


Keywords: clinical trial, creatinine clearance, dietary sodium, kidney injury molecule-1, neutrophil gelatinase-associated lipocalin, sodium chloride
Abstract
In rodents and older patients with elevated blood pressure (BP), high dietary sodium increases excretion of biomarkers of kidney injury, but it is unclear whether this effect occurs in healthy young adults. The purpose of this study was to determine whether short-term high dietary salt increases urinary excretion of the kidney injury biomarkers neutrophil gelatinase-associated lipocalin (NGAL) and kidney injury molecule-1 (KIM-1) in healthy young adults. Twenty participants participated in a double-blind, placebo-controlled, randomized crossover study. For 10 days each, participants were asked to consume salt (3,900 mg sodium) or placebo capsules. We measured BP during each visit, obtained 24-h urine samples for measurements of electrolytes, NGAL, and KIM-1, and assessed creatinine clearance. Compared with placebo, salt loading increased daily urinary sodium excretion (placebo: 130.3 ± 62.4 mmol/24 h vs. salt: 287.2 ± 72.0 mmol/24 h, P < 0.01). There was no difference in mean arterial BP (placebo: 77 ± 7 mmHg vs. salt: 77 ± 6 mmHg, P = 0.83) between conditions. However, salt loading increased the urinary NGAL excretion rate (placebo: 59.8 ± 44.4 ng/min vs. salt: 80.8 ± 49.5 ng/min, P < 0.01) and increased creatinine clearance (placebo: 110.5 ± 32.9 mL/min vs. salt: 145.0 ± 24.9 mL/min, P < 0.01). Urinary KIM-1 excretion was not different between conditions. In conclusion, in healthy young adults 10 days of dietary salt loading increased creatinine clearance and increased urinary excretion of the kidney injury biomarker marker NGAL but not KIM-1.
NEW & NOTEWORTHY In healthy young adults, 10 days of dietary salt loading increased creatinine clearance and increased urinary excretion of the kidney injury biomarker marker neutrophil gelatinase-associated lipocalin despite no change in resting blood pressure.
INTRODUCTION
The vast majority of Americans consume more than the Dietary Guidelines for Americans recommendation of no more than 2,300 mg sodium/day (1–3). Notably, high dietary sodium increases the risk of cardiovascular and kidney disease (4–6), and there is growing interest in sensitive biomarkers of kidney injury to identify individuals who may be at higher risk for developing future kidney disease with high salt consumption. Neutrophil gelatinase-associated lipocalin (NGAL) and kidney injury molecule-1 (KIM-1) are established kidney tubule injury biomarkers (7–9) and have shown promise as biomarkers of acute kidney injury and chronic kidney disease in the general population (7, 10–13). Potential mechanisms by which high dietary sodium may cause tubular injury include high salt-induced blunted renal autoregulation and increased oxidative stress in the kidney (14–16). Studies in normotensive (17) and hypertensive (18, 19) rodents have demonstrated that high dietary sodium increases urinary excretion of urinary NGAL and/or KIM-1. A recent investigation in older adults also demonstrated that a modest reduction in habitual dietary sodium intake reduced the urinary concentration of NGAL (20). However, it is unclear whether short-term changes in sodium intake influence kidney injury biomarkers in healthy young adults.
Blood pressure (BP) variability is associated with target organ damage (21–23), and preclinical data have demonstrated that high dietary salt increases BP variability (24). From a clinical standpoint, increased BP variability with salt loading could contribute to organ damage in high-flow resistance organs such as the kidney. However, the limited human data on salt and BP variability are equivocal (25, 26). Cross-sectional data indicate that salt intake is associated with BP variability in patients with hypertension (25). In contrast, a recent report demonstrated that 10 days of controlled high-salt feeding, compared with low and recommended sodium, did not increase BP variability in healthy adults (26). Additionally, in patients with hypertension, high salt intake is associated with a high glomerular filtration rate (GFR; >135 mL/min) (27), which is prognostic of future cardiovascular events (28). In otherwise healthy adults, there are data suggesting that high salt does (29–31) and does not (31, 32) influence GFR. Thus, there is a need for additional data on the effects of salt on BP variability and kidney function in healthy adults as well.
Therefore, the primary purpose of the present study was to determine whether short-term high dietary sodium increases urinary excretion of the kidney injury biomarkers NGAL and KIM-1 in healthy young adults. We recently demonstrated that high dietary salt loading caused BP dysregulation and vascular dysfunction in a cohort of healthy young adults in placebo-controlled, randomized studies (33, 34). Here, we hypothesized that short-term high dietary sodium would increase urinary excretion of NGAL and KIM-1 in this same cohort. Because it is unclear whether high dietary salt elicits changes in GFR in healthy adults, we assessed creatinine clearance, urinary cystatin C, and urinary urea as additional measures of kidney function, and we speculated that GFR would be unaffected. Finally, we sought to determine whether beat-to-beat BP variability was adversely influenced (i.e., increased) by salt manipulation. The findings of this investigation will provide additional insights into the relation between dietary salt and future kidney disease by determining whether short-term high dietary sodium increases kidney injury biomarkers, alters creatinine clearance, and increases BP variability in healthy adults.
METHODS
Study Participants
All participants provided written and verbal informed consent before engaging in any study activities. The study protocol and procedures were approved by the Institutional Review Board of the University of Delaware. They conformed with the provisions of the Declaration of Helsinki, and the trial was registered on clinicaltrials.gov (NCT03565653). The primary data reported here consisted of a post hoc analysis using frozen urine and plasma samples. Additional details of the parent study, conducted from 2017 to 2019, have been provided in two prior publications (33, 34). Participant age ranged from 18 to 34 yr. Exclusion criteria included a history of hypertension, diagnosis of cardiovascular disease, cancer, type 1 or type 2 diabetes, kidney dysfunction, current pregnancy, obesity (body mass index > 30 kg/m2), and current use of tobacco products (i.e., within the preceding 6 mo).
Sodium Intervention
We enrolled participants into a randomized double-blind, placebo-controlled crossover study. We instructed participants to consume a recommended sodium diet (2,300 mg/day) in each of two 10-day periods. Our prior publications with the salt capsule model have indicated that this duration is sufficient to elicit physiological effects (33, 34). We provided participants with instructions for interpreting nutrition labels and meeting sodium intake requirements. Participants consumed unmarked capsules each day containing either salt [Morton table salt (NaCl), 3,900 mg sodium/day] or a placebo (NOW Foods dextrose). Total sodium intake was designed to be 6,200 mg/day during the high-salt condition and 2,300 mg/day during the dextrose condition. We randomized the condition order and separated conditions by ≥2 wk for male participants and by 1 mo for female participants. Prior data have demonstrated that randomized-order diets with no washout period still result in demonstrable changes in vascular and autonomic function across diets (35, 36). All female participants (n = 8 of 20 total participants) were using oral hormonal contraceptives, and experimental visits occurred 2–6 days into their inactive week (i.e., when individuals take placebo pills rather than the contraceptive). Participants recorded their diet during the first intervention and were asked to match their diet during the second intervention via a copy of their initial diet log. We analyzed 3 days of diet records from a subset of participants (n = 13) including 2 weekdays and a weekend day with the Nutrition Data System for Research.
Twenty-Four-Hour Urine Collection
Participants were asked to collect all urine beginning after their first void on the ninth day of the intervention through their first void the morning of day 10 in a light-protected, sterile 3,500-mL container. We measured total urine volume, urine specific gravity (Goldberg Brix Refractometer, Reichert Technologies), urinary electrolyte concentrations (EasyElectrolyte Analyzer, Medica), and urinary osmolality (Advanced 3D3 Osmometer, Advanced Instruments) from a mixed aliquot of the 24-h urine collection container. The urine flow rate was derived from urine volume and self-reported time that the participant used the container (placebo: 1,357 ± 135 min and salt: 1,378 ± 192 min, P = 0.616). We also stored mixed aliquots from the 24-h collection container in cryogenic tubes at −80°C.
Experimental Visits
On the 10th day of each diet, participants were instrumented for single-lead electrocardiography and oscillometric BP measurements after ≥15 min of supine rest. Beat-to-beat BP was recorded via photoplethysmography (Finometer, Finapres Medical Systems) at the finger. Systolic BP and diastolic BP were defined as the maximum and minimum values, respectively, from the arterial BP waveform during each cardiac cycle (LabChart 8). Mean arterial BP was calculated as the integral of the BP waveform (26, 37).
We calculated beat-to-beat BP variability over 10 min of quiet rest in the laboratory. As we have previously described (26), we assessed BP variability using the standard deviation (SD) and average real variability (ARV) index. The SD method has frequently been used for BP in the literature and is associated with cardiovascular morbidity and mortality (38). The SD of systolic, diastolic, and mean BP was determined as the SD of all BPs during the 10-min recording with the following equation:
where σ is the population SD, xi is each individual value from the population (each BP value), µ is the population mean, and N is the size of the population.
However, the ARV of BP has greater prognostic value (39) and is seen as a more precise representation of BP variability than SD. Thus, we assessed BP variability using both SD and ARV. Specifically, rather than measuring total variance within an array of BP measures, the ARV expresses the absolute difference of consecutive BP measurements and was calculated with the following formula (39):
where w is the time interval between BPk and BPk + 1, N is the number of BP readings, and k indicates the order of the measurements.
Regarding the utility of beat-to-beat BP variability as opposed to ambulatory BP variability or repeated clinic measures, several studies have demonstrated that beat-to-beat BP variability has prognostic value. For example, in middle-aged adults, just ∼10 s of central BP ARV and SD were associated with first and recurrent cardiovascular events (40). Additionally, BP variability derived from short-term (e.g., 5–10 min) beat-to-beat assessment has been associated with cardiovascular events (41), poor hospital outcomes (42), and organ damage (23) in patients with a history of cerebrovascular disease and hypertension. It is also worth noting that multiple recent investigations have used beat-to-beat BP variability measures in healthy young adults, similar to our cohort, to 1) characterize participants who may be at risk for future cardiovascular disease and morbidity based in part on augmented BP variability (43) and 2) determine the influence of diet on BP variability (44).
Venipuncture and Biochemical Analysis
We obtained venous plasma (K+-EDTA) and serum samples via venipuncture and centrifugation. We assessed serum electrolytes (EasyElectrolyte Analyzer, Medica), plasma osmolality (Advanced 3D3 Osmometer, Advanced Instruments), hemoglobin (Hb 201+, HemoCue), and hematocrit (Sure prep capillary tubes, Clay Adams) spun in a microcentrifuge at 1,950 g for 5 min (Legend Micro 17, Thermo Sorvall). As previously described (45), we calculated the percent change in plasma volume (expressed as a percentage) using changes in hemoglobin and hematocrit and the following equation (34):
where Hb is hemoglobin, Hct is hematocrit, and subscripts PLA and HS are the placebo and high-salt conditions, respectively.
Assays for Kidney Function
We measured plasma NGAL and KIM-1 and urine NGAL, KIM-1, cystatin C, and urea concentrations in triplicate with ELISAs. We obtained the ELISA kits from Toronto BioScience (catalog no. 31050) to measure plasma and urine NGAL (46), from R&D Systems to measure plasma KIM-1 (DSKM100) and urine KIM-1 (DKM100), and from Abcam to measure urine cystatin C (ab179883) and urea (ab83362). The intra-assay coefficients of variation were as follows: 3.1% for plasma NGAL, 6.7% for urine NGAL, 4.6% for plasma KIM-1, 4.4% for urine KIM-1, 4.3% for cystatin C, and 3.5% for urea. We performed all of the assays with manufacturer-provided instructions except for urine KIM-1, for which we loaded 200 µL (instead of 50 μL) of sample because of low reported kit sensitivity (20) and calculated the respective concentration [i.e., (concentration) × (50 µL/200 µL)]. Based on prior publications and manufacturer recommendations (30), we used the following dilutions for urine samples: 1:10 for NGAL, 1:200 for cystatin C, and 1:5,000 for urea. For urinary KIM-1, samples from four participants were below the sensitivity of the ELISA; however, we ran additional quality control samples (QC24, R&D Systems) to ensure validity of the values from all remaining samples. All values reported here for the other ELISAs were above the minimum detectable sensitivity for each of the respective kits. In addition to the undetectable KIM-1 concentration, urinary NGAL was missing from one participant. However, sample sizes are indicated in each graph. We calculated NGAL and KIM-1 excretion rates (Fig. 1B and Fig. 2B) as concentration of the assayed sample (ng/mL) × flow rate (mL/min), resulting in units of ng/min.
Figure 1.
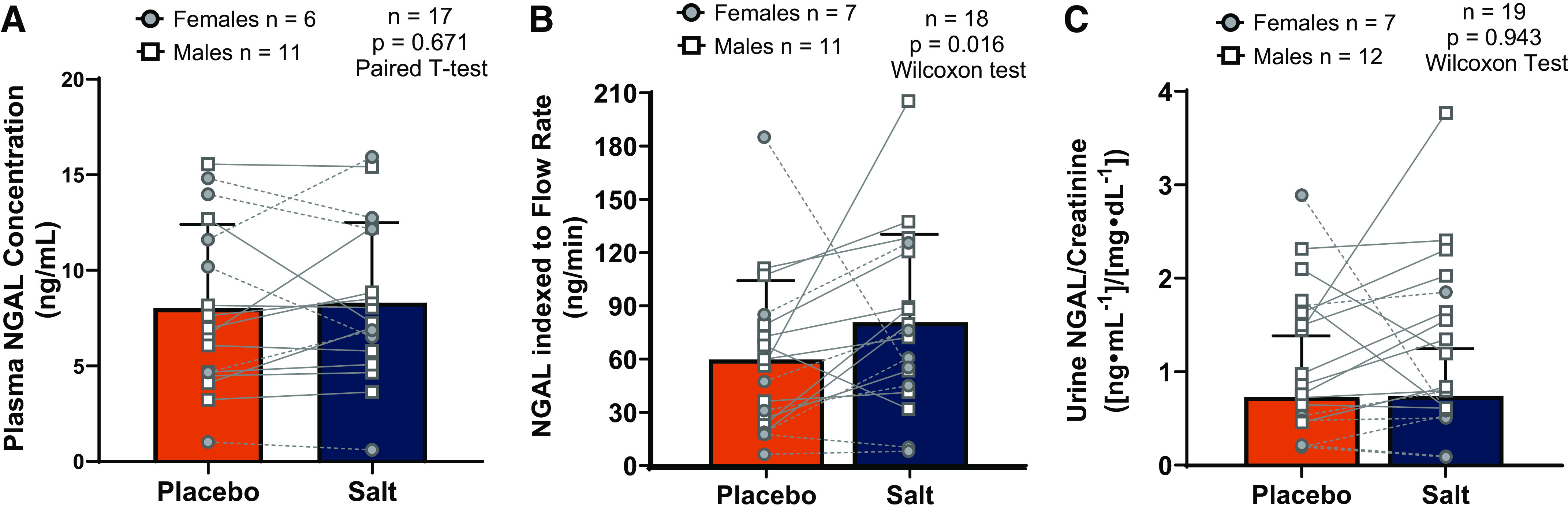
Effects of high dietary salt intake on neutrophil gelatinase-associated lipocalin (NGAL). A: plasma NGAL was not affected by salt loading. B: urine NGAL indexed to urine flow rate was increased after 10 days of dietary salt loading. C: NGAL indexed to creatinine concentration was not affected by salt loading. Sample sizes and statistical tests are reported in each graph. Statistical analyses were completed on all participants, but we have visually depicted females and males separately. All data are presented as individual data points overlaid with means ± SD.
Figure 2.
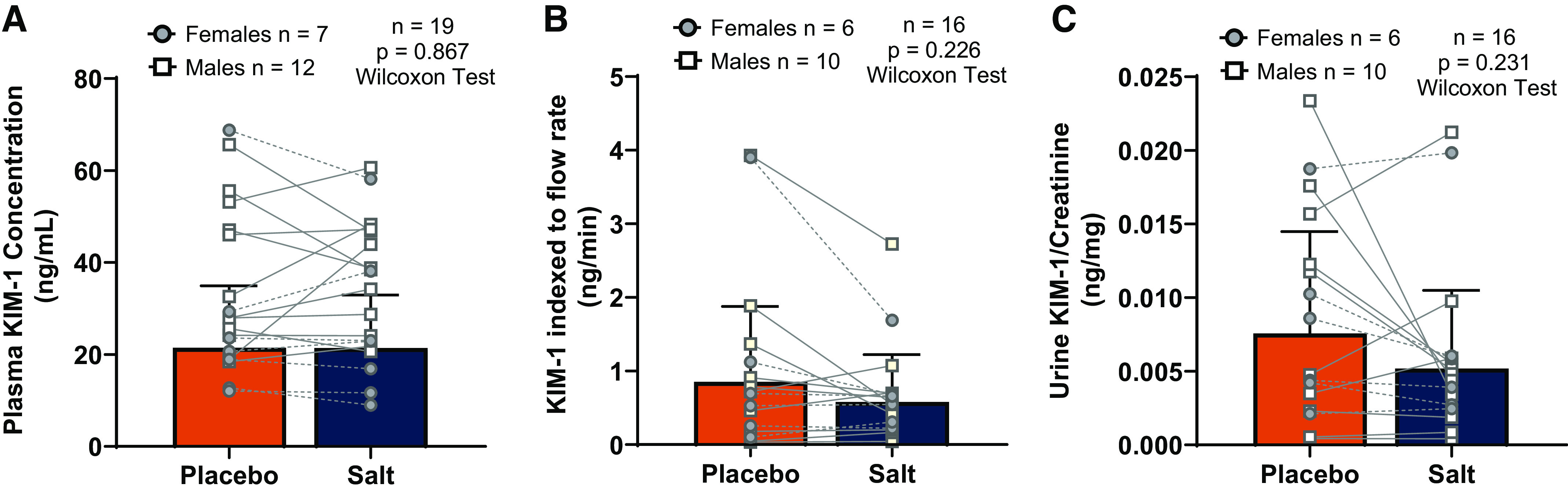
Effects of high dietary salt intake on kidney injury molecule-1 (KIM-1). A: plasma KIM-1 was not affected by salt loading. B and C: urine KIM-1 indexed to urine flow rate (B) and KIM-1 indexed to urine creatinine concentration (C) were not affected by salt loading. Sample sizes and statistical tests are reported in each graph. Statistical analyses were completed on all participants, but we have visually depicted females and males separately. All data are presented as individual data points overlaid with means ± SD.
We used frozen urine and serum samples to measure creatinine via kinetic modification of the Jaffe procedure; the measurements were performed at Christiana Care Lab Services (Newark, DE). As previously described (48), we assessed creatinine clearance, both adjusted and unadjusted for body surface area, as a measure of GFR using urine and serum creatinine concentration and urine flow rate.
We also calculated estimated GFR (eGFR) using serum creatinine with the 2009 Chronic Kidney Disease Epidemiology Collaboration equation (49). Based on prior publications, we defined a “high GFR” as >135 mL/min (48, 50). eGFR was calculated and presented to describe the cohort during screening. However, creatinine clearance data were used to estimate GFR throughout the remainder of the study when we compared the salt intervention with the placebo intervention. We also calculated the fractional excretion of sodium from sodium concentration in the serum and urine as well as the change in serum and urinary creatinine concentration (27, 46). Plasma angiotensin II concentration was measured via radioimmunoassay at the Biomarker Analytical Core at Wake Forest University. Creatinine clearance data were missing from two participants because of their self-reported urine volume resulting in a creatinine clearance of >1.5 times the interquartile range.
Statistical Analysis
We used two-tailed t tests when data were normally distributed. When data were not normally distributed, we used Wilcoxon matched-pairs signed-rank tests. We tested plasma volume change with a one-sample t test. We used simple regression analysis to assess the relation between changes of angiotensin II and creatinine clearance. Statistical significance was set at P < 0.05. We also calculated effect size using Cohen’s d for select variables to provide further context when interpreting data. Statistical analyses were completed with IBM SPSS 26.0 and GraphPad Prism 9.0. Data are presented as means ± SD
RESULTS
Participant characteristics, including age, sex, body mass index, BP, and eGFR, are shown in Table 1. Participants were nonobese and had normal to elevated BP.
Table 1.
Participant descriptive characteristics at screening
| Participant Characteristic | Value | Range |
|---|---|---|
| Age, yr | 24.2 ± 3.9 | 18–34 |
| Number of participants (female/male) | 20 (8/12) | |
| Race/ethnicity | 13 NHW, 1 BR, 4 A, 2 H | |
| Body height, cm | 172.8 ± 9.8 | 152.4–185.4 |
| Body mass, kg | 69.4 ± 12.5 | 48.6–96.8 |
| Body mass index, kg/m2 | 23.1 ± 2.6 | 18.4–28.9 |
| Brachial systolic blood pressure, mmHg | 112 ± 10 | 86–136 |
| Brachial mean blood pressure, mmHg | 80 ± 8 | 50–80 |
| Brachial diastolic blood pressure, mmHg | 64 ± 8 | 64–93 |
| Estimated glomerular filtration rate, mL/min | 126 ± 27 | 87–176 |
Data are presented as means ± SD; n = 18–20. Estimated glomerular filtration rate was derived from the Chronic Kidney Disease Epidemiology Collaboration equation (2009). A, Asian; BR, biracial; H, Hispanic; NHW, non-Hispanic white. t tests were used for analysis.
Responses to Interventions
The effects of the capsule intervention on physical and biochemical measures are shown in Table 2. Urine sodium excretion, estimated plasma volume, total body mass, and serum chloride were higher after 10 days of the salt intervention compared with the placebo intervention. In contrast, brachial systolic BP, brachial mean BP, brachial diastolic BP, plasma osmolality, and serum sodium and potassium were not different between conditions (Table 2). Beat-to-beat systolic, mean, and diastolic BP ARV were also not different between conditions (Table 2). Compared with after 10 days of the placebo intervention, the salt loading intervention resulted in a modestly yet significantly greater SD of beat-to-beat systolic BP (placebo: 5.6 ± 2.2 mmHg vs. salt loading: 6.5 ± 1.9 mmHg, P = 0.035). However, the SD of beat-to-beat mean BP (placebo: 4.5 ± 1.6 mmHg vs. salt loading: 4.9 ± 1.4 mmHg, P = 0.186) and beat-to-beat diastolic BP (placebo: 3.9 ± 1.4 mmHg vs. salt loading: 4.4 ± 1.2 mmHg, P = 0.091) were not different between conditions. Because the SD of systolic BP was the only significantly increased measure of variability, we confirmed that the effect size was also moderately large, using Cohen’s d (d = 0.44).
Table 2.
Effects of salt loading on blood pressure and biochemical measures
| Placebo | Salt | P Value | |
|---|---|---|---|
| Body mass, kg | 68.8 ± 12.8 | 69.2 ± 12.8 | 0.042* |
| Brachial systolic BP, mmHg | 108 ± 11 | 108 ± 8 | 0.902 |
| Brachial mean BP, mmHg | 77 ± 7 | 77 ± 6 | 0.830 |
| Brachial diastolic BP, mmHg | 61 ± 7 | 61 ± 6 | 0.914 |
| Systolic BP ARV, mmHg | 0.99 ± 0.40 | 1.09 ± 0.44 | 0.140 |
| Mean BP ARV, mmHg | 0.89 ± 0.29 | 0.95 ± 0.36 | 0.290 |
| Diastolic BP ARV, mmHg | 0.99 ± 0.36 | 1.03 ± 0.41 | 0.553 |
| Urine Na+ excretion, mmol/24 h | 130.3 ± 62.4 | 287.2 ± 72.0 | <0.001* |
| Plasma Osm, mosmol/kgH2O | 294 ± 5 | 294 ± 5 | 0.691 |
| Serum Na+, mmol/L | 141.0 ± 1.8 | 141.4 ± 2.1 | 0.319 |
| Serum K+, mmol/L | 4.1 ± 0.5 | 4.1 ± 0.5 | 0.505 |
| Serum Cl−, mmol/L | 104.8 ± 2.0 | 106.2 ± 1.9 | 0.017* |
| Plasma volume change, % | 7.2 ± 11.3 | 0.012* |
Data are presented as means ± SD; n = 18–20. Average real variability (ARV) was derived from beat-to-beat blood pressure (BP) readings. Osm, osmolality. *Statistical significance. t tests or Wilcoxon tests were used for continuous variables. Fisher’s exact tests were used for ordinal data.
Neutrophil Gelatinase-Associated Lipocalin
Plasma NGAL concentration was not different between conditions (placebo: 8.0 ± 4.4 ng/mL vs. salt loading: 8.3 ± 4.2 ng/mL; Fig. 1A). Salt loading significantly increased urinary NGAL excretion rate compared with placebo (placebo: 59.8 ± 44.4 ng/min vs. salt loading: 80.8 ± 49.5 ng/min; Fig. 1B). Apart from being significantly increased, the effect size for the key outcome measure, urine NGAL excretion rate (Cohen’s d = 0.46), was moderate. However, urinary NGAL concentration indexed to creatinine was not different between conditions (placebo: 0.730 ± 0.635 ng/mg vs. salt loading: 0.741 ± 0.490 ng/mg; Fig. 1C).
Kidney Injury Molecule-1
Plasma KIM-1 (placebo: 21.5 ± 13.5 ng/mL vs. salt loading: 21.4 ± 11.2 ng/mL; Fig. 2A), urinary KIM-1 concentration indexed to the flow rate (placebo: 0.85 ± 1.03 ng/min vs. salt loading: 0.58 ± 0.64 ng/min: Fig. 2B), and urinary KIM-1 concentration indexed to creatinine (placebo: 0.0076 ± 0.0069 ng/mg vs. salt loading: 0.0052 ± 0.0053 ng/mg; Fig. 2C) were also not different between conditions.
Kidney Function
Indexes of kidney function are shown in Table 3. Compared with the placebo intervention, salt loading resulted in greater creatinine clearance, both adjusted and unadjusted for body surface area, and greater fractional excretion of sodium. Compared with the placebo intervention, salt loading resulted in a higher incidence of high GFR when assessed via the creatinine clearance unadjusted, but not when adjusted, for body surface area. Urinary cystatin C excretion was greater after salt loading compared with the placebo intervention. Plasma angiotensin II concentration was reduced after salt loading compared with the placebo intervention (Table 3). Interestingly, there was a modest, but significant, relation between suppression of angiotensin II and creatinine clearance (R2 = 0.545, P < 0.001). Those with greater suppression of angiotensin II had less of an increase in creatinine clearance (Fig. 3). Urinary urea excretion was not different between conditions (Table 3). Finally, urine volume was modestly but significantly higher after 10 days of salt loading compared with the placebo intervention (placebo: 1,763 ± 812 mL/day vs. salt loading: 1,969 ± 925 mL/day, P = 0.047).
Table 3.
Effects of salt loading on measures of kidney function
| Placebo | Salt | P Value | |
|---|---|---|---|
| Urine creatinine excretion, mg/day | 1,240 ± 398 | 1,598 ± 231 | <0.001* |
| Serum creatinine, mg/dL | 0.8 ± 0.1 | 0.8 ± 0.1 | 0.552 |
| Unadjusted creatinine clearance, mL/min | 110.5 ± 32.9 | 145.4 ± 23.9 | <0.001* |
| High GFR | 6/18 (33%) | 12/18 (67%) | 0.046* |
| Adjusted creatinine clearance, per 1.73 m2 | 105.6 ± 30.8 | 137.0 ± 23.7 | <0.001* |
| High GFR | 6/18 (33%) | 10/18 (56%) | 0.180 |
| Urinary cystatin C excretion, mg/day | 0.15 ± 0.06 | 0.22 ± 0.09 | 0.015* |
| Urea excretion, g/day | 18.8 ± 13.4 | 19.9 ± 13.7 | 0.543 |
| Plasma angiotensin II, pg/mL | 20.9 ± 4.3 | 18.2 ± 3.1 | 0.006* |
| Fractional excretion of Na+, % | 0.62 ± 0.39 | 0.98 ± 0.29 | <0.001* |
Data are presented as means ± SD. GFR, glomerular filtration rate. *Statistical significance. t tests or Wilcoxon tests were used for continuous variables.
Figure 3.

Inverse relation between angiotensin II suppression and high salt-induced elevations in creatinine clearance (glomerular filtration rate). Results from simple bivariate linear regression are shown. Those with greater suppression of angiotensin II had less of a change in creatinine clearance. Statistical analysis was completed on all participants, but we have visually depicted females and males separately.
Regarding the participants’ self-reported nutrition data from their diet records, there was no difference in reported sodium intake between conditions (placebo: 2,506 ± 779 mg/day vs. salt loading: 2,752 ± 554 mg/day, P = 0.493). Based on 24-h urinary sodium excretion after 10 days of placebo (2,990 mg/day) and reported sodium intake on the placebo intervention (2,506 mg/day), it appears that the participants underreported dietary sodium by ∼16%.
DISCUSSION
The key findings of this investigation were that short-term dietary salt loading increases urinary excretion of the kidney injury biomarker NGAL in healthy young adults. Although NGAL was not increased when indexed to creatinine, this finding could be attributable to salt loading also eliciting increase in creatinine excretion (compared with placebo) in our cohort. A large body of literature demonstrates that high dietary salt is associated with the future development of chronic kidney disease (6) and adverse health outcomes associated with kidney disease, such as the initiation of dialysis and mortality (51). Our investigation provides unique insights demonstrating that even 10 days of high dietary salt elicits an increase in urinary NGAL excretion, an established clinical biomarker of kidney tubular injury.
Our observation that dietary salt results in increased urinary NGAL excretion is consistent with prior human work. A recent study in critically ill patients found that saline infusion modestly increased urinary NGAL excretion compared with balanced crystalloids (52). Importantly, intravenous saline provides water and sodium chloride (similar to dietary salt), whereas balanced crystalloids provide sodium, potassium, and chloride content closer to that of extracellular fluid concentrations and are typically associated with fewer adverse effects for acid-base balance. Our data are also consistent with another recent investigation that reported that 5 wk of dietary salt restriction resulted in a reduction in urinary NGAL concentration in older adults with moderately elevated BP (20). However, in the prior study urine NGAL concentration was reduced, but it was unclear whether urine NGAL excretion was reduced because urine volume or flow rate was not reported. In contrast to NGAL, we found that urinary excretion of the kidney injury biomarker KIM-1 was not elevated after 10 days of high dietary salt. Although there are rodent data demonstrating that high dietary salt results in increased KIM-1 excretion, it took ∼4 mo for hypertensive rats being fed a high-salt diet to exhibit elevated urinary excretion of KIM-1 (19). In contrast, normotensive rats fed a high-salt diet do not exhibit elevated urinary excretion of KIM-1 even after several weeks to months of high dietary salt (17, 18). Taken together, these findings indicate that NGAL may be a more sensitive marker of kidney injury and that even in more at-risk populations (e.g., hypertension) it may take longer-term high dietary salt to increase KIM-1 excretion. In the only other human study that we are aware of that investigated the influence of high dietary salt on KIM-1, the investigators were unable to detect KIM-1 in urine samples in all but one of the participants (20). Thus, there is a need for future studies to assess KIM-1 after longer-term habitual or experimental high dietary salt consumption and in higher-risk populations.
The molecular mechanisms underlying high dietary salt-induced renal tubular damage may include oxidative stress and activation of the local renin-angiotensin-aldosterone system (RAAS). Downregulation of endocytic receptors involved in NGAL reabsorption may also have played a role in increased NGAL excretion. Multiple rodent studies have indicated that high dietary salt increases NADPH oxidase (oxidant-producing enzyme) activity in the kidney (14, 15) and suppresses superoxide dismutase (antioxidant enzyme) activity in the kidney (14, 16). Regarding the RAAS, it is well known that salt suppresses the release of renin from the renal juxtaglomerular apparatus, causing a reduction in circulating angiotensin II. Indeed, we found that circulating angiotensin II was suppressed in our cohort after salt loading. Additionally, in the present investigation, there was a significant inverse relation between suppression of angiotensin II changes and creatinine clearance, suggesting that the plasma angiotensin II did reflect physiological regulation of kidney function. However, there is preclinical evidence that despite suppression of circulating angiotensin II, salt loading leads to increased activation of the intrarenal RAAS and increased angiotensin II content in proximal tubular fluid (53). Notably, these findings typically only occur in salt-sensitive rodent models (54). To our knowledge, there are no data supporting that prevention of oxidative stress (e.g., with antioxidants) or pharmacological manipulation of the RAAS attenuates increased excretion of kidney injury biomarkers in the context of dietary salt, but these are plausible targets to pursue in future human investigations related to dietary salt and kidney injury.
Studies of NGAL gene expression in acute renal impairment have demonstrated a rapid and large upregulation of NGAL mRNA in the distal nephron, in particular in the ascending limb of Henle’s loop and in the collecting duct (55). Extrarenal NGAL can be absorbed in the proximal tubules via endocytic receptors including megalin (low-density lipoprotein receptor-related protein 2) and cubulin (56–59). Specifically, megalin binds NGAL with high affinity, and uptake of NGAL into cultured cells is blocked by anti-megalin antibodies (58). Additionally, megalin-knockout mice, but not wild-type mice, excrete NGAL in their urine in the absence of any stimulus to promote kidney injury. Receptor-mediated endocytosis of NGAL may also occur by the 24p3 cell surface receptor (24p3R) in the distal tubule (60). Overexpression of 24p3R in multiple cell lines induces binding and uptake of NGAL (57); however, it has only been shown to localize to the distal tubule in the mammalian kidney (61). Interestingly, in salt-sensitive rodents, a high-salt diet resulted in a reduction in mRNA expression for megalin and cubulin, although the time course differed for the two receptors (19). There was an average ∼20% reduction in 24p3R, but this finding was not statistically significant. In contrast, megalin, cubulin, and 24p3R were all reduced with high dietary salt in Wistar-Kyoto rats (17). In human participant research, we are unable to measure expression of these endocytic receptors in the kidney tubules, but this could represent one pathway through which urinary NGAL could have been increased. KIM-1 is mainly upregulated in proximal tubule cells during acute kidney injury (62). Nonetheless, prior animal literature in normotensive rodents and our investigation in normotensive humans both indicate that high salt increases NGAL in the absence of an increase in KIM-1 (17). Whether or not this has to do with the location of NGAL versus KIM-1 production is unclear but is consistent with the literature that NGAL appears to be among the most rapid kidney injury biomarkers to become elevated in the context of kidney injury whereas KIM-1 may play more of a recovery role (61, 63).
Interestingly, in our cohort, high dietary salt increased urinary NGAL even though participants did not experience different resting BP between diets. We also assessed BP variability, because greater variability could also contribute to aberrant patterns of blood flow to the kidneys. BP variability is associated with target organ damage (21–23), and given our hypothesis that salt loading would elicit kidney injury, we sought to determine whether BP variability was also increased. Changes in BP variability are regulated by redundant homeostatic mechanisms, including the baroreceptor reflex, sympathetic tone, the RAAS, and the bioavailability of nitric oxide, all of which can be affected by dietary sodium (64, 65). Although the SD of systolic BP was significantly increased with salt loading in the present investigation, systolic BP ARV was not, and neither were measures of diastolic and mean BP variability. In addition to the SD of systolic BP being significantly increased with salt loading, the effect size was also moderately large. However, given that five of our six measures of BP variability were not changed with salt and that the ARV is seen as a more precise measure of beat-to-beat BP variability (as well as a stronger prognostic indicator) (39), it appears that BP variability was only minimally influenced by salt loading. Moreover, using regression analysis, we determined that the SD of systolic BP was not correlated with NGAL excretion (R2 = 0.002, P = 0.781).
In contrast to our findings in healthy humans, high dietary salt elevates nocturnal BP variability (measured over 2 h by telemetry) in salt-resistant rodents with normotensive BP (24). Additionally, 24-h urinary sodium excretion is positively correlated with 24-h ambulatory BP variability in patients with hypertension (25). Whereas the rodent study and cross-sectional study in patients measured BP over several hours, our BP variability was derived from a 10-min beat-to-beat recording. However, we also recently demonstrated that beat-to-beat and 24-h BP variability were not influenced by salt manipulation in a cohort of healthy adults who were largely salt resistant and who had normotensive BP (26). There are caveats to our largely null BP variability findings. For example, controlled salt loading interventions may result in increased BP variability in higher-risk populations (e.g., older adults or patients with hypertension) and in cohorts with a high proportion of individuals with salt-sensitive BP. Moreover, this study and our other recent investigation (26) were relatively short (i.e., 10 days). Although speculative, it could be that high dietary salt administered over longer periods of time elicits increased BP variability.
Given that our cohort was composed of healthy young adults who did not have salt-sensitive BP (i.e., a change of mean arterial BP of >5 mmHg), we did not anticipate that high dietary salt would elicit a large increase in creatinine clearance (31, 32). For example, in a prior controlled feeding study in healthy male cosmonauts, urinary creatinine excretion was not different across reported sodium intakes of ∼1,100 mg/day to greater than ∼5,500 mg/day, although the sample size was small and there was considerable overall variability (66). We also reasoned that 10 days of high salt is likely not sufficient to elicit hyperperfusion or increases in GFR. In such a scenario, the hyperperfusion would presumably injure the glomeruli and subsequently the tubules. However, there are prior investigations demonstrating that short-term (i.e., one to several weeks) of high dietary salt increased GFR in generally healthy adults (29–31). Interestingly, we also found that there was an inverse relation between suppression of the RAAS and increased creatinine clearance (Fig. 3), suggesting that inadequate suppression of the RAAS could play a role in salt-induced elevations in GFR. Importantly, a sustained elevation in GFR is an independent predictor of adverse cardiovascular events (28). Although we find it unlikely given the short-term intervention and health of the participants, the changes in creatinine clearance exhibited by our cohort suggest that there could have been an increase in kidney perfusion pressure with salt loading.
Underscoring the clinical relevance of our GFR findings, a recent cross-sectional investigation also determined that 24-h urine sodium excretion is associated with a higher prevalence of high GFR in patients with hypertension (27). The study also found that high salt intake was associated with protein catabolism as demonstrated by higher urea excretion (48). Interestingly, prior translational studies have also demonstrated that salt loading promotes protein catabolism (67, 68). These reports included speculation that high salt elicits protein catabolism to provide osmotically active urea to conserve body water. Specifically, in cosmonauts exposed to at least one month each of recommended sodium and of high sodium (68), the cosmonauts exhibited no increase in water consumption. The water-conserving mechanism of dietary salt excretion was reliant on urea transporter-driven urea recycling by the kidneys and on urea production by the liver and skeletal muscle. However, it has traditionally been thought that high dietary salt is associated with transiently increased serum sodium and osmolality, which subsequently stimulates thirst, leading to greater fluid consumption and increased plasma volume (69, 70).
In the present investigation, we did not find a difference in urea excretion between conditions, and participants did exhibit higher urine volume with salt loading. They also exhibited increased plasma volume, body mass, and fractional sodium excretion and decreased angiotensin II concentration in the plasma, all of which indicate at least some degree of water retention. Similarly, a recent secondary analysis of the Dietary Approaches to Stop Hypertension (DASH)-Sodium Trial also demonstrated that 4 wk of high dietary salt was not associated with changes in urea excretion (69). Although we did not assess thirst in our study, thirst was increased with higher dietary sodium in the DASH-Sodium Trial and there was a slight increase in body weight despite energy intake not being different between diets (69). Thus, our present findings and the DASH-Sodium findings indicate that high salt does not lead to protein catabolism or the utilization of urea as a fluid conservation mechanism. One potential reason for the discrepancy regarding high salt and protein catabolism between the cross-sectional study and cosmonaut trial and DASH-Sodium Trial and our study could be that the cosmonaut study and the cross-sectional study involved long-term high dietary sodium (several months to years), whereas our trial and the DASH-Sodium Trial were short term (i.e., ≤4 wk per condition).
In addition to creatinine clearance and urea, we also measured urine cystatin C to assess kidney function. Cystatin C is a 13-kDa cysteine protease inhibitor and is produced by all nucleated cells at a constant rate (71). In healthy participants, cystatin C is nearly freely filtered by the glomeruli and almost entirely reabsorbed and catabolized in the proximal tubules (71). Therefore, we speculated that cystatin C would likely not change with dietary sodium, as it is influenced to a lesser extent by factors such as age, biological sex, and muscle mass than creatinine is (72, 73). However, urinary cystatin C excretion was significantly increased (large effect size, Cohen’s d = 0.92). Thus, we speculate that the increase in cystatin C may be the result of kidney injury and reduced reabsorption in the proximal tubule, which has been associated with progression of nephropathies (72, 74). We did not measure plasma cystatin C, as plasma cystatin C is typically reported to be a more stable analyte than serum creatinine (73), which was unchanged across salt conditions.
Our study does have important limitations. All participants were given the same sodium load, whereas normalizing sodium to body mass or caloric intake (i.e., controlling for sodium density) may provide additional generalizability (75). Additionally, the participants’ urinary sodium excretion while on the placebo (∼2,990 mg) indicates that they consumed closer to habitual levels of dietary salt rather than the prescribed 2,300 mg. We are also aware of limitations regarding the use of creatinine clearance; however, the concomitant increase in urinary cystatin C gives us reason to believe that GFR was indeed elevated after 10 days of high dietary salt. Future investigations using gold-standard assessments of GFR (e.g., plasma iohexol clearance) are warranted. Given the significant difference between salt loading and placebo interventions and the moderate effect size, we are confident that salt loading did substantially increase NGAL excretion in this cohort of healthy adults despite the relatively modest sample size. It also remains to be determined whether habitual sodium intake is linked cross-sectionally with kidney injury markers. However, a prior study indicating that even modestly reducing habitual sodium reduces urine NGAL concentration in older adults suggests that this may be the case (20). The existing preclinical data (17–19) and the human data discussed here (20, 52) highlight the need for future investigations in large, diverse cohorts to further examine the role of dietary salt and kidney injury markers as well as underlying mechanisms.
Nonetheless, important strengths of our study include that 24-h urine collection is the gold standard for the assessment of sodium intake, as it is estimated to account for ≥90% of dietary sodium intake (76). Additionally, we used a within-participant crossover design to reduce variance. Finally, given that the participants in this investigation were generally healthy and young, our findings suggest that it is important for Americans to take preventative action to reduce sodium consumption from a cardiorenal perspective. The results may be even more pronounced with longer-term high sodium intake and in older adults, those with hypertension, and individuals with salt-sensitive BP. In conclusion, dietary salt loading elicited high GFR and increased urinary excretion of the kidney injury biomarker marker NGAL after short-term high sodium intake in healthy young adults.
DATA AVAILABILITY
Data will be made available by the corresponding author upon reasonable request.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants K01HL147998 (to A.T.R.) and R01HL128388 (to W.B.F.), American College of Sports Medicine Foundation Doctoral Student Research Grant 17-00521 (to M.C.B.), and American Heart Association Grant 18POST34060020 (to A.T.R.). This publication was also made possible by the Delaware COBRE in Cardiovascular Health, supported by NIH Grant P20GM113125.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.C.B., J.C.W., W.B.F., and A.T.R. conceived and designed research; A.M.B., M.C.B., J.C.W., K.U.M., and A.T.R. performed experiments; A.M.B., M.C.B., and A.T.R. analyzed data; A.M.B., M.C.B., J.C.W., K.U.M., O.M.G., W.B.F., and A.T.R. interpreted results of experiments; A.M.B., M.C.B., and A.T.R. prepared figures; A.M.B., M.C.B., and A.T.R. drafted manuscript; A.M.B., M.C.B., J.C.W., K.U.M., O.M.G., W.B.F., and A.T.R. edited and revised manuscript; A.M.B., M.C.B., J.C.W., K.U.M., O.M.G., W.B.F., and A.T.R. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Sofia Sanchez and Wendy Nichols for the support and assistance in this project. We also thank the individuals who participated in this study. Our graphical abstract was created with BioRender.com.
REFERENCES
- 1.Harnack LJ, Cogswell ME, Shikany JM, Gardner CD, Gillespie C, Loria CM, Zhou X, Yuan K, Steffen LM. Sources of sodium in US adults from 3 geographic regions. Circulation 135: 1775–1783, 2017. doi: 10.1161/CIRCULATIONAHA.116.024446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu JR, Sahni S, Mukamal KJ, Millar CL, Wu Y, Appel LJ, Juraschek SP. Dietary sodium intake and sodium density in the United States—estimates from NHANES 2005–2006 and 2015–2016. Am J Hypertens 33: 825–830, 2020. doi: 10.1093/ajh/hpaa104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Quader Z, Zhao L, Gillespie C, Cogswell M, Terry A, Moshfegh A, Rhodes D. Sodium intake among persons aged ≥2 years—United States, 2013–2014. Morb Mortal Wkly Rep 66: 324–338, 2017. doi: 10.15585/mmwr.mm6612a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cook NR, He FJ, MacGregor GA, Graudal N. Sodium and health-concordance and controversy. BMJ 369: m2440, 2020. doi: 10.1136/bmj.m2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cook NR, Appel LJ, Whelton PK. Sodium intake and all-cause mortality over 20 years in the trials of hypertension prevention. J Am Coll Cardiol 68: 1609–1617, 2016. doi: 10.1016/j.jacc.2016.07.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sugiura T, Ohte N, Dohi Y. Dietary salt intake is a significant determinant of impaired kidney function in the general population. Kidney Blood Press Res 43: 1245–1254, 2018. doi: 10.1159/000492406. [DOI] [PubMed] [Google Scholar]
- 7.Ko GJ, Grigoryev DN, Linfert D, Jang HR, Watkins T, Cheadle C, Racusen L, Rabb H. Transcriptional analysis of kidneys during repair from AKI reveals possible roles for NGAL and KIM-1 as biomarkers of AKI-to-CKD transition. Am J Physiol Renal Physiol 298: F1472–F1483, 2010. doi: 10.1152/ajprenal.00619.2009. [DOI] [PubMed] [Google Scholar]
- 8.Vaidya VS, Waikar SS, Ferguson MA, Collings FB, Sunderland K, Gioules C, Bradwin G, Matsouaka R, Betensky RA, Curhan GC, Bonventre JV. Urinary biomarkers for sensitive and specific detection of acute kidney injury in humans. Clin Transl Sci 1: 200–208, 2008. doi: 10.1111/j.1752-8062.2008.00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koyner JL, Vaidya VS, Bennett MR, Ma Q, Worcester E, Akhter SA, Raman J, Jeevanandam V, O’Connor MF, Devarajan P, Bonventre JV, Murray PT. Urinary biomarkers in the clinical prognosis and early detection of acute kidney injury. Clin J Am Soc Nephrol 5: 2154–2165, 2010. doi: 10.2215/CJN.00740110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhavsar NA, Köttgen A, Coresh J, Astor BC. Neutrophil gelatinase-associated lipocalin (NGAL) and kidney injury molecule 1 (KIM-1) as predictors of incident CKD stage 3: the Atherosclerosis Risk in Communities (ARIC) Study. Am J Kidney Dis 60: 233–240, 2012. doi: 10.1053/j.ajkd.2012.02.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peralta CA, Katz R, Bonventre JV, Sabbisetti V, Siscovick D, Sarnak M, Shlipak MG. Associations of urinary levels of kidney injury molecule 1 (KIM-1) and neutrophil gelatinase-associated lipocalin (NGAL) with kidney function decline in the Multi-Ethnic Study of Atherosclerosis (MESA). Am J Kidney Dis 60: 904–911, 2012. doi: 10.1053/j.ajkd.2012.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang WR, Craven TE, Malhotra R, Cheung AK, Chonchol M, Drawz P, Sarnak MJ, Parikh CR, Shlipak MG, Ix JH; SPRINT Research Group. Kidney damage biomarkers and incident CKD during blood pressure reduction: a case-control study within SPRINT. Ann Intern Med 169: 610–618, 2018. doi: 10.7326/M18-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel ML, Sachan R, Misra R, Kamal R, Sachan P, Shyam R. Prognostic significance of urinary NGAL in chronic kidney disease. Int J Nephrol Renovasc Dis 8: 139–144, 2015. doi: 10.2147/IJNRD.S87423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitiyakara C, Chabrashvili T, Chen Y, Blau J, Karber A, Aslam S, Welch WJ, Wilcox CS. Salt intake, oxidative stress, and renal expression of NADPH oxidase and superoxide dismutase. J Am Soc Nephrol 14: 2775–2782, 2003. doi: 10.1097/01.ASN.0000092145.90389.65. [DOI] [PubMed] [Google Scholar]
- 15.Fellner RC, Cook AK, O’Connor PM, Zhang S, Pollock DM, Inscho EW. High-salt diet blunts renal autoregulation by a reactive oxygen species-dependent mechanism. Am J Physiol Renal Physiol 307: F33–F40, 2014. doi: 10.1152/ajprenal.00040.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meng S, Roberts LJ 2nd, Cason GW, Curry TS, Manning RD. Jr.. Superoxide dismutase and oxidative stress in Dahl salt-sensitive and -resistant rats. Am J Physiol Regul Integr Comp Physiol 283: R732–R738, 2002. doi: 10.1152/ajpregu.00346.2001. [DOI] [PubMed] [Google Scholar]
- 17.Washino S, Hosohata K, Jin D, Takai S, Miyagawa T. Early urinary biomarkers of renal tubular damage by a high-salt intake independent of blood pressure in normotensive rats. Clin Exp Pharmacol Physiol 45: 261–268, 2018. doi: 10.1111/1440-1681.12871. [DOI] [PubMed] [Google Scholar]
- 18.De Miguel C, Sedaka R, Kasztan M, Lever JM, Sonnenberger M, Abad A, Jin C, Carmines PK, Pollock DM, Pollock JS. Tauroursodeoxycholic acid (TUDCA) abolishes chronic high salt‐induced renal injury and inflammation. Acta Physiol (Oxf) 226: e13227, 2019. doi: 10.1111/apha.13227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hosohata K, Yoshioka D, Tanaka A, Ando H, Fujimura A. Early urinary biomarkers for renal tubular damage in spontaneously hypertensive rats on a high salt intake. Hypertens Res 39: 19–26, 2016. doi: 10.1038/hr.2015.103. [DOI] [PubMed] [Google Scholar]
- 20.Wang W, Chonchol M, Seals DR, Nowak KL. Dietary sodium restriction decreases urinary NGAL in older adults with moderately elevated systolic blood pressure free from chronic kidney disease. J Investig Med 68: 1271–1275, 2020. doi: 10.1136/jim-2020-001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tatasciore A, Renda G, Zimarino M, Soccio M, Bilo G, Parati G, Schillaci G, De Caterina R. Awake systolic blood pressure variability correlates with target-organ damage in hypertensive subjects. Hypertension 50: 325–332, 2007. doi: 10.1161/HYPERTENSIONAHA.107.090084. [DOI] [PubMed] [Google Scholar]
- 22.Madden JM, O’Flynn AM, Dolan E, Fitzgerald AP, Kearney PM. Short-term blood pressure variability over 24 h and target organ damage in middle-aged men and women. J Hum Hypertens 29: 719–725, 2015. doi: 10.1038/jhh.2015.18. [DOI] [PubMed] [Google Scholar]
- 23.Wei FF, Li Y, Zhang L, Xu TY, Ding FH, Wang JG, Staessen JA. Beat-to-beat, reading-to-reading, and day-to-day blood pressure variability in relation to organ damage in untreated Chinese. Hypertension 63: 790–796, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02681. [DOI] [PubMed] [Google Scholar]
- 24.Simmonds SS, Lay J, Stocker SD. Dietary salt intake exaggerates sympathetic reflexes and increases blood pressure variability in normotensive rats. Hypertension 64: 583–589, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ozkayar N, Dede F, Ates I, Akyel F, Yildirim T, Altun B. The relationship between dietary salt intake and ambulatory blood pressure variability in non-diabetic hypertensive patients. Nefrologia 36: 694–700, 2016. doi: 10.1016/j.nefro.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 26.Migdal KU, Babcock MC, Robinson AT, Watso JC, Wenner MM, Stocker SD, Farquhar WB. The impact of high dietary sodium consumption on blood pressure variability in healthy, young adults. Am J Hypertens 33: 422–429, 2020. doi: 10.1093/ajh/hpaa014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossitto G, Maiolino G, Lerco S, Ceolotto G, Blackburn G, Mary S, Antonelli G, Berton C, Bisogni V, Cesari M, Seccia TM, Lenzini L, Pinato A, Montezano A, Touyz RM, Petrie MC, Daly R, Welsh P, Plebani M, Rossi GP, Delles C. High sodium intake, glomerular hyperfiltration and protein catabolism in patients with essential hypertension. Cardiovasc Res 117: 1372–1381, 2021. doi: 10.1093/cvr/cvaa205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reboldi G, Verdecchia P, Fiorucci G, Beilin LJ, Eguchi K, Imai Y, Kario K, Ohkubo T, Pierdomenico SD, Schwartz JE, Wing L, Saladini F, Palatini P. Glomerular hyperfiltration is a predictor of adverse cardiovascular outcomes. Kidney Int 93: 195–203, 2018. doi: 10.1016/j.kint.2017.07.013. [DOI] [PubMed] [Google Scholar]
- 29.Schmidlin O, Forman A, Tanaka M, Sebastian A, Morris RC. NaCl-induced renal vasoconstriction in salt-sensitive African Americans: antipressor and hemodynamic effects of potassium bicarbonate. Hypertension 33: 633–639, 1999. doi: 10.1161/01.HYP.33.2.633. [DOI] [PubMed] [Google Scholar]
- 30.Wenstedt EF, Verberk SG, Kroon J, Neele AE, Baardman J, Claessen N, Pasaoglu OT, Rademaker E, Schrooten EM, Wouda RD, de Winther MP, Aten J, Vogt L, Van den Bossche J. Salt increases monocyte CCR2 expression and inflammatory responses in humans. JCI Insight 4: e130508, 2019. doi: 10.1172/jci.insight.130508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krikken JA, Lely AT, Bakker SJ, Navis G. The effect of a shift in sodium intake on renal hemodynamics is determined by body mass index in healthy young men. Kidney Int 71: 260–265, 2007. doi: 10.1038/sj.ki.5002011. [DOI] [PubMed] [Google Scholar]
- 32.Burnier M, Rutschmann B, Nussberger J, Versaggi J, Shahinfar S, Waeber B, Brunner HR. Salt-dependent renal effects of an angiotensin II antagonist in healthy subjects. Hypertension 22: 339–347, 1993. doi: 10.1161/01.HYP.22.3.339. [DOI] [PubMed] [Google Scholar]
- 33.Babcock MC, Robinson AT, Watso JC, Migdal KU, Martens CR, Edwards DG, Pescatello LS, Farquhar WB. Salt loading blunts central and peripheral postexercise hypotension. Med Sci Sports Exerc 52: 935–943, 2020. doi: 10.1249/MSS.0000000000002187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Babcock MC, Robinson AT, Migdal KU, Watso JC, Martens CR, Edwards DG, Pescatello LS, Farquhar WB. High salt intake augments blood pressure responses during submaximal aerobic exercise. J Am Heart Assoc 9: e015633, 2020. doi: 10.1161/JAHA.120.015633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Babcock MC, Brian MS, Watso JC, Edwards DG, Stocker SD, Wenner MM, Farquhar WB. Alterations in dietary sodium intake affect cardiovagal baroreflex sensitivity. Am J Physiol Regul Integr Comp Physiol 315: R688–R695, 2018. doi: 10.1152/ajpregu.00002.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DuPont JJ, Greaney JL, Wenner MM, Lennon-Edwards SL, Sanders PW, Farquhar WB, Edwards DG. High dietary sodium intake impairs endothelium-dependent dilation in healthy salt-resistant humans. J Hypertens 31: 530–536, 2013. doi: 10.1097/HJH.0b013e32835c6ca8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watso JC, Robinson AT, Babcock MC, Migdal KU, Wenner MM, Stocker SD, Farquhar WB. Short-term water deprivation does not increase blood pressure variability or impair neurovascular function in healthy young adults. Am J Physiol Regul Integr Comp Physiol 318: R112–R121, 2020. doi: 10.1152/ajpregu.00149.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mehlum MH, Liestøl K, Kjeldsen SE, Julius S, Hua TA, Rothwell PM, Mancia G, Parati G, Weber MA, Berge E. Blood pressure variability and risk of cardiovascular events and death in patients with hypertension and different baseline risks. Eur Heart J 39: 2243–2251, 2018. doi: 10.1093/eurheartj/ehx760. [DOI] [PubMed] [Google Scholar]
- 39.Mena LJ, Felix VG, Melgarejo JD, Maestre GE. 24-Hour blood pressure variability assessed by average real variability: a systematic review and meta-analysis. J Am Heart Assoc 6: e006895, 2017. doi: 10.1161/JAHA.117.006895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sluyter JD, Camargo CA Jr, Scragg RK. Ten-second central SBP variability predicts first and recurrent cardiovascular events. J Hypertens 37: 530–537, 2019. doi: 10.1097/HJH.0000000000001930. [DOI] [PubMed] [Google Scholar]
- 41.Webb AJ, Mazzucco S, Li L, Rothwell PM. Prognostic significance of blood pressure variability on beat-to-beat monitoring after transient ischemic attack and stroke. Stroke 49: 62–67, 2018. doi: 10.1161/STROKEAHA.117.019107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dawson SL, Manktelow BN, Robinson TG, Panerai RB, Potter JF. Which parameters of beat-to-beat blood pressure and variability best predict early outcome after acute ischemic stroke? Stroke 31: 463–468, 2000. doi: 10.1161/01.STR.31.2.463. [DOI] [PubMed] [Google Scholar]
- 43.Vranish JR, Holwerda SW, Young BE, Credeur DP, Patik JC, Barbosa TC, Keller DM, Fadel PJ. Exaggerated vasoconstriction to spontaneous bursts of muscle sympathetic nerve activity in healthy young black men. Hypertension 71: 192–198, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chapman CL, Reed EL, Worley ML, Pietrafesa LD, Kueck PJ, Bloomfield AC, Schlader ZJ, Johnson BD. Sugar-sweetened soft drink consumption acutely decreases spontaneous baroreflex sensitivity and heart rate variability. Am J Physiol Regul Integr Comp Physiol 320: R641–R652, 2021. doi: 10.1152/ajpregu.00310.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dill DB, Costill DL. Calculation of percentage changes in volumes of blood, plasma, and red cells in dehydration. J Appl Physiol 37: 247–248, 1974. doi: 10.1152/jappl.1974.37.2.247. [DOI] [PubMed] [Google Scholar]
- 46.Chapman CL, Johnson BD, Sackett JR, Parker MD, Schlader ZJ. Soft drink consumption during and following exercise in the heat elevates biomarkers of acute kidney injury. Am J Physiol Regul Integr Comp Physiol 316: R189–R198, 2019. doi: 10.1152/ajpregu.00351.2018. [DOI] [PubMed] [Google Scholar]
- 48.Rossitto G, Maiolino G, Lerco S, Ceolotto G, Blackburn G, Mary S, Antonelli G, Berton C, Bisogni V, Cesari M, Seccia TM, Lenzini L, Pinato A, Montezano A, Touyz RM, Petrie MC, Daly R, Welsh P, Plebani M, Rossi GP, Delles C. High sodium intake, glomerular hyperfiltration and protein catabolism in patients with essential hypertension. Cardiovasc Res 117: 1372–1381, 2021. doi: 10.1093/cvr/cvaa205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wenner MM, Paul EP, Robinson AT, Rose WC, Farquhar WB. Acute NaCl loading reveals a higher blood pressure for a given serum sodium level in African American compared to Caucasian adults. Front Physiol 9: 1354, 2018. doi: 10.3389/fphys.2018.01354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sochett EB, Cherney DZ, Curtis JR, Dekker MG, Scholey JW, Miller JA. Impact of renin angiotensin system modulation on the hyperfiltration state in type 1 diabetes. J Am Soc Nephrol 17: 1703–1709, 2006. doi: 10.1681/ASN.2005080872. [DOI] [PubMed] [Google Scholar]
- 51.Malta D, Petersen KS, Johnson C, Trieu K, Rae S, Jefferson K, Santos JA, Wong MM, Raj TS, Webster J, Campbell NR, Arcand J. High sodium intake increases blood pressure and risk of kidney disease. From the Science of Salt: a regularly updated systematic review of salt and health outcomes (August 2016 to March 2017). J Clin Hypertens 20: 1654–1665, 2018. doi: 10.1111/jch.13408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Funke BE, Jackson KE, Self WH, Collins SP, Saunders CT, Wang L, Blume JD, Wickersham N, Brown RM, Casey JD, Bernard GR, Rice TW, Siew ED, Semler MW; SMART Investigators; Pragmatic Critical Care Research Group. Effect of balanced crystalloids versus saline on urinary biomarkers of acute kidney injury in critically ill adults. BMC Nephrol 22: 54, 2021. doi: 10.1186/s12882-021-02236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kobori H, Nishiyama A, Abe Y, Navar LG. Enhancement of intrarenal angiotensinogen in dahl salt-sensitive rats on high salt diet. Hypertension 41: 592–597, 2003. doi: 10.1161/01.HYP.0000056768.03657.B4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Majid D, Prieto M, Navar L. Salt-sensitive hypertension: perspectives on intrarenal mechanisms. Curr Hypertens Rev 11: 38–48, 2015. doi: 10.2174/1573402111666150530203858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Helanova K, Spinar J, Parenica J. Diagnostic and prognostic utility of neutrophil gelatinase-associated lipocalin (NGAL) in patients with cardiovascular diseases—review. Kidney Blood Press Res 39: 623–629, 2014. doi: 10.1159/000368474. [DOI] [PubMed] [Google Scholar]
- 56.Christensen EI, Birn H. Megalin and cubilin: multifunctional endocytic receptors. Nat Rev Mol Cell Biol 3: 256–266, 2002. doi: 10.1038/nrm778. [DOI] [PubMed] [Google Scholar]
- 57.Schmidt-Ott KM, Mori K, Li JY, Kalandadze A, Cohen DJ, Devarajan P, Barasch J. Dual action of neutrophil gelatinase-associated lipocalin. J Am Soc Nephrol 18: 407–413, 2007. doi: 10.1681/ASN.2006080882. [DOI] [PubMed] [Google Scholar]
- 58.Hvidberg V, Jacobsen C, Strong RK, Cowland JB, Moestrup SK, Borregaard N. The endocytic receptor megalin binds the iron transporting neutrophil-gelatinase-associated lipocalin with high affinity and mediates its cellular uptake. FEBS Lett 579: 773–777, 2005. doi: 10.1016/j.febslet.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 59.Mori K, Lee HT, Rapoport D, Drexler IR, Foster K, Yang J, Schmidt-Ott KM, Chen X, Li JY, Weiss S, Mishra J, Cheema FH, Markowitz G, Suganami T, Sawai K, Mukoyama M, Kunis C, D’Agati V, Devarajan P, Barasch J. Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury. J Clin Invest 115: 610–621, 2005. doi: 10.1172/JCI23056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Langelueddecke C, Roussa E, Fenton RA, Wolff NA, Lee WK, Thévenod F. Lipocalin-2 (24p3/Neutrophil Gelatinase-associated Lipocalin (NGAL)) receptor is expressed in distal nephron and mediates protein endocytosis. J Biol Chem 287: 159–169, 2012. doi: 10.1074/jbc.M111.308296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schrezenmeier EV, Barasch J, Budde K, Westhoff T, Schmidt-Ott KM. Biomarkers in acute kidney injury—pathophysiological basis and clinical performance. Acta Physiol (Oxf) 219: 554–572, 2017. doi: 10.1111/apha.12764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sabbisetti VS, Waikar SS, Antoine DJ, Smiles A, Wang C, Ravisankar A, Ito K, Sharma S, Ramadesikan S, Lee M, Briskin R, De Jager PL, Ngo TT, Radlinski M, Dear JW, Park KB, Betensky R, Krolewski AS, Bonventre JV. Blood kidney injury molecule-1 is a biomarker of acute and chronic kidney injury and predicts progression to ESRD in type I diabetes. J Am Soc Nephrol 25: 2177–2186, 2014. doi: 10.1681/ASN.2013070758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schlader ZJ, Hostler D, Parker MD, Pryor RR, Lohr JW, Johnson BD, Chapman CL. The potential for renal injury elicited by physical work in the heat. Nutrients 11: 2087, 2019. doi: 10.3390/nu11092087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stevens SL, Wood S, Koshiaris C, Law K, Glasziou P, Stevens RJ, McManus RJ. Blood pressure variability and cardiovascular disease: systematic review and meta-analysis. BMJ 354: i4098, 2016. doi: 10.1136/bmj.i4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rosei EA, Chiarini G, Rizzoni D. How important is blood pressure variability? Eur Heart J Suppl 22: E1–E6, 2020. doi: 10.1093/eurheartj/suaa061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lerchl K, Rakova N, Dahlmann A, Rauh M, Goller U, Basner M, Dinges DF, Beck L, Agureev A, Larina I, Baranov V, Morukov B, Eckardt KU, Vassilieva G, Wabel P, Vienken J, Kirsch K, Johannes B, Krannich A, Luft FC, Titze J. Agreement between 24-hour salt ingestion and sodium excretion in a controlled environment. Hypertension 66: 850–857, 2015. doi: 10.1161/HYPERTENSIONAHA.115.05851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kitada K, Daub S, Zhang Y, Klein JD, Nakano D, Pedchenko T, Lantier L, LaRocque LM, Marton A, Neubert P, Schröder A, Rakova N, Jantsch J, Dikalova AE, Dikalov SI, Harrison DG, Müller DN, Nishiyama A, Rauh M, Harris RC, Luft FC, Wassermann DH, Sands JM, Titze J. High salt intake reprioritizes osmolyte and energy metabolism for body fluid conservation. J Clin Invest 127: 1944–1959, 2017. doi: 10.1172/JCI88532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rakova N, Kitada K, Lerchl K, Dahlmann A, Birukov A, Daub S, Kopp C, Pedchenko T, Zhang Y, Beck L, Johannes B, Marton A, Müller DN, Rauh M, Luft FC, Titze J. Increased salt consumption induces body water conservation and decreases fluid intake. J Clin Invest 127: 1932–1943, 2017. doi: 10.1172/JCI88530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Juraschek SP, Miller ER 3rd, Chang AR, Anderson CA, Hall JE, Appel LJ. Effects of sodium reduction on energy, metabolism, weight, thirst, and urine volume. Hypertension 75: 723–729, 2020. doi: 10.1161/HYPERTENSIONAHA.119.13932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Stachenfeld NS. Acute effects of sodium ingestion on thirst and cardiovascular function. Curr Sports Med Rep 7: S7–S13, 2008. doi: 10.1249/JSR.0b013e31817f23fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Abrahamson M, Olafsson I, Palsdottir A, Ulvsbäck M, Lundwall A, Jensson O, Grubb A. Structure and expression of the human cystatin C gene. Biochem J 268: 287–294, 1990. doi: 10.1042/bj2680287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim SS, Song SH, Kim IJ, Jeon YK, Kim BH, Kwak IS, Lee EK, Kim YK. Urinary cystatin C and tubular proteinuria predict progression of diabetic nephropathy. Diabetes Care 36: 656–661, 2013. doi: 10.2337/dc12-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kar S, Paglialunga S, Islam R. Cystatin C is a more reliable biomarker for determining eGFR to support drug development studies. J Clin Pharmacol 58: 1239–1247, 2018. doi: 10.1002/jcph.1132. [DOI] [PubMed] [Google Scholar]
- 74.Conti M, Moutereau S, Zater M, Lallali K, Durrbach A, Manivet P, Eschwège P, Loric S. Urinary cystatin C as a specific marker of tubular dysfunction. Clin Chem Lab Med 44: 288–291, 2006. doi: 10.1515/CCLM.2006.050. [DOI] [PubMed] [Google Scholar]
- 75.Murtaugh MA, Beasley JM, Appel LJ, Guenther PM, McFadden M, Greene T, Tooze JA. Relationship of sodium intake and blood pressure varies with energy intake: secondary analysis of the DASH (Dietary Approaches to Stop Hypertension)-Sodium Trial. Hypertension 71: 858–865, 2018. doi: 10.1161/HYPERTENSIONAHA.117.10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lucko AM, Doktorchik C, Woodward M, Cogswell M, Neal B, Rabi D, Anderson C, He FJ, MacGregor GA, L’Abbe M, Arcand J, Whelton PK, McLean R, Campbell NR; TRUE Consortium. Percentage of ingested sodium excreted in 24-hour urine collections: a systematic review and meta-analysis. J Clin Hypertens (Greenwich) 20: 1220–1229, 2018. doi: 10.1111/jch.13353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available by the corresponding author upon reasonable request.