

Abstract
Aims
End‐stage heart failure necessitating evaluation for heart transplantation is increasingly recognized in arrhythmogenic right ventricular cardiomyopathy (ARVC). These patients present unique challenges in pre‐transplant and peri‐transplant management given their predominantly right ventricular (RV) failure and propensity for ventricular arrhythmias. We sought to utilize a tertiary ARVC referral and heart transplant centre experience to describe management of a series of patients with ARVC undergoing heart transplantation at our centre.
Methods and results
We queried the Johns Hopkins ARVC Registry for all patients who underwent heart transplantation and further studied the subset undergoing transplantation at the Johns Hopkins Hospital. Patient demographics, clinical characteristics, and pre‐transplant clinical course were obtained from the registry and electronic medical records. Of the 532 patients in the ARVC Registry, 63 (12%) underwent heart transplantation. Nine (six male) of these patients both had known ARVC prior to transplant and were transplanted at Johns Hopkins Hospital between 2006 and 2020 at a mean age of 42 ± 14 years old. Pathogenic ARVC genetic variants were identified in six (67%) patients, all of whom had variants in the plakophilin‐2 (PKP2) gene. RV failure was universal with median right atrial to pulmonary capillary wedge pressure (RA/PCWP) ratio of 1.4 [interquartile range (IQR) 1.2–1.5] and median right ventricular stroke work index (RVSWI) of 0 g·m/m2/beat (IQR 0–0.3). Six had a history of catheter ablation for ventricular arrhythmia with five of the six having at least three ablations. Transplant evaluation was initiated an average of 344 ± 407 days after first developing heart failure symptoms. The most common bridge to transplant support included inotropes (n = 3) and extracorporeal membrane oxygenation (ECMO) (n = 2). Contraindication to inotropes or mechanical support was common due to ventricular arrhythmia and RV predominant cardiomyopathy.
Conclusions
Heart transplantation is a curative treatment for ARVC, but due to frequent ventricular arrhythmias and RV predominant pathology, patients require unique considerations in regard to timing of evaluation, haemodynamic support options, and wait listing qualification.
Keywords: Arrhythmogenic right ventricular cardiomyopathy, Heart transplantation, Mechanical circulatory support, Genetics, Arrhythmogenic cardiomyopathy, Right ventricular failure
Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited cardiomyopathy characterized by fibrofatty replacement of the myocardium, leading to ventricular arrhythmia and progressive heart failure (HF). The diagnosis is based on established Task Force Criteria (TFC) 1 and carries an estimated prevalence of 1/2000 to 1/5000. 2 End‐stage HF is increasingly recognized in these patients with greater disease recognition via genetic testing and advanced cardiac imaging prompting earlier risk stratification and placement of implantable cardioverter defibrillators (ICD). 3 Progressive right ventricular (RV) failure may necessitate heart transplantation (HT) but requires unique management considerations as compared with traditional predominantly left ventricular (LV) systolic dysfunction cardiomyopathies. More recently, the heart transplant allocation system in the USA was modified in an attempt to improve waitlist survival and account for specific HF phenotypes such as restrictive, hypertrophic, and congenital cardiomyopathies, which may warrant different considerations than LV systolic cardiomyopathies. 4 Early studies post‐allocation system change have demonstrated shorter waitlist times and improved waitlist survival for higher acuity patients as well as increased number of transplants bridged from temporary mechanical circulatory support (MCS). 5 , 6 However, the significant RV dilatation and dysfunction seen in HF due to ARVC represents an additional unique subset of patients that are underrepresented in the current United Network of Organ Sharing allocation system. ARVC patients thus present a distinct set of haemodynamic and end‐organ support challenges as they await HT. There has been limited experience reported on bridge to transplantation strategies and outcomes in ARVC patients. 7 , 8 , 9 We therefore sought to utilize our institutional ARVC referral and heart transplant experience to describe pre‐transplantation and peri‐transplantation strategies in patients with ARVC.
Methods
The Johns Hopkins ARVC Registry was established in 1999 and enrols patients affected or at risk for ARVC. The registry includes all patients evaluated by the ARVC Program at Johns Hopkins who consented to participate or those patients who contacted the program and consented to be part of the registry. Medical records, with a focus on arrhythmia events, imaging, and clinic visit notes, are obtained from treating physicians and continuously updated as new information becomes available in addition to standard annual updates. Participants include patients meeting diagnostic criteria for ARVC, at risk family members, and patients who carry an ARVC‐associated genetic variant but do not meet current diagnostic criteria for ARVC. We identified registry subjects who underwent HT from 1995 to 2020. We then included only those transplanted at Johns Hopkins Hospital for further analysis in the current study in order to ensure clinical data completeness and ability to address the study aim. The investigation conforms with the principles outlined in the Declaration of Helsinki. The Johns Hopkins Institutional Review Board approved the study protocol, and written, informed consent was obtained.
Demographic and clinical characteristics including diagnostic ARVC TFC, imaging studies, pathology results, and genetic testing results were obtained from the registry. Time of ARVC diagnosis was defined as when the patient was evaluated for and met ARVC TFC. Genetic testing including at a minimum 50 genes was performed in each patient including desmosomal genes encoding plakophilin‐2 (PKP2), desmoplakin (DSP), desmoglein‐2 (DSG2), desmocollin‐2 (DSC2), and plakoglobin (JUP) as well as nondesmosomal gene analysis including transmembrane protein 43 (TMEM43) and phospholamban (PLN). The electronic medical record was queried for peri‐transplant course. Indication for HT was adjudicated based on clinical documentation by the referring transplant physician and recorded as refractory ventricular arrhythmia, HF, or both. Transthoracic echocardiography data at the time of transplant evaluation was used for assessment of left ventricular ejection fraction (LVEF), degree of RV dilation, and RV dysfunction. Patients with evidence of RV dysfunction were categorized as RV failure only (LVEF > 50%), predominantly RV failure (LVEF ≥ 40% but ≤50%), or biventricular (BiV) failure (LVEF < 40%). Invasive haemodynamics were obtained from pre‐transplant right heart catheterization closest to the time of transplant evaluation. Cardiopulmonary exercise testing (CPET) results performed closest to time of transplant were attained. Pre‐transplant acute management (including use of intravenous inotropes and MCS), waitlist allocation, time to transplant, and outcomes were ascertained. Initial waitlist status was defined as the status at time of initial listing, while terminal waitlist status was defined as status at time of transplant.
Descriptive statistical analysis was performed as follows: continuous variables were expressed as mean ± standard deviation (normally distributed) or median [interquartile range (IQR)] (skewed) and categorical variables as numbers (percentages).
Results
As of March 2021, the Johns Hopkins ARVC Registry has 532 unique patients meeting 2010 TFC. Of these, 63 (12%) underwent HT between the years 1995 and 2020 at any institution, with 11 (17%) occurring at Johns Hopkins Hospital. Two of the 11 patients were diagnosed with ARVC retrospectively after HT. Thus, nine patients underwent HT for pre‐transplant diagnosis of ARVC, all confirmed on post‐transplant examination of heart pathology. Pathological analysis demonstrated minimal to small amount of LV involvement in all but one patient who had patchy, marked fibrofatty infiltrate of the LV myocardium. The transplants occurred between 2006 and 2020. Notably, three patients in the registry transplanted at other institutions were diagnosed instead with cardiac sarcoidosis (n = 2) or myocarditis (n = 1) based on heart explant pathology. Figure 1 illustrates the inclusion criteria for patients in this present analysis.
Figure 1.
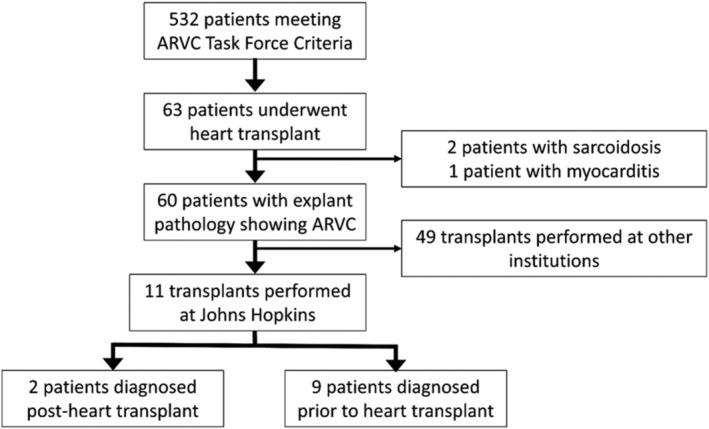
Methodology for inclusion in analysis of ARVC heart transplantation at the Johns Hopkins Hospital. ARVC, arrhythmogenic right ventricular cardiomyopathy.
Table 1 depicts patient demographics, comorbidities, anti‐arrhythmic medications, symptoms, and clinical time course leading up to HT in the nine patients with known ARVC (serially numbered Patient #1 through #9). The TFC met by each patient prior to transplant is shown in Supporting Information, Table S1 . Hypothyroidism was the most common comorbidity (n = 4, 44%). All patients were on beta‐blockers for either arrhythmia prevention or HF at some point during their disease course, but five (56%) stopped prior to transplant admission for lack of arrhythmia suppression or concerns they were contributing to worsening RV HF. All patients had an ICD prior to transplant and six (67%) patients underwent catheter ablation for ventricular tachycardia (VT); five of the six patients underwent three or more ablations. Half of the patients with history of VT ablation (n = 3) developed HF >10 years after most recent VT ablation. The other three patients had already developed HF symptoms prior to or concomitant with their last VT ablation. Patient #3 had prior cardiac surgery for anomalous right coronary artery and three (33%) patients had prior epicardial access for either VT ablation or ICD lead placement. HT evaluation was typically initiated within 1 year of the onset of first HF symptoms (mean 344 ± 407 days).
Table 1.
Patient demographics, symptom timeline, and pre‐transplant waitlist progression and support
| Pt #, sex Blood type, BMI | Variant | Comorbidities | 1st symptoms (age) | Age: ARVC Dx, HF onset | # VT ablations (age last) | Anti‐arrhythmic meds a | HF phenotype | HT eval indication (age) | Time eval to list (days) | Initial, terminal waitlist status | Support at HT | Total waitlist time (time at terminal) | Ischaemic time (min) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1, M | PKP2 | hypoTHY, GERD, IBS | VT (25) | 25, 31 | 3 (33) | Metoprolol, amio | pRV | HF (32) | 62 | 2, 1Ae | ‐ | 148 | 161 |
| O+, 25.7 | |||||||||||||
| 2, M | ‐ | ‐ | Orthopnoea/PND/oedema (40) | 40, 40 | βB, amio | RV | HF (40) | 153 | 2 b , 2 | ‐ | 308 b (‐) | 168 | |
| O−, 26.3 | |||||||||||||
| 3, M | PKP2 | ‐ | Palpitations (12) | 15, 16 | 3 (17) | Amio, βB | pRV | HF + VT (17) | 56 | 2, 1B | Milrin | 195 (104) | 196 |
| B+, 18.3 | |||||||||||||
| 4, M | ‐ | CKD | VT (14) | 14, 32 | ‐ | Amio, βB | RV | HF (32) | 391 | 2, 1Ae | ‐ | 93 (90) | 212 |
| O+, 19.2 | |||||||||||||
| 5, F | PKP2 | GERD, atrial tachycardia | VT (21) | 21, 34 | 3 (25) | Flec, βB | RV | HF (35) | 75 | 2e, 2e | ‐ | 4 (‐) | 154 |
| O−, 32.1 | |||||||||||||
| 6, F c | ‐ | Cirrhosis, atrial flutter | VT (18) | 18, 47 | 4 (35) | Sotalol | RV | HF (47) | 76 | 6, 2e | Dopa | 244 (90) | 191 |
| A+, 18.5 | |||||||||||||
| 7, F | PKP2 | hypoTHY, CKD, afib | HF (37) | 58, 37 d | ‐ | Amio, βB | BiV | HF (61) | 407 | 6, 2e | Dobut | 175 (7) | 249 |
| B+, 21.8 | |||||||||||||
| 8, M | PKP2 | hypoTHY, recent PE | Palpitations/dyspnoea (44) | 44, 65 | 1 (65) | Flec, metoprolol | RV | VT > HF (65) | 17 | 1, 1 | ECMO | 7 (‐) | 118 |
| A+, 20.2 | |||||||||||||
| 9, M | PKP2 | hypoTHY | SCA (28) | 29, 42 | 4 (30) | Dofetilide, metoprolol, sotalol, amio | RV | HF (42) | 53 | 6, 1e | ECMO | 66 (8) | 175 |
| A+, 24.1 |
Afib, atrial fibrillation; ARVC, arrhythmogenic right ventricular cardiomyopathy; BiV, biventricular failure; CKD, chronic kidney disease; dobut, dobutamine; dopa, dopamine; Dx, diagnosis; ECMO, extracorporeal membrane oxygenation; eval, evaluation; GERD, gastroesophageal reflux disease; HF, heart failure; HT, heart transplant; Hx, history; hypoTHY, hypothyroidism; IABP, intra‐aortic balloon pump; IBS, irritable bowel syndrome; milrin, milrinone; PKP2, plakophilin‐2 gene; PND, paroxysmal nocturnal dyspnoea; pRV, predominant right ventricular failure; RV, isolated right ventricular failure; SCA, sudden cardiac arrest; VT, ventricular tachycardia.
Patients are listed in heart transplant date chronological order. Grey rows represent heart transplants performed prior to the October 2018 heart transplant allocation system revision in the USA. The patient in the orange row underwent heart transplant as a paediatric patient under the paediatric heart transplant allocation system.
Italicized medications were previously tried but were discontinued because of ineffectiveness, side effects, or intolerance.
Status 2 five years prior to heart transplant but improved leading to Status 7 listing. Elapsed time in table only represents relisting as Status 2.
Patient underwent simultaneous liver transplant at the time of heart transplant.
Heart failure symptoms resolved for 10+ years before recurring.
Representative pre‐transplant haemodynamics, cardiac imaging, and explant heart pathology are shown in Figure 2 . Pre‐transplant right heart catheterization was available in eight of nine patients prior to escalation of haemodynamic support; one patient (Patient #8) underwent right heart catheterization after urgent extracorporeal membrane oxygenation (ECMO) cannulation. In the eight patients, right heart catheterization haemodynamics were consistent with RV failure. Median right atrial to pulmonary capillary wedge pressure (RA/PCWP) ratio was 1.4 (IQR 1.2–1.5). Median right ventricular stroke work index (RVSWI) was 0 g·m/m2/beat (IQR 0–0.3) compared with a median left ventricular stroke work index (LVSWI) of 22.9 g·m/m2/beat (n = 7, IQR 19.2–40.1). Median pulmonary vascular resistance (PVR) was 1.3 Woods units (IQR 0.9–1.5). Median cardiac index (CI) was 1.8 L/min/m2 (IQR 1.5–2.3). Six patients underwent pre‐transplant CPET consistent with advanced cardiac limitation as follows: median peak oxygen consumption (pVO2) 11.7 mL/kg/min (IQR 10.3–12.0) at maximal exercise (RER ≥ 1.05), median per cent predicted pVO2 34.9% (n = 5, IQR 32.8–39.1), and median ventilatory efficiency (Ve/VCO2 slope) of 46.2 (n = 5, IQR 30.5–56.3).
Figure 2.
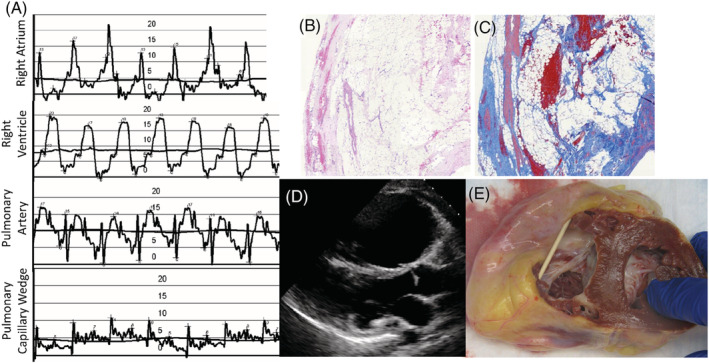
Representative sample of pre‐transplant testing results and explant pathology. (A) Right heart catheterization showing elevated right‐sided filling pressures, low pulmonary artery pulse pressure, and low left atrial filling pressures. Patient had a low cardiac output and cardiac index (not shown). The vertical scale shown is in millimetres of mercury (mmHg). (B) Haematoxylin and eosin stain showing fibrofatty replacement of right ventricular myocardium at ×2 magnification. (C) Masson trichrome stain showing increased fibrosis of right ventricular myocardium at ×2 magnification. (D) Parasternal long‐axis (PLAX) echocardiography image showing dilation of right ventricle. (E) Native heart explant showing dilated right ventricle and wall thinning with fibrofatty replacement.
Four patients were transplanted under the prior allocation system, all waitlisted as Status 2 (old allocation system) at initial listing. One of four was transplanted while Status 2 and the other three progressed to needing inpatient hospitalization with subsequent upgrade in waitlist status. Of the five patients transplanted under the revised allocation system, three patients were initially listed as Status 6, with all five requiring escalation to either Status 1 or 2 qualifying support at time of transplant. Haemodynamic and mechanical support at time of transplant are shown in Table 1 . Exceptions under both the prior and revised allocation systems commonly included barriers to indwelling pulmonary artery catheter guided inotropic support and lack of benefit of intra‐aortic balloon pump (IABP) in severe primary RV failure. For these reasons, patients were approved at similar urgency to other patients qualifying based on indwelling pulmonary artery catheter guided inotropic support or, for example, IABP support under old and revised allocation systems, respectively. Two of these patients (Patients #6 and #8) underwent attempted percutaneous right ventricular assist device (RVAD), which were both unsuccessful given severely dilated RV. The fixed distance between the inflow and outflow caused the outflow to be displaced out from the pulmonary artery even after successful initial placement in a massively dilated RV. Additionally, indwelling pulmonary artery catheter placement as required for certain waitlist statuses for duration of wait time was often limited in these patients given catheter arrhythmogenicity and positioning challenges with severely dilated RV. Left ventricular assist device (LVAD) was contraindicated in all patients because of significant RV dysfunction in addition to VT and normal LV function observed in the majority.
Two patients (Patients #8 and #9) required MCS and were supported with ECMO (one peripheral and one central). Patient #8 was originally admitted for refractory VT after recent VT ablation, thus prompting urgent HT evaluation. Significant and rapidly progressive HF became evident within days necessitating ECMO support from which he could not be weaned. Given that he did not have his full evaluation completed, we placed him initially on peripheral femoral veno‐arterial ECMO for stabilization and immediate haemodynamic support. After a period of reversal of end‐organ damage and to allow for full work‐up and mitigate deconditioning, we moved his peripheral ECMO cannulation to a central configuration with a dual lumen internal jugular venous drainage catheter and a partial upper sternotomy with direct aortic cannulation. Patient #9 was admitted for worsening low output HF symptoms despite diuretic escalations at home. He was trialled on multiple intravenous (IV) inotropes but had increasing ventricular arrhythmia burden, prompting ECMO support. These patients were supported on ECMO for 20 and 8 days, respectively, prior to transplant. Patient #8 required transfusions for bleeding; Patient #9 had significant epistaxis requiring transfusion and embolization. All transplants were performed using a standard bicaval technique. Excluding two patients that never left the intensive care unit (ICU), the median post‐transplant ICU length of stay was 6 (IQR 4–10); the two patients supported on ECMO pre‐transplant were discharged on Post‐operative Days 33 and 10. Patient #7 expired in the ICU after experiencing primary graft dysfunction (PGD) requiring ECMO support; prior to transplant, she had worsening haemodynamics despite escalating inotropes. Patient #4 had a prolonged ICU course before expiring as a consequence of technical complications of the initial operation. As of 10/1/2021, the remaining patients are alive.
The two patients diagnosed with ARVC retrospectively after HT were a father/son pair. The father had recurrent ventricular arrhythmias and LV systolic dysfunction, which was initially diagnosed as dilated cardiomyopathy. He required mechanical support with an IABP that was transitioned to a Novacor (Novacor Corp, Oakland, CA, USA) LV assist system before undergoing HT in 1995. His son developed chest pain and was found to have LV systolic dysfunction. He had recurrent arrhythmias before undergoing HT in 2008 with native explant pathology consistent with ARVC leading to diagnosis in both him and his father. Whole exome sequencing was negative for identifiable variant to explain cardiomyopathy.
Discussion
We describe the pre‐transplant course of patients with RV predominant phenotype ARVC undergoing HT and describe haemodynamic support and wait listing strategies unique to this patient population. Despite ARVC being a rare disease, we were able to leverage our high volume ARVC referral centre, which accounts for a significant proportion of ARVC heart transplants performed in the USA, to provide more granular data on peri‐transplant characteristics. We found that ARVC presents unique challenges in pre‐transplant management with significant ventricular arrhythmias limiting use of inotropes and RV morphology plus dysfunction limiting mechanical support options. The current allocation system, though modified to account for special populations, does not include RV predominant conditions, thus resulting in higher need for waitlist exceptions. However, despite these challenges, we found that with appropriate anticipation and early recognition of worsening HF, timely referral for HT evaluation, and multidisciplinary discussion of haemodynamic support options, ARVC patients have favourable HT candidacy and outcomes.
Predominant RV failure seen in ARVC requires specific diagnostic considerations. First, progressive HF may be under recognized in these patients. 3 Typical LV HF signs and symptoms such as dyspnoea and orthopnoea may be lacking, and rather more predominant may be abdominal bloating, jugular venous distention, and fatigue. In our cohort, one patient required dual organ transplantation with heart and liver due to cirrhosis in the setting of chronic right‐sided congestion. Patients are also younger and disproportionately competitive or endurance athletes prior to the diagnosis of ARVC, which often results in cessation of exercise to prevent progression. 10 Thus, a significant decline from prior peak cardiac performance needs to occur before they notice activity limitations, a hallmark of HF. Lastly, symptoms of fatigue due to deconditioning and high beta‐blocker use must be distinguished from low cardiac output.
Several clinical and diagnostic markers may herald progression of cardiomyopathy in ARVC and aid clinicians in risk stratification. For example, increased HF risk based on gene variant has been demonstrated with DSG2 variants as compared with PKP2. 11 This was not seen in our cohort with all patients with an identified variant existing in PKP2, perhaps due to the North American predominance of PKP2. Patients with DSP variants more often have significant LV involvement and more traditional dilated cardiomyopathy phenotype. 11 Depolarization abnormalities on electrocardiogram (ECG) are associated with RV progression and other ECG findings may signal LV involvement. 12 Additionally, recurrent arrhythmias can signal progression, 13 but their relation to HF may be more complex as shown in the current cohort. Of those patients who had arrhythmias, half were still having recurrent arrhythmias when they developed HF but the other half had gone >10 years without significant arrhythmia issues. Non‐invasive imaging like echocardiography provides a readily available modality for monitoring patients, and recent work has demonstrated a relationship between RV strain and progression. 14 CPET is traditionally used to predict transplant free survival in HF patients, but utilization is low in ARVC due to presumed risk of exercise‐induced arrhythmia or ICD therapies. Our group has previously demonstrated the safety of CPET as well as utility of Ve/VCO2 slope even on submaximal exercise tests to predict transplant free survival in ARVC. 15 Further investigation of serial CPET measurements and HF outcomes are needed in this population. Finally, invasive haemodynamics showing low pulmonary artery pulse pressure, low cardiac output, and right‐sided congestion are also concerning for advanced RV cardiomyopathy. Taken together, these objective markers can help supplement careful clinical assessment to identify patients who may benefit from HT.
Our practice is to refer for advanced HF assessment if patient undergoes repeat VT ablation, has progression of cardiomyopathy on imaging, or develops congestive and/or low output symptoms as shown in Figure 3 . In RV affected phenotypes like this cohort, primary HF treatment consists of diuretic therapy to maintain euvolaemia, and, if tolerated, angiotensin‐converting enzyme inhibitor (ACEi) or angiotensin receptor blocker (ARB) therapy from extrapolated human LV and animal model data on beneficial role in ventricular remodelling. Beta‐blockers may be concurrently or previously prescribed for arrhythmia suppression. In the presence of significant RV dysfunction, especially in the absence of LV dysfunction, beta‐blockers are typically not initiated for a primary indication of cardiomyopathy. In the setting of low output HF, beta‐blocker dose may be reduced or discontinued, however in conjunction with awareness of patient's arrhythmia history.
Figure 3.
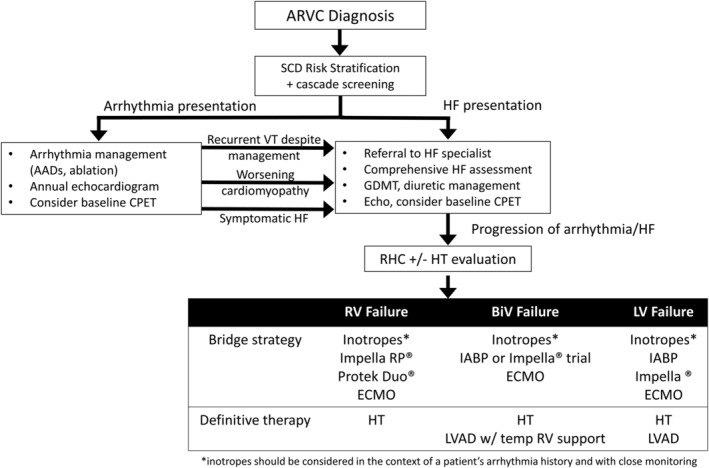
Stepwise progression and triggers for escalation in patients diagnosed with ARVC. Impella RP ® & Impella ® (Abiomed, Danvers, MA, USA), Protek Duo ® (TandemLife, Pittsburgh, PA, USA). ARVC, arrhythmogenic right ventricular cardiomyopathy; CPET, cardiopulmonary exercise test; echo, echocardiogram; ECMO, extracorporeal membrane oxygenation; GDMT, guideline‐directed medical therapy; HF, heart failure; HT, heart transplant; IABP, intra‐aortic balloon pump; LVAD, left ventricular assist device; RV, right ventricle; SCD, sudden cardiac death; VT, ventricular tachycardia.
As seen in the present cohort, there may be fairly rapid progression when overt HF symptoms manifest. We have a low threshold to obtain invasive haemodynamics partially based on the degree of symptoms at time of referral and subsequent trajectory under close monitoring (Figure 3 ). Poor haemodynamics despite volume optimization (or recurrent arrhythmias) combined with significant limiting symptoms should immediately trigger an HT evaluation. Under the revised HT allocation system, Status 6 patients account for only 3.9% of all HT performed. 5 As cardiac function deteriorates, additional support is needed to avoid end‐organ dysfunction. Inotropes are traditionally first‐line options in this situation, but use may be limited in patients with recent recurrent ventricular arrhythmias, particularly in the arrhythmogenic substrate of ARVC. Even if tolerated at low dose, patients infrequently tolerate high enough inotrope doses to qualify for more urgent waitlist status. Only three of the nine patients in our cohort were safely bridged to transplant with inotropes alone.
The final option for clinical worsening is MCS. While many of these patients have some degree of LV failure, RV failure is nearly always present and at least moderate if not severe. Durable LVADs have been used successfully in select patients with ARVC but frequently are not an option. 16 To the best of our knowledge, none of the patients in the registry have undergone durable LVAD placement. Although a plethora of temporary MCS options exists, the majority are designed to aid the LV. Percutaneous options for providing RV only support include the Impella RP (Abiomed, Danvers, MA, USA) (inserted via femoral vein) as well as the Tandem ProTek Duo (TandemLife, Pittsburgh, PA, USA) (dual lumen catheter inserted via the internal jugular vein). They unfortunately tend to be positional and are dependent on optimal RV geometry. The major limitation in ARVC is the significant RV dilation, as shown in Figure 2 , which often precludes effective positioning. Patient #8 had an attempted percutaneous RVAD to enable weaning of ECMO, but it could not be positioned adequately given RV dilation. Placement of a surgical RVAD was contemplated by utilizing a right‐sided CentriMag (Abbott, Chicago, IL, USA). However, the unknown effects of providing full support for a failing RV in the setting of unpredictability of the LV to handle the sudden changes in haemodynamics combined with a previous poor outcome in a surgically implanted RVAD in a patient with ARVC made us appropriately weary of this as a viable option. In actuality, the prior surgically implanted RVAD was carried out while the patient was acutely ill and was complicated by significant pulmonary regurgitation ultimately requiring ECMO prior to the patient expiring. In this situation and in our cohort, ECMO represents the primary modality to provide adequate haemodynamic support in those patients who remain under supported. In addition, ECMO has become a more frequent and reliable method to bridge patients with biventricular support to HT in all aetiologies of HF. 5 As Status 1, the time to transplant in these patients is decreased at the cost of complications associated with ECMO combined with immobility that may hamper post‐HT recovery. The latter could be partially overcome by alternative central ECMO configuration via the internal jugular vein for venous drainage and either the axillary artery or direct aortic cannulation for outflow. This allows for ambulation and was employed in Patient #8, who had already had an extensive ICU hospitalization for refractory VT and ablation. The consequences of under‐utilization and hesitancy in employing ECMO support in this population are evident in Patient #7 who quickly developed PGD post‐operatively. She had the confounding factor of a longer ischaemic time (249 min; Table 1 ) in addition to being under‐supported going into transplantation with evidence of liver congestion and abnormal kidney function. Expected post‐operative RV dysfunction exacerbated pre‐existing hepatic congestion with rapidly worsening hepatic function, vasoplegia, and eventual bleeding diathesis complicating ECMO cannulation. Ensuring optimal circulatory support and RV decongestion prior to HT helps avoid passive liver congestion, which could otherwise lead to a dysfunctional coagulation profile and worse vasoplegia perioperatively.
In regard to donor selection, typically younger donors are preferred given younger recipient age; however, there is less need for donor oversizing as patients with ARVC tend to have favourable pulmonary haemodynamics and thus lower risk of pulmonary hypertension‐related PGD.
Limitations of our study include its relatively small sample size and single‐centre retrospective design with risk of referral bias. However, ARVC is a rare disease, and as a referral centre, we leverage our ability to contribute what we hope will be important knowledge in the management of these patients globally. Finally, it should be noted that this study was limited to patients meeting the 2010 ARVC TFC, which has known limitations in diagnosing patients with left dominant forms of ARVC (arrhythmogenic cardiomyopathy or arrhythmogenic left ventricular cardiomyopathy). The aim of this study was to describe peri‐transplant management of those with predominant RV disease though as those with LV cardiomyopathy are often managed in a more traditional manner with use of inotropes and left‐sided MCS.
We describe contemporary strategies in patients on the waitlist and undergoing more expedited evaluation for HT with the diagnosis of ARVC. HF is becoming increasingly prevalent in ARVC and some patients may require HT. Regular clinical assessment emphasizing signs and symptoms of RV failure can be combined with both non‐invasive and invasive objective markers of progression such as echocardiography, CPET, and right heart catheterization to identify patients who would benefit from early referral to an ARVC advanced HF and transplant centre. Timely referral allows patient and clinician anticipation and shared decision making, as well as multidisciplinary planning for HT evaluation, wait listing, and operative management, with patient‐specific considerations such as arrhythmia risk. If these factors are adequately addressed, HT is a curative treatment for ARVC and can substantially prolong life. Ongoing clinical experience and future multicentre prospective analyses of ARVC HT outcomes under the revised HT allocation system will be paramount to identifying necessary considerations for this unique patient population.
Conflict of interest
P.S., K.G., D.C., T.F., S.H., J.V., S.L., P.U., H.T., K.S., C.W.C., and A.K. have no declarations. H.C. has no relevant declarations; H.C. is a consultant for Medtronic Inc, Biosense Webster, and St. Jude Medical/Abbott and receives research support from Medtronic, Biosense Webster, Farapulse, Adagio, and Boston Scientific Corp. C.T. and C.J. receive salary support from this grant (Boston Scientific Corp); C.T. and C.J. have no relevant declarations. B.M. has no relevant declarations. B.M. is a consultant for MyGeneCounsel. N.G. has no relevant declarations. N.G. is on the advisory board and speaking honorarium from scPharmaceuticals and receives research funding from Intra‐cellular Therapies, Inc.
Funding
This work and the Johns Hopkins ARVD/C Program is supported by the Leonie‐Wild Charitable Foundation (Heidelberg, Germany), the Dr. Francis P. Chiaramonte Private Foundation (Alexandria, VA, USA), the Leyla Erkan Family Fund for ARVD Research, the Dr. Satish, Rupal, and Robin Shah ARVD Fund at Johns Hopkins (Baltimore, MD, USA), the Bogle Foundation, the Healing Hearts Foundation, the Campanella Family, the Patrick J. Harrison Family, the Peter French Memorial Foundation, and the Wilmerding Endowments. The authors also wish to acknowledge a grant from the Fondation Leducq (HC) (Paris, France).
This publication was made possible by the Johns Hopkins Institute for Clinical and Translational Research (ICTR) which is funded in part by Grant Number UL1 TR003098 from the National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the Johns Hopkins ICTR, NCATS, or NIH.
Study data were collected and managed using REDCap electronic data capture tools hosted at Johns Hopkins University. REDCap (Research Electronic Data Capture) is a secure, web‐based software platform designed to support data capture for research studies, providing (i) an intuitive interface for validated data capture; (ii) audit trails for tracking data manipulation and export procedures; (iii) automated export procedures for seamless data downloads to common statistical packages; and (iv) procedures for data integration and interoperability with external sources.
Supporting information
Table S1. ARVC Task Force Criteria (TFC) categories met by each patient prior to heart transplant.
Acknowledgements
We are grateful to the ARVC/D patients and families who have made this work possible.
Scheel, P. J. III , Giuliano, K. , Tichnell, C. , James, C. , Murray, B. , Tandri, H. , Carter, D. , Fehr, T. , Umapathi, P. , Vaishnav, J. , Lewsey, S. C. , Hsu, S. , Calkins, H. , Sharma, K. , Choi, C. W. , Gilotra, N. A. , and Kilic, A. (2022) Heart transplantation strategies in arrhythmogenic right ventricular cardiomyopathy: a tertiary ARVC centre experience. ESC Heart Failure, 9: 1008–1017. 10.1002/ehf2.13757.
References
- 1. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MGPJ, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Yoerger Sanborn DM, Steinber JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the Task Force Criteria. Circulation 2010; 121: 1533–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Corrado D, Thiene G. Arrhythmogenic right ventricular cardiomyopathy/dysplasia: clinical impact of molecular genetic studies. Circulation 2006; 113: 1634–1637. [DOI] [PubMed] [Google Scholar]
- 3. Gilotra NA, Bhonsale A, James CA, te Riele ASJ, Murray B, Tichnell C, Swant A, Ong CS, Judge DPJ, Russell SD, Calkins H, Tedford RJ. Heart failure is common and under‐recognized in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Circ Heart Fail 2017; 10: 1–9. [DOI] [PubMed] [Google Scholar]
- 4. OPTN Board Briefing . Proposal to modify the adult heart allocation December 2016. https://optn.transplant.hrsa.gov/media/2006/thoracic_brief_201612.pdf. Accessed March 3, 2021.
- 5. Kilic A, Mathier MA, Hickey GW, Sultan I, Morell VO, Mulukulta SR, Keebler ME. Evolving trends in adult heart transplant with the 2018 heart allocation policy change. JAMA Cardiol 2021; 6: 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parker WF, Chung K, Anderson AS, Siegler M, Huang ES, Churpek MM. Practice changes at U.S. transplant centers after the new adult heart allocation policy. J Am Coll Card 2020; 75: 2906–2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Depasquale EC, Cheng RK, Deng MC, Nsair A, McKenna WJ, Fonarow GC, Jacoby DL. Survival after heart transplantation in patients with arrhythmogenic right ventricular cardiomyopathy. J Cardiac Fail 2017; 23: 107–112. [DOI] [PubMed] [Google Scholar]
- 8. Gilljam T, Haugaa KH, Jensen HK, Svensson A, Bundgaard H, Hansen J, Dellgren G, Gustafsson F, Eiskjær H, Andreassen AK, Sjögren J, Edvardsen T, Holst AG, Svendsen JH, Platonov PG. Heart transplantation in arrhythmogenic right ventricular cardiomyopathy—experience from the Nordic ARVC Registry. Int J Cardiol 2018; 250: 201–206. [DOI] [PubMed] [Google Scholar]
- 9. Yoda M, Minami K, Fritzsche D, Tendrich G, Schulte‐Eistrup S, Koerfer R. Three cases of orthotopic heart transplantation for arrhythmogenic right ventricular cardiomyopathy. Ann Thorac Surg 2005; 80: 2358–2360. [DOI] [PubMed] [Google Scholar]
- 10. James CA, Bhonsale A, Tichnell C, Murray B, Russell SD, Tandri H, Tedford RJ, Judge DP, Calkins H. Exercise increases age‐related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy‐associated desmosomal mutation carriers. J Am Coll Card 2013; 62: 1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gandjbakhch E, Reheuil A, Pousset F, Charron P, Frank R. Clinical diagnosis, imaging, and genetics of arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Am Coll Card 2018; 72: 784–804. [DOI] [PubMed] [Google Scholar]
- 12. Mast TP, James CA, Calkins H, Teske AJ, Tichnell C, Murray B, Loh P, Russell SD, Velthuis BK, Judge DP, Dooijes D, Tedford RJ, van der Heijden JF, Tandri H, Nauer RN, Abraham TP, Doevendans PA, te Riele ASJM, Cramer MJ. Evaluation of structural progression in arrhythmogenic right ventricular dysplasia/cardiomyopathy. JAMA Cardiol 2017; 2: 293–302. [DOI] [PubMed] [Google Scholar]
- 13. Groeneweg JA, Bhonsale A, James CA, te Riele AS, Dooijes D, Tichnell C, Murray B, Wiesfeld AC, Sawant AS, Kassamali B, Atsma DE, Volders PG, de Groot NM, de Boer K, Zimmerman SL, Kamel IR, van der Heijden JF, Russell SD, Jan Cramer M, Tedford RJ, Doevendans PA, van Veen TA, Tandri H, Wilde AA, Judge DP, van Tintelen JP, Hauer RN, Calkins H. Clinical presentation, long‐term follow‐up, and outcomes of 1001 arrhythmogenic right ventricular dysplasia/cardiomyopathy patients and family members. Circ Cardiovasc Genet 2015; 8: 437–446. [DOI] [PubMed] [Google Scholar]
- 14. Malik N, Win S, James CA, Kutty S, Mukherjee M, Gilotra NA, Tichnell C, Murray B, Agafonova J, Tandri H, Calkins H, Hays AG. Right ventricular strain predicts structural disease progression in patients with arrhythmogenic right ventricular cardiomyopathy. J Am Heart Assoc 2020; 9: e015016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scheel PJ 3rd, Florido R, Hsu S, Murray B, Tichnell C, James CA, Agafonova J, Tandri H, Judge DP, Russell SD, Tedford RJ, Calkins H, Gilotra NA. Safety and utility of cardiopulmonary exercise testing in arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Am Heart Assoc 2020; 9: e013695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Minegishi S, Kinoshita O, Hoshino Y, Komae H, Kimura M, Shimada S, Yamauchi H, Nawata K, Ono M. Long‐term support by left ventricular assist device for arrhythmogenic right ventricular cardiomyopathy. Artif Organs 2019; 43: 909–912. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. ARVC Task Force Criteria (TFC) categories met by each patient prior to heart transplant.