

Abstract
Aims
Heart failure is an increasingly recognized later stage manifestation of arrhythmogenic right ventricular cardiomyopathy (ARVC) that can require heart transplantation (HT) to appropriately treat. We aimed to study contemporary ARVC HT outcomes in a national registry.
Methods and results
The United Network for Organ Sharing registry was queried for HT recipients from 1/1994 through 2/2020. ARVC patients were compared with non‐ARVC dilated, restrictive, and hypertrophic cardiomyopathy HT patients (HT for ischaemic and valvular disease was excluded from analysis). Post‐HT survival was assessed using Kaplan–Meier estimates. A total of 189 of 252 (75%) waitlisted ARVC patients (median age 48 years, 65% male) underwent HT, representing 0.3% of the total 65 559 HT during the study time period. Annual frequency of HT for ARVC increased significantly over time. ARVC patients had less diabetes (5% vs. 17%, P < 0.001), less cigarette use (15% vs. 23%, P < 0.001), lower pulmonary artery and pulmonary capillary wedge pressures, and lower cardiac output than the 33 659 non‐ARVC patients (P < 0.001). Ventricular assist device use was significantly lower in ARVC patients (8% vs. 32%, P < 0.001); 1 and 5 year post‐HT survival was 97% and 93% for ARVC vs. 95% and 82% for non‐ARVC HT recipients (P < 0.001). On adjusted multivariable Cox regression, ARVC had decreased risk of post‐HT death compared with non‐ARVC aetiologies (hazard ratio 0.48, 95% confidence interval 0.28–0.82, P = 0.008). Patients with ARVC also had lower risk of death or graft failure than non‐ARVC patients (hazard ratio 0.51, 95% confidence interval 0.32–0.81, P = 0.004).
Conclusions
In the largest series of HT in ARVC, we found that HT is increasingly performed in ARVC, with higher survival compared with other cardiomyopathy aetiologies. The right ventricular predominant pathophysiology may require unique considerations for heart failure management, including HT.
Keywords: Arrhythmogenic right ventricular cardiomyopathy, Heart transplantation, Heart failure, Post‐transplant outcomes
Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited cardiomyopathy (CM) characterized by fibrofatty infiltration of the myocardium, resulting in ventricular arrythmias and predominantly right ventricular (RV) dysfunction. 1 , 2 The left ventricle may be involved in at least 50% of cases, with some forms having a predominant left ventricular phenotype, leading to increasing use of the broader term arrhythmogenic CM. 3 , 4 , 5 , 6 , 7 , 8 Diagnostic criteria based on structural abnormalities, fatty or fibrofatty replacement of the RV myocardium, electrocardiographic changes, arrhythmias of RV origin, and familial disease were initially defined in 1994 and revised as the 2010 Task Force Criteria to reflect technical advances in diagnostic imaging. 2 , 9 Nonetheless, ARVC can still be challenging to diagnose. 10 , 11
Fatal ventricular arrhythmia has traditionally been the primary cause of death in these patients; however, increased early recognition of ARVC and improvement in arrhythmic risk stratification and treatment with exercise avoidance, antiarrhythmics, ablation, and implantable cardioverter defibrillator implantation have substantially improved survival. 1 , 12 ARVC is, however, a progressive disease, ultimately leading to RV and late‐stage biventricular failure. 13 , 14 Previous studies have estimated the incidence of heart failure (HF) in patients with ARVC to be between 5% and 20%, 12 , 15 , 16 , 17 , 18 but a recent study from our group found that it is more prevalent, occurring in nearly 50% of a 289‐patient ARVC cohort. 19 Heart transplantation (HT) can be utilized in this population for end‐stage HF or refractory ventricular arrythmias, with outcomes reported primarily in case reports, single‐institution case series, and a Nordic registry. 20 , 21 , 22 , 23 An analysis of the United Network for Organ Sharing (UNOS) registry was undertaken for ARVC patients between 1994 and 2011, largely prior to the revised ARVC diagnostic criteria established in 2010. 24
Overall, ARVC remains a rare disease with poorly studied HF outcomes despite likely under‐recognized high prevalence of HF. We therefore sought to describe waitlisting and HT practices as well as outcomes inclusive of the current ARVC diagnostic criteria and HT era using a large, national database. We hypothesized that ARVC patients would have more favourable post‐HT outcomes as compared with non‐ARVC patients.
Methods
After institutional review board approval, we surveyed the UNOS database for HT recipients from January 1994 through February 2020. Paediatric patients, multi‐organ transplant recipients, and candidates listed for retransplantation were included (Figure 1 ). We assessed the characteristics of waitlisted patients with the primary diagnosis of ARVC, including demographics, co‐morbidities, duration on waitlist, waitlist inactivity, and any mechanical circulatory support (MCS). We compared ARVC patients listed for HT before (1/1994 to 17/10/2018) vs. after (18/10/2018 to 2/2020) the UNOS adult heart allocation policy change. 25 We then compared the characteristics of transplanted ARVC patients to those of patients transplanted for non‐ARVC, including dilated, restrictive, and hypertrophic CM. Patients transplanted for ischaemic CM, valvular heart disease, or congenital heart disease were excluded. Continuous and categorical variables were compared using Student's t‐test and χ 2 test, respectively. For all statistical tests, a P‐value of <0.05 was considered statistically significant.
Figure 1.

Study population of heart transplantation patients from the Organ Procurement and Transplantation Network database. ARVC, arrhythmogenic right ventricular cardiomyopathy; CM, cardiomyopathy.
Kaplan–Meier survival analysis of ARVC and non‐ARVC patients was performed, and survival was compared with log‐rank testing. Cox proportional hazards model was utilized to assess the impact of primary diagnosis and other patient and transplant variables on survival. A multivariable analysis was also performed, controlling for factors found to be significant on univariable analysis and those known to predict outcomes after HT. 26 Kaplan–Meier analysis and Cox proportional hazards model was also performed for survival free of graft failure. In this model, a failure event was either death (from any cause) or graft failure requiring retransplantation.
Results
Characteristics of waitlisted arrhythmogenic right ventricular cardiomyopathy patients
Between January 1994 and February 2020, a total of 252 patients were listed for HT for a primary diagnosis of ARVC (Supporting Information, Table S1 ). Their median age was 48 [inter‐quartile range (IQR) 30–58] years, 65.1% (n = 164) were men, and the majority were Caucasian (79.4%, n = 200). Their incidence of co‐morbidities was overall low, with 5.6% (n = 14) of patients with diabetes mellitus, 2.4% (n = 6) with cerebrovascular disease, and 15.9% (n = 40) with cigarette use. The majority (85.3%, n = 215) had an implantable cardioverter defibrillator. Eleven patients (4.4%) were listed for multi‐organ transplant, six for heart–liver and five for heart–kidney.
Outcomes of waitlisted arrhythmogenic right ventricular cardiomyopathy patients
The median number of days on the waitlist for ARVC patients was 111 (IQR 28–312) (Supporting Information, Table S2 ). At a median follow‐up time (from listing to last follow‐up) of 3.4 years (IQR 1.3–7.3), 75% (n = 189) of the listed ARVC patients had undergone transplantation. Of these, eight (4.2%) underwent multi‐organ transplant, four with a simultaneous kidney and four with a simultaneous liver. The number of HTs performed per year for ARVC significantly increased over the study period (P < 0.001; Figure 2 ). Specifically, one transplant was performed in 1994, increasing to 26 transplants in 2019. Concomitantly, the percentage of all HTs performed for the primary indication of ARVC increased from 0.04% in 1994 to 0.73% in 2019.
Figure 2.
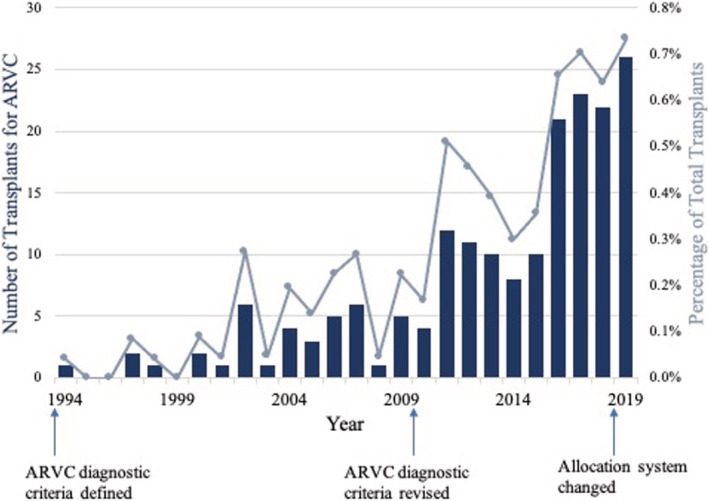
The number of transplants performed per year for arrhythmogenic right ventricular cardiomyopathy (ARVC) (columns) and the percentage of total transplants (line graph) have significantly increased over the study period (P < 0.001).
Just over 15% (n = 39) of patients experienced a period of inactivity while on the waitlist, with the most common reason being that they were temporarily too sick (14 patients). Thirty‐seven patients (14.7%) were removed from the waitlist; 11 patients (4.4%) died while on the waitlist.
Utilization of mechanical circulatory support
At listing, a total of eight ARVC patients (3.2%) were on extracorporeal membrane oxygenation (ECMO), three of whom were transplanted and alive on last follow‐up (Supporting Information, Table S1 ). Ten patients (5.3% of 189 transplanted ARVC patients) had an intra‐aortic balloon pump (IABP) at the time of transplant (Table 1 ). Six patients (2.4%) had ventricular assist devices (VADs) on listing—one with a left VAD (LVAD), two with a total artificial heart (TAH), and three with both an LVAD and a right VAD (RVAD). By the time of transplantation, a total of 15 patients (7.9%) had VADs—six with LVADs, two with RVADs, four with TAHs, and three with LVAD + RVAD.
Table 1.
Characteristics of the 189 patients who underwent heart transplantation for ARVC compared with those undergoing transplantation for dilated, restrictive, or hypertrophic CM, 1/1994 to 2/2020
| ARVC transplants | Non‐ARVC transplants | P | |
|---|---|---|---|
| N = 189 | N = 33 659 | ||
| Age (years) | 48 (30–58) | 50 (34–59) | 0.422 |
| Paediatric patients (<18 years old) | 19 (10.1%) | 4447 (13.2%) | 0.201 |
| Male gender | 116 (61.4%) | 22 068 (65.6%) | 0.227 |
| Race | <0.001 | ||
| Caucasian | 156 (82.5%) | 20 542 (61.0%) | |
| African American | 13 (6.9%) | 8523 (25.3%) | |
| Hispanic | 8 (4.2%) | 3064 (9.1%) | |
| Asian | 12 (6.3%) | 1027 (3.1%) | |
| Other/multiracial | 0 (0%) | 503 (1.5%) | |
| BMI (kg/m2) | 25.1 ± 5.2 | 25.7 ± 5.8 | 0.161 |
| Co‐morbidities | |||
| Cerebrovascular disease | 4 (2.1%) | 1394 (4.1%) | 0.177 |
| Diabetes mellitus | 9 (4.8%) | 5552 (16.5%) | <0.001 |
| End‐stage renal disease on haemodialysis | 5 (2.6%) | 1222 (3.6%) | 0.450 |
| Prior malignancy | 11 (5.8%) | 2231 (6.6%) | 0.199 |
| History of cigarette use | 29 (15.3%) | 7736 (23.0%) | <0.001 |
| Implantable cardiac defibrillator | 161 (85.6%) | 17 635 (52.4%) | <0.001 |
| Previous transplant | 1 (0.5%) | 177 (0.5%) | 0.995 |
| Multi‐organ transplant | 8 (4.2%) | 1141 (3.4%) | 0.523 |
| Circulatory support (at transplant) | |||
| ECMO | 3 (1.6%) | 466 (1.4%) | 0.812 |
| IABP | 10 (5.3%) | 2212 (6.6%) | 0.478 |
| VAD | 15 (7.9%) | 10 640 (31.6%) | <0.001 |
| LVAD | 6 (3.2%) | 8085 (24.0%) | |
| RVAD | 2 (1.1%) | 53 (0.2%) | |
| TAH | 4 (2.1%) | 252 (0.7%) | |
| LVAD + RVAD | 3 (1.6%) | 828 (2.5%) | |
| Unspecified | 0 (0%) | 1422 (4.2%) | |
| IV inotropes | 74 (39.2%) | 15 016 (44.6%) | 0.132 |
| Haemodynamics (at transplant) | |||
| Cardiac output (L/min) | 3.9 ± 1.6 | 4.3 ± 1.6 | <0.001 |
| PA systolic (mmHg) | 25 ± 8 | 41 ± 14 | <0.001 |
| PA diastolic (mmHg) | 13 ± 6 | 21 ± 9 | <0.001 |
| PA pulse pressure (mmHg) | 12 ± 6 | 20 ± 10 | <0.001 |
| PCW (mmHg) | 12 ± 6 | 19 ± 9 | <0.001 |
| Waitlist status at transplant | <0.001 | ||
| Pre‐allocation change (1/1994 to 9/2018) | |||
| Status 1A | 72 (38.2%) | 14 807 (44.0%) | |
| Status 1B | 65 (34.4%) | 8030 (23.9%) | |
| Status 1, A/B unspecified | 4 (2.1%) | 3621 (10.8%) | |
| Status 2 | 16 (8.5%) | 4177 (12.4%) | |
| Post‐allocation change (10/2018 to 2/2020) | |||
| Status 1A (peds patients) | 2 (1.1%) | 217 (0.6%) | |
| Status 1B (peds patients) | 1 (0.5%) | 49 (0.1%) | |
| Old Status 2 (peds patients) | 0 (0%) | 9 (0.03%) | |
| Status 1 | 1 (0.5%) | 224 (0.7%) | |
| Status 2 | 12 (6.3%) | 1352 (4.0%) | |
| Status 3 | 8 (4.2%) | 582 (1.7%) | |
| Status 4 | 7 (3.7%) | 455 (1.4%) | |
| Status 5 | 0 (0%) | 12 (0.04%) | |
| Status 6 | 1 (0.5%) | 93 (0.3%) | |
| Unknown | 0 (0%) | 31 (0.09%) | |
| Mean days on waitlist | 199 ± 367 | 196 ± 335 | 0.899 |
| Post‐transplant complications | |||
| Stroke | 3 (1.6%) | 793 (2.4%) | 0.101 |
| Dialysis | 18 (9.5%) | 2941 (8.9%) | 0.376 |
| Permanent pacemaker | 4 (2.1%) | 1037 (3.1%) | 0.113 |
| Follow‐up time, from transplant to last follow‐up (years) | 3.3 (1.7–7.1) | 5.6 (2.1–10.4) | <0.001 |
| Renal function on follow‐up after HT | |||
| Creatinine (mg/dL) | 1.3 ± 1.0 | 1.7 ± 1.5 | <0.001 |
| Chronic dialysis | 2 (1.1%) | 2042 (6.1%) | 0.015 |
| Subsequent renal transplant | 3 (1.6%) | 355 (1.1%) | 0.689 |
ARVC, arrhythmogenic right ventricular cardiomyopathy; BMI, body mass index; CM, cardiomyopathy; ECMO, extracorporeal membrane oxygenation; HT, heart transplantation; IABP, intra‐aortic balloon pump; IV, intravenous; LVAD, left ventricular assist device; PA, pulmonary artery (PA pulse pressure = PA systolic − PA dialysis); PCW, pulmonary capillary wedge; RVAD, right ventricular assist device; TAH, total artificial heart; VAD, ventricular assist device.
Categorical variables are listed as frequency (%), and continuous variables are listed as mean ± standard deviation or median (inter‐quartile range), as appropriate. Bolded values indicate significant differences, with P < 0.05.
Impact of 2018 heart allocation policy
Of the total 252 ARVC patients listed for HT, 209 (82.9%) were listed before 18/10/2018 under the prior allocation system [of which 162 (77.5%) were transplanted], and 43 (17.1%) were listed after the revised allocation system [of which 27 (62.8%) have been transplanted] (Supporting Information, Table S2 ). In the old allocation system, the most common initial listing status was Status 2 (in 58.9%, n = 123). In the new system, the most common initial listing status was Status 4 (in 37.2%, n = 16). The two groups (pre‐allocation change vs. post‐allocation change) were overall similar in terms of their demographics, co‐morbidities, and haemodynamics, with the following exceptions: more patients waitlisted with ARVC post‐allocation change had diabetes (14.0% vs. 4.3%, P = 0.029) and cerebrovascular disease (7.0% vs. 1.4%, P = 0.0008) than those with ARVC pre‐allocation change (Supporting Information, Table S3 ). There was no significant difference in the use of ECMO, IABP, VADs, or intravenous inotropes in pre‐allocation vs. post‐allocation change. Patients with ARVC post‐allocation change were transplanted, on average, sooner from time of waitlisting than patients with ARVC pre‐allocation change [70 (SD 88) days vs. 220 (SD 391) days, P < 0.001].
Transplanted arrhythmogenic right ventricular cardiomyopathy patients vs. non‐arrhythmogenic right ventricular cardiomyopathy cardiomyopathy patients
During the study period, a total of 65 559 HTs were performed—189 (0.3%) for the primary diagnosis of ARVC and 33 659 for non‐ARVC including 29 216 (86.8%) for dilated CM, 1905 (6.7%) for restrictive CM, and 1353 (4.0%) for hypertrophic CM (Figure 1 ). ARVC and non‐ARVC transplanted patient characteristics are compared in Table 1 .
Arrhythmogenic right ventricular cardiomyopathy patients were more commonly Caucasian (82.5% vs. 61.0%, P < 0.001). They had lower incidence of diabetes (4.8% vs. 16.5%, P < 0.001) and cigarette use (15.3% vs. 23.0%, P < 0.001) compared with non‐ARVC patients. More ARVC patients had implantable cardioverter defibrillators (85.6% vs. 53.9%, P < 0.001). The use of ECMO, IABP, and intravenous inotropes did not differ between the groups, but ARVC patients less often had a VAD at the time of transplant (7.9% vs. 31.6%, P < 0.001). This difference is due primarily to a lower use of LVADs in the ARVC population (3.2% vs. 24.0%, P < 0.001). ARVC patients had lower pulmonary artery (PA) systolic (25 vs. 41 mmHg, P < 0.001), PA diastolic (13 vs. 21 mmHg, P < 0.001), PA pulse pressure (12 vs. 20 mmHg, P < 0.001), and pulmonary capillary wedge pressures (12 vs. 19 mmHg, P < 0.001) than non‐ARVC patients. Their cardiac outputs were also lower (3.9 vs. 4.3 L/min, P < 0.001).
Post‐transplant survival
Median follow‐up time (from transplant to last follow‐up) was 3.3 (IQR 1.7–7.1) years for the ARVC and 5.6 (IQR 2.1–10.4) years for non‐ARVC patients (Table 1 ). Median survival for the entire cohort of HT patients (ARVC and non‐ARVC) was 14.7 years. ARVC patients had significantly higher survival after transplant compared with non‐ARVC patients (Figure 3A ) (P < 0.001). Specifically, 1 and 5 year survival after transplant was 96.8% and 93.4% for ARVC vs. 94.9% and 82.2% for non‐ARVC recipients.
Figure 3.

Kaplan–Meier survival estimates demonstrated (A) improved survival and (B) improved survival free of graft failure after heart transplantation for patients with primary diagnosis of arrhythmogenic right ventricular cardiomyopathy (ARVC) vs. non‐ARVC dilated, restrictive, and hypertrophic cardiomyopathy aetiologies (both P < 0.001 by log‐rank test).
Table 2 summarizes the univariable and multivariable Cox proportional hazards model for factors associated with post‐transplant mortality for all HT patients. On univariable analysis, the primary HT indication of ARVC was associated with a reduced risk of post‐transplant death compared with non‐ARVC patients [hazard ratio (HR) 0.45, 95% confidence interval (CI) 0.28–0.74, P = 0.002]. After adjusting for age, gender, race, listing status, support at time of transplant (ECMO, VAD, IABP, inotropes, ventilator, inhaled nitric oxide, or prostaglandin), and post‐operative complications (stroke, haemodialysis, or need for a permanent pacemaker), ARVC was still significantly associated with reduced risk of post‐HT death (HR 0.48, 95% CI 0.28–0.82, P = 0.008). Increasing age (HR 1.01, 95% CI 1.01–1.01, P < 0.001), African American race (HR 1.62, 95% CI 1.54–1.70, P < 0.001 vs. Caucasian), increasing body mass index (HR 1.02, 95% CI 1.02–1.02, P < 0.001), a previous transplant (HR 1.64, 95% CI 1.17–2.27, P = 0.003), medical co‐morbidities (including cerebrovascular disease, diabetes, end‐stage renal disease, prior malignancy, and history of cigarette use), and the occurrence of a post‐transplant complication (stroke, dialysis, or need for a permanent pacemaker) also significantly increased the risk of post‐transplant mortality on multivariable analysis (Table 2 ).
Table 2.
Factors associated with post‐transplant mortality on univariable and multivariable Cox regression for entire study cohort of n = 33 848 heart transplantation patients
| Univariable | P | Multivariable | P | |
|---|---|---|---|---|
| Hazard ratio (95% CI) | Hazard ratio (95% CI) | |||
| Age | 1.01 (1.01–1.01) | <0.001 | 1.01 (1.01–1.01) | <0.001 |
| Male gender | 1.02 (0.98–1.06) | 0.337 | 1.01 (0.97–1.06) | 0.615 |
| Race | ||||
| Caucasian | Ref | Ref | ||
| African American | 1.53 (1.46–1.60) | <0.001 | 1.62 (1.54–1.70) | <0.001 |
| Hispanic | 0.92 (0.51–1.00) | 0.053 | 1.04 (0.96–1.13) | 0.353 |
| Asian | 0.78 (0.67–0.91) | 0.002 | 0.85 (0.72–0.99) | 0.047 |
| Co‐morbidities | ||||
| Cerebrovascular disease | 1.06 (0.95–1.19) | 0.310 | 1.14 (1.00–1.29) | 0.046 |
| Diabetes mellitus | 2.70 (2.48–2.93) | <0.001 | 2.27 (2.06–2.51) | <0.001 |
| ESRD on HD | 1.27 (1.14–1.43) | <0.001 | 1.29 (1.13–1.48) | <0.001 |
| Prior malignancy | 1.11 (1.01–1.21) | 0.025 | 1.12 (1.01–1.24) | 0.032 |
| History of cigarette use | 1.42 (1.33–1.51) | <0.001 | 1.36 (1.25–1.47) | <0.001 |
| BMI | 1.02 (1.02–1.03) | <0.001 | 1.02 (1.02–1.02) | <0.001 |
| Defibrillator | 0.92 (0.82–1.04) | 0.193 | 0.93 (0.80–1.08) | 0.326 |
| Previous transplant | 1.23 (0.94–1.62) | 0.137 | 1.64 (1.17–2.27) | 0.003 |
| Multi‐organ transplant | 0.96 (0.84–1.10) | 0.547 | 0.92 (0.78–1.07) | 0.279 |
| Days on waitlist | 1.00 (1.00–1.00) | 0.165 | 1.00 (1.00–1.00) | 0.948 |
| Post‐transplant complications | ||||
| Stroke | 1.57 (1.37–1.80) | <0.001 | 1.59 (1.36–1.87) | <0.001 |
| Dialysis | 1.94 (1.80–2.09) | <0.001 | 2.05 (1.87–2.24) | <0.001 |
| Permanent pacemaker | 1.19 (1.07–1.32) | 0.001 | 1.21 (1.07–1.37) | 0.002 |
| Indication: ARVC | 0.45 (0.28–0.74) | 0.002 | 0.48 (0.28–0.82) | 0.008 |
ARVC, arrhythmogenic right ventricular cardiomyopathy; BMI, body mass index; CI, confidence interval; ECMO, extracorporeal membrane oxygenation; ESRD, end‐stage renal disease; HD, haemodialysis; IABP, intra‐aortic balloon pump; IV, intravenous; NO, nitric oxide; PGE, prostaglandin E; VAD, ventricular assist device.
Multivariable regression controls for age, gender, race, listing status, support (VAD, ECMO, IABP, IV inotropes, ventilator, inhaled NO, or PGE), and any post‐op complication (stroke, HD, or pacemaker). Bolded values indicate significant differences, with P < 0.05.
Graft function after transplant
Kaplan–Meier analysis demonstrated improved survival free of graft failure for ARVC vs. non‐ARVC patients (Figure 3 B, P < 0.001). Specifically, 1 and 5 year survival without graft failure (i.e. alive and without experiencing a graft failure requiring retransplantation) was 96.1% and 91.6% for ARVC vs. 94.8% and 80.9% for non‐ARVC HT recipients. On univariable Cox regression, ARVC as a primary indication for HT was associated with a reduced risk of death or graft failure when compared with other indications for HT (HR 0.47, 95% CI 0.29–0.74, P = 0.001). After controlling for age, gender, ethnicity, listing status, support, and post‐op complications on multivariable regression, ARVC patients continued to have significantly lower risk of death or graft failure than non‐ARVC patients (HR 0.51, 95% CI 0.32–0.81, P = 0.004). Risk of developing coronary allograft vasculopathy was similar (HR 1.30, 95% CI 0.69–2.4, P = 0.416). Average post‐transplant left ventricular ejection fraction of ARVC patients was 61% (±7%), vs. 57% (±13%) in non‐ARVC patients (P < 0.001). In follow‐up after HT, ARVC patients had lower average serum creatinine (1.3 ± 1.0 vs. 1.7 ± 1.5 mg/dL, P < 0.001) and less need for chronic dialysis (1.1% vs. 6.1%, P = 0.015) compared with non‐ARVC HT patients (Table 1 ).
Discussion
In this contemporary analysis of a large, national database of HT recipients, we demonstrate a temporal increase in HT for ARVC, key clinical differences of ARVC patients undergoing HT compared with other CM aetiologies, and favourable post‐transplant outcomes in patients with ARVC. Although ARVC remains a relatively rare indication for HT, comprising only 0.3% of all HTs during this study period, there has been a significant increase in both the absolute number of and percentage of total transplants performed for ARVC between 1994 and 2020. While some of this increase may be due to updated diagnostic criteria, increased recognition, and genetic testing in recent years, numbers have continued to rise even in the most recent years. With continued increasing recognition and improvements in risk stratification to reduce the burden of arrhythmic sudden cardiac death in ARVC, HF prevalence is anticipated to increase in this patient population, with a subsequent increase in HT. 2 , 19
Arrhythmogenic right ventricular cardiomyopathy is an inherited CM, with diagnosis often made in otherwise young patients with few co‐morbidities. 1 Initial presentation may be one of ventricular arrhythmia—or even sudden cardiac death in young people or athletes 27 , 28 —or a diagnosis identified via familial cascade genetic screening. Consistent with this, we found that ARVC HT patients had a median age of 48 years, and compared with non‐ARVC HT transplants, they had fewer co‐morbidities (significantly less diabetes and tobacco use) and were more likely to have implantable cardioverter defibrillators. Reflective of their intrinsic RV‐dominant pathology, ARVC patients in this study had significantly lower PA pressures compared with non‐ARVC patients while having significantly narrower PA pulse pressures (an indirect measure of the combined effects of RV contractility and pulmonary vascular distensibility 29 ) and significantly impaired cardiac output, likely driving need for HT. Likewise, we have demonstrated that patients with ARVC can have impaired ventilatory efficiency, which is in turn associated with HF and need for HT. 30 These haemodynamic differences highlight the unique pathophysiological considerations that drive pre‐transplant mechanical support strategies in patients with predominantly RV cardiomyopathies.
Overall, the use of MCS was rare in the ARVC population, with the most common MCS utilization in ARVC being ECMO. Rates of ECMO, IABP, and intravenous inotrope use did not differ compared with non‐ARVC patients; however, only 3% of ARVC patients had left‐sided VAD support, and the majority of patients with VAD support required RV devices (60%). As the predominant pathology in ARVC is RV failure, the utility of durable LVAD therapy is limited in this population. Additionally, the lack of Food and Drug Administration‐approved durable RV MCS devices, historically poor outcomes in transplanted TAH patients (compared with LVAD or non‐MCS patients), and our findings of favourable post‐transplant outcomes support transplantation as the definitive treatment for patients with advanced ARVC. 19 , 31 , 32 Given that the average age of ARVC diagnosis is ~30 years, 6 , 12 , 18 , 19 the current data—with ARVC patients listed for HT at an average of 44 (±17) years—suggest that the need for transplantation occurs approximately one to two decades after diagnosis.
Our data demonstrate that ARVC patients have excellent post‐transplant survival with improvement compared with previous reports (post‐HT survival for ARVC patients previously reported as 87% and 71% at 1 and 5 years, respectively, in a UNOS cohort from 1993 to 2011). 24 This is most likely due to a parallel improvement in post‐transplant survival for all‐comers as has been reported in the most recent era. 33 Prior single ARVC registry HT cohorts demonstrated 1 year survival of 94% in North America 20 and a 5 year survival of 91% in Europe. 23 After adjustment, we found that the primary diagnosis of ARVC was associated with a 50% reduced risk of post‐HT mortality compared with other dilated, restrictive, and hypertrophic CM indications. ARVC patients also had a close to 50% reduced risk of death or graft failure compared with non‐ARVC patients, after controlling for age, gender, ethnicity, listing status, support, and post‐operative complications. These improved outcomes can potentially be attributed to the unique haemodynamic profile of ARVC patients, as well as their relative lack of medical co‐morbidities, especially for graft loss, where co‐morbidities such as diabetes and history of smoking increase risk. 34 , 35
In October 2018, the adult heart allocation policy was modified in an attempt to improve prioritization of specific CM phenotypes (such as restrictive and congenital) and of more severely ill patients, ultimately to reduce waitlist mortality. 25 Studies to date have demonstrated an increased use of temporary MCS devices such as ECMO and IABP and a concomitant decreased use of durable VADs since the allocation change. 25 , 36 , 37 , 38 We found that after the allocation policy change, ARVC patients waitlisted for HT had a non‐significant increase in the use of ECMO and IABP. The overall small number of patients and low frequency use of MCS in ARVC patients may have limited detection of significant changes in MCS use thus far. Notably, ARVC patients undergoing HT under the revised allocation system did have shorter average waitlist durations than those listed prior to the change (70 vs. 220 days), similar to previously reported data in all‐comers for HT. 39 Longer duration follow‐up is needed to make further conclusions regarding peri‐transplant management of ARVC patients.
While the use of a national database allowed us to study the largest cohort of ARVC HT patients to date, this study has the inherent limitations of database research, including lack of granularity and missing/unknown variables. Notably, diagnosis in UNOS is based on listing diagnosis and does not take into consideration explanted heart pathology. Furthermore, diagnosis is defined and input by the listing centre. Haemodynamic data did not include right‐sided heart pressures. Additionally, the number of patients undergoing HT for ARVC was relatively small, thus limiting some statistical analyses, and with increased numbers in more recent years, decreasing the follow‐up duration in comparison with the non‐ARVC study patients. Specific to ARVC, the UNOS database does not include information on genetic mutations, which have been shown to impact the clinical course and expression of the disease, particularly whether patients have RV or left ventricular predominant phenotypes. 17
In conclusion, while ARVC remains overall a rare indication for HT, the number of HTs performed for this disease has steadily increased over time, which likely represents a combination of increased recognition and diagnosis as well as a true use of HT for management of these patients. We demonstrated excellent post‐transplant survival for ARVC patients, which may reflect their lower burden of co‐morbid conditions and a favourable pre‐transplant haemodynamic profile. Results from this large UNOS registry, combined with the limited MCS options for this patient population, support the use and early consideration of HT in patients with ARVC. However, larger, longer‐term studies are required to further assess the role of MCS as a bridge to transplant strategy in ARVC.
Conflict of interest
H.T. is a consultant for Abbott. K.S. is a consultant and advisory board member and received honoraria from Novartis, Janssen, Bayer, Novo Nordisk, and Bristol Myers Squibb. H.C. is a consultant for Medtronic Inc., Biosense Webster, Pfizer, StrideBio, and Abbott. N.A.G. is a consultant and advisory board member for scPharmaceuticals, Inc. The remaining authors have no disclosures or possible conflicts of interest to report.
Funding
The authors wish to acknowledge funding from Leducq Foundation (Fondation Leducq). The Johns Hopkins ARVD/C Program is supported by the Leonie Wild Foundation, the Leyla Erkan Family Fund for ARVD Research, the Dr Francis P. Chiramonte Private Foundation, the Dr Satish, Rupal, and Robin Shah ARVD Fund at Johns Hopkins, the Bogle Foundation, the Healing Hearts Foundation, the Campanella family, the Patrick J. Harrison family, the Peter French Memorial Foundation, and the Wilmerding Endowments.
Supporting information
Table S1. Characteristics of 252 patients listed for heart transplantation with the primary diagnosis of arrhythmogenic right ventricular cardiomyopathy (1/1994‐2/2020).
Table S2. Outcomes of the 252 patients with ARVC waitlisted for heart transplantation, 1/1994‐2/2020.
Table S3. Characteristics of patients listed for heart transplantation with the primary diagnosis of arrhythmogenic right ventricular cardiomyopathy before (1/1994‐10/17/2018) versus after (10/18/2018‐2/2020) the heart allocation policy change.
Giuliano, K. , Scheel, P. III , Etchill, E. , Fraser, C. D. III , Suarez‐Pierre, A. , Hsu, S. , Wittstein, I. S. , Kasper, E. K. , Florido, R. , Tandri, H. , Calkins, H. , Choi, C. W. , Sharma, K. , Kilic, A. , and Gilotra, N. A. (2022) Heart transplantation outcomes in arrhythmogenic right ventricular cardiomyopathy: a contemporary national analysis. ESC Heart Failure, 9: 988–997. 10.1002/ehf2.13687.
References
- 1. Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. The Lancet. 2009; 373: 1289‐1300. [DOI] [PubMed] [Google Scholar]
- 2. Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, Fontaine G, Gear K, Hauer R, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri H, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010; 121: 1533‐1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Calkins H, Tandri H. Left Ventricular Involvement in ARVD/C. Circ Arrhythm Electrophysiol. 2015; 8: 1311‐1312. [DOI] [PubMed] [Google Scholar]
- 4. Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy: Dysplasia, dystrophy, or myocarditis?. Circulation. 1996; 94: 983‐991. [DOI] [PubMed] [Google Scholar]
- 5. Thiene G, Basso C. Arrhythmogenic right ventricular cardiomyopathy: An update. Cardiovasc Pathol. 2001; 10: 109‐117. [DOI] [PubMed] [Google Scholar]
- 6. Corrado D, Basso C, Thiene G, McKenna W.J., Davies M.J., Fontaliran F., Nava A., Silvestri F., Blomstrom‐Lundqvist C., Wlodarska E.K., Fontaine G., Camerini F.. Spectrum of clinicopathologic manifestations of arrhythmogenic right ventricular cardiomyopathy/dysplasia: A multicenter study. J Am Coll Cardiol. 1997; 30: 1512‐1520. [DOI] [PubMed] [Google Scholar]
- 7. Rastegar N, Zimmerman SL, Te Riele ASJM, te Riele ASJM, James C, Burt JR, Bhonsale A, Murray B, Tichnell C, Judge D, Calkins H, Tandri H, Bluemke DA, Kamel IR. Spectrum of biventricular involvement on CMR among carriers of ARVD/C‐associated mutations. JACC Cardiovasc Imaging. 2015; 8: 863‐864. [DOI] [PubMed] [Google Scholar]
- 8. Bennett RG, Haqqani HM, Berruezo A, Della Bella P, Marchlinski FE, Hsu CJ, Kumar S. Arrhythmogenic cardiomyopathy in 2018–2019: ARVC/ALVC or Both?. Heart Lung Circ. 2019; 28: 164‐177. [DOI] [PubMed] [Google Scholar]
- 9. McKenna WJ, Thiene G, Nava A, Fontaliran F, Blomstrom‐Lundqvist C, Fontaine G, Camerini F. Diagnosis of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Task force of the working group myocardial and pericardial disease of the european Society of Cardiology and of the scientific council on cardiomyopathies of the international society and Federation of Cardiology. Br Heart J. 1994; 71: 215‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Antonini‐Canterin F, Di Nora C. Arrhythmogenic right ventricular cardiomyopathy or athlete's heart? Challenges in assessment of right heart morphology and function. Monaldi Arch Chest Dis. 2019; 89. [DOI] [PubMed] [Google Scholar]
- 11. Pinamonti B, De Luca A. Challenge of early identification of arrhythmogenic (Right Ventricular) cardiomyopathy. Circ Cardiovasc Imaging. 2019; 12: e009084. [DOI] [PubMed] [Google Scholar]
- 12. Dalal D, Nasir K, Bomma C, Prakasa K, Tandri H, Piccini J, Roguin A, Tichnell C, James C, Russell SD, Judge DP, Abraham T, Spevak PJ, Bluemke DA, Calkins H. Arrhythmogenic right ventricular dysplasia: A United States experience. Circulation. 2005; 112: 3823‐3832. [DOI] [PubMed] [Google Scholar]
- 13. Mast TP, James CA, Calkins H, Teske AJ, Tichnell C, Murray B, Loh P, Russell SD, Velthuis BK, Judge DP, Dooijes D, Tedford RJ, van der Heijden JF, Tandri H, Hauer RN, Abraham TP, Doevendans PA, te Riele ASJM, Cramer MJ. Evaluation of structural progression in arrhythmogenic right ventricular Dysplasia/Cardiomyopathy. JAMA Cardiol. 2017; 2: 293‐302. [DOI] [PubMed] [Google Scholar]
- 14. James CA, Calkins H. Arrhythmogenic right ventricular cardiomyopathy: Evidence for progression increases. Eur Heart J. 2020; 41: 1411‐1413. [DOI] [PubMed] [Google Scholar]
- 15. Peters S, Trümmel M, Meyners W. Prevalence of right ventricular dysplasia‐cardiomyopathy in a non‐referral hospital. Int J Cardiol. 2004; 97: 499‐501. [DOI] [PubMed] [Google Scholar]
- 16. Peters S, Peters H, Thierfelder L. Heart failure in arrhythmogenic right ventricular dysplasia‐cardiomyopathy. Int J Cardiol. 1999; 71: 251‐256. [DOI] [PubMed] [Google Scholar]
- 17. Bhonsale A, Groeneweg JA, James CA, Dooijes D, Tichnell C, Jongbloed JDH, Murray B, te Riele ASJM, van den Berg MP, Bikker H, Atsma DE, de Groot NM, Houweling AC, van der Heijden JF, Russell SD, Doevendans PA, van Veen TA, Tandri H, Wilde AA, Judge DP, van Tintelen JP, Calkins H, Hauer RN. Impact of genotype on clinical course in arrhythmogenic right ventricular dysplasia/cardiomyopathy‐associated mutation carriers. Eur Heart J. 2015; 36: 847‐855. [DOI] [PubMed] [Google Scholar]
- 18. Hulot J‐S.. Natural history and risk stratification of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circulation. 2004; 110: 1879‐1884. [DOI] [PubMed] [Google Scholar]
- 19. Gilotra NA, Bhonsale A, James CA, te Riele ASJ, Murray B, Tichnell C, Sawant A, Ong CS, Judge DP, Russell SD, Calkins H, Tedford RJ. Heart failure is common and under‐recognized in patients with arrhythmogenic right ventricular Cardiomyopathy/Dysplasia. Circ Heart Fail. 2017; 10. [DOI] [PubMed] [Google Scholar]
- 20. Tedford RJ, James C, Judge DP, Tichnell C, Murray B, Bhonsale A, Philips B, Abraham T, Dalal D, Halushka MK, Tandri H, Calkins H, Russell SD. Cardiac transplantation in arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2012; 59: 289‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fiorelli AI, Coelho GHB, Oliveira JL, Nascimento CN, Boas LV, Napolitano CF, Bacal F, Bochi EA, Stolf NA. Heart transplantation in arrhythmogenic right ventricular dysplasia: case reports. Transplant Proc. 2009; 41: 962‐964. [DOI] [PubMed] [Google Scholar]
- 22. Yoda M, Minami K, Fritzsche D, Tendrich G, Schulte‐Eistrup S, Koerfer R. Three cases of orthotopic heart transplantation for arrhythmogenic right ventricular cardiomyopathy. Ann Thorac Surg. 2005; 80: 2358‐2360. [DOI] [PubMed] [Google Scholar]
- 23. Gilljam T, Haugaa KH, Jensen HK, Svensson A, Bundgaard H, Hansen J, Dellgren G, Gustafsson F, Eiskjær H, Andreassen AK, Sjögren J, Edvardsen T, Holst AG, Svendsen JH, Platonov PG. Heart transplantation in arrhythmogenic right ventricular cardiomyopathy ‐ experience from the nordic ARVC registry. Int J Cardiol. 2018; 250: 201‐206. [DOI] [PubMed] [Google Scholar]
- 24. DePasquale EC, Cheng RK, Deng MC, Nsair A, McKenna WJ, Fonarow GC, Jacoby DL. Survival after heart transplantation in patients with arrhythmogenic right ventricular cardiomyopathy. J Cardiac Failure 2017; 23: 107–112. [DOI] [PubMed] [Google Scholar]
- 25.Adult heart allocation ‐ OPTN. Accessed January 14, 2021. https://optn.transplant.hrsa.gov/learn/professional‐education/adult‐heart‐allocation/
- 26. Medved D, Ohlsson M, Höglund P, Andersson B, Nugues P, Nilsson J. Improving prediction of heart transplantation outcome using deep learning techniques. Sci Rep. 2018; 8: 3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maron BJ. Right ventricular cardiomyopathy: Another cause of sudden death in the young. N Engl J Med. 1988; 318: 178‐180. [DOI] [PubMed] [Google Scholar]
- 28. Furlanello F, Bettini R, Bertoldi A, Vergara G, Visona L, Durante GB, Inama G, Frisanco L, Antolini R, Zanuttini D. Arrhythmia patterns in athletes with arrhythmogenic right ventricular dysplasia. Eur Heart J 1989; 10: 16‐19. [DOI] [PubMed] [Google Scholar]
- 29. Mwansa H, Bilchick KC, Parker AM, Harding W, Ruth B, Kennedy JLW, Mysore M, Kwon Y, Mihalek A, Mazimba S. Decreased pulmonary arterial proportional pulse pressure is associated with increased mortality in group 1 pulmonary hypertension. Clin Cardiol. 2017; 40: 988‐992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Scheel PJ III, Florido R, Hsu S, Murray B, Tichnell C, James CA, Agafonova J, Tandri H, Judge DP, Russell SD, Tedford RJ. Safety and utility of cardiopulmonary exercise testing in arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Am Heart Assoc. 2020; 9: e013695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Caldeira CCB, Machado RC, Caldeira DCB. Implantation of short‐term and long‐term right ventricular assist devices. Braz J Cardiovasc Surg. 2017; 32: 435‐437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Morshuis M, Rojas SV, Hakim‐Meibodi K, Razumov A, Gummert JF, Schramm R. Heart transplantation after SynCardia® total artificial heart implantation. Ann Cardiothorac Surg. 2020; 9: 98‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lund LH, Khush KK, Cherikh WS, Goldfarb S, Kucheryavaya AY, Levvey BJ, Meiser B, Rossano JW, Chambers DC, Yusen RD, Stehlik J, International Society for Heart and Lung Transplantation . The registry of the International Society for Heart and Lung Transplantation: Thirty‐fourth adult heart transplantation Report‐2017; focus theme: Allograft ischemic time. J Heart Lung Transplant Off Publ Int Soc Heart Transplant. 2017; 36: 1037‐1046. [DOI] [PubMed] [Google Scholar]
- 34. Drakos SG, Kfoury AG, Gilbert EM, Long JW, Stringham JC, Hammond EH, Jones KW, Bull DA, Hagan MBE, Folsom JW, Horne BD, Renlund DG. Multivariate predictors of heart transplantation outcomes in the era of chronic mechanical circulatory support. Ann Thorac Surg. 2007; 83: 62‐67. [DOI] [PubMed] [Google Scholar]
- 35. Fluschnik N, Geelhoed B, Becher PM, Schrage B, Brunner FJ, Knappe D, Bernhardt AM, Blankenberg S, Kobashigawa J, Reichenspurner H, Schnabel RB, Magnussen C. Non‐immune risk predictors of cardiac allograft vasculopathy: Results from the U.S. Organ procurement and transplantation network. Int J Cardiol. 2021; 331: 57‐62. [DOI] [PubMed] [Google Scholar]
- 36. Varshney AS, Berg DD, Katz JN, Baird‐Zars VM, Bohula EA, Carnicelli AP, Chaudhry SP, Guo J, Lawler PR, Nativi‐Nicolau J, Sinha SS, Teuteberg JJ, van Diepen S, Morrow DA, for the Critical Care Cardiology Trials Network Investigators . Use of temporary mechanical circulatory support for Management of Cardiogenic Shock before and after the united network for organ sharing donor heart allocation system changes. JAMA Cardiol. 2020; 5: 703‐708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hanff TC, Harhay MO, Kimmel SE, Molina M, Mazurek JA, Goldberg LR, Birati EY. Trends in mechanical support use as a bridge to adult heart transplant under new allocation rules. JAMA Cardiol. 2020; 5: 728‐729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Parker WF, Chung K, Anderson AS, Siegler M, Huang ES, Churpek MM. Practice changes at U.S. Transplant centers after the new adult heart allocation policy. J Am Coll Cardiol. 2020; 75: 2906‐2916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cogswell R, John R, Estep JD, Duval S, Tedford RJ, Pagani FD, Martin CM, Mehra MR. An early investigation of outcomes with the new 2018 donor heart allocation system in the United States. J Heart Lung Transplant. 2020; 39: 1‐4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Characteristics of 252 patients listed for heart transplantation with the primary diagnosis of arrhythmogenic right ventricular cardiomyopathy (1/1994‐2/2020).
Table S2. Outcomes of the 252 patients with ARVC waitlisted for heart transplantation, 1/1994‐2/2020.
Table S3. Characteristics of patients listed for heart transplantation with the primary diagnosis of arrhythmogenic right ventricular cardiomyopathy before (1/1994‐10/17/2018) versus after (10/18/2018‐2/2020) the heart allocation policy change.