

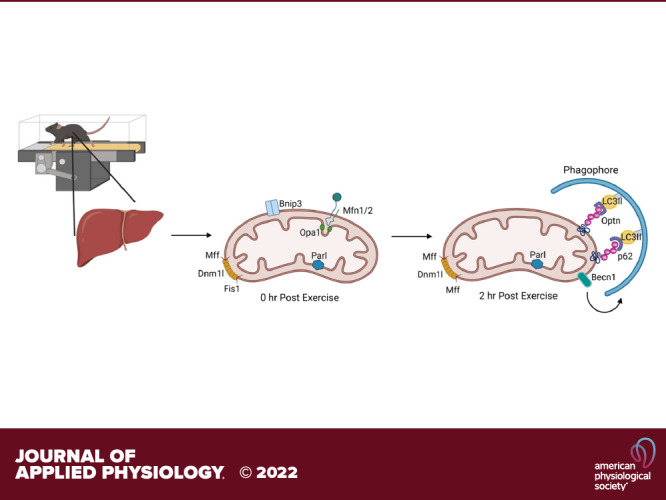
Keywords: exercise, leupeptin, liver, mitophagic flux, mitophagy
Abstract
Exercise is critical for improving metabolic health and putatively maintains or enhances mitochondrial quality control in metabolic tissues. Although previous work has shown that exercise elicits hepatic mitochondrial biogenesis, it is unknown if acute exercise activates hepatic mitophagy, the selective degradation of damaged or low-functioning mitochondria. We tested if an acute bout of treadmill running increased hepatic mitophagic flux both right after and 2-h postexercise in 15- to 24-wk-old C57BL/6J female mice. Acute exercise did not significantly increase markers of autophagic flux, however, mitophagic flux was activated 2-h post-treadmill running as measured by accumulation of both LC3-II and p62 in isolated mitochondria in the presence of leupeptin, an inhibitor of autophagosome degradation. Furthermore, mitochondrial-associated ubiquitin, which recruits the autophagy receptor protein p62, was also significantly increased at 2 h. Further examination via Western blot and proteomics analysis revealed that acute exercise elicits a time-dependent, dynamic activation of mitophagy pathways. Moreover, the results suggest that exercise-induced hepatic mitophagy is likely mediated by both polyubiquitination and receptor-mediated signaling pathways. Overall, we provide evidence that acute exercise activates hepatic mitophagic flux while also revealing specific receptor-mediated proteins by which exercise maintains mitochondrial quality control in the liver.
NEW & NOTEWORTHY This study provides evidence that acute exercise activates hepatic mitophagic flux and mitochondrial polyubiquitination while additionally revealing specific receptor-mediated proteins by which exercise maintains mitochondrial quality control in the liver.
INTRODUCTION
Exercise exerts numerous benefits and is a powerful treatment for hepatic steatosis, with effects linked to enhanced hepatic mitochondrial function (1). During exercise, hepatic mitochondria serve as a metabolic engine that burns lipids and processes substrates to fuel hepatic gluconeogenesis and maintain euglycemia (2). Numerous processes putatively exist to remodel mitochondria homeostasis after exercise including mitochondrial biogenesis (3, 4). However, the coordinated breakdown and recycling of dysfunctional mitochondria may also be critical for adaptations to exercise. A loss in efficiency or function of mitochondria (loss of membrane potential or aberrant reactive oxygen species production) leads to targeted degradation (5, 6). Autophagy is the general process of recycling damaged cellular materials whereas mitophagy is specific targeting of mitochondria for autophagic degradation. Mitophagy is dynamic and requires coordination of numerous proteins to target, recruit, engulf, fuse with lysosome, and ultimately degrade mitochondria. Mitophagy works in parallel or in direct coordination with mitochondrial fission and fusion with the same goal of maintaining a highly functioning mitochondrial pool. Mitophagic flux studies require chemical inhibitors that block mitochondrial autophagic degradation, and studies examining these phenomena in vivo are limited. Studies without inhibitors fail to definitively conclude whether mitophagy has been activated or inhibited, limiting interpretation (7). Leupeptin is a serine, threonine, and cysteine protease inhibitor that is potently effective in blocking hepatic macroautophagy in vivo (8). Leupeptin has been used to show that fasting induces hepatic mitophagic flux through lysosomal accumulation of microtubule-associated proteins 1A/1B light chain 3B (LC3-II) (8). We have previously used leupeptin to investigate the acute impact of heat (9) and chronic exercise (10) on hepatic mitophagy.
Exercise induces activation of mitophagic flux in skeletal muscle (11, 12), but its effect on hepatic mitophagic flux is unknown. Previously, we showed that female mice have reduced hepatic mitophagic flux compared with males after chronic exercise (10), but mitophagy adaptations to chronic exercise are difficult to interpret. Most exercise studies are measuring mitophagic flux after chronic exercise thus missing a direct assessment of whether each acute bout stimulates mitophagy. Herein, we utilize wild-type (WT) female C57BL/6J mice subjected to a single acute bout of treadmill exercise to determine if acute exercise activates hepatic mitophagic flux using a variety of methodological tools including leupeptin, immunofluorescent labeling, and proteomics of isolated mitochondria. We hypothesized that acute exercise activates hepatic mitophagic flux as seen through an accumulation of LC3-II and p62 proteins on the mitochondria (in the presence of leupeptin) and the capturing of fluorescently tagged mitochondrial complex IV protein subunit (Cox8) in lysosomes. Furthermore, we utilized proteomics approaches to examine exercise-induced pathways driving hepatic mitophagy.
MATERIALS AND METHODS
Ethical Approval
The animal protocol was approved by the Institutional Animal Care and Use Committee at the University of Kansas Medical Center and Kansas City Veterans Affairs Medical Center (animal Protocol Number 2019-2539). All experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Guide, 8th ed., 2011). Mice were anesthetized with pentobarbital sodium (100 mg·kg−1) before a terminal procedure.
Animals
Female C57Bl/6J mice (15 to 23 wk old) (No. 000664, The Jackson Laboratory) were housed near thermoneutrality (∼30°C) with ad libitum access to water and food (Teklad 8604). Mice were held on a reverse light cycle with lights dark from 10:00 AM to 10:00 PM so that daytime running would occur during the mouse active phase. C57Bl/6J mice have mutations in the nicotinamide nucleotide transhydrogenase protein (NNT) that catalyzes the reduction of NADH to NAD+. Despite this known mutation, we have elected to study this model to directly relate the effects seen in this current study to our previously published mitophagy and mitophagic flux results in mice undergoing chronic exercise or high-fat diets.
Leupeptin Time Course
Twenty-four mice (n = 3/group) received a 250 µL intraperitoneal injection of either saline (SAL) (0.9%) or leupeptin (LEU) (4 µg·µL−1 or 40 mg·kg−1 total) ∼1 h after lights went off (11:00 AM). Mice were given access to food for an additional 15 min to reduce potential confounding impacts of fasting on autophagy and mitophagy. Mice were euthanized at 30-, 60-, 120-, and 240-min postinjection of leupeptin or saline, and livers were harvested for mitochondrial isolation.
Acute Exercise
Seventy-seven mice were first treadmill acclimated running at 10 m⋅min−1 for 5 min before increasing the incline to 5° incline for 10 more min 72 h before acute exercise. Mice were randomly separated into sedentary (SED) and treadmill running (EX). On study day, mice received 250 µL of either SAL or LEU (4 µg·µL−1) (n = 8–11/each of 8 treatment group) at 10:30 AM, food was removed at 10:45 AM, and treadmill running began at 11 AM. EX mice performed the following: 8 min at 10 m·min−1 at 0°, increase speed to 12 m·min−1 for 2 min, increase incline to 5° for 8 min, increase speed to 12.5 m·min−1 for 2 min, increase incline to 10° for 10 min, increase incline to 15° for 10 min, increase incline to 20° for 10 min, increase incline to 25° for 10 min. The treadmill was then stopped, and animals were euthanized quickly at 0 h or 2-h postexercise. SED controls remained on separate, idle treadmill for the 1 h of treatment and euthanized at the same timepoints.
Mitochondrial Isolation
Following exercise, livers were excised and half of the liver was snap frozen in liquid N2 while the other half was placed in 8 mL ice-cold mitochondria isolation buffer (220 mM mannitol, 70 mM sucrose, 10 mM Tris, 1 mM EDTA, pH adjusted to 7.4 with KOH) and homogenized with a Teflon pestle to create freshly isolated hepatic mitochondria, as described previously (13). Briefly, homogenates were centrifuged (4°C, 10 min, 1,500 g) and supernatant was transferred to a fresh conical tube and spun again (4°C, 10 min, 8,000 g) to generate a pellet. Pellet was resuspended in 6 mL isolation buffer using glass-on-glass Dounce homogenizer and centrifuged again (4°C, 10 min, 6,000 g). The pellet was resuspended in 4 mL isolation buffer, Dounce homogenized, and spun at (4°C, 10 min, 4,000 g). The final pellet was resuspended in 0.75–1 mL of isolation buffer. Protein concentrations were determined by bicinchoninic acid assay (BCA) and frozen at −80°C. For whole cell lysate samples, frozen livers were powdered on liquid nitrogen, homogenized, and prepared for Western blotting as reported previously (13). Briefly, 1 mL of homogenization buffer was added to 100 mg of frozen tissue. Tissue was homogenized using Qiagen TissueLyser II (2 Hz for 2 min, 10 min on ice, 2 Hz for 2 min). Samples were centrifuged at 4°C, 15,000 g for 25 min. Supernatant was removed, protein concentration was measured via BCA, and prepared for Western blot.
Quantitative Determination of Autophagic and Mitophagic Flux
Western‐ready Laemmli samples were produced separately from both liver-isolated mitochondria and tissue homogenate, separated by SDS-PAGE, quantified with Image Laboratory software (Bio-Rad Laboratories), and all values were normalized to total mitochondrial protein using 0.1% amido black as described previously (13). We used the following primary antibodies from Cell Signaling Technology at 1:2,000: Bnip3 (3769S) (14), Mfn2 (9482S) (15), Parkin (4211S) (16), Ubiquitin (3933) (17), sequestosome 1 (SQSTM1/p62; 5114) (18), microtubule-associated protein 1 light chain 3 A/B (LC3A/B; 12741S) (19), 5′-AMP-activated protein kinase catalytic subunit alpha-1 (AMPKα) (5831) (20), phospho-AMPKα (Thr172) (2535) (21), Akt (4691) (22), phospho-Akt (Ser473) (4060) (23), Mammalian target of rapamycin (mTOR) (2983) (24), phospho-mTOR (Ser2448) (5536) (25), acetyl-CoA carboxylase (ACC) (3662S) (26), and phospho-ACC (Ser79) (3661S) (27). Cell Signaling Technology goat anti-mouse (7076) (28) and goat anti-rabbit (7074) (29) secondary antibodies were used at a concentration of 1:10,000.
Citrate Synthase
Citrate synthase was determined from hepatic whole homogenate as previously described (30). Briefly, samples were freeze thawed three times to fracture mitochondrial membranes. Homogenates were diluted in dH2O to ensure linear reaction rate without depleting the substrate. In a 96-well plate, 10 µL of diluted sample was incubated with 170 µL of reaction media (100 mM Tris, 1 mM DTNB dissolved in 100 mM Tris, 10 mM oxaloacetate, pH = 8.3) and read for 2 min at 405 nm. Quickly added 30 µL of 3 mM acetyl-CoA and read again on a BioTek Epoch 2 plate reader for 7 min to generate Vmax values calculated from BioTek Gen5 software.
Confocal Microscopy for Mitophagy
Ten to fourteen days before acute treadmill running bout, a cohort of mice were injected via tail vein with an Adenoviral (Ad) Cox8-GFP-mCherry [5 × 108 plaque-forming units (PFU)/mouse diluted in 200 µL saline] mitophagy reporter assay (31, 32). As the Cox8 (cytochrome c oxidase or complex IV subunit) protein is labeled with both GFP and mCherry, mitochondria will display a yellow color. As mitochondria are targeted to the autolysosome, the GFP will be quenched and only the mCherry red fluorescence should be visible. At euthanasia, a small portion of liver tissue was fixed in 4% paraformaldehyde and processed for fluorescent image analysis using a Leica TCS SPE Confocal Microscope (9).
Mitochondrial Proteomics
A subset (n = 5/group) of isolated mitochondrial fractions were used for proteomics (33). Proteins were reduced, alkylated, and purified by chloroform/methanol extraction before digestion with sequencing grade modified porcine trypsin (Promega). Tryptic peptides were then separated by reverse phase XSelect CSH C18 2.5 μm resin (Waters) on an in-line 150 × 0.075 mm column using an UltiMate 3000 RSLCnano system (Thermo). Peptides were eluted using a 60-min gradient from 98:2 to 65:35 buffer A:B ratio (buffer A = 0.1% formic acid, 0.5% acetonitrile; buffer B = 0.1% formic acid, 99.9% acetonitrile). Eluted peptides were ionized by electrospray (2.2 kV) followed by mass spectrometric analysis on an Orbitrap Exploris 480 mass spectrometer (Thermo). To assemble a chromatogram library, six gas-phase fractions were acquired on the Orbitrap Exploris with 4 m/z DIA spectra (4 m/z precursor isolation windows at 30,000 resolution, normalized AGC target 100%, maximum inject time 66 ms) using a staggered window pattern from narrow mass ranges using optimized window placements. Precursor spectra were acquired after each DIA duty cycle, spanning the m/z range of the gas-phase fraction (i.e., 496–602 m/z, 60,000 resolution, normalized AGC target 100%, maximum injection time 50 ms). For wide-window acquisitions, the Orbitrap Exploris was configured to acquire a precursor scan (385–1,015 m/z, 60,000 resolution, normalized AGC target 100%, maximum injection time 50 ms) followed by 50 times 12 m/z DIA spectra (12 m/z precursor isolation windows at 15,000 resolution, normalized AGC target 100%, maximum injection time 33 ms) using a staggered window pattern with optimized window placements. Precursor spectra were acquired after each DIA duty cycle.
Mitochondrial Proteomic Data Analysis
Proteomic data were searched using an empirically corrected library and performed a quantitative analysis to obtain a comprehensive proteomic profile. Proteins were identified and quantified using EncyclopeDIA (34) and visualized with Scaffold DIA using 1% false discovery thresholds at both the protein and peptide level.
Protein exclusive intensity values were assessed for quality using our in-house ProteiNorm app, a user-friendly tool for a systematic evaluation of normalization methods, imputation of missing values, and comparisons of different differential abundance methods (35). Popular normalization methods were evaluated including log2 normalization (Log2), median normalization (Median), mean normalization (Mean), variance stabilizing normalization (VSN) (36), quantile normalization (Quantile) (37), cyclic loess normalization (Cyclic Loess) (38), global robust linear regression normalization (RLR) (39), and global intensity normalization (Global Intensity) (39). The individual performance of each method was evaluated by comparison of the following metrices: total intensity, intragroup coefficient of variation (PCV), pooled intragroup median absolute deviation (PMAD), pooled intragroup estimate of variance (PEV), intragroup correlation, sample correlation heatmap (Pearson), and log2-ratio distributions.
Proteomics data were cross-referenced with MitoCarta3.0 (40) gene inventory to identify proteins with strong evidence of mitochondria localization. All proteins not included within the MitoCarta3.0 gene inventory were removed from analysis. Qiagen’s Ingenuity pathway analysis (IPA) (41) was performed to determine changes in mitochondrial pathways. Using expression core analysis with expression log ratio (log fold change) incorporated for each respective protein, directionality (Z-score) of pathway regulation was determined.
Statistics
The main effects of exercise (E; SED vs. EX) and time (T; 0 and 2 h) were examined using two-way ANOVAs within either the saline (SAL) or the leupeptin (LEU) treatment groups utilizing GraphPad Prism 9 software. Upon discovering significant main effects, post hoc analyses were performed using Fisher’s least-significant difference (LSD) test. Statistical significance was set at P < 0.05. Significant outliers for all data sets were removed using the Grubb’s method. Data are presented as means ± standard error. For proteomics analysis, the normalized data were used to perform statistical analysis using linear models for microarray data (limma) with empirical Bayes (eBayes) smoothing to the standard errors (38). Proteins with a P value <0.05 were considered significant.
RESULTS
Leupeptin Inhibits Mitophagy for at Least 4 h
Although LEU has previously been shown effective in inhibiting macroautophagic flux in hepatic lysosomal-enriched fractions in vivo (8) through measuring the accumulation of LC3-II (marker of autophagosomes) and p62 (a ubiquitin-binding autophagy receptor protein), we initially sought to determine its utility in studying mitophagic flux in isolated hepatic mitochondria. We first used intraperitoneal LEU injections (n = 3/group) to confirm inhibition of mitophagy pathways in SED mice (Fig. 1). After injecting LEU both LC3-II and p62 accumulated in isolated hepatic mitochondria. Within 30 min, LEU elicited a 45% and 394% increase in LC3-II and p62 on hepatic mitochondria, respectively, compared with SAL controls (Fig. 1, A and B). This accumulation in LC3-II and p62 continued at 60, 120, and 240 min postinjection (P < 0.05 at 120 and 240 min, Fig. 1B). As depicted in Fig. 1C, we next tested if leupeptin treatment would elicit a greater accumulation of LC3-II and p62 in hepatic mitochondria in treadmill-run mice compared with sedentary controls (Fig. 1C).
Figure 1.

Leupeptin (Leu) rapidly inhibits hepatic mitophagic flux. Mitophagy proteins microtubule-associated proteins 1A/1B light chain 3B (LC3-II) (A) and p62 (B) were assessed via Western blotting from isolated hepatic mitochondria at 30-, 60-, 120-, and 240 min postinjection of either 250 µL saline or leupeptin (4 mg/mL). All proteins quantified were normalized to total protein staining using amido black. Data are presented as means ± SE (n = 3/group). C: schematic showing hypothesized effects of leupeptin on blocking lysosomal mitochondrial degradation in treadmill-run mice resulting in accumulation of mitochondria. Image created with BioRender and published with permission. L, significant main effect of leupeptin; T, significant main effect of time; *P < 0.05 within condition time effect (vs. 30 min); #P < 0.05 within condition time effect (vs. 60 min).
Acute Exercise Does Not Increase Hepatic Autophagy Flux within 2 h
Since LEU blocks both hepatic autophagy and mitophagy, we sought to investigate the impact of acute EX on markers of hepatic autophagic flux via Western blotting in liver whole homogenate post-EX. EX had no impact on LC3-II or p62 (Fig. 2, A and B, respectively), although LEU increased p62 accumulation in both SED and EX (P < 0.05, Fig. 2B). Because autophagy is activated in response to a metabolic challenges, such as starvation (42, 43), we examined the phosphorylation status and total protein content for the critical energy pathway proteins AMPKα, ACC, and mTOR (Fig. 2, C–E and, Supplemental Fig. S1, see https://doi.org/10.6084/m9.figshare.19027718) and found no relevant significant effects of treatment (44).
Figure 2.

Acute exercise does not increase hepatic autophagic flux. Protein content for autophagy proteins microtubule-associated proteins 1A/1B light chain 3B (LC3-II) (A) and p62 (B) were assessed in liver whole homogenate at 0- and 2-h post-sedentary (SED) or treadmill running (EX) from wild-type (WT) female mice receiving either saline or leupeptin. All proteins quantified were normalized to total protein staining using amido black. Data are presented as means ± SE (n = 8–11/group). T, P < 0.05 main effect for time (2 h vs. 0 h); t, P < 0.05 within condition time effect (vs. 0 h); SAL, saline condition; LEU, leupeptin condition. Representative blots showing no significant changes in either phospho- or total ACC, AMPKα, and mTOR (C, D, and E).
Acute Exercise Induces Hepatic Mitophagic Flux
We next determined if mitophagic flux was engaged using multiple methodologies. We utilized isolated mitochondria (freshly obtained at time of euthanasia) and assessed for LC3-II and p62 protein content via Western blotting at 0 and 2 h post-EX. LEU treatment blunted lysosomal degradation resulting in accumulation of LC3-II and p62 in isolated mitochondria (Fig. 3, A, B, and E). EX did not elicit an increased accumulation of either LC3-II or p62 compared with SED at 0 h. However, at 2-h postexercise LEU treatment captured a significant EX induced increase in both LC3-II (45%) and p62 (65%) (P < 0.05, Fig. 3, A, B, and E) compared with SED. Comparison of EX versus SED in the SAL groups showed no effects. Since p62 was increased with acute EX and is a ubiquitin-binding receptor protein we next examined whether exercise increased mitochondrial ubiquitin content. Acute EX increased ubiquitin content by 20% at 2 h (P < 0.05, Fig. 3, C and E). To visually confirm activation of mitophagic flux by EX, we employed the use of a fluorescent adenovirus reporter for Cox8 (Cox8-GFP-mCherry), a mitochondrial protein. Upon mitochondria entry into the lysosome the GFP fluorescent signal is quenched resulting in red punctae. Used in conjunction with leupeptin, we confirmed increased red punctae 2 h post-EX compared with SED (Fig. 3D) further demonstrating acute EX increases hepatic mitophagic flux.
Figure 3.

Acute exercise increases hepatic mitophagic flux and mitochondrial ubiquitination. Protein content for mitophagy markers microtubule-associated proteins 1A/1B light chain 3B (LC3-II) (A) and p62 (B), and ubiquitin (C) were examined in liver mitochondrial isolates from wild-type (WT) female mice 0 and 2 h after sedentary (SED) or treadmill running (EX) and injected with either SAL or LEU. All quantified proteins (E) were normalized to total protein utilizing amido black staining. Qualitative visual confirmation of mitophagy was obtained via confocal imaging of livers from WT animals exposed to the Ad-Cox8-GFP-mCherry fluorescent mitophagy reporter (D). E, P < 0.05 main effect for exercise; T, P < 0.05 main effect for time (2 h vs. 0 h); E × T, P < 0.05 exercise by time interaction; e, P < 0.05 within condition exercise effect (vs. SED); t, P < 0.05 within condition time effect (vs. 0 h).
Acute Exercise Rapidly Elicits Mitochondrial Accumulation of Mitophagy Proteins
Western blot analysis of mitochondrial fractions was determined in only the SAL-treated groups as the impacts of LEU treatment on mitophagy protein interactions with mitochondria are unknown. At 0-h post-EX, BCL2/adenovirus E1B 19-kDa protein-interacting protein 3 (Bnip3), a receptor-mediated mitophagy protein, and mitofusin-2 (Mfn2), a mitochondrial fusion protein, were significantly increased 66% and 81% in the mitochondrial fractions compared with SED (P < 0.05 for both, Fig. 4, B and C), however this increase was gone by 2 h post-EX. Dynamin-1-like protein (Drp1), a mitochondrial fission protein, was not influenced by EX but decreased over time in both EX and SED (P < 0.05, Fig. 4D). Parkin, a mitochondrial E3 ubiquitin ligase, was also not changed by EX (Fig. 4A). Citrate synthase, a marker of mitochondrial content, was similar across all SAL-treated groups (Fig. 4E).
Figure 4.

Acute exercise rapidly increases mitochondrial mitophagy proteins. Protein content for mitophagy proteins Parkin (A), BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) (B), mitofusin-2 (Mfn2) (C), and dynamin-1-like protein (Drp1) (D), as well as citrate synthase (E) were examined in mitochondria isolates from wild-type (WT) female mice 0- and 2-h post-condition. Quantified proteins (F) were normalized to total protein using amido black staining for quantitation. T, P < 0.05 main effect for time (2 h vs. 0 h); E × T, P < 0.05 exercise by time interaction; x, P < 0.05 within condition exercise effect (vs. sedentary, SED); t, P < 0.05 within condition time effect (vs. 0 h).
Proteomics Analysis of Isolated Mitochondria
To further investigate which proteins and pathways are enriched with acute exercise, we used untargeted proteomics on isolated hepatic mitochondrial samples (n = 5/group). The proteins revealed by proteomics were then cross-referenced to MitoCarta3.0, an evolving library of mitochondrial-specific proteins. We estimate the mitochondrial enrichment fraction (MEF), or purity of our isolated samples using a technique previously described (45). Our findings revealed that 56% of the proteins are known mitochondrial proteins (45). Briefly, we quantitatively compared the summed number of mitochondrial proteins in our samples (found in MitoCarta3.0) to total protein abundance (based on peak intensity values). In total, 3,241 proteins were identified across all samples through proteomics analysis and 700 were identified as being mitochondrial. Although our final MEF value was lower than that published previously there are a couple of important methodological differences, including differences in MitoCarta versions and our isolation procedure has a lower final centrifugation speed to generate the mitochondrial pellet compared with previously reported methodology. Only proteins that appeared in both the isolated mitochondrial proteomics and in MitoCarta3.0 were further analyzed (700 proteins out of 1,140 MitoCarta proteins). Examination of our SAL-injected animal samples via IPA analysis showed reduced mitophagy activation in the EX compared with the SED cohorts at both 0 and 2 h (Fig. 5A). Similar to our Western blot results, IPA showed an increased quantity of BNIP3 at 0 h post-EX. (Fig. 5A, first panel). EX in the LEU-treated mice increased the content of several key mitochondrial fission/fusion proteins including Drp1 (Dnm1l), Mfn1, mitochondrial fission factor (Mff), and BNIP3. This effect was magnified at 2 h post-EX. The changes in these mitophagy-related proteins culminated in predicted activation of “Autophagy of Mitochondria” (mitophagy—Fig. 5A, right side under leupeptin designation). The predicted mitophagy activation was enhanced at the 2 h timepoint (bottom portion of figure) compared with the 0 h (top portion). Log fold change (logFC) analysis of the proteomics data revealed increased mitochondrial p62 (Sqstm1), optineurin, and LC3-II in the EX-group at 0 h compared with SED with SAL treatment (Fig. 5B, top left). Proteomics revealed BNIP3 content was modestly increased in the SAL + EX group compared with the SAL + SED cohort at 0 h. Interestingly, proteomics data showed a 0.22 logFC decrease in Mfn2 with SAL + EX at 0 h when compared with the SAL + SED group which was not observed by Western blot. Proteomics revealed a significant 0.57 logFC increase in Drp1/Dnm1l at 2 h in the SAL + EX cohort compared with SAL + SED, a result also not observed by Western analysis (P < 0.05, Fig. 5B, bottom left). These differences may result from different sensitivities between W and proteomics methods in addition to only a portion of all samples from every group being sent for proteomics analysis compared with Western blot in which every sample was analyzed. LEU + EX elicited the largest log-fold increases in Bnip3, Presenilins-associated rhomboid-like protein (Parl—mitochondrial intramembrane serine protease), and LC3-II (0.59, 0.27, and 0.23 logFC, respectively) at 0 h (Fig. 5B, top right). At 2 h post, LEU + EX elicited an increase in Beclin-1 (0.90 logFC), Optineurin (0.47 logFC), p62 (0.37 logFC), and Bnip3 (0.35 logFC) compared with SED group (Fig. 5B, bottom right).
Figure 5.

Mitochondrial proteomics further reveals acute exercise increases several key mitophagy regulating proteins. Ingenuity pathway analysis (IPA) (n = 5/group) reveals changes in mitophagy protein abundance to predict activation or inhibition of the mitophagy pathway (A). Log-fold changes for specific mitophagy proteins were compared between sedentary (SED), treadmill running (EX), saline (SAL), and leupeptin (LEU) treatment groups (B). *P < 0.05 when comparing EX to SED condition.
Comparison of the time effect within exercise treatment (2-h post-EX to 0-h post-EX) revealed a few notable differences in mitophagy proteins (Fig. 6). IPA analysis revealed that 2-h SAL + EX mitochondria had increased measurement of a number of mitophagy proteins including SLC25A46, a fission protein, MFN1, Mff, Fis1, and DNM1L compared with the 0-h SAL + EX cohort (Fig. 6A, top left) and reduced BNIP3. When examining the logFC changes, there was a robust 1.9 logFC reduction in Bnip3 at 2 h (P < 0.05, Fig. 6A) with SAL + EX (Fig. 6B, bottom left). Interestingly, IPA showed the opposite effect wherein the 2-h LEU + EX group had a reduction in a number of mitophagy proteins including MFN1, Fis1, and DNM1L when compared with 0 h (Fig. 6A, top right). LEU + EX treatment elicited a nonsignificant 0.69 logFC increase in Bnip3 at 2 h (Fig. 6B, bottom right). LEU + EX treated mice elicited increases in Mff (0.89 logFC), Becn1 (1.03 logFC), and a significant increase in optineurin (Optn) (1.81 logFC, P < 0.05, Fig. 6B, bottom right) at 2 h compared with 0. Finally, LEU + EX treated mice showed significant increases in hepatic mitochondrial LC3-II (1.19 logFC) and p62 (2.71 logFC) (P < 0.05 for both, Fig. 6B, bottom right). Leveraging proteomics, LEU treatment compared with SAL revealed numerous changes in mitochondrial-associated proteins confirming exercise induction of mitophagic flux.
Figure 6.

Dynamic mitophagy protein changes after exercise. Ingenuity pathway analysis (IPA) comparing the 2 h and 0 h exercise cohorts (n = 5/group) (A). Log-fold changes in mitophagy proteins were compared between the 2-h and 0-h postexercise cohorts (B). A log-fold increase represents enhanced protein content in the 2 h compared to the 0 h groups. *P < 0.05 when comparing 2 h treadmill running (EX) to 0 h EX.
DISCUSSION
Exercise is a powerful regulator of hepatic mitochondrial function (10, 13, 46, 47) but it is unknown if this effect is dependent on mitophagy. Herein, we used various tools to test if acute exercise modulates hepatic mitophagy in mice. Our primary findings are: 1) acute exercise rapidly activates hepatic mitophagy and mitophagic flux, 2) this activation of mitophagy flux coincides with increased mitochondrial ubiquitination and likely receptor-mediated events, and 3) changes in mitophagy-associated proteins induced by exercise are time dependent (i.e., variable across time).
Measuring mitophagy “flux” requires the use of chemical inhibitors of autophagosome formation or lysosomal processing (48, 49). Leupeptin, a serine/threonine protease inhibitor, has been shown to be effective to cease in vivo hepatic mitophagy within 1 h (8). In the current study, we confirmed that leupeptin elicits a stronger hepatic accumulation of p62 than LC3-II with effects lasting up to at least 4 h in sedentary mice, providing an opportunity to determine if hepatic mitophagy flux is activated after a 1-h acute bout of treadmill running.
Numerous studies have examined how exercise influences autophagy in skeletal muscle. Results from mice and humans in the absence of chemical inhibitors show that exercise increases skeletal muscle autophagy via increased protein content of LC3-II (50, 51). Mice subjected to chronic exercise and a chemical inhibitor (colchicine) further confirm that exercise drives increased autophagic flux in skeletal muscle (52). A recent mouse study showed that an acute bout of treadmill exercise altered hepatic autophagy as measured through reductions in LC3-I protein, a transient and unstable protein (43). In contrast, our data suggest that acute exercise does not elicit activation of autophagic flux in the liver (no change in whole homogenate hepatic LC3-II or p62 post-EX in leupeptin treated mice). There are a number of critical methodological factors that could account for this difference. First, our mice were housed near thermoneutrality to reduce known effects of room temperature housing on thermogenesis and our acclimation protocol required only one brief treadmill run 72 h before study day. Furthermore, the lack of increased autophagic flux in our study may attribute to differences in exercise variables (duration, speed, and intensity) and the amount of time food was withdrawn before exercise stimulus.
Studies have also used isolated mitochondria from muscle to show exercise can activate mitophagy flux. A rat study utilizing chronic contractile activity (electrical pulse) plus an inhibitor of autophagy for 3 days resulted in markers of increased mitochondrial quality (53). These results are in agreement with the hepatic response seen in our previous study of chronic exercise (wheel running) where leupeptin injections in the last 24 h revealed that chronically exercising mice had lower activation of overall mitophagic flux compared with sedentary mice (10). We interpreted these findings to indicate that daily exercise lowered baseline levels of mitophagy flux paired with enhanced mitochondrial respiratory function following chronic exercise. These results caused us to question if a single bout of exercise in naïve, sedentary mice activates measurable hepatic mitophagy flux. Our results showed no increase in LC3-II and p62 right after exercise, but a significant accumulation of both proteins in the 2 h after recovery. Importantly, this only occurred in the leupeptin-treated condition where autophagy flux was inhibited, suggesting these proteins and their cargo are degraded in the saline control samples. In addition, we saw increased red punctae utilizing the mitochondrial-specific COX8-mCherry assay providing a confirmatory assessment of enhanced flux. Together, these results suggest that mitophagic flux is not immediately turned on during, but that the machinery is initiated and transiently increases in the early period following exercise.
Our results also suggest that mitochondrial polyubiquitination is one mechanism that may drive exercise-induced mitophagy in the liver, similar to what we recently reported with heat therapy (9). In support, exercise has been shown to increase mitochondrial ubiquitination in skeletal muscle of mice (12). p62 provides a molecular link between ubiquitin stress-induced degradation and selective mitophagy. p62 binds to ubiquitin/polyubiquitin chains marking them for autophagosomal degradation (54). Overexpression of ubiquitin alone can activate p62-dependent autophagy (55) whereas loss of hepatocyte p62 reduces mitochondrial ubiquitination (56). p62 contains two critical regions, a ubiquitin-associated domain allowing it to noncovalently bind to ubiquitin proteins and an LC3-interacting region for binding to LC3-II, which is bound to the phagophore membrane (57). Thus, having a significant increase in LC3-II, p62, and ubiquitin at 2-h post-EX in our isolated mitochondria strongly supports the notion that ubiquitination of mitochondrial proteins plays a critical role for exercise-induced mitophagy in the liver.
Hepatic energy metabolism is tightly modulated by nutrients, hormones, and neuronal signals (58) and these same signals may regulate hepatic autophagy or mitophagy. In the postprandial state, insulin blocks or slows autophagy through mTOR activation (59). In contrast, exercise reduces insulin secretion and increases glucagon, and these combined effects are necessary to drive increased hepatic glucose output (60). At the same time, glucagon is known to increase hepatic autophagy (61, 62) with effects seen in as little as 30 min (63). Recent studies have shown that glucagon can specifically activate hepatic mitophagy marked by mitochondria moving to lysosomes (64), and hepatic mitophagy being activated in a BNIP3-dependent manner (65). Although not studied here, future studies should determine the role of these powerful hormonal signals to modulate mitophagy during exercise. Autophagy serves to liberate stored amino acids and lipids within the liver for gluconeogenic precursors or energy utilization, respectively. In a similar manner, exercise is also known to liberate amino acid and lipid stores in the liver due to the livers role in maintaining euglycemia (hepatic glucose output) due to the significant increase in energy expenditure (66). It is unknown if these same signals mediate mitophagy or if different pathways mediated by mitochondrial signals are at play (i.e., ROS emission and membrane polarization).
Although results are inconsistent, likely due to different exercise modalities and different muscle groups, acute exercise seems to consistently increase the mitophagy proteins Parkin and Drp1 in skeletal muscle (18, 67, 68). One chronic exercise study showed significant changes in hepatic mitochondrial Drp1 and Parkin with both endurance treadmill training and wheel running (69). However, in the liver, our Western data showed no rapid or transient increase in Drp1 and Parkin after exercise. Again, these differences may be due to exercise parameters or simply due to studying liver versus muscle. Our results did show an increase in BNIP3 and Mfn2 right after exercise. Although our data suggests BNIP3 may be important for exercise-induced receptor-mediated hepatic mitophagy, we have previously shown that BNIP3 knockouts have no change in measures of hepatic mitophagic flux after chronic exercise, suggestive of redundant pathway regulating hepatic mitophagy (10).
We further leveraged proteomics in our isolated liver mitochondrial samples in various conditions (sedentary vs. exercise and saline vs. leupeptin). Comparing our isolated mitochondria to MitoCarta 3.0 (40), we had 60% of the MitoCarta proteins in our mitochondrial isolation preparations, in agreement with a recent study using equivalent techniques (45). Although IPA analysis of our proteomics data confirmed mitophagy pathways were activated at 0 h and further increased at 2-h postexercise, we also saw an accumulation of Dnml1 at 2 h, an effect not revealed by Western analysis. Our data also revealed a nonsignificant accumulation of Beclin1 (Becn1, autophagosome formation), Optineurin (Optn, adaptor protein for LC3-II and ubiquitin), and Bnip3 (receptor-mediated mitophagy protein) 2-h postexercise. Optineurin is an autophagy/mitophagy adaptor protein that binds to ubiquitinated cargo, serving a role similar or redundant to p62 (70). Comparison of saline-treated animals versus leupeptin-treated animals postexercise along with untargeted proteomics provides a unique platform to determine what proteins would normally be removed postexercise (saline treated) versus those preserved due to autophagosome inhibition (leupeptin treated), respectively. Overall, these results provide further supporting evidence that activation of hepatic metabolic flux postexercise likely involves the dual recruitment of the receptor-mediated protein BNIP3 and ubiquitination of mitochondrial proteins.
In conclusion, this is the first study to show acute exercise activates hepatic mitophagy, an important mechanism regulating mitochondrial turnover and function. Furthermore, utilizing proteomics, we elucidate regulatory proteins that are engaged acutely postexercise. Identifying the mechanisms by which exercise promotes hepatic mitochondrial recycling will further inform of the importance of exercise in human metabolic health, while potentially providing therapeutic targets to regulate degradation of damaged or low-functioning mitochondria.
SUPPLEMENTAL DATA
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.19027718.
GRANTS
This work was supported in part by a VA-Merit Grant 1I01BX002567 (J. P. Thyfault), NIH R01 DK121497 (J. P. Thyfault), Center for Children’s Healthy Lifestyles and Nutrition Pilot Grant Award (C. S. McCoin). The Ingenuity Pathways Analysis (IPA) software used in this publication was supported by the Biostatistics and Informatics Shared Resource, funded by the National Cancer Institute Cancer Center Support Grant P30 CA168524, and the Kansas IDeA Network of Biomedical Research Excellence Bioinformatics Core, supported in part by the National Institute of General Medical Science award P20GM103418. We acknowledge the IDeA National Resource for Quantitative Proteomics and Grants R24GM137786 and P20GM121293 and the Exploris instrument Grant S10OD026736.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.S.M. and J.P.T. conceived and designed research; C.S.M. and E.F. performed experiments; C.S.M., E.F., F.D., and D.P. analyzed data; C.S.M., E.F., W.-X.D., and J.P.T. interpreted results of experiments; C.S.M., E.F., and F.D. prepared figures; C.S.M. drafted manuscript; C.S.M., E.F., F.D., D.P., W.-X.D., and J.P.T. edited and revised manuscript; C.S.M., E.F., F.D., D.P., W.-X.D., and J.P.T. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Samuel G. Mackintosh and Dr. Stephanie Byrum at The University of Arkansas Medical Campus Proteomics Core (NIH R24GM137786 and P20GM121293) for contribution of mitochondrial proteomics. Graphical images created with BioRender.com and published with permission.
REFERENCES
- 1.Thyfault JP, Bergouignan A. Exercise and metabolic health: beyond skeletal muscle. Diabetologia 63: 1464–1474, 2020. doi: 10.1007/s00125-020-05177-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thyfault JP, Rector RS. Exercise combats hepatic steatosis: potential mechanisms and clinical implications. Diabetes 69: 517–524, 2020. doi: 10.2337/dbi18-0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 92: 829–839, 1998. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 4.Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 98: 115–124, 1999. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- 5.Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, Ruonala MO, Priault M, Salin B, Reichert AS. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta 1823: 2297–2310, 2012. doi: 10.1016/j.bbamcr.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Narendra D, Tanaka A, Suen D-F, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803, 2008. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12: 1–222, 2016. [Erratum in Autophagy 12: 443, 2016]. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haspel J, Shaik RS, Ifedigbo E, Nakahira K, Dolinay T, Englert JA, Choi AMK. Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Autophagy 7: 629–642, 2011. doi: 10.4161/auto.7.6.15100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Von Schulze AT, Deng F, Fuller KNZ, Franczak E, Miller J, Allen J, McCoin CS, Shankar K, Ding W-X, Thyfault JP, Geiger PC. Heat treatment improves hepatic mitochondrial respiratory efficiency via mitochondrial remodeling. Function 2: zqab001, 2021. doi: 10.1093/function/zqab001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schulze AV, McCoin CS, Onyekere C, Allen J, Geiger P, Dorn GW 2nd, Morris EM, Thyfault JP. Hepatic mitochondrial adaptations to physical activity: impact of sexual dimorphism, PGC1α and BNIP3-mediated mitophagy. J Physiol 596: 6157–6171, 2018. doi: 10.1113/JP276539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balan E, Schwalm C, Naslain D, Nielens H, Francaux M, Deldicque L. Regular endurance exercise promotes fission, mitophagy, and oxidative phosphorylation in human skeletal muscle independently of age. Front Physiol 10: 1088, 2019. doi: 10.3389/fphys.2019.01088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen CCW, Erlich AT, Crilly MJ, Hood DA. Parkin is required for exercise-induced mitophagy in muscle: impact of aging. Am J Physiol Endocrinol Metab 315: E404–E415, 2018. doi: 10.1152/ajpendo.00391.2017. [DOI] [PubMed] [Google Scholar]
- 13.McCoin CS, Schulze AV, Allen J, Fuller KNZ, Xia Q, Koestler DC, Houchen CJ, Maurer A, Dorn GW 2nd, Shankar K, Morris EM, Thyfault JP. Sex modulates hepatic mitochondrial adaptations to high-fat diet and physical activity. Am J Physiol Endocrinol Metab 317: E298–E311, 2019. doi: 10.1152/ajpendo.00098.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nobis S, Goichon A, Achamrah N, Guérin C, Azhar S, Chan P, Morin A, Bôle-Feysot C, do Rego JC, Vaudry D, Déchelotte P, Belmonte L, Coëffier M. Alterations of proteome, mitochondrial dynamic and autophagy in the hypothalamus during activity-based anorexia. Sci Rep 8: 7233, 2018. doi: 10.1038/s41598-018-25548-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zheng L, Rao Z, Guo Y, Chen P, Xiao W. High-intensity interval training restores glycolipid metabolism and mitochondrial function in skeletal muscle of mice with type 2 diabetes. Front Endocrinol (Lausanne) 11: 561, 2020. doi: 10.3389/fendo.2020.00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen CCW, Erlich AT, Hood DA. Role of Parkin and endurance training on mitochondrial turnover in skeletal muscle. Skelet Muscle 8: 10, 2018. doi: 10.1186/s13395-018-0157-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schiattarella GG, Altamirano F, Kim SY, Tong D, Ferdous A, Piristine H, Dasgupta S, Wang X, French KM, Villalobos E, Spurgin SB, Waldman M, Jiang N, May HI, Hill TM, Luo Y, Yoo H, Zaha VG, Lavandero S, Gillette TG, Hill JA. Xbp1s-FoxO1 axis governs lipid accumulation and contractile performance in heart failure with preserved ejection fraction. Nat Commun 12: 1684, 2021. doi: 10.1038/s41467-021-21931-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore TM, Zhou Z, Cohn W, Norheim F, Lin AJ, Kalajian N, Strumwasser AR, Cory K, Whitney K, Ho T, Ho T, Lee JL, Rucker DH, Shirihai O, van der Bliek AM, Whitelegge JP, Seldin MM, Lusis AJ, Lee S, Drevon CA, Mahata SK, Turcotte LP, Hevener AL. The impact of exercise on mitochondrial dynamics and the role of Drp1 in exercise performance and training adaptations in skeletal muscle. Mol Metab 21: 51–67, 2019. doi: 10.1016/j.molmet.2018.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Challa TD, Wueest S, Lucchini FC, Dedual M, Modica S, Borsigova M, Wolfrum C, Blüher M, Konrad D. Liver ASK1 protects from non-alcoholic fatty liver disease and fibrosis. EMBO Mol Med 11: e10124, 2019. doi: 10.15252/emmm.201810124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loh K, Tam S, Murray-Segal L, Huynh K, Meikle PJ, Scott JW, van Denderen B, Chen Z, Steel R, LeBlond ND, Burkovsky LA, O'Dwyer C, Nunes JRC, Steinberg GR, Fullerton MD, Galic S, Kemp BE. Inhibition of adenosine monophosphate-activated protein kinase-3-hydroxy-3-methylglutaryl coenzyme A reductase signaling leads to hypercholesterolemia and promotes hepatic steatosis and insulin resistance. Hepatol Commun 3: 84–98, 2019. doi: 10.1002/hep4.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim SY, Jeong JM, Kim SJ, Seo W, Kim MH, Choi WM, Yoo W, Lee JH, Shim YR, Yi HS, Lee YS, Eun HS, Lee BS, Chun K, Kang SJ, Kim SC, Gao B, Kunos G, Kim HM, Jeong WI. Pro-inflammatory hepatic macrophages generate ROS through NADPH oxidase 2 via endocytosis of monomeric TLR4-MD2 complex. Nat Commun 8: 2247, 2017. doi: 10.1038/s41467-017-02325-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Z, Shen H, Sun C, Yin L, Tang F, Zheng P, Liu Y, Brink R, Rui L. Myeloid cell TRAF3 promotes metabolic inflammation, insulin resistance, and hepatic steatosis in obesity. Am J Physiol Endocrinol Metab 308: E460–E469, 2015. doi: 10.1152/ajpendo.00470.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corbit KC, Camporez JPG, Edmunds LR, Tran JL, Vera NB, Erion DM, Deo RC, Perry RJ, Shulman GI, Jurczak MJ, Weiss EJ. Adipocyte JAK2 regulates hepatic insulin sensitivity independently of body composition, liver lipid content, and hepatic insulin signaling. Diabetes 67: 208–221, 2018. doi: 10.2337/db17-0524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tran M, Lee SM, Shin DJ, Wang L. Loss of miR-141/200c ameliorates hepatic steatosis and inflammation by reprogramming multiple signaling pathways in NASH. JCI Insight 2: e96094, 2017. doi: 10.1172/jci.insight.96094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shu Y, Hassan F, Ostrowski MC, Mehta KD. Role of hepatic PKCβ in nutritional regulation of hepatic glycogen synthesis. JCI Insight 6: e149023, 2021. doi: 10.1172/jci.insight.149023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nemazanyy I, Montagnac G, Russell RC, Morzyglod L, Burnol AF, Guan KL, Pende M, Panasyuk G. Class III PI3K regulates organismal glucose homeostasis by providing negative feedback on hepatic insulin signalling. Nat Commun 6: 8283, 2015. doi: 10.1038/ncomms9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinds TD Jr, Burns KA, Hosick PA, McBeth L, Nestor-Kalinoski A, Drummond HA, AlAmodi AA, Hankins MW, Vanden Heuvel JP, Stec DE. Biliverdin reductase A attenuates hepatic steatosis by inhibition of glycogen synthase kinase (GSK) 3β phosphorylation of serine 73 of peroxisome proliferator-activated receptor (PPAR) α. J Biol Chem 291: 25179–25191, 2016. doi: 10.1074/jbc.M116.731703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Magalhães-Novais S, Bermejo-Millo JC, Loureiro R, Mesquita KA, Domingues MR, Maciel E, Melo T, Baldeiras I, Erickson JR, Holy J, Potes Y, Coto-Montes A, Oliveira PJ, Vega-Naredo I. Cell quality control mechanisms maintain stemness and differentiation potential of P19 embryonic carcinoma cells. Autophagy 16: 313–333, 2020. doi: 10.1080/15548627.2019.1607694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peshkova IO, Aghayev T, Fatkhullina AR, Makhov P, Titerina EK, Eguchi S, Tan YF, Kossenkov AV, Khoreva MV, Gankovskaya LV, Sykes SM, Koltsova EK. IL-27 receptor-regulated stress myelopoiesis drives abdominal aortic aneurysm development. Nat Commun 10: 5046, 2019. doi: 10.1038/s41467-019-13017-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Srere P.A. [1] Citrate synthase: [EC 4.1.3.7. Citrate oxaloacetate-lyase (CoA-acetylating)], In: Methods in Enzymology, edited by Lowenstein JM. New York: Academic Press, 1969, vol. 13, p. 3–11, ISSN 0076-6879, ISBN 9780121818708, 10.1016/0076-6879(69)13005-0. [DOI] [Google Scholar]
- 31.Wang H, Ni H-M, Chao X, Ma X, Rodriguez YA, Chavan H, Wang S, Krishnamurthy P, Dobrowsky R, Xu D-X, Jaeschke H, Ding W-X. Double deletion of PINK1 and Parkin impairs hepatic mitophagy and exacerbates acetaminophen-induced liver injury in mice. Redox Biol 22: 101148–101148, 2019. doi: 10.1016/j.redox.2019.101148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma X, Ding WX. A fluorescence imaging based-assay to monitor mitophagy in cultured hepatocytes and mouse liver. Liver Res 5: 16–20, 2021. doi: 10.1016/j.livres.2020.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Byrum SD, Washam CL, Tackett AJ, Garcia-Rill E, Bisagno V, Urbano FJ. Proteomic measures of γ oscillations. Heliyon 5: e02265, 2019. doi: 10.1016/j.heliyon.2019.e02265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Searle BC, Pino LK, Egertson JD, Ting YS, Lawrence RT, MacLean BX, Villén J, MacCoss MJ. Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat Commun 9: 5128, 2018. doi: 10.1038/s41467-018-07454-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Graw S, Tang J, Zafar MK, Byrd AK, Bolden C, Peterson EC, Byrum SD. proteiNorm: a user-friendly tool for normalization and analysis of TMT and label-free protein quantification. ACS Omega 5: 25625–25633, 2020. doi: 10.1021/acsomega.0c02564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huber W, von Heydebreck A, Sültmann H, Poustka A, Vingron M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18 Suppl 1: S96–S104, 2002. doi: 10.1093/bioinformatics/18.suppl_1.s96. [DOI] [PubMed] [Google Scholar]
- 37.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19: 185–193, 2003. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 38.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43: e47, 2015. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chawade A, Alexandersson E, Levander F. Normalyzer: a tool for rapid evaluation of normalization methods for omics data sets. J Proteome Res 13: 3114–3120, 2014. doi: 10.1021/pr401264n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, Goodman RP, Grabarek Z, Haas ME, Hung WHW, Joshi PR, Jourdain AA, Kim SH, Kotrys AV, Lam SS, McCoy JG, Meisel JD, Miranda M, Panda A, Patgiri A, Rogers R, Sadre S, Shah H, Skinner OS, To TL, Walker MA, Wang H, Ward PS, Wengrod J, Yuan CC, Calvo SE, Mootha VK. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res 49: D1541–D1547, 2021. doi: 10.1093/nar/gkaa1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alhamdoosh M, Ng M, Wilson NJ, Sheridan JM, Huynh H, Wilson MJ, Ritchie ME. Combining multiple tools outperforms individual methods in gene set enrichment analyses. Bioinformatics 33: 414–424, 2017. doi: 10.1093/bioinformatics/btw623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab 13: 495–504, 2011. doi: 10.1016/j.cmet.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kristensen CM, Jessen H, Ringholm S, Pilegaard H. Muscle PGC-1α in exercise and fasting-induced regulation of hepatic UPR in mice. Acta Physiol (Oxf) 224: e13158, 2018. doi: 10.1111/apha.13158. [DOI] [PubMed] [Google Scholar]
- 44.Dunlop EA, Tee AR. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol 36: 121–129, 2014. doi: 10.1016/j.semcdb.2014.08.006. [DOI] [PubMed] [Google Scholar]
- 45.McLaughlin KL, Hagen JT, Coalson HS, Nelson MAM, Kew KA, Wooten AR, Fisher-Wellman KH. Novel approach to quantify mitochondrial content and intrinsic bioenergetic efficiency across organs. Sci Rep 10: 17599, 2020. doi: 10.1038/s41598-020-74718-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fletcher JA, Meers GM, Linden MA, Kearney ML, Morris EM, Thyfault JP, Rector RS. Impact of various exercise modalities on hepatic mitochondrial function. Med Sci Sports Exerc 46: 1089–1097, 2014. doi: 10.1249/MSS.0000000000000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lima FD, Stamm DN, Della-Pace ID, Dobrachinski F, de Carvalho NR, Royes LFF, Soares FA, Rocha JB, González-Gallego J, Bresciani G. Swimming training induces liver mitochondrial adaptations to oxidative stress in rats submitted to repeated exhaustive swimming bouts. PLoS One 8: e55668, 2013. doi: 10.1371/journal.pone.0055668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 3: 542–545, 2007. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- 49.Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, Abdellatif M, Abdoli A, Abel S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 17: 1–382, 2021. doi: 10.1080/15548627.2020.1797280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lira VA, Okutsu M, Zhang M, Greene NP, Laker RC, Breen DS, Hoehn KL, Yan Z. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J 27: 4184–4193, 2013. doi: 10.1096/fj.13-228486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brandt N, Gunnarsson TP, Bangsbo J, Pilegaard H. Exercise and exercise training-induced increase in autophagy markers in human skeletal muscle. Physiol Rep 6: e13651, 2018. doi: 10.14814/phy2.13651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ju J-S, Jeon S-I, Park J-y, Lee J-y, Lee S-C, Cho K-J, Jeong J-M. Autophagy plays a role in skeletal muscle mitochondrial biogenesis in an endurance exercise-trained condition. J Physiol Sci 66: 417–430, 2016. doi: 10.1007/s12576-016-0440-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carter HN, Kim Y, Erlich AT, Zarrin-Khat D, Hood DA. Autophagy and mitophagy flux in young and aged skeletal muscle following chronic contractile activity. J Physiol 596: 3567–3584, 2018. doi: 10.1113/JP275998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu HL, Yang C, Liu HF. p62 links the autophagy pathway and the ubiqutin–proteasome system upon ubiquitinated protein degradation. Cell Mol Biol Lett 21: 29, 2016. doi: 10.1186/s11658-016-0031-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peng H, Yang J, Li G, You Q, Han W, Li T, Gao D, Xie X, Lee B-H, Du J, Hou J, Zhang T, Rao H, Huang Y, Li Q, Zeng R, Hui L, Wang H, Xia Q, Zhang X, He Y, Komatsu M, Dikic I, Finley D, Hu R. Ubiquitylation of p62/sequestosome1 activates its autophagy receptor function and controls selective autophagy upon ubiquitin stress. Cell Res 27: 657–674, 2017. doi: 10.1038/cr.2017.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamada T, Murata D, Adachi Y, Itoh K, Kameoka S, Igarashi A, Kato T, Araki Y, Huganir RL, Dawson TM, Yanagawa T, Okamoto K, Iijima M, Sesaki H. Mitochondrial stasis reveals p62-mediated ubiquitination in parkin-independent mitophagy and mitigates nonalcoholic fatty liver disease. Cell Metab 28: 588–604.e5, 2018. doi: 10.1016/j.cmet.2018.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell 34: 259–269, 2009. doi: 10.1016/j.molcel.2009.04.026. [DOI] [PubMed] [Google Scholar]
- 58.Rui L. Energy metabolism in the liver. Compr Physiol 4: 177–197, 2014. doi: 10.1002/cphy.c130024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frendo-Cumbo S, Tokarz VL, Bilan PJ, Brumell JH, Klip A. Communication between autophagy and insulin action: at the crux of insulin action-insulin resistance? Front Cell Dev Biol 9: 708431, 2021. doi: 10.3389/fcell.2021.708431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trefts E, Williams AS, Wasserman DH. Exercise and the Regulation of Hepatic Metabolism. Prog Mol Biol Transl Sci 135: 203–225, 2015. doi: 10.1016/bs.pmbts.2015.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ashford TP, Porter KR. Cytoplasmic components in hepatic cell lysosomes. J Cell Biol 12: 198–202, 1962. doi: 10.1083/jcb.12.1.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kanasaki K, Kawakita E, Koya D. Relevance of autophagy induction by gastrointestinal hormones: focus on the incretin-based drug target and glucagon. Front Pharmacol 10: 476–476, 2019. doi: 10.3389/fphar.2019.00476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Deter RL, De Duve C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol 33: 437–449, 1967. doi: 10.1083/jcb.33.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rodriguez-Enriquez S, Kim I, Currin RT, Lemasters JJ. Tracker dyes to probe mitochondrial autophagy (mitophagy) in rat hepatocytes. Autophagy 2: 39–46, 2006. doi: 10.4161/auto.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Springer MZ, Poole LP, Drake LE, Bock-Hughes A, Boland ML, Smith AG, Hart J, Chourasia AH, Liu I, Bozek G, Macleod KF. BNIP3-dependent mitophagy promotes cytosolic localization of LC3B and metabolic homeostasis in the liver. Autophagy, 17: 3530–3517, 2021. doi: 10.1080/15548627.2021.1877469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shimomura Y, Honda T, Shiraki M, Murakami T, Sato J, Kobayashi H, Mawatari K, Obayashi M, Harris RA. Branched-chain amino acid catabolism in exercise and liver disease. J Nutr 136: 250s–253s, 2006. doi: 10.1093/jn/136.1.250S. [DOI] [PubMed] [Google Scholar]
- 67.Yoo S-Z, No M-H, Heo J-W, Park D-H, Kang J-H, Kim J-H, Seo D-Y, Han J, Jung S-J, Kwak H-B. Effects of acute exercise on mitochondrial function, dynamics, and mitophagy in rat cardiac and skeletal muscles. Int Neurourol J 23: S22–S31, 2019. doi: 10.5213/inj.1938038.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vainshtein A, Tryon LD, Pauly M, Hood DA. Role of PGC-1α during acute exercise-induced autophagy and mitophagy in skeletal muscle. Am J Physiol Cell Physiol 308: C710–C719, 2015. doi: 10.1152/ajpcell.00380.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Santos-Alves E, Marques-Aleixo I, Rizo-Roca D, Torrella JR, Oliveira PJ, Magalhães J, Ascensão A. Exercise modulates liver cellular and mitochondrial proteins related to quality control signaling. Life Sci 135: 124–130, 2015. doi: 10.1016/j.lfs.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 70.Ryan TA, Tumbarello DA. Optineurin: a coordinator of membrane-associated cargo trafficking and autophagy. Front Immunol 9: 1024, 2018. doi: 10.3389/fimmu.2018.01024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1: https://doi.org/10.6084/m9.figshare.19027718.