

Abstract
Aims
Heart failure (HF) represents a clinical syndrome resulting from different aetiologies and degrees of heart diseases. Among these, a key role is played by primary heart muscle disease (cardiomyopathies), which are the combination of multifactorial environmental insults in the presence or absence of a known genetic predisposition. The aim of the Maastricht Cardiomyopathy registry (mCMP‐registry; NCT04976348) is to improve (early) diagnosis, risk stratification, and management of cardiomyopathy phenotypes beyond the limits of left ventricular ejection fraction (LVEF).
Methods and results
The mCMP‐registry is an investigator‐initiated prospective registry including patient characteristics, diagnostic measurements performed as part of routine clinical care, treatment information, sequential biobanking, quality of life and economic impact assessment, and regular follow‐up. All subjects aged ≥16 years referred to the cardiology department of the Maastricht University Medical Center (MUMC+) for HF‐like symptoms or cardiac screening for cardiomyopathies are eligible for inclusion, irrespective of phenotype or underlying causes. Informed consented subjects will be followed up for 15 years. Two central approaches will be used to answer the research questions related to the aims of this registry: (i) a data‐driven approach to predict clinical outcome and response to therapy and to identify clusters of patients who share underlying pathophysiological processes; and (ii) a hypothesis‐driven approach in which clinical parameters are tested for their (incremental) diagnostic, prognostic, or therapeutic value. The study allows other centres to easily join this initiative, which will further boost research within this field.
Conclusions
The broad inclusion criteria, systematic routine clinical care data‐collection, extensive study‐related data‐collection, sequential biobanking, and multi‐disciplinary approach gives the mCMP‐registry a unique opportunity to improve diagnosis, risk stratification, and management of HF and (early) cardiomyopathy phenotypes beyond the LVEF limits.
Keywords: Registry, Heart failure, Cardiomyopathies, Diagnosis, Prognosis
Introduction
Heart failure (HF) is a heterogeneous, multifactorial, and rising epidemic syndrome. It currently affects over 50 million patients worldwide, causing a significant societal, clinical, and economic burden. 1 , 2 HF symptoms are often non‐specific, making the diagnosis—particularly during early stages—challenging. 3 , 4 The difficulty in diagnosing HF is reflected by the multitude of proposed reference standards, which often include different clinical variables and biomarkers with varying cut‐off values. 3 , 5 , 6 , 7 , 8
Left ventricular ejection fraction (LVEF) is one of the cornerstones within these reference standards, mainly because major therapeutic progress has been made in patients with a reduced LVEF. 9 , 10 HF patients are nowadays usually categorized in HF with reduced (HFrEF), mildly reduced (HFmrEF), and preserved ejection fraction (HFpEF). 3 While categorizing HF based on LVEF provided valuable insight into the pathophysiology of HF, it results in an enormous oversimplification of this complex syndrome. 9 , 10 , 11
Heart failure represents a clinical syndrome resulting from different aetiologies and degrees of heart diseases. Among these, a key role is played by primary heart muscle disease (cardiomyopathies), which are the combination of multifactorial environmental insults in the presence or absence of a known genetic predisposition. 2 , 9 , 10 , 12 The usage of guideline‐based and LVEF‐based inclusion criteria for registries seriously hampers the possibility to better understand the clinical course of early to overt cardiomyopathy phenotypes. A better understanding of the (early) cardiomyopathy phenotypes, their underlying pathophysiological processes, and their related (future) disease burden and progression towards overt HF is essential in order to pave the path for novel targeted prevention and intervention studies and is the objective of the Maastricht Cardiomyopathy registry (mCMP‐registry).
Study design
Objectives
The aim of the mCMP‐registry is to improve (early) diagnosis, risk stratification, and management of cardiomyopathy phenotypes in individuals that are referred to the cardiology department for HF‐like symptoms or cardiac screening for cardiomyopathies (Figure 1 ). Specific aims are to (i) improve (early) diagnosis of cardiomyopathy phenotypes and aetiologies in (a) symptomatic individuals; (ii) improve (early) risk stratification of (a) symptomatic individuals with and without an overt cardiac phenotype that are referred for HF‐like symptoms or cardiac screening for cardiomyopathies (e.g. because of known familial cardiomyopathy); (iii) develop a better understanding of the societal and economic impact of (early) cardiomyopathies; (iv) develop a better understanding of pathophysiological processes involved in the development and progression of (early) cardiomyopathies; and (v) develop novel treatment strategies based on these pathophysiological processes.
Figure 1.
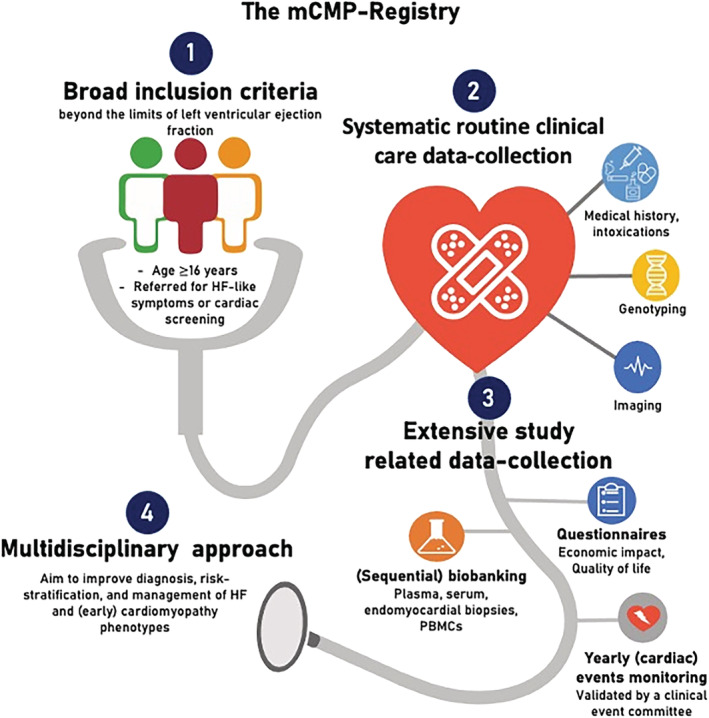
The Maastricht Cardiomyopathy registry (mCMP‐registry) includes subjects referred to the cardiology clinic of the Maastricht University Medical Centre (MUMC+) for HF‐like symptoms or cardiac screening (e.g. because of known familial cardiomyopathy). The broad inclusion criteria, systematic routine clinical care data‐collection, extensive study‐related data‐collection, and multi‐disciplinary approach gives the mCMP‐registry a unique opportunity to improve diagnosis, risk stratification, and management of HF and (early) cardiomyopathy phenotypes beyond the left ventricular ejection fraction limits. HF, heart failure; PBMCs, peripheral blood mononuclear cells.
Study design
The mCMP‐registry (NCT04976348) is an investigator‐initiated single‐centre prospective observational registry founded in July 2021. It includes patient characteristics, diagnostic measurements (Figure 2 ) performed as part of routine clinical care, treatment information, standardized sequential biobanking, yearly questionnaires (including quality of life and economic impact assessment), and long‐term clinical follow‐up (the data dictionary and overview of samples collected are available at www.cardiomyopathyresearch.eu). Each patient is followed up for 15 years or until death or withdrawal of consent. The study does not interfere with routine clinical practice at any time point, and all patients are treated at the discretion of their physician in accordance with the latest guidelines and consensus statements. All subjects included in the registry provide written informed consent. Additionally, to improve future risk stratification (such as to determine which patients would benefit from an implanted defibrillator) and to minimize selection bias for such analyses, patients who died before informed consent was signed will be included in the registry if the deceased person was eligible for inclusion and did not object to the use of their medical data for research purposes (opt‐out approach). The study is performed in accordance with the principles of the Declaration of Helsinki and the European Union General Data Protection Regulation (GDPR). An independent Medical Ethics Committee of the Maastricht University Medical Center (MUMC+) has approved this registry.
Figure 2.
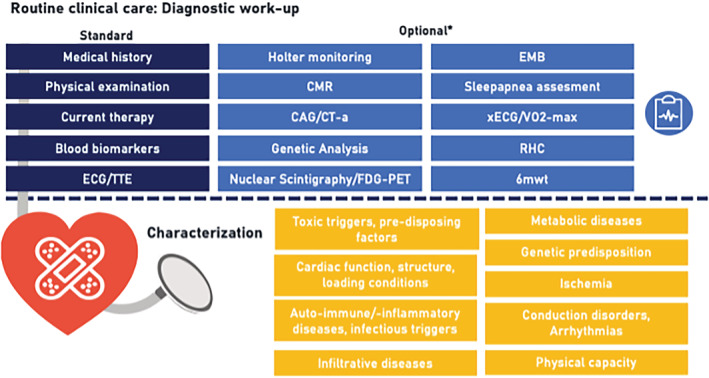
Standard care protocol for the diagnostic workup of individuals referred to the cardiology department of the Maastricht University Medical Center for heart failure‐like symptoms or cardiac screening. *The treating cardiologist may decide to perform additional diagnostic measurements beyond this protocolled diagnostic workup based on the medical indication at baseline or during follow‐up. Additional information (such as medication usage at follow‐up and cardiac interventions) is stored within the electronic online case‐record forms (the data dictionary is available at www.cardiomyopathyresearch.eu). 6MWT, 6 min walking test; CAG, invasive coronary angiography; CT‐a, computed tomography angiography; CMR, cardiac magnetic resonance imaging; ECG, electrocardiography; EMB, endomyocardial biopsy; FDG‐PET, fluorodeoxyglucose‐positron emission tomography; RHC, right heart catheterization; TTE, transthoracic echocardiography; VO2‐max, maximal oxygen consumption test; xECG, exercise electrocardiography.
Inclusion and exclusion criteria
All individuals aged ≥16 years referred to the cardiology department of the MUMC+ for HF‐like symptoms 3 or cardiac screening for cardiomyopathies (heart muscle diseases, including but not limited to dilated cardiomyopathy) are eligible for inclusion. Individuals will not be prospectively included in the registry if they are not willing to participate or unable to provide written informed consent.
Clinical data
All subjects referred to our clinic receive the study information of this registry and can provide written informed consent during the upcoming appointment (more information is provided at www.cardiomyopathyresearch.eu). All hospital visits will take place according to regular clinical procedures. At baseline, a standard care protocol is used for the clinical diagnostic workup of individuals referred to the cardiology department of the MUMC+, including medical/family history assessment, physical examination, blood analysis (including but not limited to creatinine and N‐terminal pro‐B‐type natriuretic peptide), electrocardiography, and echocardiography. The treating cardiologist may decide to perform additional diagnostic measurements beyond this protocolled diagnostic workup (e.g. genetic testing is offered in patients with dilated cardiomyopathy, hypertrophic cardiomyopathy, arrhythmogenic cardiomyopathy, or in arrhythmic/conduction disorders) based on the medical indication at baseline or during follow‐up (Figure 2 ). 13 , 14 The measurements performed during clinical visits are collected in standardized forms within the patient's electronic medical record (EMR) for routine clinical care purposes (also including medication usage). Subsequently, the data are uploaded pseudo‐anonymized to a database using standardized electronic online case‐record forms (eCRF) for subjects included within the mCMP‐registry. More details are provided in the Supporting Information.
Regular clinical follow‐up periods will be at 6 and 12 months, and finally yearly unless the treating cardiologist decides otherwise (Figure 3 ). Measurements performed for clinical purposes, diagnosis of cardiac and non‐cardiac comorbidities, treatment, and serious adverse events (such as death and hospitalizations) will be monitored for all subjects included in this study. A clinical event committee (CEC; existing of at least three physicians who are part of the study team) will discuss, sign, and lock the occurrence of clinical events three times a year.
Figure 3.
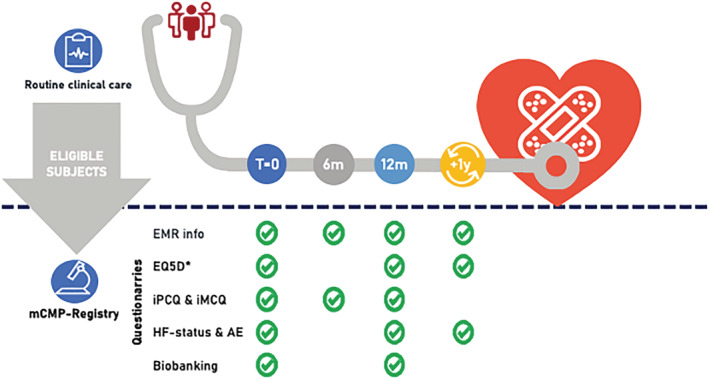
Subjects included in the mCMP‐registry undergo clinical care as usual. Regular clinical visits will be at baseline, 6 and 12 months, and finally yearly unless the treating cardiologist decides otherwise. Upon inclusion in the mCMP‐registry, subjects are asked for additional consent for yearly surveying short questionnaires for a period of 15 years and sequential biobanking. AE, adverse events; EMR, electronic medical records; *EQ‐5D, EuroQol 5D questionnaire (obtained at baseline, and after 1, 3, 5, 10, and 15 years in informed consented subjects); HF, heart failure; iMCQ, iMTA Medical Consumption Questionnaire; iPCQ, iMTA Productivity Cost Questionnaire; m, months; mCMP‐registry, Maastricht Cardiomyopathy registry; T, time.
Longitudinal questionnaires and events
Upon inclusion, subjects are asked for additional consent for yearly surveying questionnaires for a period of 15 years (Figure 3 ). These questionnaires include (i) a yearly questionnaire that focuses on the occurrence of (adverse) events and current signs/symptoms. If the questionnaire reveals that (cardiac) events have occurred outside the MUMC+, the study subjects will be contacted by telephone to determine the nature of the event and date of occurrence (both of which are necessary for the development of valid prediction models and time‐to‐event analysis). If no event occurred, these data are semi‐automatically updated in the eCRF; (ii) a questionnaire at baseline and 1, 3, 5, 10, and 15 years after inclusion that focuses on the quality of life (EuroQol 5D) 15 ; and (iii) two questionnaires at baseline, 6, and 12 months after inclusion that focus on the productivity [iMTA Productivity Cost Questionnaire (iPCQ) 16 ] and the medical consumption [iMTA Medical Consumption Questionnaire (iMCQ) 17 ] to allow economic impact evaluation of (early) cardiomyopathies and HF.
Biobanking for the Maastricht Cardiomyopathy registry
Upon inclusion in the mCMP‐registry, subjects are asked consent for additional biobanking. During routine blood sampling, an additional amount of 60 mL will be obtained at baseline and 1 year follow‐up in informed consented subjects (Figure 3 ). Blood samples are stored as serum, plasma, buffy‐coat, and peripheral blood mononuclear cells (PBMCS) for downstream analysis (such as pluripotent stem cell‐related research). Additional consent is asked for the usage of biomaterial left‐overs collected for routine clinical practice purposes (including blood, urine, and endomyocardial biopsies if available) and for the performance of genetic analysis on the stored biomaterial. All samples will be stored coded at the MUMC+ Biobank.
Data collection and management
A systematic approach for study management, data collection, data cleaning, and data availability was developed to ensure the sustainability of the mCMP‐registry, which allows reproducibility and scalability of the study structure and procedures in line with the FAIR Data Principles. 18 Procedures are elaborated on in the Supporting Information. Briefly, patient inclusion information and study logistics (including automatically sending of questionnaires and related reminders) are recorded in an online web‐based tool developed for this purpose (developed by MEMIC; Center for Data and Information Management, Faculty of Health, Medicine and Life Sciences, Maastricht University, and MUMC+). Separately, research data are systematically collected from standardized forms within the EMR and stored with a pseudo‐anonymized study ID in the eCRF of CASTOR EDC (Ciwit B.V., Amsterdam, the Netherlands). All events and additional‐diagnostics information are stored with corresponding dates of occurrence, allowing to answer multiple research questions with divergent baseline moments (T = 0) within this registry. Source data such as echocardiographic images or electrocardiography recordings are stored with the pseudo‐anonymized study ID in the research facility. Importantly, the developed data infrastructures and processes allow easy implementation of other centres in the near future.
Data availability
The data dictionary and procedures for data sharing with external researchers are available through our website (www.cardiomyopathyresearch.eu).
Statistical approach
This registry aims to include 10 000 subjects. The mCMP‐registry Steering Committee will review all statistical plans. Two central approaches will be used to answer the research questions related to the aims of this registry: (i) a data‐driven approach to predict clinical outcome – such as HF hospitalization, (sudden) cardiac death, and changes in quality of life – and response to therapy, and to identify clusters of patients who share underlying pathophysiological processes, in order to pave the path for precision medicine; and (ii) a hypothesis‐driven approach in which clinical parameters are tested for their (incremental) diagnostic, prognostic, or therapeutic value.
Discussion
The mCMP‐registry is an ongoing registry including all subjects (≥16 years of age) referred to the cardiology clinic of the MUMC+ for HF‐like symptoms or cardiac screening for cardiomyopathies, irrespective of diagnosis or LVEF. The registry enables a unique opportunity to improve diagnosis, risk stratification, and management of HF and (early) cardiomyopathy phenotypes, which is achieved by (Figure 1 ) (i) the broad inclusion criteria; (ii) the systematic routine clinical care at fixed time points, which is documented in standardized EMR forms, allowing semi‐automatic data collection within the eCRF; (iii) the extensive study‐related data collection, including yearly automatically sent questionnaires for a period of 15 years, sequential biobanking, and yearly (cardiac) events monitoring validated by a CEC; and (iv) the multi‐disciplinary approach within and beyond our centre, including both pre‐clinical and clinical researchers from multiple departments (including the department of immunology, pathology, clinical genetics, medical microbiology, and cardiology) and supporting staff (including research nurses, lab technicians, bio‐statisticians, and IT support).
The years of experience with large‐scale cohort studies in different HF phenotypes of our group, particularly in HFpEF 19 , 20 and non‐ischaemic non‐valvular cardiomyopathy, 21 , 22 , 23 , 24 formed the foundation of the current registry. The study logistics and eCRF have been set up to allow other centres to easily join this initiative, which will optimize the process of external validations and opens possibilities to study less prevalent cardiomyopathies.
The registry will allow data‐driven (e.g. with the use of machine learning 23 ) and hypothesis‐driven approaches (e.g. to assess the incremental value of novel diagnostic and prognostic biomarkers) by the extensive clinical data and biobank materials. It will allow testing of the hypothesis that challenge LVEF as the cornerstone for HF classification, for example, by introducing and combining alternative cardiac function measurements (such as left atrial function parameters 25 and global longitudinal strain 26 ), by biomarkers and corresponding biological pathways measured at multiple time points, 27 or by introducing alternative multi‐organ cardiomyopathy classifications (e.g. MOGES‐like classifications). 10 , 11 , 28 Because this registry will provide real‐world data, it even allows the performance of registry‐based trials. Moreover, the mCMP‐registry allows the creation of a virtual waiting room for future (interventional) studies.
Due to the close collaboration with the department of clinical genetics, there is access to extensive and large‐scale genotyping of subjects in the registry. The genetic predisposition of a patient with HF is increasingly receiving attention, partly due to the first published polygenic risk scores that explain HF risk beyond the monogenic dogma. 29 , 30 Genetic testing for monogenic causes is incorporated in routine clinical care in our centre and offered to all patients with a dilated, hypertrophic, arrhythmogenic, and non‐compaction cardiomyopathy irrespective of aetiology or family history. Subjects included in the registry give permission to perform genetic testing on their stored biomaterials, which allows monogenic and polygenic testing beyond these phenotypes. Genetic testing in our centre includes Sanger sequencing, whole exome and genome sequencing (WES, WGS), RNA sequencing, and panel analysis using single‐molecular Molecular Inversion Probes (smMIP). The last method also grants the possibility to perform genetic testing on paraffine‐embedded material of deceased subjects, which opens new collaborative possibilities with the department of pathology and The Netherlands Heart Tissue Bank (www.hearttissuebank.nl). 31
Study limitations
This study has some challenges that should be addressed. First, the clinical follow‐up data of subjects without an (overt) cardiac phenotype who are referred to the general practitioner will be limited because all clinical (diagnostic) measurements and follow‐up are performed as part of routine clinical care. However, the yearly questionnaires—with subsequently telephone contact if indicated—still allow us to monitor health status and the occurrence of (cardiac) events in these patients. Second, the collected clinical data originate from daily clinical practice. Although HF care is standardized in our centre as much as possible, clinical variations due to physician or patient preference or logistical limitations may influence variability in factors such as timing and type of additional diagnostics and initiation of HF therapy (because the treatment of patients and performance of additional diagnostics is part of routine clinical care). Nonetheless, this variation itself can also result in clinically relevant insights. Moreover, while the observational design of the registry limits to draw definite conclusions on causal relationships regarding, for example, treatment effects, the mCMP‐registry provides real‐life data that provides an important basis to execute randomized registry‐based clinical trials in the (near) future.
Conclusions
The broad inclusion criteria, systematic routine clinical care data‐collection, extensive study‐related data‐collection, sequential biobanking, and multi‐disciplinary approach gives the mCMP‐registry a unique opportunity to improve diagnosis, risk stratification, and management of HF and (early) cardiomyopathy phenotypes beyond the LVEF limits.
Conflict of interest
None related to the content of the manuscript or foundation of the registry.
Funding
The authors are supported by the Netherlands Cardiovascular Research Initiative: an initiative with the support of the Dutch Heart Foundation, CVON2015‐RECONNECT, CVON2016‐Early HFPEF, and CVON 2017‐ShePREDICTS, Double‐Dose (2020B005).
Supporting information
Data S1. Supporting Information.
Acknowledgements
We acknowledge the Dutch Cardiovascular Alliance, an initiative with support of the Dutch Heart Foundation, and Stichting Hartedroom. The authors would also like to acknowledge all the involved healthcare workers who are crucial for the clinical care provided to the subjects. Additionally, we would like to thank all team members who are supporting the research processes of this study and all individuals that (will) participate in this study.
Henkens, M. T. H. M. , Weerts, J. , Verdonschot, J. A. J. , Raafs, A. G. , Stroeks, S. , Sikking, M. A. , Amin, H. , Mourmans, S. G. J. , Geraeds, C. B. G. , Sanders‐van Wijk, S. , Barandiarán Aizpurua, A. , Uszko‐Lencer, N. H. M. K. , Krapels, I. P. C. , Wolffs, P. F. G. , Brunner, H. G. , van Leeuwen, R. E. W. , Verhesen, W. , Schalla, S. M. , van Stipdonk, A. W. M. , Knackstedt, C. , Li, X. , Abdul Hamid, M. A. , van Paassen, P. , Hazebroek, M. R. , Vernooy, K. , Brunner‐La Rocca, H.‐P. , van Empel, V. P. M. , and Heymans, S. R. B. (2022) Improving diagnosis and risk stratification across the ejection fraction spectrum: the Maastricht Cardiomyopathy registry. ESC Heart Failure, 9: 1463–1470. 10.1002/ehf2.13833.
References
- 1. Group NCHFGW , Atherton JJ, Sindone A, De Pasquale CG, Driscoll A, MacDonald PS, Hopper I, Kistler PM, Briffa T, Wong J, Abhayaratna W, Thomas L, Audehm R, Newton P, O'Loughlin J, Branagan M, Connell C. National Heart Foundation of Australia and Cardiac Society of Australia and New Zealand: guidelines for the prevention, detection, and management of heart failure in Australia 2018. Heart Lung Circ 2018; 27: 1123–1208. [DOI] [PubMed] [Google Scholar]
- 2. Ziaeian B, Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol 2016; 13: 368–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, Falk V, Gonzalez‐Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GMC, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P, Group ESCSD . 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC)Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 2016; 37: 2129–2200. [DOI] [PubMed] [Google Scholar]
- 4. Groenewegen A, Rutten FH, Mosterd A, Hoes AW. Epidemiology of heart failure. Eur J Heart Fail 2020; 22: 1342–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McMurray JJ, Adamopoulos S, Anker SD, Auricchio A, Bohm M, Dickstein K, Falk V, Filippatos G, Fonseca C, Gomez‐Sanchez MA, Jaarsma T, Kober L, Lip GY, Maggioni AP, Parkhomenko A, Pieske BM, Popescu BA, Ronnevik PK, Rutten FH, Schwitter J, Seferovic P, Stepinska J, Trindade PT, Voors AA, Zannad F, Zeiher A, Guidelines ESCCfP . ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: the Task Force for the diagnosis and treatment of acute and chronic heart failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J 2012; 33: 1787–1847. [DOI] [PubMed] [Google Scholar]
- 6. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL, American College of Cardiology F , American Heart Association Task Force on Practice G . 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol 2013; 62: e147–e239. [DOI] [PubMed] [Google Scholar]
- 7. Okuda S, Yano M. Guidelines for treatment of chronic heart failure (JCS 2010). Nihon Rinsho 2011; 69: 595–604. [PubMed] [Google Scholar]
- 8. Ezekowitz JA, O'Meara E, McDonald MA, Abrams H, Chan M, Ducharme A, Giannetti N, Grzeslo A, Hamilton PG, Heckman GA, Howlett JG, Koshman SL, Lepage S, McKelvie RS, Moe GW, Rajda M, Swiggum E, Virani SA, Zieroth S, Al‐Hesayen A, Cohen‐Solal A, D'Astous M, De S, Estrella‐Holder E, Fremes S, Green L, Haddad H, Harkness K, Hernandez AF, Kouz S, LeBlanc MH, Masoudi FA, Ross HJ, Roussin A, Sussex B. 2017 comprehensive update of the Canadian cardiovascular society guidelines for the management of heart failure. Can J Cardiol 2017; 33: 1342–1433. [DOI] [PubMed] [Google Scholar]
- 9. De Keulenaer GW, Brutsaert DL. Systolic and diastolic heart failure are overlapping phenotypes within the heart failure spectrum. Circulation 2011; 123: 1996–2004 discussion 2005. [DOI] [PubMed] [Google Scholar]
- 10. Triposkiadis F, Butler J, Abboud FM, Armstrong PW, Adamopoulos S, Atherton JJ, Backs J, Bauersachs J, Burkhoff D, Bonow RO, Chopra VK, de Boer RA, de Windt L, Hamdani N, Hasenfuss G, Heymans S, Hulot JS, Konstam M, Lee RT, Linke WA, Lunde IG, Lyon AR, Maack C, Mann DL, Mebazaa A, Mentz RJ, Nihoyannopoulos P, Papp Z, Parissis J, Pedrazzini T, Rosano G, Rouleau J, Seferovic PM, Shah AM, Starling RC, Tocchetti CG, Trochu JN, Thum T, Zannad F, Brutsaert DL, Segers VF, De Keulenaer GW. The continuous heart failure spectrum: moving beyond an ejection fraction classification. Eur Heart J 2019; 40: 2155–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lam CSP, Solomon SD. Classification of heart failure according to ejection fraction: JACC review topic of the week. J Am Coll Cardiol 2021; 77: 3217–3225. [DOI] [PubMed] [Google Scholar]
- 12. McKenna WJ, Judge DP. Epidemiology of the inherited cardiomyopathies. Nat Rev Cardiol 2021; 18: 22–36. [DOI] [PubMed] [Google Scholar]
- 13. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Bohm M, Burri H, Butler J, Celutkiene J, Chioncel O, Cleland JGF, Coats AJS, Crespo‐Leiro MG, Farmakis D, Gilard M, Heymans S, Hoes AW, Jaarsma T, Jankowska EA, Lainscak M, Lam CSP, Lyon AR, McMurray JJV, Mebazaa A, Mindham R, Muneretto C, Francesco Piepoli M, Price S, Rosano GMC, Ruschitzka F, Kathrine Skibelund A, Group ESCSD . 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 2021; 42: 3599–3726. [DOI] [PubMed] [Google Scholar]
- 14. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006; 113: 1807–1816. [DOI] [PubMed] [Google Scholar]
- 15. EuroQol Group . EuroQol—a new facility for the measurement of health‐related quality of life. Health Policy 1990; 16: 199–208. [DOI] [PubMed] [Google Scholar]
- 16. Bouwmans C, Krol M, Severens H, Koopmanschap M, Brouwer W, Hakkaart‐van RL. The iMTA productivity cost questionnaire: a standardized instrument for measuring and valuing health‐related productivity losses. Value Health 2015; 18: 753–758. [DOI] [PubMed] [Google Scholar]
- 17. iMTA Productivity and Health Research Group. Handleiding iMTA MedicalCost Questionnaire (iMCQ). Rotterdam; 2018. [10‐02‐2021] Available from: www.imta.nl.
- 18. Wilkinson MD, Dumontier M, Aalbersberg IJ, Appleton G, Axton M, Baak A, Blomberg N, Boiten JW, da Silva Santos LB, Bourne PE, Bouwman J, Brookes AJ, Clark T, Crosas M, Dillo I, Dumon O, Edmunds S, Evelo CT, Finkers R, Gonzalez‐Beltran A, Gray AJ, Groth P, Goble C, Grethe JS, Heringa J, 't Hoen PAC, Hooft R, Kuhn T, Kok R, Kok J, Lusher SJ, Martone ME, Mons A, Packer AL, Persson B, Rocca‐Serra P, Roos M, van Schaik R, Sansone SA, Schultes E, Sengstag T, Slater T, Strawn G, Swertz MA, Thompson M, van der Lei J, van Mulligen E, Velterop J, Waagmeester A, Wittenburg P, Wolstencroft K, Zhao J, Mons B. The FAIR Guiding Principles for scientific data management and stewardship. Sci Data 2016; 3: 160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sanders‐van Wijk S, Barandiaran Aizpurua A, Brunner‐La Rocca HP, Henkens M, Weerts J, Knackstedt C, Uszko‐Lencer N, Heymans S, van Empel V. The HFA‐PEFF and H2FPEF scores largely disagree in classifying patients with suspected heart failure with preserved ejection fraction. Eur J Heart Fail 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barandiaran Aizpurua A, Sanders‐van Wijk S, Brunner‐La Rocca HP, Henkens M, Heymans S, Beussink‐Nelson L, Shah SJ, van Empel VPM. Validation of the HFA‐PEFF score for the diagnosis of heart failure with preserved ejection fraction. Eur J Heart Fail 2020; 22: 413–421. [DOI] [PubMed] [Google Scholar]
- 21. Hazebroek MR, Henkens M, Raafs AG, Verdonschot JAJ, Merken JJ, Dennert RM, Eurlings C, Abdul Hamid MA, Wolffs PFG, Winkens B, Brunner‐La Rocca HP, Knackstedt C, van Paassen P, van Empel VPM, Heymans SRB. Intravenous immunoglobulin therapy in adult patients with idiopathic chronic cardiomyopathy and cardiac parvovirus B19 persistence: a prospective, double‐blind, randomized, placebo‐controlled clinical trial. Eur J Heart Fail 2020; 23: 302–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Verdonschot JAJ, Hazebroek MR, Derks KWJ, Barandiaran Aizpurua A, Merken JJ, Wang P, Bierau J, van den Wijngaard A, Schalla SM, Abdul Hamid MA, van Bilsen M, van Empel VPM, Knackstedt C, Brunner‐La Rocca HP, Brunner HG, Krapels IPC, Heymans SRB. Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long‐term life‐threatening arrhythmias. Eur Heart J 2018; 39: 864–873. [DOI] [PubMed] [Google Scholar]
- 23. Verdonschot JAJ, Merlo M, Dominguez F, Wang P, Henkens M, Adriaens ME, Hazebroek MR, Mase M, Escobar LE, Cobas‐Paz R, Derks KWJ, van den Wijngaard A, Krapels IPC, Brunner HG, Sinagra G, Garcia‐Pavia P, Heymans SRB. Phenotypic clustering of dilated cardiomyopathy patients highlights important pathophysiological differences. Eur Heart J 2021; 42: 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hazebroek MR, Moors S, Dennert R, van den Wijngaard A, Krapels I, Hoos M, Verdonschot J, Merken JJ, de Vries B, Wolffs PF, Crijns HJ, Brunner‐La Rocca HP, Heymans S. Prognostic relevance of gene‐environment interactions in patients with dilated cardiomyopathy: applying the MOGE(S) classification. J Am Coll Cardiol 2015; 66: 1313–1323. [DOI] [PubMed] [Google Scholar]
- 25. Bisbal F, Baranchuk A, Braunwald E, Bayes de Luna A, Bayes‐Genis A. Atrial failure as a clinical entity: JACC review topic of the week. J Am Coll Cardiol 2020; 75: 222–232. [DOI] [PubMed] [Google Scholar]
- 26. Verdonschot JAJ, Merken JJ, Brunner‐La Rocca HP, Hazebroek MR, Eurlings C, Thijssen E, Wang P, Weerts J, van Empel V, Schummers G, Schreckenberg M, van den Wijngaard A, Lumens J, Brunner HG, Heymans SRB, Krapels IPC, Knackstedt C. Value of speckle tracking‐based deformation analysis in screening relatives of patients with asymptomatic dilated cardiomyopathy. JACC Cardiovasc Imaging 2020; 13: 549–558. [DOI] [PubMed] [Google Scholar]
- 27. Ferreira JP, Verdonschot J, Wang P, Pizard A, Collier T, Ahmed FZ, Brunner‐La‐Rocca HP, Clark AL, Cosmi F, Cuthbert J, Diez J, Edelmann F, Girerd N, Gonzalez A, Grojean S, Hazebroek M, Khan J, Latini R, Mamas MA, Mariottoni B, Mujaj B, Pellicori P, Petutschnigg J, Pieske B, Rossignol P, Rouet P, Staessen JA, Cleland JGF, Heymans S, Zannad F, HOMAGE (Heart Omics in AGEing) Consortium . Proteomic and mechanistic analysis of spironolactone in patients at risk for HF. JACC Heart Fail 2021. Apr; 9: 268–277. [DOI] [PubMed] [Google Scholar]
- 28. Arbustini E, Narula N, Tavazzi L, Serio A, Grasso M, Favalli V, Bellazzi R, Tajik JA, Bonow RO, Fuster V, Narula J. The MOGE(S) classification of cardiomyopathy for clinicians. J Am Coll Cardiol 2014; 64: 304–318. [DOI] [PubMed] [Google Scholar]
- 29. Pirruccello JP, Bick A, Wang M, Chaffin M, Friedman S, Yao J, Guo X, Venkatesh BA, Taylor KD, Post WS, Rich S, Lima JAC, Rotter JI, Philippakis A, Lubitz SA, Ellinor PT, Khera AV, Kathiresan S, Aragam KG. Analysis of cardiac magnetic resonance imaging in 36,000 individuals yields genetic insights into dilated cardiomyopathy. Nat Commun 2020; 11: 2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tadros R, Francis C, Xu X, Vermeer AMC, Harper AR, Huurman R, Kelu Bisabu K, Walsh R, Hoorntje ET, Te Rijdt WP, Buchan RJ, van Velzen HG, van Slegtenhorst MA, Vermeulen JM, Offerhaus JA, Bai W, de Marvao A, Lahrouchi N, Beekman L, Karper JC, Veldink JH, Kayvanpour E, Pantazis A, Baksi AJ, Whiffin N, Mazzarotto F, Sloane G, Suzuki H, Schneider‐Luftman D, Elliott P, Richard P, Ader F, Villard E, Lichtner P, Meitinger T, Tanck MWT, van Tintelen JP, Thain A, McCarty D, Hegele RA, Roberts JD, Amyot J, Dube MP, Cadrin‐Tourigny J, Giraldeau G, L'Allier PL, Garceau P, Tardif JC, Boekholdt SM, Lumbers RT, Asselbergs FW, Barton PJR, Cook SA, Prasad SK, O'Regan DP, van der Velden J, Verweij KJH, Talajic M, Lettre G, Pinto YM, Meder B, Charron P, de Boer RA, Christiaans I, Michels M, Wilde AAM, Watkins H, Matthews PM, Ware JS, Bezzina CR. Shared genetic pathways contribute to risk of hypertrophic and dilated cardiomyopathies with opposite directions of effect. Nat Genet 2021; 53: 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. The Netherlands Heart Tissue Bank . www.hearttissuebank.nl [Date Accessed: 27‐01‐2022].
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information.
Data Availability Statement
The data dictionary and procedures for data sharing with external researchers are available through our website (www.cardiomyopathyresearch.eu).