

Abstract
The manuscript deals with cost-effective synthesis, structural characterization and in silico SARS-CoV-2 screening activity of 5-membered heterocycle-substituted benzimidazole derivatives, 1-((1H-pyrrol-2-yl)methyl)-2-(1H-pyrrol-2-yl)-1H-benzo[d]imidazole (L1), 2-(furan-2-yl)-1-(furan-2-ylmethyl)-1H-benzo[d]imidazole (L2), 2-(thiophen-2-yl)-1-(thiophen-2-ylmethyl)-1H-benzo[d]imidazole (L3). The benzimidazole compounds were synthesized through a green-synthetic approach by coupling of 5-membered heterocyclic-carboxaldehyde and o-phenylenediamine in water under an aerobic condition. The compounds were characterized by various spectroscopic methods and X-ray structural analysis. The suitable single-crystals of the methyl derivative of L3 were grown as L3′ which crystallized in a monoclinic system and the thiophene groups co-existed in a nearly a perpendicular orientation. Further, in silico anti-SARS-CoV-2 proficiency of the synthetic derivatives is evaluated against main protease (Mpro) and non-structural proteins (nsp2 and nsp7) of SARS-CoV-2. Molecular docking and molecular dynamics analysis of the ligands (L1-L3) against Mpro and nsp2 and nsp7 for 50 ns reveal that L3 turns out to be the superlative antiviral candidate against Mpro, nsp2 and nsp7 of SARS-CoV-2 as evident from the binding score and stability of the ligand-docked complexes with considerable binding energy changes.
Keywords: 1,2-disubstituted benzimidazole; Crystal structure; In silico anti-SARS-CoV-2 screening activity; MD simulations; Molecular docking; Green synthesis
Graphical abstract

1. Introduction
The appearance of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and their uncontrolled expansion explodes the social and economic situations of the modern civilization to a great extent [1]. The living and non-living world are also seriously affected by the corona virus infection in direct and indirect ways. Literature survey proposes that 2019-nCoV virus belongs to the family of Coronaviridae (Scheme 1 ) and the beta-coronaviruses are believed to be originated from bats [2], [3], [4]. This disease exhibits various symptoms, from mild flu to lethal conditions [5], [6], [7], [8], [9]. Thus, World Health Organization (WHO) avowed this COVID-19 a pandemic on 12th March 2020 (WHO, 2020).
Scheme 1.

Schematic diagram of SARS-CoV-2.
Moreover, the corona virus infection spectacles more worsening starting from acute respiratory failure to sepsis and death [10], [11], [12], [13]. Nevertheless, critical issues like aggravated seditious reaction, tenacious viral load, organ-specific indices, and flawed antiviral resistance routes [14,15] are accountable to monitor the COVID-19 disease. Therefore, investigations on the underlying cellular mechanisms at molecular grabs a great deal of interests to the scientists for the development of physiopathology and consequent progress of suitable therapeutics. During the last two years, various scientific groups made a significant effort to design and development of molecular therapeutics and vaccines including treatment procedures to prevent, control, and cure this disease is praiseworthy [14,15]. In the true sense, this immense effort results in significant progress of medical science to combat human coronaviruses, although, the huge population, consciousness, and inadequate therapeutics enforce a restriction in recovering from this pandemic quickly.
Typically, the rational design of cost-effective antibiotic compounds is crucial to the development of suitable therapeutics. Noteworthy, benzimidazole derivatives are a widely recommended functional pharmacophores used in a large number of drugs (Scheme 2 ) [16], [17], [18], [19]. Benzimidazole based drugs have also been used as potent inhibitors for reverse transcriptase of HIV-1 virus [20], NSSB polymerase of Hepatitis C virus [21], thrombin inhibitors [22] and as antibacterial agents [23]. Although mono-substituted benzimidazole derivatives are mostly used in drug design but the use of di-substituted benzimidazoles are limited. Only few structure of the di-substituted benzimidazole derivatives are also reported in the literature [24], [25], [26]. Nevertheless, 5-membered heterocycle substituted benzimidazoles have a great impact in therapeutic actions against bacterial, fungal and viral species [27], [28], [29]. Like 2-(2-Furyl) benzimidazole (FBD) showed an excellent antifungal activity [27]. The compound (E)-(2-(2 (furan-2-yl)vinyl)-1H-benzo[d]imidazol-6yl)(phenyl)methanone and (E)-phenyl(2-(2-(thiophe-2-yl)vinyl)-1H-benzo[d]imidazole-6-yl)methanone compounds turn out to show noteworthy antitubercular activity against M. tuberculosis H37Rv strain [28,29]. 2-(5′-Cyano-2,2′-bithiophen-5-yl)-1H-benzimidazole-5(6)-carbonitrile compound showed a remarkable antiparasitic activity against T. brucei rhodesiense [29].
Scheme 2.

Available benzimidazole drugs comprising heterocyclic benzimidazoles.
Very recently, we have designed and synthesized one hydrated phenazine chloride explored their therapeutic characteristics with computational aided drug design and in vitro SARS-CoV-2 screening activities against Vero cell lines [30]. In this context, we have designed and developed three 5-membered heterocycle-substituted benzimidazoles and characterized them with a suite of spectroscopic methods and X-ray structural analysis. We also evaluated the effect of 5-member heterocycle-substituted benzimidazole derivatives against Mpro, nsp2 and nsp7 through in silico SARS-CoV-2 screening to reveal the bio-potency of the compounds.
2. Experimental
2.1. Preparation of the benzimidazole
(a) Chemicals, solvents and starting materials
o-phenylenediamine (Sigma Aldrich, USA), 3,4-diaminotoluene (TCI, Japan) furfural (Sigma Aldrich, USA), pyrrole-2-aldehyde (Sigma Aldrich, USA), thiophene-2-aldehyde (Sigma Aldrich, USA) were purchased from the respective outlets. An analytical grade chemicals and solvents was used in this work.
(b) Synthesis of the benzimidazole compounds
The benzimidazole derivatives, 1-((1H-pyrrol-2-yl)methyl)-2-(1H-pyrrol-2-yl)-1H-benzo[d]imidazole (L1), 2-(furan-2-yl)-1-((furan-2-yl)methyl)-1H-benzo[d]imidazole (L2) and 2-(thiophen-2-yl)-1-((thiophen-2-yl)methyl)-1H-benzo[d]imidazole (L3) and were synthesized following our established synthetic strategy with a little modification [31]. Aromatic amine, o-phenylene diamine (1 mmol, 0.108 g) was taken in 15 mL test tubes. Aromatic aldehydes, pyrrole-2-aldehyde (2 mmol, 0.190 g), furan-2-aldehyde (2 mmol, 0.192 g) and thiophene-2-aldehyde (2 mmol, 0.224 g) were added separately to the amine with the addition of 2 mL 10% ethanol-water mixture. The compound 6-methyl-2-(thiophen-2-yl)-1-((thiophen-2-yl)methyl)-1H-benzo[d]imidazole (L3′) was synthesized by following the same synthetic procedure through a coupling of 3,4-diaminotoluene (1 mmol, 0.123 g) with thiophene-2-aldehyde (2 mmol, 0.224 g). The solution mixtures were charged into an oil-bath and heated with 75 °C. A syringe attached with an aquarium air pump was immersed into the reaction mixture with the controlled airflow of 300 bubbles/min. This solution was allowed to stir for 12 h at 75 °C. After 12 h. Ethyl acetate (3 × 3 mL) was added and the organic part was collected and dried the solution over sodium sulfate for 3, 4 h followed by product purification with column chromatography.
Yield of L1: 0.253 g (85%). M.P. : 260-261 °C. Anal. Calc. for C16H14N (L1): C, 73.26; H, 5.38; N, 21.36; Found: C, 73.24; H, 5.42; N, 21.40. IR (KBr, cm–1; Fig. S2): 3432 (νN—H), 1615(νC = N); UV-Vis (λmax, nm; Fig. S1): 240, 300; 1HNMR (DMSO): δ 11.83–11.80 [1H, NH-Pyrrole], δ 10.95 [1H, NH-Pyrrole], δ 7.62–5.91 [10H, Ar-H], δ 5.68–5.57 [2H, -CH 2]. 13C{1H}(DMSO): δ 147.20 (H— C in imidazole ring), δ 142.86–111.29 (PhC & PyrroleC), δ 43.50 (CH2).
Yield of L2: 0.258 g (86%). M.P. : 93-96 °C. Anal. Calc. for C16H12N2O2 (L2): C, 72.72; H, 4.58; N, 10.60; O, 12.11; Found: C, 72.69; H, 4.63; N, 10.57; O, 12.16. IR (KBr, cm–1; Fig. S2): 1620(νC = N); UV-Vis (λmax, nm; Fig. S1): 246, 305; 1HNMR (CDCl3): δ 7.68–7.67 [1H, H-Furan], δ 7.36–7.33 [1H, H-Furan], δ 7.33–7.31 [1H, H-Furan], δ 7.28 [3H, H-Furan], δ 7.25–7.24 [1H, Ph-H], δ 6.64–6.63 [1H, Ph-H], 6.31–6.30 [1H, Ph-H], 6.27 [1H, Ph-H], δ 5.67 [2H, -CH 2]. 13C{1H}(CDCl3): δ 149.60 (H— C in imidazole ring), δ 145.40–108.37 (PhC & FuranC), δ 41.70 (CH2).
Yield of L3: 0.234 g (79%). M.P. : 146–149 °C. Anal. Calc. for C16H12N2S2 (L3): C, 64.83; H, 4.08; N, 9.45; S, 21.64; Found: C, 64.79; H, 4.10; N, 9.47; S, 21.61. IR (KBr, cm–1; Fig. S2): 3102 (νAr-C-H), 1622(νC = N); UV-Vis (λmax, nm; Fig. S1): 234, 309; 1HNMR (DMSO): δ 7.83–7.82 [1H, Ph-H], δ 7.74–7.66 [3H,H-2-substituted Thio], δ 7.41–7.40 [1H, Ph-H], δ 7.39–7.24 [3H,H-2-substituted Thio], δ 7.03 [1H, Ph-H], 6.97–6.95 [1H, Ph-H], δ 5.94 [2H, CH 2], δ 13C{1H}(CDCl3): δ 147.20 (H— C in imidazole ring), δ 142.86–111.29 (PhC & ThioC), δ 43.50 (CH2).
Yield of L3′: 0.295 g (85%). Anal. Calc. for C17H14N2S2 (L3ʹ): C, 65.77; H, 4.55; N, 9.02; S, 20.66; Found: C, 65.74; H, 4.60; N, 9.07; S, 20.61. 1HNMR (CDCl3): δ 7.71–7.69 [1H, Ph-H], δ 7.50–7.49 [1H, Ph-H], δ 7.44–7.43[1H, Ph-H], δ 7.26–7.24 [1H, H-2-substituted Thio], δ 7.16 [3H, H-2-substituted Thio], 7.14–7.12 [1H, H-2-substituted Thio], δ 6.87–6.86 [1H, H-2-substituted Thio], δ 5.68 [2H, CH 2], δ 2.48 [3H, CH 3]. 13C{1H}(CDCl3): δ 147.15 (H— C in imidazole ring), δ 141.11–109.43 (PhC & ThioC), δ 44.05 (CH2), δ 21.93–21.61(CH3).
2.2. Physical measurements
FTIR-8400S SHIMADZU spectrometer (Shimadzu, Kyoto, Japan) was used for recording the IR spectra (KBr) of the synthetic benzimidazoles. The NMR spectra of the benzimidazole products were obtained on a Bruker Advance 400 MHz spectrometer (Bruker, Massachusetts, USA) in CDCl3 and DMSO at 25 ºC. Steady-state absorption and other spectral data were obtained on a JASCO V-730 UV-Vis spectrophotometer (Jasco, Tokyo, Japan).A Perkin Elmer 2400 CHN microanalyzer (Perkin Elmer, Waltham, USA) was used to perform the elemental analysis. The electron spray ionization mass spectral (ESI-MS) measurement was carried out on a Q-tof-micro quadruple mass spectrometer.
2.3. Structural refinement of L3′
Suitable single-crystals of L3′ was selected for single crystal X-ray diffraction studies. The structural diffraction data of L3′ were collected on a Bruker-Kappa APEX II CCD diffractometer equipped with a 1 K charge-coupled device (CCD) area detector employing a graphite monochromated Mo-Ka radiation (k ¼ 0.71073 Å) at 100.0(2) K. The cell parameters and the reduction and correction of the collected data were determined by SMART SAINTPLus software, respectively [32] followed by SADABS absorption corrections [33]. Finally, the structure was solved by direct method with SHELXL-97 program package [34]. The refinement by full-matrix least-squares method was executed on all F2 data with SHELXL-97. For all non-hydrogen atoms, anisotropic refinement was performed. Subsequently the additional hydrogen atoms were positioned by riding model.
2.4. In silico SARS-CoV-2 screening activity
2.4.1. Protein and ligand preparation
The therapeutic potency of the synthetic benzimidazole derivatives was examined against main protease and non-structural proteins (Covid-19 main protease, NSP2 and NSP7) by molecular docking and dynamic simulations studies. Prior to carry out the docking study, initially 3D structures of the target proteins of interest were retrieved from protein data bank (https://www.rcsb.org/). Later, the obtained structures corresponding to each of the proteins were prepared and pre-processed to carry out further analysis. This step was performed using Flare protein preparation option available in the Flare version 4.0 of cresset software (https://www.cresset-group.com/software/flare/). The steps involve addition of missing hydrogen's, assigning optimal ionization states to residues, optimizing spatial positions of polar hydrogen's to improve hydrogen bonding, minimizing the steric strain and reconstructing unresolved side chains [35]. Followed by protein preparation, structures of each of the compounds to be analyzed for molecular docking analysis were prepared and minimized using ‘Flare minimize selection calculation’ option to remove clashes among atoms of the ligand and to develop a reasonable starting pose.
2.4.2. Molecular docking
Following protein and ligand preparation, each compound was docked with each of the prepared target proteins mentioned above. For carrying the docking analysis, Flare module of Cresset with default settings was used and the docking mode was set as “Very accurate but slow” . The grid box was created around the selected key residues within the active binding site. The best docking pose for each of the compound was selected based on LF rank score, LF dG score and LF LE score . As per the literature, amino acids Asp 5, Thr 9, Ala 65, Asp 67 and Glu 74 are crucial to facilitate the inhibition of NSP 7 by inhibitors. Therefore, considering these reports, grid was generated around these key residues [36]. Herein for carrying the docking analysis PDB ID: 7JLT was selected. In case of NSP2, PDB ID: 7EXM was selected because a x-ray crystallographer [37] suggested that Lys111, Lys112, Lys113 were key residues which are required for interacting with nucleic acid and further regulating intracellular signaling pathways. Based on Ma and co-workers study, these three positively charged residues were considered for grid generation and molecular docking [37]. For the third target, PDB ID: 6LU7 was considered and docking was carried out by choosing some of the important amino acids i.e. Asn 142, Gly 143, Ser 144 and Cys 145 [38]. For docking analysis, Lead Finder™ which is a dedicated algorithm and scoring function for virtual screening was employed.
2.4.3. Molecular dynamics study
The selected docked complexes of each target protein with the designed compounds based on best docking scores were further subjected to molecular dynamic simulations for a period of 50 ns using dynamic option available with Flare module of cresset software. To carry out the dynamic simulations, AMBER force-field and version GAFF2 was employed. AM1-BCC charge method was set up while system was build using explicit solvent with 10 Å × 10 Å × 10 Å dimensions. The build system was initially equilibrated for 200 ps and 2fs was kept as time step [39]. The whole calculations were run on GPU Nvidia Tesla V100, 16 GB.
2.4.4. Electrostatic complementarity
Generally, small molecules interacts with their respective receptors via non-covalent interactions including hydrogen bonding, ionic, cation-π, lone-pair sigma hole (halogen bonding), π-π, and orthogonal multipolar interactions (fluorine bonding). These non-covalent interactions are important contributors to the binding free energy ΔG (enthalpic term) of each protein-ligand complexes. We performed electrostatic complementarity analysis using Flare module of Cresset software to assess the match between each of the designed molecules and binding pockets in terms of electrostatics. This type of analysis provided deep insight into how these ligands bind within each of these three targets associated with SARS-CoV2.
3. Results and discussion
3.1. Synthesis and spectroscopic characterization of the benzimidazole derivatives (L1-L3)
The five-membered heterocycle-substituted benzimidazole compounds were synthesized following a straightforward synthetic approach by coupling of aromatic amines and aldehydes. We have established this synthetic approach to a large number of benzimidazole derivatives in aqueous medium and aerobic condition. Here, we modified this synthetic method and used 10% ethanol-water mixture instead of pure aqueous medium to obtain the product in more yield. The benzimidazole derivatives (L1 - L3) were synthesized by the coupling of o-phenylenediamine as the amine source with pyrrole-2-aldehyde, furan-2-aldehyde and thiophene-2-aldehyde as the aldehyde sources in a 1:2 mole ratio at 75 °C for 12 h. We made several attempts to produce single-crystals of L1-L3 compounds employing different crystallization methods (slow evaporation, diffusion technique, solvothermal) as well as various solvent mixtures like chloroform-pet ether, dichloromethane-methanol, ethanol, etc., however, we could not able to develop the single-crystals for those compounds. Although, we successfully prepared the single-crystals of L3′, a methyl derivative of L3 with slow-evaporation technique at room temperature. All the compounds are soluble in MeOH, EtOH, DMSO etc. The synthetic procedure for the benzimidazole derivatives is given in Scheme 3 .
Scheme 3.

Preparative route for benzimidazoles, L1-L3 and L3ʹ.
The structural formulations of the 1,2-disubstituted benzimidazole compounds (L1-L3) were assessed with FT-IR, UV-Vis, 1H and 13C NMR spectral analysis. The absorption profiles of the benzimidazole compounds L1-L3 were recorded in methanol medium. L1-L3 displayed high energetic electronic transitions in the ultra-violet region. All the compounds showed two characteristics absorbance bands in 230–250 nm and 300–310 nm at room temperature (Fig. S1). The appearence of these higher energetic electronic bands corresponds to the π→π or n→π electronic transitions of the 5-membered heterocyclea in all the benzimidazole scaffolds. The notable electronic bands of L1-L3 agree with the literature values [40,41]. FT-IR spectral analysis of the benzimidazoles exhibited characteristics peaks at 1615, 1620 and 1622 cm–1 for L1, L2 and L3, respectively attributing the stretching frequencies of azomethine groups,–CH=N– groups of benzimidazoles (Fig. S2). Other characteristic stretching frequencies in the range from 3105 to 2900 cm–1 suggest the characteristic stretching frequencies of R1–CH2 groups. These IR spectral values resemble well with the reported values [23,30]. The solution stabilities of the compounds were assessed by time-dependent UV-Vis spectra in methanol at room temperature for 24 h. The electronic band positions were unaffected during this period and suggests the stability of the molecules in methanol.
The 1H NMR spectral analyses of the benzimidazoles help to understand the molecular geometries of the 1,2-disubstituted benzimidazoles. All the compounds showed the presence of a characteristic peak at δ 5.57, 5.67 5.94 and 5.68 ppm for L1, L2, L3 and L3ʹ, respectively revealing the methylene protons of the 5-membered heterocycles substituted benzimidazoles. The signals for the aromatic protons of the L1-L3 and L3ʹ appeared in the same region ranging from δ7.8 to 6.2 ppm, respectively. The chemical shift values at δ 11.83–11.80 ppm and 10.95 ppm represent the –NH protons of the pyrrole heterocycle in L1 and the peak at δ 2.48 ppm represents the methyl protons of L3ʹ (Figs. S3, S5, S7, S9). The 13C NMR spectra were also recorded to confirm the structural composition of the C-atoms in L1-L3 and L3ʹ. The signals at δ 147.20, 149.60, 147.20 and 147.15 ppm attributed the presence of C-atom in the imidazole ring (L1-L3 and L3ʹ) while the charcteristic signals at δ 43.50, 41.70, 43.50 and 44.05 ppm correspond to the methylene-C in the 1,2-disubstituted benzimidazoles L1, L2, L3 and L3ʹ, respectively. The chemical shift values of the aromatic-C in L1, L2, L3 and L3ʹ appeared in the range δ142.86–111.29, 145.40–108.37, 142.86–111.29 and 141.11–109.43 ppm, respectively (Figs. S4, S6, S8, S10). The reported values of the benzimidazoles correlate well to the reported literature values [23,31].
3.2. Description of crystal structure and non-covalent interactions
The crystal structural analysis reveals that the methyl derivative of thiophene substituted-benzimidazole (L3′) crystallized in a monoclinic system with a P21/c space group. The ORTEP diagram of the compound is displayed in Fig. 1 (a). The crystallographic refinement parameter is summarized in Table 1 and bond angles and bond distances are noted in Table S1. In the structure of L3′, one thiophene directly links with the benzimidazole group while the second thiophene attaches to the benzimidazole moiety through a methylene-C. Furthermore, two thiophene rings co-exist in a perpendicular orientation.
Fig. 1.

(a) ORTEP diagram of the L3′ with 30% probability; (b) Formation of a 1D framework of L3′ based on N…H, S…H hydrogen bonding and C–H…π (cyan dotted) along b axis; (c) Formation of the 3D crystalline architecture.
Table 1.
Crystallographic data and structure refinement parameters for L3.
| Parameters | L3 |
|---|---|
| Empirical formula | C17.54H14N4S2 |
| Formula weight | 316.39 |
| Temperature (K) | 100 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| a (Å) | 15.4110(9) |
| b (Å) | 6.2845(3) |
| c (Å) | 15.6534(9) |
| Volume (Å3) | 1468.10(14) |
| Z | 4 |
| ρ (gcm–3) | 1.434 |
| μ (mm–1) | 0.367 |
| F (000) | 661 |
| Rint | 0.063 |
| θ ranges (º) | 3.5–30.5 |
| Number of unique reflections | 3552 |
| Total number of reflections | 13,979 |
| Final R indices | 0.0727, 0.2049 |
| Largest peak and hole (eA˚−3) | 1.66, -1.22 |
Self-assembly analysis of L3′ reveals that the thiophene-substituted benzimidazole grows through moderate distant C–H…π, S…H and N…H interactions in crystalline phase (Fig. 1b) leading to a 1D supramolecular frameworks along b axis. The supramolecular 1D chain interacts with another supramolecular 1D framework to develop a 3D supramolecular architecture (Fig. 1c). The interconnecting 1D frameworks are further stabilized by C–H…π interactions. The details of the non-covalent interactions for 3 are summarized in Table S2.
3.4. Molecular docking studies
To predict the best matching mode of the designed ligand to each of the selected target, molecular docking analysis was carried out. This approach assisted in analyzing all possible non-covalent interactions that may facilitate the nsp7 inhibition. It was observed that the synthetic heterocycle substituted benzimidazoles manifested crucial pi-pi stacking and hydrogen bond interactions with key residues His 41, Leu 141, Gly 143, Ser 144 and Cys 145 in case of main protease (6LU7). The docking scores corresponding to each of the designed molecule have been displayed in Table 2 . The scores were obtained in terms of LF rank, dG value, LF VSscore and LF LE. Herein, LF rank and dG indicate correct energy-ranking of each docked ligand poses and accurate binding energy, respectively. On the other hand, VSscore and LE indicate correct rank-ordering of active and inactive compounds in virtual screening experiments and estimated ligand efficiency, respectively. Among these three designed compounds; thiophene substituted benzimidazole (L3) displayed good rank score, dG, VSscore and LF-LE score with values -10.523, -6.148, -7.777 and -0.307, respectively. It was observed that L3 displayed 2-pi-pi stacking interactions with His 41 and 1- Hydrogen bonding with Gly 143. The pi-pi stacking interactions were observed owing to aromatic thiophene and benzimidazole rings with imidazole ring in His 41.
Table 2.
Docking results for designed molecules and Covid-19 main protease (6LU7) complexes.
| S.no | Compound | LF Rank Score | LF dG | LF VSscore | LF LE | Interactions |
|---|---|---|---|---|---|---|
| 1. | 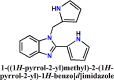 |
-5.956 | -5.795 | -7.184 | -0.290 | 2-pi-pi stacking interactions with His 41, 3-Hydrogen bonding with Gly 143, Ser 144 and Cys 145, 1-steric clashes with Leu 141 |
| 2. | 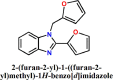 |
-5.922 | -5.747 | -6.871 | -0.287 | 2-pi-pi stacking interactions with His 41, 3-Hydrogen bonding with Gly 143, Ser 144 and Cys 145, 1-steric clashes with Leu 141 |
| 3. | 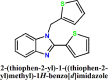 |
-10.523 | -6.148 | -7.777 | -0.307 | 2-pi-pi stacking interactions with His 41, 1- Hydrogen bonding with Gly 143 |
⁎⁎⁎LF-Rank score indicates correct energy-ranking of docked ligand poses.
* LF-dG score symbolizes accurate binding energy predictions.
⁎⁎ LF-VScore indicates correct rank-ordering of active and inactive compounds in virtual screening experiments.
⁎⁎⁎⁎LF-LE score signifies estimated ligand efficiency.
In the case of docking the designed compounds with nsp2, L3 also conferred the highest docking scores with -5.075, -2.548, -3.757 and -0.127 values (Table S3) corresponding to Rank, dG, VSscore and LE score, respectively. However, these scores were found to be slightly lower than docking scores observed with main-protease of the same designed compounds. This compound harbored 3 - Hydrogen bonding interaction with key residues including Lys 113 and Glu 110, which are very crucial for demonstrating nsp2 inhibitory activity. The pyrrole substituted benzimidazole (L1) manifested very low scores with none of the crucial interactions.
The designed compounds were also observed for interactions with target nsp7, it was observed that all the designed compounds were found to interact with Asp 67 via hydrogen bonding within the active site of nsp7. Among all the compounds, L3 could manage to score highest in docking analysis with Rank, dG, VSscore and LE scores as -6.639, -4.420, -5.426 and -0.221, respectively (Table S4). Overall, it was observed that L3 manifested highest docking scores with Covid-19 main-protease than other two targets including nsp2 and nsp7. This observation directly suggests main-protease as the main target of these benzimidazole based molecules. Similarly rests of the two compounds were also observed to be promising compounds for main-protease. The 3D docked poses for L3 in each of the target including main protease, nsp2 and nsp7 have been displayed in Fig. 2 . The estimated molecular docking results are also compared with previously reported work [42,43]. Hosseini et al. reported the binding energy for Ramelteon, Levomefolic acid, Ketoprofen etc. with main protease of SARS-CoV-2, revealing –6.0 to –6.66 kcal/mol energy score [43]. We found the binding score for the synthetic benzimidazoles against the main protease and non-structural proteins to lie in the range of –5.0 - –11.0 kcal/mol, suggesting the promising inhibition actions of these derivatives against SARS-CoV-2. It is noticed that in silico screening studies against nsp-2 are limited; therefore, the study against nsp2 inhibition is a novel addition.
Fig. 2.

3D docked poses for L3 in the active site of (A). main-protease; 6LU7, (B). nsp2; 7EXM and (C) nsp7; 7JLT.
3.5. Electrostatic complementarity analysis
The best scored L3 was analyzed for electrostatic complementarity (EC scores) analysis. Among three of the selected targets, this compound could show better EC scores for target Covid-19 main protease (PDB ID: 6LU7) in comparison to other two targets nsp2 and nsp7. The obtained EC scores can be classified into EC, EC r and EC rho, where EC indicates first computed score, a normalized surface integral of the complementarity score that effectively display the average value of the score over the ligand's surface. Meanwhile, EC r and EC rho indicate pearson's correlation coefficient and the spearman rank correlation coefficient, respectively. These scores generally compute the ligand and protein electrostatic potential sampled on the surface vertices. These measures of these scores generally range from 1 (perfect complementarity) to -1 (perfect clash) but have different characteristics. Herein, the L3 with best docking scores was found to manifest better EC scores while this compound couldn't display favorable EC scores with nsp2 and nsp7, as observed in Table 3 . The negative values for EC r and EC rho indicate some steric clashes in case of docked complexes of L3 each with nsp2 and nsp7.
Table 3.
Electrostatic complementarity analysis results for L3 with each of the selected targets.
| Compounds | EC | EC r | EC rho |
|---|---|---|---|
| L3-6LU7 (Covid-19-main-protease) | 0.21 | 0.377 | 0.254 |
| L3- 7EXM (nsp2) | 0.17 | -0.045 | -0.106 |
| L3- 7JLT (nsp 7) | 0.068 | -0.229 | -0.204 |
Thus L3 is a potential inhibitor of main-protease, while partial inhibition could be expected in case of nsp2 and nsp7. Fig. 3.A & C, displayed high electrostatic complementarity within the active site of main-protease with slight steric clashes observed near thiophene moiety. This can be explained by understanding the electrostatic potential surfaces; it could be observed that positive potential around the thiophene moiety shown clashes with the positive electrostatic surfaces generated around the His 41 and His 163. The electrostatic potential surfaces generated around the ligand and protein can be observed in Fig. 3B & D. Both these imidazole containing amino acids exerted face-edge steric clashes with the thiophene moieties present in the ligand ‘L3’.
Fig. 3.

Electrostatic complementarity (3A and 3C) and potential analysis (3B and 3D) considering the complex of L3 and main protease.
3.6. Molecular dynamics
As observed in bothdocking and electrostatic analyses that L3 exhibited not only good docking scores but also good complementarity especially with the target main-protease. Furthermore, to analyze the stability of compound-3 within the active site of each target protein including main protease, nsp2 and nsp7, molecular dynamic analysis was carried out. In each of the case, only slight RMSD fluctuations were observed i.e. within the limit of 1 Å. These RMSD fluctuations can be observed in Fig. 4 . The docking interactions retained after MD simulations were further analyzed and noted some key observations. It can be observed in Table 4 that compound-3 manifested most prominent interactions in terms of sulfur-lone pair and aromatic-aromatic interactions with His 41 by 21.4% and 20.1%, respectively. Though this compound mange to retain some interactions with target nsp2 and nsp7, however the percentage contact with crucial amino acids were lower than the 15%, as observed in Tables S5 and S6. From all the considered in-silico analysis, it would be concluded that L3 is most promising anti-SARs-CoV2 and has a potential to inhibit main-protease more effectively than nsp2 and nsp7. The 3D interactions after MD for a period of 50 ns considering each of the complex for compound-3 with main-protease, nsp2 and nsp7 are displayed in Fig. 5 .
Fig. 4.

RMSD plots considering the docked complexes for MD simulations at a period of 50 ns (A) L3-main protease (B) L3-nsp2 (C) L3-nsp7.
Table 4.
Interactions after carrying Molecular dynamics for the L3 and main protease 6LU7 complex.
| Bond type | Ligand atom | Protein atom | %Frames present |
|---|---|---|---|
| Sulfur-Ion Pair | Compound-3 Atom-S2 | A HIS 41 ND1 | 21.4% |
| Aromatic-Aromatic | Compound-3 Atom-H12 | A HIS 41 CE1 | 20.1% |
| Aromatic-Aromatic | Compound-3 Atom-H8 | A HIS 41 CE1 | 11.2% |
| Sulfur-Ion Pair | Compound-3 Atom-S1 | A ASP 187 O | 9.5% |
| Aromatic-Aromatic | Compound-3 Atom-C12 | A HIS 41 HD2 | 8.8% |
| Sulfur-Ion Pair | Compound-3 Atom-S1 | A ARG 188 O | 8.2% |
| Aromatic-Aromatic | Compound-3 Atom-H7 | A HIS 41 CE1 | 8.1% |
| Aromatic-Aromatic | Compound-3 Atom-C12 | A HIS 41 CE1 | 6.3% |
| Aromatic-Aromatic | Compound-3 Atom-H9 | A HIS 41 CE1 | 4.4% |
| Sulfur-lone Pair | Compound-3 Atom-S1 | A MET 49 O | 4.2% |
| Sulfur-Ion Pair | Compound-3 Atom-S1 | A HIS 164 O | 3.6% |
Fig. 5.

3D interaction diagram after MD considering docked complexes (A) L3-main protease (B) nsp2 and (C) nsp7.
Currently, structural modification based on chromophore-based drug design gains a paramount attention among the scientific community [44], [45], [46]. Culletta et al. [47] made an in silico inhibition activities of the designed pharmacophores against the different proteins of SARS-CoV-2. A significant change of dG Bind energy was observed (-35 to -90 kcal/mol) for those compounds against Mpro and nsp proteins. Badavath et al. [48] performed the computer-aided anti-SARS-CoV-2 screening activity for 118 isatin derivatives and evaluated their binding propensities against main. Furthermore, Purwati et al. [49] ratiometrically evaluated the time-dependent inhibition properties of a series of dual combinatory drugs like Lopinavir-Ritonavir-Clarithromycin, Lopinavir-Ritonavir-Azithromycin, Lopinavir-Ritonavir-Doxycycline, Hydroxychloroquine-Azithromycin etc. against Vero cell lines. In comparing the reported binding scores of the reported compounds calculated through molecular docking and MD simulation, the synthetic ligand L3 displayed an excellent binding preference against main protease of SARS-CoV-2 through His 41 which was further driven by π…π interactions of the thiophene ring of L3 with histidine.
4. Conclusions
In conclusion, we present the synthesis and spectroscopic characterization of a series of 5-membered heterocycle substituted benzimidazoles employing a straightforward synthetic approach. We also prepared the single-crystals of methyl derivative of L3 and analyzed the structural and long-range supramolecular frameworks of L3′ through X-ray crystallography. The therapeutic properties of the designed compounds were evaluated through in silico screening activities with molecular docking, electrostatic complementarity analysis and molecular dynamics studies against main protease (Mpro) and non-structural proteins (nsp2 and nsp7). Molecular docking predicted the highest binding scores for L3-Mpro, L3-nsp2 and L3-nsp7 complexes as −10.52, −5.07 and −6.63 kcal/mol. The MD simulation studies for 50 ns strongly recommend good binding propensity of L3 with Mpro attributing the conformationally stable docked complex. Moreover, the docking interactions observed for L3-Mpro complex were also evident to be retained after an exhaustive MD simulations. Thus, it clearly suggest that L3 compound is a promising molecule that can be further analyzed in future for its in-vitro activity. Though there are large number of report on computation aided drug design in the scientific database; however, this cost-effective straightforward synthesis and significant pharmaocokinetic properties of the synthetic benzimidazoles may turn out to be a potent therapeutic agent.
Supplementary data
Supplementary crystallographic data are available free of charge from The Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK (fax: +44-1223-336033; E-mail: deposit@ccdc.cam.ac.uk or www: http://www.ccdc.cam.ac.uk) upon request, quoting deposition number CCDC 2020847. Experimental information such FT-IR, UV-Vis, 1H &13C NMR, and ADME properties etc. are given here.
CRediT authorship contribution statement
Prafullya Kumar Mudi: Conceptualization, Formal analysis, Methodology, Investigation. Rajani Kanta Mahato: Formal analysis, Methodology, Investigation. Himanshu Verma: Formal analysis, Methodology, Investigation, Validation. Subhra Jyoti Panda: Formal analysis, Methodology, Investigation. Chandra Sekhar Purohit: Formal analysis, Visualization. Om Silakari: Software, Validation. Bhaskar Biswas: Conceptualization, Writing – review & editing, Supervision.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgment
Dr. Bhaskar Biswas gratefully acknowledge the financial support received from the University of North Bengal, Darjeeling 734013.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.molstruc.2022.132869.
Appendix. Supplementary materials
References
- 1.Behera B.C., Mishra R.R., Thatoi H. Recent biotechnological tools for diagnosis of corona virus disease: a review. Biotechnol. Prog. 2021;37(1):e3078. doi: 10.1002/btpr.3078. [DOI] [PubMed] [Google Scholar]
- 2.Kahn J.S., McIntosh K. History and recent advances in corona virus discovery. Pediatr. Infect. Dis. J. 2005;24(11 Suppl):S223–S226. doi: 10.1097/01.inf.0000188166.17324.60. [DOI] [PubMed] [Google Scholar]
- 3.Monto A.S. Medical reviews: coronaviruses. Yale J. Biol. Med. 1974;47(4):234–251. [PMC free article] [PubMed] [Google Scholar]
- 4.Coelho C., Gallo G., Campos C.B., Hardy L., Würtele M. Biochemical screening for SARS-CoV-2 main protease inhibitors. PLoS One. 2020;15(10) doi: 10.1371/journal.pone.0240079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yuan Y., Zhao Y.J., Zhang Q.E., Zhang L., Cheung T., Jackson T., Jiang G.Q., Xiang Y.T. COVID-19-related stigma and its sociodemographic correlates: a comparative study. Glob. Health. 2021;17:54. doi: 10.1186/s12992-021-00705-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pedersen S.F., Ho Y.C. SARS-CoV-2: a storm is raging. J. Clin. Investig. 2020;130(5):2202–2205. doi: 10.1172/JCI137647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li R., Pei S., Chen B., Song Y., Zhang T., Yang W., Shaman J. Substantial undocumented infection facilitates the rapid dissemination of novel coronavirus (SARS-CoV-2) Science. 2020;368(6490):489–493. doi: 10.1126/science.abb3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peiris J.S., Yuen K.Y., Osterhaus A.D., Stöhr K. The severe acute respiratory syndrome. N. Engl. J. Med. 2003;349(25):2431–2441. doi: 10.1056/NEJMra032498. [DOI] [PubMed] [Google Scholar]
- 10.Wang F., Hou H., Luo Y., Tang G., Wu S., Huang M., Liu W., Zhu Y., Lin Q., Mao L., Fang M., Zhang H., Sun Z. The laboratory tests and host immunity of COVID-19 patients with different severity of illness. JCI Insight. 2020;5(10) doi: 10.1172/jci.insight.137799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444–1448. doi: 10.1126/science.bb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C., Breillat P., Carlier N., Gauzit R., Morbieu C., Pène F., Marin N., Roche N., Szwebel T.A., Merkling S.H., Treluyer J.M., Terrier B. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369(6504):718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olagnier D., Farahani E., Thyrsted J., Blay-Cadanet J., Herengt A., Idorn M., Hait A., Hernaez B., Knudsen A., Iversen M.B., Schilling M., Jørgensen S.E., Thomsen M., Reinert L.S., Lappe M., Hoang H.D., Gilchrist V.H., Hansen A.L., Ottosen R., Nielsen C.G., Holm C.K. SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 2020;11(1):4938. doi: 10.1038/s41467-020-18764-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saichi M., Ladjemi M.Z., Korniotis S., Rousseau C., AitHamou Z., Massenet-Regad L., Amblard E., Noel F., Marie Y., Bouteiller D., Medvedovic J., Pène F., Soumelis V. Single-cell RNA sequencing of blood antigen-presenting cells in severe COVID-19 reveals multi-process defects in antiviral immunity. Nat. Cell Biol. 2021;23(5):538–551. doi: 10.1038/s41556-021-00681-2. [DOI] [PubMed] [Google Scholar]
- 15.Mahato R.K., Mahanty A.K., Kotakonda M., Prasad S., Bhattacharjee S., Biswas B. A hydrated 2,3-diaminophenazinium chloride: a promising building block against SARS-CoV-2. Sci. Rep. 2021;11:23122. doi: 10.1038/s41598-021-02280-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobayashi S., Mori Y., Fossey J.S., Salter M.M. Catalytic enantioselective formation of C-C bonds by addition to imines and hydrazones: a ten-year update. Chem. Rev. 2011;111(4):2626–2704. doi: 10.1021/cr100204f. [DOI] [PubMed] [Google Scholar]
- 17.Layer R.W. The chemistry of imines. Chem. Rev. 1963;63(5):479–510. doi: 10.1021/cr60225a003. [DOI] [Google Scholar]
- 18.Gnanaprakasam B., Zhang J., Milstein D. Direct Synthesis of imines from alcohols and amines with liberation of H2. Angew. Chem. Int. Ed. 2010;49:1468–1471. doi: 10.1002/anie.200907018. [DOI] [PubMed] [Google Scholar]
- 19.Cano R., Ramón D.J., Yus M. Transition-metal-free O-,S -, and N-arylation of alcohols, thiols, amides, amines, and related heterocycles. J. Org. Chem. 2011;76(2):654–660. doi: 10.1021/jo1022052. [DOI] [PubMed] [Google Scholar]
- 20.Chen B., Wang L., Gao S. Recent advances in aerobic oxidation of alcohols and amines to imines. ACS Catal. 2015;5:5851–5876. doi: 10.1021/acscatal.5b01479. [DOI] [Google Scholar]
- 21.Wang J., Lu S., Cao X., Gu H. Common metal of copper(0) as an efficient catalyst for preparation of nitriles and imines by controlling additives. Chem. Commun. 2014;50:5637–5640. doi: 10.1039/C4CC01389A. [DOI] [PubMed] [Google Scholar]
- 22.Sonobe T., Oisaki K., Kanai M. Catalytic aerobic production of imines en route to mild, green, and concise derivatizations of amines. Chem. Sci. 2012;3:3249–3255. doi: 10.1039/C2SC20699D. [DOI] [Google Scholar]
- 23.Mudi P.K., Mahato R.K., Joshi M., Shit M., Choudhury A.R., Das H.S., Biswas B. Copper(II) complexes with a benzimidazole functionalized Schiff base: synthesis, crystal structures, and role of ancillary ions in phenoxazinone synthase activity. Appl. Organomet. Chem. 2021;35:e6211. doi: 10.1002/aoc.6211. [DOI] [Google Scholar]
- 24.Yeap C.S., Kargar H., Kia R., Jamshidvand A., Fun H.K. 2-(1H-Benzoimidazol-2-yl)-6-ethoxyphenol. Acta Crystallogr. E Crystallogr. Commun. 2009;E65:o745–o746. doi: 10.1107/S20569890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kia R., Fun H.K., Kargar H. 4-[1-(4-Cyanobenzyl)-1H-benzimidazol-2-yl]benzonitrile. Acta Crystallogr. E Crystallogr. Commun. 2009;E65:o660–o661. doi: 10.1107/S1600536809006989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kargar H., Kargar K., Mehrjardi M.F., Munawar K.S. Syntheses, characterization, and catalytic potential of novel vanadium and molybdenum Schiff base complexes for the preparation of benzimidazoles, benzoxazoles, and benzothiazoles under thermal and ultrasonic conditions. Monatshefte für Chem. Chem. Mon. 2021;152:593–605. doi: 10.1007/s00706-021-02780-0. [DOI] [Google Scholar]
- 27.Dhanamjayulu P., Boga R.B., Mehta A. Inhibition of aflatoxin B1 biosynthesis and down regulation of aflR and aflB genes in presence of benzimidazole derivatives without impairing the growth of Aspergillus flavus. Toxicon. 2019;170:60–67. doi: 10.1016/j.toxicon.2019.09.018. [DOI] [PubMed] [Google Scholar]
- 28.Anguru M.R., Taduri K., Bhoomireddy R.D., Jojula M., Gunda S.K. Novel drug targets for mycobacterium tuberculosis: 2-heterostyrylbenzimidazoles as inhibitors of cell wall protein synthesis. Chem. Cent. J. 2017;11:68. doi: 10.1186/s13065-017-0295-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Farahata A.A., Ismailc M.A., Kumara A., Wenzlerd T., Brund R., Paula A., Wilsona W.D., Boykin D.W. Indole and benzimidazole bichalcophenes: synthesis, DNA binding and antiparasitic activity. Eur. J. Med. Chem. 2018;143:1590–1596. doi: 10.1016/j.ejmech.2017.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mudi P.K., Mahanty A.K., Kotakonda M., Prasad S., Bhattacharjee S., Biswas B. A benzimidazole scaffold as a promising inhibitor against SARS-CoV-2. J. Biomol. Struct. Dyn. 2022 doi: 10.1080/07391102.2021.2024448. [DOI] [PubMed] [Google Scholar]
- 31.Mahato R.K., Mudi P.K., Deb M., Biswas B. A direct metal-free synthetic approach for the efficient production of privileged benzimidazoles in water medium under aerobic condition. Asian J. Org. Chem. 2021;10:2954–2963. doi: 10.1002/ajoc.202100477. [DOI] [Google Scholar]
- 32.Bruker. SMART (Version 5.0) and SAINT (Version 6.02), Bruker AXS Inc., Madison, WI, USA (2000).
- 33.Sheldrick G.M. University of Gottingen; Germany: 2000. SADABS, Program For Empirical Correction of Area Detector Data. [Google Scholar]
- 34.Sheldrick G.M. University of Gottingen; Germany: 1997. SHELXS97 and SHELXL97, Program For Crystal Structure Refinement. [Google Scholar]
- 35.Zhang D., Hamdoun S., Chen R., Yang L., Ip C.K., Qu Y., Li R., Jiang H., Yang Z., Chung S.K. Identification of natural compounds as SARS-CoV-2 entry inhibitors by molecular docking-based virtual screening with bio-layer interferometry. Pharmacol. Res. 2021;172 doi: 10.1016/j.phrs.2021.105820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yi Y., Li J., Lai X., Zhang M., Kuang Y., Bao Y.O., Yu R., Hong W., Muturi E., Xue H., Wei H., Li T., Zhuang H., Qiao X., Xiang K., Yang H., Ye M. Natural triterpenoids from licorice potently inhibit SARS-CoV-2 infection. J. Adv. Res. 2022;36:201–210. doi: 10.1016/j.jare.2021.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma J., Chen Y., Wu W., Chen Z. Structure and function of N-terminal zinc finger domain of SARS-CoV-2 NSP2. Virol. Sin. 2021;36:1104–1112. doi: 10.1007/s12250-021-00431-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thuy B.T.P., My T.T.A., Hai N.T.T., Hieu L.T., Hoa T.T., Loan H.T.P., Triet N.T., Anh T.T.V., Quy P.T., Tat P.V. Investigation into SARS-CoV-2 resistance of compounds in garlic essential oil. ACS Omega. 2020;5:8312–8320. doi: 10.1021/acsomega.0c00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eastman P., Swails J., Chodera J.D., McGibbon R.T., Zhao Y., Beauchamp K.A., Wang L.P., Simmonett A.C., Harrigan M.P., Stern C.D. OpenMM 7: rapid development of high performance algorithms for molecular dynamics. PLOS Comput. Biol. 2017;13 doi: 10.1371/journal.pcbi.1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roy S., Paul P., Karar M., Joshi M., Paul S., Choudhury A.R., Biswas B. Cascade detection of fluoride and bisulphate ions by newly developed hydrazine functionalised Schiff bases. J. Mol. Liq. 2021;326 doi: 10.1016/j.molliq.2021.115293. [DOI] [Google Scholar]
- 41.Mahato S., Meheta N., Kotakonda M., Joshi M., Shit M., Choudhury A.R., Biswas B. Synthesis, structure, polyphenol oxidase mimicking and bactericidal activity of a zinc-Schiff base complex. Polyhedron. 2020;194 doi: 10.1016/j.poly.2020.11493. [DOI] [Google Scholar]
- 42.Hosseini M., Chen W., Xiao D., Wang C. Computational molecular docking and virtual screening revealed promising SARS-CoV-2 drugs. Precis. Clin. Med. 2021;4(1):1–16. doi: 10.1093/pcmedi/pbab001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Halder U.C. Predicted antiviral drugs Darunavir, Amprenavir, Rimantadine and Saquinavir can potentially bind to neutralize SARS-CoV-2 conserved proteins. J. Biol. Res. 2021;28(1):18. doi: 10.1186/s40709-021-00149-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jamshidvand A., Sahihi M., Mirkhani V., Moghadam M., -Baltork I.M., Tangestaninejad S., Rudbari H.A., Kargar H., Keshavarzi R., Gharaghani S. Studies on DNA binding properties of new Schiff base ligands using spectroscopic, electrochemical and computationalmethods: influence of substitutions on DNA-binding. J. Mol. Liq. 2018;253:61–71. doi: 10.1016/j.molliq.2018.01.029. [DOI] [Google Scholar]
- 45.Kargar H., -Ardakani R.B., Torabi V., Kashani M., -Natanzi Z.C., Kazemi Z., Mirkhani V., Sahraei A., Tahir M.N., Ashfaq M., Munawar K.S. Synthesis, characterization, crystal structures, DFT, TD-DFT, moleculardocking and DNA binding studies of novel copper(II) and zinc(II) complexes bearing halogenated bidentate N,O-donor Schiff base ligands. Polyhedron. 2021;195 doi: 10.1016/j.poly.2020.114988. [DOI] [Google Scholar]
- 46.Kargar H., -Ardakani R.B., Torabi V., Sarvian A., Kazemi Z., -Natanzi Z.C., Mirkhani V., Sahraei A., Tahir M.N., Ashfaq M. Novel copper(II) and zinc(II) complexes of halogenated bidentate N,Odonor Schiff base ligands: synthesis, characterization, crystal structures, DNA binding, molecular docking, DFT and TD-DFT computational studies. Inorg. Chim. Acta. 2021;514 doi: 10.1016/j.ica.2020.120004. [DOI] [Google Scholar]
- 47.Culletta G., Gulotta M.R., Perricone U., Zappalà M., Almerico A.M., Tutone M. Exploring the SARS-CoV-2 proteome in the search of potential inhibitors via structure-based pharmacophore modeling/docking approach. Computation. 2020;8(3):77. doi: 10.3390/computation8030077. [DOI] [Google Scholar]
- 48.Badavath V.N., Kumar A., Samanta P.K., Maji S., Das A., Blum G., Jha A., Sen A. Determination of potential inhibitors based on isatin derivatives against SARS-CoV-2 main protease (Mpro): a molecular docking, molecular dynamics and structure-activity relationship studies. J. Biomol. Struct. Dyn. Stud. 2020 doi: 10.1080/07391102.2020.1845800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Purwati, Miatmoko A., Nasronudin, Hendrianto E., Karsari D., Dinaryanti A., Ertanti N., Ihsan I.S., Purnama D.S., Asmarawati T.P., Marfiani E., Yulistiani, Rosyid A.N., Wulaningrum P.A., Setiawan H.W., Siswanto I., Puspaningsih N.N.T. An in vitro study of dual drug combinations of anti-viral agents, antibiotics, and/or hydroxychloroquine against the SARS-CoV-2 virus isolated from hospitalized patients in Surabaya, Indonesia. PLoS One. 2021;16 doi: 10.1371/journal.pone.0252302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.