

Abstract
Objective:
We aimed to determine the population-based incidence, prevalence, and mortality of dermatomyositis (DM) using European Alliance of Associations for Rheumatology (EULAR)/American College of Rheumatology (ACR) criteria.
Methods:
This population-based cohort study included incident DM from 1/1/1995 to 12/31/2019. We manually reviewed all individuals with at least one code for DM or polymyositis to determine if they met EULAR/ACR criteria, subspecialty physician diagnosis, and/or Bohan & Peter criteria. We age- and sex-adjusted incidence and prevalence estimates to the United States Non-Hispanic White year 2000 population and estimated prevalence on 1/1/2015. Standardized mortality ratios (SMR) with 95% confidence intervals (CI) compared observed to expected mortality adjusting for age, sex, and year.
Results:
We identified 40 cases of verified DM, with 29 cases incident in Olmsted County from 1995 to 2019. Mean age was 57 years, 26 (90%) were female, and 12 (41%) had clinically amyopathic DM (CADM). Median follow-up time was 8.2 years. The overall adjusted incidence of DM was 1.1 (95% CI 0.7 to 1.5) per 100,000 person-years, and prevalence was 13 (95% CI 6 to 19) per 100,000. The standardized mortality ratio was significantly elevated among the myopathic DM cases (3.1, 95% CI 1.1 to 6.8) but not CADM cases (1.1, 95% CI 0.2 to 3.3). The positive predictive value of two or more DM codes was only 40/82 (49%).
Conclusion:
This population-based study found DM incidence and prevalence were higher than previously reported. Mortality was significantly elevated for myopathic DM but not for CADM.
Keywords: dermatomyositis, incidence, prevalence, mortality, epidemiology, cohort
Dermatomyositis (DM) is a systemic autoimmune disease characterized by a distinct rash and muscle inflammation, which often leads to muscle weakness. The subset of DM without clinical muscle weakness is termed clinically amyopathic DM (CADM) (1). While its precise etiology remains unknown, DM has several known risk factors including female sex (2, 3), genetics (4–6), ultraviolet radiation (7–9), preceding respiratory diseases (10, 11), and other geographic environmental factors (12, 13). Current DM incidence estimates range from 1.0 to 15 per million (2, 14–23), while prevalence estimates range from 1.2 to 21 per 100,000 (2, 3, 17–21, 23, 24). Importantly, both of these estimates are based largely on diagnosis codes rather than verified DM criteria.
While DM is relatively rare, it has profound impacts on both quality of life and life expectancy. Individuals with DM have reduced quality of life in all domains compared to their peers (25). Furthermore, the standardized mortality ratios and hazard ratios for DM range from 2.4 to 7.5 compared to matched controls (16, 26–33). This high mortality stems primarily from its association with malignancy (26, 28, 34), interstitial lung disease (ILD) (34, 35), and cardiovascular disease (27).
Understanding the epidemiology of DM is crucial to uncovering its pathogenesis and how to treat it, yet several gaps remain. First, no population-based study of DM incidence has used established DM classification criteria, and only one population-based study of DM prevalence has done so (3). Second, no studies of incidence, prevalence, or mortality have used the new European Alliance of Associations for Rheumatology (EULAR)/American College of Rheumatology (ACR) 2017 DM classification criteria (36). These new criteria have high sensitivity and specificity for DM (37), and outperform the prior Bohan & Peter criteria (38–41). Finally, most DM studies use diagnosis codes, which are generated for billing rather than clinical or research purposes. Therefore, ascertaining their positive predictive value using the gold standard new classification criteria has widespread importance and utility for informing the design of future DM studies.
To address these three gaps, we leveraged a population-based cohort of manually verified DM cases. First, we aimed to investigate the incidence and prevalence of DM in the study population. Second, we aimed to determine the mortality rate in patients with DM compared to general population mortality rates. Third, we aimed to define the predictive value of diagnostic codes for DM using the EULAR/ACR 2017 criteria. Given the observed rise in idiopathic inflammatory myositis in recent years (42, 43), we hypothesized that the incidence and prevalence of DM is now higher than previous reports. We also hypothesized that the mortality of individuals with DM is over three-fold greater than expected, and that the positive predictive value of one or two codes for DM will be low (<80%), as it was for the prior Bohan & Peter criteria (44).
PATIENTS AND METHODS
Study Design and Population
This retrospective population-based cohort study falls within the Rochester Epidemiology Project, a longitudinal cohort of over 500,000 unique individuals who resided and received health care in Olmsted County between 1966 and the present (45). The Rochester Epidemiology Project collects mortality data using multiple sources, including electronic health records, electronic Minnesota State Death Certificates and the National Death Index. This study included all individuals with incident, diagnosed DM between 1/1/1995 and 12/31/2019. We defined index date and started follow-up at the date of EULAR/ACR 2017 criteria fulfilment (36). This study received approval from both Mayo Clinic (20-008534) and Olmsted County (046-OMC-20) Institutional Review Boards and complied with the Declaration of Helsinki.
Dermatomyositis
To identify cases of DM (including CADM) within this population, we compiled a list of all adults age 18 and older with at least one International Classification of Diseases (ICD) 9 or 10 code from any provider for DM (710.3, M33.0-1, M33.9) or polymyositis (710.4, M33.2) (see Supplementary Table S1) within the study dates above. Using manual medical record review, we then evaluated for the presence of adult-onset DM using established criteria. We elected not to include polymyositis in this study because of increasing recognition that it may not represent its own standalone entity (46, 47). The primary DM criteria for this study was the EULAR/ACR 2017 classification criteria, which seeks to identify a well-defined, relatively homogenous population of individuals with true myositis. By these criteria, any individual with probable DM as indicated by a score of ≥5.5 with no muscle biopsy or ≥6.7 with muscle biopsy, along with presence of at least one of the three skin criteria, met criteria for DM (36).
Secondary DM criteria for this study included subspecialist (rheumatologist, neurologist and/or dermatologist) physician diagnosis as well as the Bohan & Peter 1975 criteria for DM (40, 41). For the latter, we included both definite and probable DM as meeting criteria for DM, as done previously (3, 13). For all three of the above DM criteria, we collected date of each criteria fulfillment. We excluded individuals with DM if they did not have residency in Olmsted County at index date. We also excluded individuals if they met dermatomyositis criteria prior to age 18 years, as juvenile DM may represent a different disease given its disparate clinical features including ILD, malignancy, and calcinosis (48).
Dermatomyositis Characteristics
During manual medical record review, we collected all DM characteristic information required by any of the above three DM criteria. Using REDCap, we recorded the presence or absence of each DM characteristic, as documented by a physician, along its date of onset. These characteristics included age of onset of first symptom, objective symmetric weakness of proximal upper and lower extremities, neck flexors relatively weaker than neck extensors, proximal leg muscles relatively weaker than distal muscles, heliotrope rash, Gottron’s papules, Gottron’s sign, dysphagia or esophageal dysmotility, anti-Jo-1 positivity, elevated serum muscle enzymes (CK, LDH, AST, or ALT), muscle biopsy status and findings, and EMG findings. We also collected Mi-2 positivity, P155/140 (TIF-1 gamma) positivity, NXP-2 positivity, and MDA5 positivity as reported by the Myomarker Panel 3 test (RDL Reference Laboratories, Inc.).
Covariates
We collected covariate information for each individual with DM as of index date. These included age (years), sex (male versus female), race/ethnicity (Non-Hispanic White versus other), body mass index (BMI; kg/m2), and smoking status (never, past, and current). We also evaluated for presence of ILD and recorded its date of onset. We defined ILD as diagnosis by a pulmonologist and two out of three of the following: (1) ILD observed on CT scan or chest radiograph, (2) Restrictive pattern observed on PFT (TLC <=80% predicted), and (3) Bronchoscopic or surgical lung biopsy results consistent with ILD (49).
Statistical Analysis
We used Fischer’s exact tests to compare categorical variables and Wilcoxon rank-sum tests to compare continuous variables. To ensure that the characteristics of recent and historical cases would be similar, we considered DM characteristics to be present if they occurred within two years of index date. Time from index date to date of onset of each DM characteristic used this two-year cutoff as well. We calculated DM incidence rates per 100,000 person-years and DM prevalence per 100,000 as of January 1, 2015 using the number of cases as the numerator and age-, sex- and calendar year-specific denominators from the Rochester Epidemiology Project census. We age- and sex-adjusted both incidence and prevalence estimates to the United States Non-Hispanic White year 2000 population and provided 95% confidence intervals (CI) for these estimates based on the Poisson distribution. For mortality calculations, we compared the survival of our DM cohort to expected survival rates from Minnesota life tables by age, sex, and year using standardized mortality ratios. Positive predictive value calculations divided all individuals with verified DM by the number having either one or two DM codes. As a sensitivity analysis, we compared the overlap in criteria fulfilment status and date of criteria fulfillment across the three sets of DM criteria. No participants had missing data except for certain DM antibodies, as noted in the results. We pre-specified all analyses in a protocol, set statistical significance as two-sided alpha<0.05, and analyses were performed using SAS version 9.4 (SAS Institute Inc., Cary, NC) and R 3.6.2 (R Foundation for Statistical Computing, Vienna, Austria).
RESULTS
Dermatomyositis Characteristics
From the 161 individuals screened for DM, we identified 40 cases of verified DM by at least one of the three DM criteria. All 40 also met the primary EULAR/ACR 2017 criteria. We then excluded 11 individuals for lack of residency status or date of DM incidence outside the study dates. Thus, we identified 29 cases of incident DM in Olmsted County from 1995 to 2019.
Of the 29 DM cases, 17 (59%) had clinically myopathic DM, and 12 (41%) had CADM. Median follow-up time was 8.2 years with interquartile range (IQR) 3.5 to 16.3 years. Demographic characteristics of the myopathic and amyopathic subgroups were similar, with both having mean age 57 years and predominately female, Non-Hispanic White individuals who never smoked (Table 1). Interstitial lung disease occurred only in the myopathic subgroup (18%), though this difference was not statistically significant (Table 1).
Table 1.
Demographic characteristics of the 29 incident dermatomyositis cases in Olmsted County 1995 to 2019
| Number (%) |
||||
|---|---|---|---|---|
| Characteristic* | Amyopathic (N=12) | Myopathic (N=17) | Total (N=29) | p-value |
| Age, mean (SD), years | 57 (19) | 57 (17) | 57 (17) | 0.79 |
| Female sex | 10 (83) | 16 (94) | 26 (90) | 0.55 |
| White, non-Hispanic | 11 (92) | 17 (100) | 28 (97) | 0.41 |
| BMI, mean (SD), kg/m2 | 30 (7) | 27 (5) | 28 (6) | 0.25 |
| Smoking history | 0.7 | |||
| Never | 8 (67) | 11 (65) | 19 (66) | |
| Former | 4 (33) | 4 (24) | 8 (28) | |
| Current | 0 (0) | 2 (12) | 2 (7) | |
| Interstitial lung disease | 0 (0) | 3 (18) | 3 (10) | 0.25 |
BMI = body mass index, SD = standard deviation
As of incidence date, defined as fulfillment of EULAR/ACR criteria
DM characteristics of the myopathic and amyopathic subgroups were also similar except that the myopathic DM subgroup had a higher prevalence of weakness, muscle enzyme elevation, and dysphagia or esophageal dysmotility (Table 2). Overall, Gottron’s sign was the most common DM characteristic, occurring in 93% of the DM cases (Table 2). However, in myopathic DM, proximal lower extremity weakness was also observed in 88% of cases, and muscle enzyme elevation had an even higher sensitivity of 94% (Table 2). Rash was often the first observed DM characteristic, with weakness following by a median of one to two weeks (Table 2). Most DM cases did not have antibodies other than Jo-1 tested (Table 2).
Table 2.
Dermatomyositis characteristics of the 29 incident dermatomyositis cases in Olmsted County 1995 to 2019
| Number (%) |
Median (IQR) |
||||
|---|---|---|---|---|---|
| Dermatomyositis characteristic* | Amyopathic (N=12) | Myopathic (N=17) | Total (N=29) | p-value | Days from index to onset |
| Proximal upper extremity weakness | 0 (0) | 14 (82) | 14 (48) | <0.001 | 5.5 (0,82) |
| Proximal lower extremity weakness | 0 (0) | 15 (88) | 15 (52) | <0.001 | 2 (0,82) |
| Neck flexors weaker than extensors | 0 (0) | 7 (41) | 7 (24) | 0.02 | 16 (11,82) |
| Proximal leg muscles weaker than distal | 0 (0) | 15 (88) | 15 (52) | <0.001 | 11 (0,122) |
| Heliotrope rash | 9 (75) | 12 (71) | 21 (72) | 1.00 | 0 (0,57) |
| Gottron’s papules | 11 (92) | 11 (65) | 22 (76) | 0.19 | 0 (0,0) |
| Gottron’s sign | 12 (100) | 15 (88) | 27 (93) | 0.50 | 0 (0,0) |
| Dysphagia or esophageal dysmotility | 1 (8) | 8 (47) | 9 (31) | 0.04 | 36 (27,82) |
| Anti-Jo-1 positivity | 0 (0) | 1 (6) | 1 (3) | 1.00 | 82 (82,82) |
| Mi-2 positivity | 0 (out of 4) | 0 (out of 4) | 0 (out of 8) | 0.68 | N/A |
| P155/140 (TIF-1 gamma) positivity | 0 (out of 4) | 0 (out of 3) | 0 (out of 7) | 0.40 | N/A |
| NXP-2 positivity | 0 (out of 4) | 0 (out of 3) | 0 (out of 7) | 0.40 | N/A |
| MDA5 positivity | 1 (25) | 0 (out of 3) | 1 (14) | 0.48 | N/A |
| Elevated serum CK, LDH, AST, or ALT | 2 (17) | 16 (94) | 18 (62) | <0.001 | −0.5 (−4.0,1.0) |
| Muscle biopsy performed | 0 (0) | 6 (35) | 6 (21) | 0.03 | 21 (3,66) |
| Perimysial/vascular infiltration of MNCs | N/A | 5 (83) | 5 (83) | N/A | N/A |
| Perifascicular atrophy | N/A | 3 (50) | 3 (50) | N/A | N/A |
| EMG performed | 6 (50) | 15 (88) | 21 (72) | 0.02 | 9.5 (0,28) |
| Short, polyphasic motor unit potentials | 3 (50) | 13 (87) | 16 (76) | N/A | N/A |
BMI = body mass index, IQR = interquartile range, MNCs = mononuclear cells, N/A = not applicable, SD = standard deviation
Within 2 years of incidence date, defined as fulfillment of EULAR/ACR criteria
Incidence and Prevalence
DM incidence increased with each decade of life, peaking in the 80+ year age group at 3.2 per 100,000 person-years (Table 3). The age- and sex-adjusted overall incidence of DM was 1.1 (95% CI 0.7 to 1.5) per 100,000 person-years, corresponding to 2,858 new cases of DM in the United States each year. The incidence of DM was higher among females, at 1.9 (95% CI 1.1 to 2.6) per 100,000 person-years. For myopathic DM the overall incidence was 0.7 (95% CI 0.3 to 1.0), and for CADM the incidence was 0.5 (95% CI 0.2 to 0.7) per 100,000 person-years. Analysis of the incidence in 1995-2007 vs 2008-2019 revealed no evidence of a change over time (1.2/100,000; 95%CI 0.5-1.8/100,000 vs 1.1/100,000; 95% CI 0.6-1.7/100,000).
Table 3.
Age- and sex-specific dermatomyositis incidence rates in Olmsted County from 1995 to 2019
| Female | Male | Total | ||||
|---|---|---|---|---|---|---|
|
|
||||||
| Age | N | Rate (per 100,000) | N | Rate (per 100,000) | N | Rate (per 100,000) |
| 18-39 | 5 | 0.8 | 1 | 0.2 | 6 | 0.5 |
| 40-59 | 10 | 2.1 | 1 | 0.2 | 11 | 1.2 |
| 60-79 | 7 | 2.7 | 1 | 0.5 | 8 | 1.7 |
| 80+ | 4 | 5.1 | 0 | 0.0 | 4 | 3.2 |
| Total | 26 | 1.9 (1.1,2.6)* | 3 | 0.2 (0.0,0.5)* | 29 | 1.1 (0.7,1.5)** |
Age-adjusted to the US White 2000 population
Age- and sex-adjusted to the US White 2000 population
DM prevalence was also highest among females, peaking in the 60-79 year age group (Table 4). The age- and sex-adjusted overall prevalence of DM on 1/1/2015 was 13 (95% CI 6 to 19) per 100,000, corresponding to 34,061 individuals in the United States. Among females, the prevalence of DM was 20 (95% CI 9.2 to 31) per 100,000. For myopathic DM the overall prevalence was 5.8 (95% CI 1.5 to 10), and for CADM the prevalence was 7.0 (95% CI 2.1 to 12) per 100,000.
Table 4.
Age- and sex-specific dermatomyositis prevalence rates in Olmsted County on 1 January 2015
| Female | Male | Total | ||||
|---|---|---|---|---|---|---|
|
|
||||||
| Age | N | Rate (per 100,000) | N | Rate (per 100,000) | N | Rate (per 100,000) |
| 18-39 | 1 | 3.9 | 0 | 0 | 1 | 2.1 |
| 40-59 | 4 | 20 | 1 | 5.4 | 5 | 13 |
| 60-79 | 6 | 46 | 1 | 8.9 | 7 | 29 |
| 80+ | 2 | 56 | 0 | 0 | 2 | 34 |
| Total | 13 | 20 (0.9,31)* | 2 | 3.7 (0,8.7.0)* | 15 | 13 (6,19)** |
Age-adjusted to the US White 2000 population
Age- and sex-adjusted to the US White 2000 population
Mortality
During the study period, 6 individuals with myopathic DM and 3 with CADM died, which was higher than expected for population referents (Figure 1). The standardized mortality ratio among the myopathic DM cases was significantly elevated at 3.1 (95% CI 1.1 to 6.8). The mortality of the CADM cases was not statistically significantly elevated at 1.1, though the confidence interval was wide (95% CI 0.2 to 3.3). Overall, the standardized mortality ratio for this cohort was 2.0 (95% CI 0.9 to 3.7). Five-year survival was 74% (95% CI 59% to 93%) with expected survival of 95%, and 10-year survival was only 69% (95% CI 53% to 90%) with expected survival of 89%.
Figure 1.
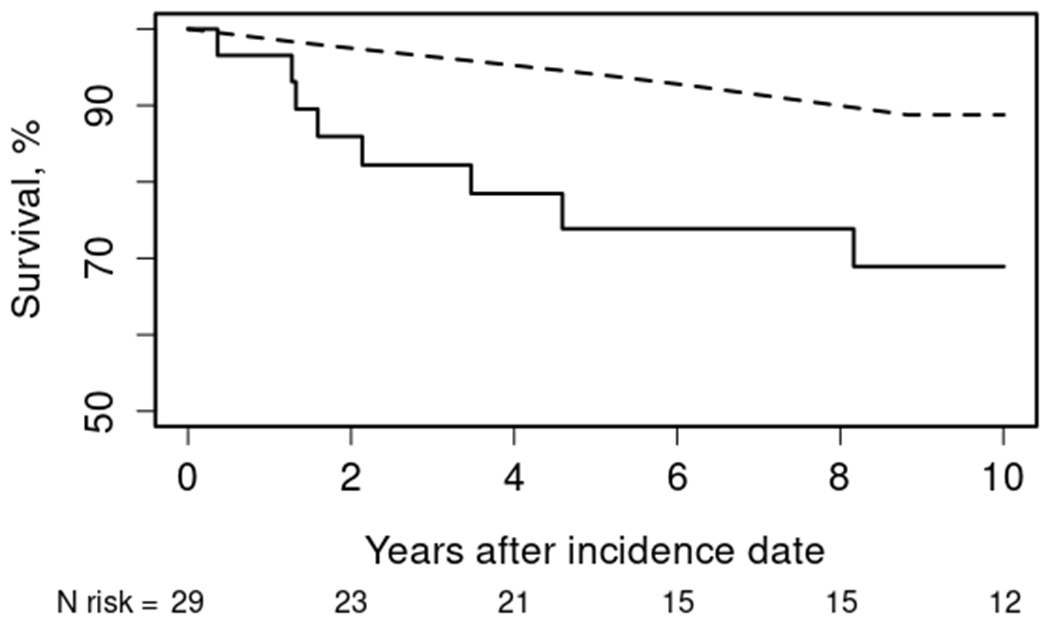
Overall survival of Olmsted county residents with index date of dermatomyositis in 1995-2019 (solid) compared to expected rates from Minnesota lifetables (dashed)
Predictive Value
All 40 individuals with verified DM by EULAR/ACR 2017 criteria had at least one code specifically for DM. The positive predictive value of one or more DM code for validated DM was 40 out of 90, or 44%. The positive predictive value of having two or more DM codes was slightly higher at 40 out of 82, or 49%.
Sensitivity Analyses
By the end of study follow-up, all 29 cases were considered “definite” by EULAR/ACR 2017 criteria, and all had been diagnosed by a rheumatologist, neurologist or dermatologist. However, 9 (75%) CADM cases did not meet Bohan & Peter criteria, corresponding to 31% of the overall DM cases. At index date of EULAR/ACR 2017 criteria, these percentages were even higher, with 1 (7%) not having a rheumatologist, neurologist, and/or dermatologist physician diagnosis of DM and 12 (41%) not meeting Bohan & Peter 1975 criteria (Table 5). Even among those eventually fulfilling the Bohan & Peter 1975 criteria, they met those criteria later than the EULAR/ACR 2017 criteria by a median of 6.5 days (IQR 0.0 to 107).
Table 5.
Comparison of the three dermatomyositis criteria as of index date*
| Number (%) |
||||
|---|---|---|---|---|
| Criteria Fulfilled | Amyopathic (N=12) | Myopathic (N=17) | Total (N=29) | p-value |
| EULAR/ACR 2017 criteria | 12 (100) | 17 (100) | 29 (100) | 1.00 |
| Definite | 10 (83) | 11 (65) | 21 (72) | 0.41 |
| Probable | 2 (17) | 6 (35) | 8 (28) | |
| Bohan & Peter 1975 criteria | 1 (8) | 16 (94) | 17 (59) | 1.00 |
| Definite | 0 (0) | 6 (35) | 6 (21) | |
| Probable | 1 (8) | 10 (59) | 11 (38) | |
| Subspecialty physician diagnosis | 12 (100) | 16 (88) | 28 (93) | 1.00 |
Within 2 years of incidence date, defined as fulfillment of EULAR/ACR criteria
ACR = American College of Rheumatology, EULAR = European League Against Rheumatism
DISCUSSION
This population-based cohort study established the incidence, prevalence, and mortality of dermatomyositis using validated DM criteria. In particular, the incidence and prevalence estimates are on the higher end of what has been previously reported, which may reflect either increased capture of CADM cases or a rise in this disease over time. The mortality for myopathic DM was over three-fold compared to referents, but not increased for CADM. Overall, these findings provide an important, foundational understanding of this debilitating disease and highlight key targets for future research, ideally with larger populations.
A significant contribution of this study is its population-based estimates of DM incidence and prevalence using EULAR/ACR 2017 criteria. In this first study to do so, the overall incidence of DM was 1.1 per 100,000 person-years. This estimate falls on the upper end of previous reports, where incidence estimates ranged from 0.1 to 1.5 per 100,000 person-years (2, 14–23). Similarly, our estimated prevalence of 13 per 100,000 was higher than previous prevalence estimates, which ranged from 1.2 to 9.2 per 100,000 (3, 17–21, 23, 24).
One reason for the higher DM incidence and prevalence estimates could be our population-based methods using established criteria, though these rigorous requirements should decrease rather than increase estimates. Another reason could be the geographical region, as the one study with a higher prevalence estimate than ours came from the same region (2). Alternatively, incidence of DM may be increasing over time (42, 43). However, other studies have not shown this (15, 20), and we were not able to assess due to sample size. Most likely, the higher estimates resulted from the large proportion of CADM included in the study population. Many previous studies of incidence and prevalence do not report CADM, perhaps because very few CADM cases met criteria for DM by the previous Bohan & Peter criteria, which rely heavily on muscle symptoms and findings (40). Future studies using the newer EULAR/ACR criteria and assessing DM incidence and prevalence over time will be important.
Another key finding from this study was that the mortality of individuals with myopathic DM was three-fold higher than peers, but not increased for CADM. This mortality ratio for myopathic DM falls within but on the lower end of previous estimates, which ranged from 2.4 to 7.5 (16, 26–33). Our estimate may have been lower due this study’s relatively long follow-up period, as recent studies have shown that DM mortality declines with time after diagnosis (31, 33). Alternatively, this cohort had a relatively low prevalence of ILD in the first two years after index date, and ILD causes a significant portion of DM deaths (35). A third possibility is that mortality of DM may be declining over time, which we were not able to assess due to sample size. Importantly, this is the first study to report CADM mortality. Although we found no increase in mortality for CADM, the wide confidence intervals make replication important. Future, larger studies should examine DM mortality over time, evaluate causes of death, and seek to understand the mechanism behind the different clinical and mortality manifestations of myopathic DM and CADM.
This study also found that using the EULAR/ACR 2017 criteria, the predictive value of DM codes was poor for identifying true DM cases. Even when requiring two DM codes, the positive predictive value remained less than 50%. One reason for this could be that the EULAR/ACR criteria were not published until 2017, near the end of this study. However, this finding is similar to a previous study examining DM code validity using Bohan & Peter 1975 criteria, where positive predictive value was 35% for outpatient codes and 46% for secondary inpatient codes (44). These results are not surprising, as diagnosis codes were designed for billing. In contrast, classification criteria like the EULAR/ACR 2017 criteria were designed for homogenous study populations with true disease, and physician diagnoses aim for accurate and optimal patient care. Thus, using diagnostic codes to capture DM cases is likely inaccurate, and manual review should remain the gold standard for studies of DM. Importantly, DM-specific codes captured all validated cases of DM in this study, which may reduce workload for future DM studies.
Finally, sensitivity analyses showed that EULAR/ACR 2017 criteria out-performed the Bohan & Peter 1975 criteria in two ways. First, its sensitivity for capturing DM was higher than the Bohan & Peter criteria. Second, the EULAR/ACR criteria established the diagnosis of DM sooner than the Bohan & Peter 1975 criteria, sometimes by several months. These findings support previous studies demonstrating superior performance of the EULAR/ACR 2017 criteria (38, 39). A unique contribution of our study, however, was the finding that sensitivity of Bohan & Peter 1975 criteria for DM was especially poor for CADM (only 25%), which may comprise a sizeable portion of DM cases.
Unique strengths of this study include its population-based design, manual verification of cases using the EULAR/ACR 2017 DM criteria, and comparison to prior criteria. This study also has several important limitations. First, because it was population-based within one geographical region, it may not generalize to other populations or locations. In particular, the race/ethnicity of the population was nearly all Non-Hispanic White, and geographical factors such as ultraviolet radiation and latitude may also play an important role in DM pathogenesis (5, 7–9, 12, 13). In addition, we relied on clinically diagnosed DM, rather than active surveillance for DM in a sample of the population. Our approach will miss undiagnosed cases of DM, thus underestimating the true prevalence/incidence of DM in our population. We also expect to miss mild cases of DM that do not come to medical attention. This approach could also lead to ascertainment bias, perpetuating assumed stereotypes of disease such as the female preponderance in DM. However, DM is a serious, progressive clinical disorder with significant adverse health impacts. Thus, we may have missed some persons in early stages of the disease, but would be less likely to miss progressive cases. Third, the retrospective data review relied on physician documentation of DM characteristics, which may not have occurred in a uniform fashion. Similarly, we were not able to separate ICD codes coming from rheumatologists or dermatologists versus other types of providers, which likely lowers the specificity of the codes. Fourth, for comparability of cases we reported clinical data only for two years after index date, and earlier DM cases often did not have myositis antibody data collected. Fifth, the small sample size limited the statistical reliability of mortality estimates. They also limited our ability to ascertain whether the mortality of CADM is significantly elevated, assess time trends in DM incidence and mortality, and evaluate causes of death. Because of the small sample size, results from this study should be considered preliminary estimates. Future studies on cause of death are essential for informing cancer screening practices. Finally, this study did not include juvenile DM patients, though future epidemiological studies of incidence, prevalence, and mortality of these patients using the new EULAR/ACR criteria is also important.
In conclusion, this population-based study using validated DM criteria found incidence and prevalence on the higher end of previous reports, and mortality of myopathic DM significantly elevated compared to peers. Future, larger studies of validated DM cases should examine incidence and mortality over time and causes of death to better understand disease pathogenesis and treatment.
Supplementary Material
SIGNIFICANCE AND INNOVATIONS.
This study represents the first population-based study of dermatomyositis (DM) using the new European Alliance of Associations for Rheumatology (EULAR)/American College of Rheumatology (ACR) criteria.
DM incidence and prevalence estimates fell on the higher end of what has been previously reported, which may reflect either increased capture of clinically amyopathic DM (CADM) cases or a rise in this disease over time.
The mortality for myopathic DM was over three-fold greater than expected, but not increased for CADM.
Predictive value of DM codes with the new classification criteria remained poor.
Funding:
This study was made possible using the resources of the Rochester Epidemiology Project, which is supported by the National Institute on Aging of the National Institutes of Health under Award Number R01AG034676, and Grant Number UL1 TR002377 from the National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosures:
Dr. Davis reports research funding from Pfizer unrelated to this work. Dr. Ernste reports research funding from Octapharma, unrelated to this work.
REFERENCES
- 1.Gerami P, Schope JM, McDonald L, Walling HW, Sontheimer RD. A systematic review of adult-onset clinically amyopathic dermatomyositis (dermatomyositis siné myositis): a missing link within the spectrum of the idiopathic inflammatory myopathies. J Am Acad Dermatol. 2006;54(4):597–613. [DOI] [PubMed] [Google Scholar]
- 2.Bendewald MJ, Wetter DA, Li X, Davis MD. Incidence of dermatomyositis and clinically amyopathic dermatomyositis: a population-based study in Olmsted County, Minnesota. Arch Dermatol. 2010;146(1):26–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dobloug C, Garen T, Bitter H, Stjärne J, Stenseth G, Grøvle L, et al. Prevalence and clinical characteristics of adult polymyositis and dermatomyositis; data from a large and unselected Norwegian cohort. Ann Rheum Dis. 2015;74(8):1551–6. [DOI] [PubMed] [Google Scholar]
- 4.Rider LG, Shamim E, Okada S, Pandey JP, Targoff IN, O’Hanlon TP, et al. Genetic risk and protective factors for idiopathic inflammatory myopathy in Koreans and American whites: a tale of two loci. Arthritis Rheum. 1999;42(6):1285–90. [DOI] [PubMed] [Google Scholar]
- 5.Werth VP, Callen JP, Ang G, Sullivan KE. Associations of tumor necrosis factor alpha and HLA polymorphisms with adult dermatomyositis: implications for a unique pathogenesis. J Invest Dermatol. 2002;119(3):617–20. [DOI] [PubMed] [Google Scholar]
- 6.Che WI, Westerlind H, Lundberg IE, Hellgren K, Kuja-Halkola R, Holmqvist M. Familial aggregation and heritability: a nationwide family-based study of idiopathic inflammatory myopathies. Ann Rheum Dis. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okada S, Weatherhead E, Targoff IN, Wesley R, Miller FW. Global surface ultraviolet radiation intensity may modulate the clinical and immunologic expression of autoimmune muscle disease. Arthritis Rheum. 2003;48(8):2285–93. [DOI] [PubMed] [Google Scholar]
- 8.Love LA, Weinberg CR, McConnaughey DR, Oddis CV, Medsger TA Jr., Reveille JD, et al. Ultraviolet radiation intensity predicts the relative distribution of dermatomyositis and anti-Mi-2 autoantibodies in women. Arthritis Rheum. 2009;60(8):2499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parks CG, Wilkerson J, Rose KM, Faiq A, Noroozi Farhadi P, Long CS, et al. Association of Ultraviolet Radiation Exposure With Dermatomyositis in a National Myositis Patient Registry. Arthritis Care Res (Hoboken). 2020;72(11):1636–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christensen ML, Pachman LM, Schneiderman R, Patel DC, Friedman JM. Prevalence of Coxsackie B virus antibodies in patients with juvenile dermatomyositis. Arthritis Rheum. 1986;29(11):1365–70. [DOI] [PubMed] [Google Scholar]
- 11.Svensson J, Holmqvist M, Lundberg IE, Arkema EV. Infections and respiratory tract disease as risk factors for idiopathic inflammatory myopathies: a population-based case-control study. Ann Rheum Dis. 2017;76(11):1803–8. [DOI] [PubMed] [Google Scholar]
- 12.Hengstman GJ, van Venrooij WJ, Vencovsky J, Moutsopoulos HM, van Engelen BG. The relative prevalence of dermatomyositis and polymyositis in Europe exhibits a latitudinal gradient. Ann Rheum Dis. 2000;59(2):141–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patrick M, Buchbinder R, Jolley D, Dennett X, Buchanan R. Incidence of inflammatory myopathies in Victoria, Australia, and evidence of spatial clustering. J Rheumatol. 1999;26(5):1094–100. [PubMed] [Google Scholar]
- 14.Findlay GH, Whiting DA, Simson IW. Dermatomyositis in the Transvaal and its occurrence in the Bantu. S Afr Med J. 1969;43(22):694–7. [PubMed] [Google Scholar]
- 15.Vargas-Leguás H, Selva-O’Callaghan A, Campins-Martí M, Hermosilla Pérez E, Grau-Junyent JM, Martínez Gómez X, et al. [Polymyositis-dermatomyositis: incidence in Spain (1997-2004)]. Med Clin (Barc). 2007;129(19):721–4. [DOI] [PubMed] [Google Scholar]
- 16.Kuo CF, See LC, Yu KH, Chou IJ, Chang HC, Chiou MJ, et al. Incidence, cancer risk and mortality of dermatomyositis and polymyositis in Taiwan: a nationwide population study. Br J Dermatol. 2011;165(6):1273–9. [DOI] [PubMed] [Google Scholar]
- 17.Furst DE, Amato AA, Iorga ŞR, Gajria K, Fernandes AW. Epidemiology of adult idiopathic inflammatory myopathies in a U.S. managed care plan. Muscle Nerve. 2012;45(5):676–83. [DOI] [PubMed] [Google Scholar]
- 18.Smoyer-Tomic KE, Amato AA, Fernandes AW. Incidence and prevalence of idiopathic inflammatory myopathies among commercially insured, Medicare supplemental insured, and Medicaid enrolled populations: an administrative claims analysis. BMC Musculoskelet Disord. 2012;13:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.See LC, Kuo CF, Chou IJ, Chiou MJ, Yu KH. Sex- and age-specific incidence of autoimmune rheumatic diseases in the Chinese population: a Taiwan population-based study. Semin Arthritis Rheum. 2013;43(3):381–6. [DOI] [PubMed] [Google Scholar]
- 20.Tan JA, Roberts-Thomson PJ, Blumbergs P, Hakendorf P, Cox SR, Limaye V. Incidence and prevalence of idiopathic inflammatory myopathies in South Australia: a 30-year epidemiologic study of histology-proven cases. Int J Rheum Dis. 2013;16(3):331–8. [DOI] [PubMed] [Google Scholar]
- 21.Balci MA, Donmez S, Saritas F, Bas V, Pamuk ON. The epidemiology of dermatomyositis in northwestern Thrace region in Turkey: epidemiology of dermatomyositis in Turkey. Rheumatol Int. 2017;37(9):1519–25. [DOI] [PubMed] [Google Scholar]
- 22.Madu PN, Williams VL, Noe MH, Omech BG, Kovarik CL, Wanat KA. Autoimmune skin disease among dermatology outpatients in Botswana: a retrospective review. Int J Dermatol. 2019;58(1):50–3. [DOI] [PubMed] [Google Scholar]
- 23.Cho SK, Kim H, Myung J, Nam E, Jung SY, Jang EJ, et al. Incidence and Prevalence of Idiopathic Inflammatory Myopathies in Korea: a Nationwide Population-based Study. J Korean Med Sci. 2019;34(8):e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Couchman KG, Wigley RD. The distribution of the systemic connective tissue diseases, ulcerative colitis and Crohn’s disease in New Zealand: an analysis of hospital admission statistics. N Z Med J. 1971;74(473):231–3. [PubMed] [Google Scholar]
- 25.Ponyi A, Borgulya G, Constantin T, Váncsa A, Gergely L, Dankó K. Functional outcome and quality of life in adult patients with idiopathic inflammatory myositis. Rheumatology (Oxford). 2005;44(1):83–8. [DOI] [PubMed] [Google Scholar]
- 26.Sigurgeirsson B, Lindelöf B, Edhag O, Allander E. Risk of cancer in patients with dermatomyositis or polymyositis. A population-based study. N Engl J Med. 1992;326(6):363–7. [DOI] [PubMed] [Google Scholar]
- 27.Limaye V, Hakendorf P, Woodman RJ, Blumbergs P, Roberts-Thomson P. Mortality and its predominant causes in a large cohort of patients with biopsy-determined inflammatory myositis. Intern Med J. 2012;42(2):191–8. [DOI] [PubMed] [Google Scholar]
- 28.Airio A, Kautiainen H, Hakala M. Prognosis and mortality of polymyositis and dermatomyositis patients. Clin Rheumatol. 2006;25(2):234–9. [DOI] [PubMed] [Google Scholar]
- 29.Dobloug GC, Garen T, Brunborg C, Gran JT, Molberg Ø. Survival and cancer risk in an unselected and complete Norwegian idiopathic inflammatory myopathy cohort. Semin Arthritis Rheum. 2015;45(3):301–8. [DOI] [PubMed] [Google Scholar]
- 30.Nuño L, Joven B, Carreira P, Maldonado V, Larena C, Llorente I, et al. Multicenter registry on inflammatory myositis from the Rheumatology Society in Madrid, Spain: Descriptive Analysis. Reumatol Clin. 2017;13(6):331–7. [DOI] [PubMed] [Google Scholar]
- 31.Li L, D’Silva KM, Lu N, Huang K, Esdaile JM, Choi HK, et al. Mortality trends in polymyositis and dermatomyositis: A general population-based study. Semin Arthritis Rheum. 2020;50(5):834–9. [DOI] [PubMed] [Google Scholar]
- 32.Kridin K, Kridin M, Amital H, Watad A, Khamaisi M. Mortality in Patients with Polymyositis and Dermatomyositis in an Israeli Population. Isr Med Assoc J. 2020;22(10):623–7. [PubMed] [Google Scholar]
- 33.D’Silva KM, Li L, Lu N, Ogdie A, Avina-Zubieta JA, Choi HK. Persistent premature mortality gap in dermatomyositis and polymyositis: a United Kingdom general population-based cohort study. Rheumatology (Oxford). 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bronner IM, van der Meulen MF, de Visser M, Kalmijn S, van Venrooij WJ, Voskuyl AE, et al. Long-term outcome in polymyositis and dermatomyositis. Ann Rheum Dis. 2006;65(11):1456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamasaki Y, Yamada H, Ohkubo M, Yamasaki M, Azuma K, Ogawa H, et al. Longterm survival and associated risk factors in patients with adult-onset idiopathic inflammatory myopathies and amyopathic dermatomyositis: experience in a single institute in Japan. J Rheumatol. 2011;38(8):1636–43. [DOI] [PubMed] [Google Scholar]
- 36.Lundberg IE, Tjärnlund A, Bottai M, Werth VP, Pilkington C, Visser M, et al. 2017 European League Against Rheumatism/American College of Rheumatology classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups. Ann Rheum Dis. 2017;76(12):1955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parker MJS, Oldroyd A, Roberts ME, Lilleker JB, Betteridge ZE, McHugh NJ, et al. The performance of the European League Against Rheumatism/American College of Rheumatology idiopathic inflammatory myopathies classification criteria in an expert-defined 10 year incident cohort. Rheumatology (Oxford). 2019;58(3):468–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jinnin M, Ohta A, Ishihara S, Amano H, Atsumi T, Fujimoto M, et al. First external validation of sensitivity and specificity of the European League Against Rheumatism (EULAR)/American College of Rheumatology (ACR) classification criteria for idiopathic inflammatory myopathies with a Japanese cohort. Ann Rheum Dis. 2020;79(3):387–92. [DOI] [PubMed] [Google Scholar]
- 39.Zhang X, Yang X, Ji L, Zhang Z. Validation of 2017 classification criteria for adult and juvenile idiopathic inflammatory myopathies proposed by EULAR/ACR in Chinese patients. Int J Rheum Dis. 2019;22(7):1278–82. [DOI] [PubMed] [Google Scholar]
- 40.Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med. 1975;292(7):344–7. [DOI] [PubMed] [Google Scholar]
- 41.Bohan A, Peter JB. Polymyositis and dermatomyositis (second of two parts). N Engl J Med. 1975;292(8):403–7. [DOI] [PubMed] [Google Scholar]
- 42.Meyer A, Meyer N, Schaeffer M, Gottenberg JE, Geny B, Sibilia J. Incidence and prevalence of inflammatory myopathies: a systematic review. Rheumatology (Oxford). 2015;54(1):50–63. [DOI] [PubMed] [Google Scholar]
- 43.Ohta A, Nagai M, Nishina M, Tomimitsu H, Kohsaka H. Prevalence and incidence of polymyositis and dermatomyositis in Japan. Mod Rheumatol. 2014;24(3):477–80. [DOI] [PubMed] [Google Scholar]
- 44.Kwa MC, Ardalan K, Laumann AE, Nardone B, West DP, Silverberg JI. Validation of International Classification of Diseases Codes for the Epidemiologic Study of Dermatomyositis. Arthritis Care Res (Hoboken). 2017;69(5):753–7. [DOI] [PubMed] [Google Scholar]
- 45.St Sauver JL, Grossardt BR, Yawn BP, Melton LJ 3rd, Pankratz JJ, Brue SM, et al. Data resource profile: the Rochester Epidemiology Project (REP) medical records-linkage system. Int J Epidemiol. 2012;41(6):1614–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vilela VS, Prieto-González S, Milisenda JC, Selva OCA, Grau JM. Polymyositis, a very uncommon isolated disease: clinical and histological re-evaluation after long-term follow-up. Rheumatol Int. 2015;35(5):915–20. [DOI] [PubMed] [Google Scholar]
- 47.Mariampillai K, Granger B, Amelin D, Guiguet M, Hachulla E, Maurier F, et al. Development of a New Classification System for Idiopathic Inflammatory Myopathies Based on Clinical Manifestations and Myositis-Specific Autoantibodies. JAMA Neurol. 2018;75(12):1528–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tansley SL, McHugh NJ, Wedderburn LR. Adult and juvenile dermatomyositis: are the distinct clinical features explained by our current understanding of serological subgroups and pathogenic mechanisms? Arthritis Res Ther. 2013;15(2):211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bongartz T, Nannini C, Medina-Velasquez YF, Achenbach SJ, Crowson CS, Ryu JH, et al. Incidence and mortality of interstitial lung disease in rheumatoid arthritis: a population-based study. Arthritis Rheum. 2010;62(6):1583–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.