 52:18–44. [DOI] [PubMed] [Google Scholar]
- 34. Hackett P.B., Largaespada D.A., Cooper L.J.N.. A transposon and transposase system for human application. Mol. Ther. 2010; 18:674–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mates L., Chuah M.K., Belay E., Jerchow B., Manoj N., Acosta-Sanchez A., Grzela D.P., Schmitt A., Becker K., Matrai J.et al.. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 2009; 41:753–761. [DOI] [PubMed] [Google Scholar]
- 36. Boehme P., Doerner J., Solanki M., Jing L., Zhang W., Ehrhardt A.. The sleeping beauty transposon vector system for treatment of rare genetic diseases: an unrealized hope?. Curr. Gene Ther. 2015; 15:255–265. [DOI] [PubMed] [Google Scholar]
- 37. Hudecek M., Ivics Z.. Non-viral therapeutic cell engineering with the Sleeping Beauty transposon system. Curr. Opin. Genet. Dev. 2018; 52:100–108. [DOI] [PubMed] [Google Scholar]
- 38. Hudecek M., Izsvak Z., Johnen S., Renner M., Thumann G., Ivics Z.. Going non-viral: the Sleeping Beauty transposon system breaks on through to the clinical side. Crit. Rev. Biochem. Mol. Biol. 2017; 52:355–380. [DOI] [PubMed] [Google Scholar]
- 39. Di Matteo M., Belay E., Chuah M.K., Vandendriessche T.. Recent developments in transposon-mediated gene therapy. Expert. Opin. Biol. Ther. 2012; 12:841–858. [DOI] [PubMed] [Google Scholar]
- 40. Amberger M., Ivics Z.. Latest advances for the sleeping beauty transposon system: 23 years of insomnia but prettier than ever: refinement and recent innovations of the sleeping beauty transposon system enabling novel, nonviral genetic engineering applications. Bioessays. 2020; 42:e2000136. [DOI] [PubMed] [Google Scholar]
- 41. Kawakami K., Largaespada D.A., Ivics Z.. Transposons as tools for functional genomics in vertebrate models. Trends Genet. 2017; 33:784–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yant S.R., Wu X., Huang Y., Garrison B., Burgess S.M., Kay M.A.. High-resolution genome-wide mapping of transposon integration in mammals. Mol. Cell. Biol. 2005; 25:2085–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Grabundzija I., Irgang M., Mates L., Belay E., Matrai J., Gogol-Doring A., Kawakami K., Chen W., Ruiz P., Chuah M.K.et al.. Comparative analysis of transposable element vector systems in human cells. Mol. Ther. 2010; 18:1200–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Moldt B., Miskey C., Staunstrup N.H., Gogol-Doring A., Bak R.O., Sharma N., Mates L., Izsvak Z., Chen W., Ivics Z.et al.. Comparative genomic integration profiling of Sleeping Beauty transposons mobilized with high efficacy from integrase-defective lentiviral vectors in primary human cells. Mol. Ther. 2011; 19:1499–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ammar I., Gogol-Doring A., Miskey C., Chen W., Cathomen T., Izsvak Z., Ivics Z.. Retargeting transposon insertions by the adeno-associated virus Rep protein. Nucleic Acids Res. 2012; 40:6693–6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Voigt K., Gogol-Doring A., Miskey C., Chen W., Cathomen T., Izsvak Z., Ivics Z.. Retargeting sleeping beauty transposon insertions by engineered zinc finger DNA-binding domains. Mol. Ther. 2012; 20:1852–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li X., Ewis H., Hice R.H., Malani N., Parker N., Zhou L., Feschotte C., Bushman F.D., Atkinson P.W., Craig N.L.. A resurrected mammalian hAT transposable element and a closely related insect element are highly active in human cell culture. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:E478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vigdal T.J., Kaufman C.D., Izsvák Z., Voytas D.F., Ivics Z.. Common physical properties of DNA affecting target site selection of Sleeping Beauty and other Tc1/mariner transposable elements. J. Mol. Biol. 2002; 323:441–452. [DOI] [PubMed] [Google Scholar]
- 49. Liu G., Geurts A.M., Yae K., Srinivasan A.R., Fahrenkrug S.C., Largaespada D.A., Takeda J., Horie K., Olson W.K., Hackett P.B.. Target-site preferences of Sleeping Beauty transposons. J. Mol. Biol. 2005; 346:161–173. [DOI] [PubMed] [Google Scholar]
- 50. Arinkin V., Smyshlyaev G., Barabas O.. Jump ahead with a twist: DNA acrobatics drive transposition forward. Curr. Opin. Struct. Biol. 2019; 59:168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Abrusan G., Yant S.R., Szilagyi A., Marsh J.A., Mates L., Izsvak Z., Barabas O., Ivics Z.. Structural determinants of sleeping beauty transposase activity. Mol. Ther. 2016; 24:1369–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Voigt F., Wiedemann L., Zuliani C., Querques I., Sebe A., Mates L., Izsvak Z., Ivics Z., Barabas O.. Sleeping Beauty transposase structure allows rational design of hyperactive variants for genetic engineering. Nat. Commun. 2016; 7:11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. van Zundert G.C.P., Rodrigues J., Trellet M., Schmitz C., Kastritis P.L., Karaca E., Melquiond A.S.J., van Dijk M., de Vries S.J., Bonvin A.. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016; 428:720–725. [DOI] [PubMed] [Google Scholar]
- 54. Passos D.O., Li M., Yang R., Rebensburg S.V., Ghirlando R., Jeon Y., Shkriabai N., Kvaratskhelia M., Craigie R., Lyumkis D.. Cryo-EM structures and atomic model of the HIV-1 strand transfer complex intasome. Science. 2017; 355:89–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kesselring L., Miskey C., Zuliani C., Querques I., Kapitonov V., Lauko A., Feher A., Palazzo A., Diem T., Lustig J.et al.. A single amino acid switch converts the Sleeping Beauty transposase into an efficient unidirectional excisionase with utility in stem cell reprogramming. Nucleic Acids Res. 2020; 48:316–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Querques I., Mades A., Zuliani C., Miskey C., Alb M., Grueso E., Machwirth M., Rausch T., Einsele H., Ivics Z.et al.. A highly soluble Sleeping Beauty transposase improves control of gene insertion. Nat. Biotechnol. 2019; 37:1502–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen S., Zhou Y., Chen Y., Gu J.. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018; 34:i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cattoglio C., Pellin D., Rizzi E., Maruggi G., Corti G., Miselli F., Sartori D., Guffanti A., Di Serio C., Ambrosi A.et al.. High-definition mapping of retroviral integration sites identifies active regulatory elements in human multipotent hematopoietic progenitors. Blood. 2010; 116:5507–5517. [DOI] [PubMed] [Google Scholar]
- 60. Holstein M., Mesa-Nunez C., Miskey C., Almarza E., Poletti V., Schmeer M., Grueso E., Ordonez Flores J.C., Kobelt D., Walther W.et al.. Efficient non-viral gene delivery into human hematopoietic stem cells by minicircle sleeping beauty transposon vectors. Mol. Ther. 2018; 26:1137–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Akalin A., Franke V., Vlahovicek K., Mason C.E., Schubeler D.. Genomation: a toolkit to summarize, annotate and visualize genomic intervals. Bioinformatics. 2015; 31:1127–1129. [DOI] [PubMed] [Google Scholar]
- 62. Ernst J., Kellis M.. ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods. 2012; 9:215–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hoffman M.M., Buske O.J., Wang J., Weng Z., Bilmes J.A., Noble W.S.. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nat. Methods. 2012; 9:473–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sadelain M., Papapetrou E.P., Bushman F.D.. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer. 2012; 12:51–58. [DOI] [PubMed] [Google Scholar]
- 65. Kaplan N., Moore I.K., Fondufe-Mittendorf Y., Gossett A.J., Tillo D., Field Y., LeProust E.M., Hughes T.R., Lieb J.D., Widom J.et al.. The DNA-encoded nucleosome organization of a eukaryotic genome. Nature. 2009; 458:362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Abeel T., Saeys Y., Bonnet E., Rouze P., Van de Peer Y.. Generic eukaryotic core promoter prediction using structural features of DNA. Genome Res. 2008; 18:310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vlahovicek K., Kajan L., Pongor S.. DNA analysis servers: plot.it, bend.it, model.it and IS. Nucleic Acids Res. 2003; 31:3686–3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Serrao E., Ballandras-Colas A., Cherepanov P., Maertens G.N., Engelman A.N.. Key determinants of target DNA recognition by retroviral intasomes. Retrovirology. 2015; 12:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang Y., Pryputniewicz-Dobrinska D., Nagy E.E., Kaufman C.D., Singh M., Yant S., Wang J., Dalda A., Kay M.A., Ivics Z.et al.. Regulated complex assembly safeguards the fidelity of Sleeping Beauty transposition. Nucleic Acids Res. 2017; 45:311–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Monjezi R., Miskey C., Gogishvili T., Schleef M., Schmeer M., Einsele H., Ivics Z., Hudecek M.. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia. 2017; 31:186–194. [DOI] [PubMed] [Google Scholar]
- 71. De Ravin S.S., Su L., Theobald N., Choi U., Macpherson J.L., Poidinger M., Symonds G., Pond S.M., Ferris A.L., Hughes S.H.et al.. Enhancers are major targets for murine leukemia virus vector integration. J. Virol. 2014; 88:4504–4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Cavazza A., Moiani A., Mavilio F.. Mechanisms of retroviral integration and mutagenesis. Hum. Gene Ther. 2013; 24:119–131. [DOI] [PubMed] [Google Scholar]
- 73. Roadmap Epigenomics C., Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J.et al.. Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Papapetrou E.P., Lee G., Malani N., Setty M., Riviere I., Tirunagari L.M., Kadota K., Roth S.L., Giardina P., Viale A.et al.. Genomic safe harbors permit high beta-globin transgene expression in thalassemia induced pluripotent stem cells. Nat. Biotechnol. 2011; 29:73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Guo Y., Park J.M., Cui B., Humes E., Gangadharan S., Hung S., FitzGerald P.C., Hoe K.L., Grewal S.I., Craig N.L.et al.. Integration profiling of gene function with dense maps of transposon integration. Genetics. 2013; 195:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gangadharan S., Mularoni L., Fain-Thornton J., Wheelan S.J., Craig N.L.. DNA transposon Hermes inserts into DNA in nucleosome-free regions in vivo. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21966–21972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Schones D.E., Cui K., Cuddapah S., Roh T.Y., Barski A., Wang Z., Wei G., Zhao K.. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008; 132:887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Baldi P., Chauvin Y., Brunak S., Gorodkin J., Pedersen A.G.. Computational applications of DNA structural scales. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998; 6:35–42. [PubMed] [Google Scholar]
- 79. Nelson H.C., Finch J.T., Luisi B.F., Klug A.. The structure of an oligo(dA).oligo(dT) tract and its biological implications. Nature. 1987; 330:221–226. [DOI] [PubMed] [Google Scholar]
- 80. Yakovchuk P., Protozanova E., Frank-Kamenetskii M.D.. Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Res. 2006; 34:564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. de Jong J., Akhtar W., Badhai J., Rust A.G., Rad R., Hilkens J., Berns A., van Lohuizen M., Wessels L.F., de Ridder J.. Chromatin landscapes of retroviral and transposon integration profiles. PLoS Genet. 2014; 10:e1004250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Guo Y., Zhang Y., Hu K.. Sleeping Beauty transposon integrates into non-TA dinucleotides. Mobile DNA. 2018; 9:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Olson W.K., Gorin A.A., Lu X.J., Hock L.M., Zhurkin V.B.. DNA sequence-dependent deformability deduced from protein-DNA crystal complexes. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:11163–11168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Montano S.P., Pigli Y.Z., Rice P.A.. The mu transpososome structure sheds light on DDE recombinase evolution. Nature. 2012; 491:413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Pribil P.A., Haniford D.B.. Target DNA bending is an important specificity determinant in target site selection in Tn10 transposition. J. Mol. Biol. 2003; 330:247–259. [DOI] [PubMed] [Google Scholar]
- 86. Ghanim G.E., Kellogg E.H., Nogales E., Rio D.C.. Structure of a P element transposase-DNA complex reveals unusual DNA structures and GTP-DNA contacts. Nat. Struct. Mol. Biol. 2019; 26:1013–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chen Q., Luo W., Veach R.A., Hickman A.B., Wilson M.H., Dyda F.. Structural basis of seamless excision and specific targeting by piggyBac transposase. Nat. Commun. 2020; 11:3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Liu C., Yang Y., Schatz D.G.. Structures of a RAG-like transposase during cut-and-paste transposition. Nature. 2019; 575:540–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bellen H.J., Levis R.W., Liao G., He Y., Carlson J.W., Tsang G., Evans-Holm M., Hiesinger P.R., Schulze K.L., Rubin G.M.et al.. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics. 2004; 167:761–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liao G.C., Rehm E.J., Rubin G.M.. Insertion site preferences of the P transposable element in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:3347–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Spradling A.C., Stern D.M., Kiss I., Roote J., Laverty T., Rubin G.M.. Gene disruptions using P transposable elements: an integral component of the Drosophila genome project. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:10824–10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Roth S.L., Malani N., Bushman F.D.. Gammaretroviral integration into nucleosomal target DNA in vivo. J. Virol. 2011; 85:7393–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Muller H.P., Varmus H.E.. DNA bending creates favored sites for retroviral integration: an explanation for preferred insertion sites in nucleosomes. EMBO J. 1994; 13:4704–4714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pryciak P.M., Varmus H.E.. Nucleosomes, DNA-binding proteins, and DNA sequence modulate retroviral integration target site selection. Cell. 1992; 69:769–780. [DOI] [PubMed] [Google Scholar]
- 95. Baller J.A., Gao J., Stamenova R., Curcio M.J., Voytas D.F.. A nucleosomal surface defines an integration hotspot for the Saccharomyces cerevisiae Ty1 retrotransposon. Genome Res. 2012; 22:704–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Hacein-Bey-Abina S., Von Kalle C., Schmidt M., McCormack M.P., Wulffraat N., Leboulch P., Lim A., Osborne C.S., Pawliuk R., Morillon E.et al.. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003; 302:415–419. [DOI] [PubMed] [Google Scholar]
- 97. Hacein-Bey-Abina S., Garrigue A., Wang G.P., Soulier J., Lim A., Morillon E., Clappier E., Caccavelli L., Delabesse E., Beldjord K.et al.. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008; 118:3132–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ott M.G., Schmidt M., Schwarzwaelder K., Stein S., Siler U., Koehl U., Glimm H., Kuhlcke K., Schilz A., Kunkel H.et al.. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Medicine. 2006; 12:401–409. [DOI] [PubMed] [Google Scholar]
- 99. Howe S.J., Mansour M.R., Schwarzwaelder K., Bartholomae C., Hubank M., Kempski H., Brugman M.H., Pike-Overzet K., Chatters S.J., de Ridder D.et al.. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig. 2008; 118:3143–3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Robert X., Gouet P.. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014; 42:W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main article and in Supplementary Information and Supplementary Datasets.