

Abstract
Objective
Distinct dominant mutations in the calcium‐permeable ion channel TRPV4 (transient receptor potential vanilloid 4) typically cause nonoverlapping diseases of either the neuromuscular or skeletal systems. However, accumulating evidence suggests that some patients develop mixed phenotypes that include elements of both neuromuscular and skeletal disease. We sought to define the genetic and clinical features of these patients.
Methods
We report a 2‐year‐old with a novel R616G mutation in TRPV4 with a severe neuropathy phenotype and bilateral vocal cord paralysis. Interestingly, a different substitution at the same residue, R616Q, has been reported in families with isolated skeletal dysplasia. To gain insight into clinical features and potential genetic determinants of mixed phenotypes, we perform in‐depth analysis of previously reported patients along with functional and structural assessment of selected mutations.
Results
We describe a wide range of neuromuscular and skeletal manifestations and highlight specific mutations that are more frequently associated with overlap syndromes. We find that mutations causing severe, mixed phenotypes have an earlier age of onset and result in more marked elevations of intracellular calcium, increased cytotoxicity, and reduced sensitivity to TRPV4 antagonism. Structural analysis of the two mutations with the most dramatic gain of ion channel function suggests that these mutants likely cause constitutive channel opening through disruption of the TRPV4 S5 transmembrane domain.
Interpretation
These findings demonstrate that the degree of baseline calcium elevation correlates with development of mixed phenotypes and sensitivity to pharmacologic channel inhibition, observations that will be critical for the design of future clinical trials for TRPV4 channelopathies.
1. Introduction
Mutations in the transient receptor vanilloid 4 gene (TRPV4), encoding a polymodal, mechanically sensitive Ca2+ channel, are associated with several disease phenotypes that can be classified into two groups: neuropathies and skeletal disorders. 1 The former includes a spectrum of hereditary neuropathies with either sensorimotor or exclusively motor involvement and that are frequently associated with vocal cord paresis, diaphragmatic weakness, scapular winging, and sensorineural hearing loss. 2 , 3 , 4 , 5 Individual disease phenotypes include Charcot–Marie–Tooth disease type 2C (CMT2C), 2 scapuloperoneal spinal muscular atrophy (SPSMA), 5 and congenital distal spinal muscular atrophy (CDSMA). 6 TRPV4‐mediated skeletal diseases include a heterogeneous group of skeletal dysplasias (spondylometaphyseal dysplasia Kozlowski type (SMD‐K), metatropic dysplasia (MD), and spondyloepimetaphyseal dysplasia Missouri type (SEMD‐M)) with overlapping features including short stature, short trunk with associated platyspondyly, scoliosis, and brachydactyly. 3 , 7 , 8 An additional musculoskeletal disease is familial digital arthropathy brachydactyly (FDAB), a progressive, painful, and deforming osteoarthropathy of the hand and feet9. Whereas there are clear genotype–phenotype correlations in TRPV4‐mediated skeletal dysplasias, TRPV4‐related neuropathies demonstrate marked phenotypic heterogeneity in age of onset, disease severity, and phenotypic manifestations, even among patients with the same mutation. 10 , 11 Given clear demonstration of mutation‐dependent gain of ion channel function and reversibility of toxicity with TRPV4 antagonism in cell and fly models, 12 , 13 , 14 , 15 TRPV4 channelopathies may be treatable.
Whereas disease‐causing mutations affect amino acid residues localized throughout multiple domains of the TRPV4 ion channel, some structure‐phenotype correlations have emerged. 3 TRPV4 ion channels are functional tetramers of four TRPV4 subunits, with each subunit consisting of large N‐ and C‐terminal cytoplasmic domains connected by six transmembrane segments. 16 Notably, most but not all neuropathy‐causing mutations in TRPV4 affect highly conserved arginine residues within the exposed surface of the cytosolic N‐terminal ankyrin repeat domain (ARD), which is predicted to function as a protein interaction domain, 16 , 17 , 18 whereas skeletal dysplasia mutations are more widespread and tend to affect residues involved in inter‐subunit interactions or critical ion channel gating regions. 3
Although a strict separation between neuropathy and skeletal dysplasias was initially applied, cases of “combined” or “mixed” disease phenotypes have been increasingly recognized, blurring the distinction between these clinical entities. In fact, various skeletal malformations are commonly encountered among patients with TRPV4‐associated polyneuropathy, although often not fulfilling clinical criteria for a diagnosis of skeletal dysplasia. 19 The opposite occurrence, peripheral neuropathy co‐occurring in a patient with primary skeletal dysplasia, is less frequently reported. 9 Despite increasing reports of patients with mixed phenotypes, the genetic and functional determinants of these conditions have been largely unexplored.
Here, we describe a 2‐year‐old patient with a novel c.1846C > G missense mutation (p.R616G) in TRPV4 resulting in a congenital CMT2C neuropathy phenotype with bilateral vocal cord paralysis, and no definitive evidence of skeletal involvement. Interestingly, a different amino acid substitution at the R616 position, R616Q, has been reported to cause pure skeletal dysplasia phenotypes. 20 In the setting of this unusual finding of two different substitutions at the same amino acid residue with the potential to cause either neuropathy or skeletal involvement, we review the literature of patients with TRPV4 mutations displaying mixed neuropathy and skeletal phenotypes. We further perform functional analysis of selected TRPV4 mutations, demonstrating that mutations with severe and/or mixed phenotypes, including the R616G mutation, show markedly elevated baseline calcium levels and cytotoxicity when expressed in heterologous systems. Finally, we show that specific TRPV4 mutations display substantial heterogeneity in dose–response characteristics of ion channel inhibition with a small molecule antagonist. These findings suggest that baseline calcium elevation may predict the likelihood of severe and/or mixed phenotypes as well as sensitivity to pharmacologic channel inhibition. As the potential of therapeutic channel inhibition with available TRPV4 antagonists has been made clearer, 15 these results will have important implications for patient selection and outcome measure determination in future clinical trials.
2. Materials and Methods
2.1. Clinical data
Clinical information on the proband's case was collected after informed consent by both parents. Genetic testing was performed by whole exosome sequencing (GeneDx XomeDxPlus) on the proband and by targeted sequencing on the parents.
2.2. Literature review of combined phenotypes
We reviewed published literature from inception to February 2021. A PubMed review of English language literature using the search terms “TRPV4,” “neuropathy,” “skeletal dysplasia,” “Charcot–Marie‐Tooth,” “scapuloperoneal spinal muscular atrophy,” and “congenital distal spinal muscular atrophy” was performed. We also reviewed relevant reference lists from published cases series and literature reviews. Cases of combined phenotypes were selected using three processes. We included (1) cases with neuropathy and a formal diagnosis of skeletal dysplasia, including SMD‐K, MD, and SEMD‐M; (2) cases with neuropathy and a constellation of skeletal abnormalities to which authors had assigned a potential skeletal dysplasia diagnosis; and (3) cases with neuropathy and a constellation of skeletal abnormalities that correlated with potential skeletal dysplasia, but without a named diagnosis in the paper. When selecting combined phenotypes, we did not consider abnormal skeletal characteristics commonly associated with hereditary neuropathy phenotypes, such as isolated scoliosis, kyphoscoliosis, pes cavus, and hip dysplasia.
2.3. Antibodies and reagents
Primary antibodies used were rabbit anti‐GFP (Cell Signaling Technology, 2555), mouse anti‐GFP (Thermo Fisher Scientific, A‐11120), rabbit anti‐TRPV4 (Cosmo Bio USA, KAL‐KM119). Secondary antibody used was HRP‐conjugated monoclonal mouse anti‐rabbit IgG, light chain specific (Jackson ImmunoResearch, 211–032‐171). Reagents used include HC067047 (Sigma‐Aldrich, SML0143), GSK2193874 (Sigma‐Aldrich, SML0942), and AlexFluor 555 Phalloidin (ThermoFisher Scientific).
2.4. Mammalian expression plasmids
C‐terminally tagged TRPV4‐GFP has been previously described. 18 GFP‐tagged TRPV4‐R269C, TRPV4‐D333G, TRPV4‐S542Y, TRPV4‐R616Q, TRPV4‐R616G, TRPV4‐L619P, and TRPV4‐W785C were generated by site‐directed mutagenesis using the Quikchange XL‐II kit (Agilent, 200,521) according to manufacturer protocols.
2.5. Cell culture and western blotting
MN‐1, HEK293T, and MDCK cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% (vol/vol) fetal calf serum (FCS) and penicillin/streptomycin at 37°C with 6% CO2. For western blots, MN‐1 and HEK293T cells were transfected with Lipofectamine LTX with Plus Reagent (Thermo Fisher Scientific), lysed 24 h after transfection in RIPA buffer (150 mM NaCl, 1.0% IGEPAL CA‐630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0, Millipore Sigma) supplemented with EDTA‐free Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific), and processed as previously described. 18
2.6. Immunofluorescence
MN‐1 and MDCK cells were grown on glass coverslips and transfected with either Lipofectamine LTX (ThermoFisher Scientific) for MN‐1 cells or GenJet version II (SignaGen Laboratories) for MDCK cells. Cells were then fixed and permeabilized as previously described. 18 Cells were incubated with phalloidin‐555 (1:200) for 30 minutes at room temperature in blocking buffer. Coverslips were imaged with a Zeiss 800 LSM confocal laser scanning microscope with a 63x oil objective.
2.7. Calcium imaging
MN‐1 cells were transfected with GFP‐tagged TRPV4 (WT or mutant) and calcium imaging was performed as previously described. 18 For hypotonic saline treatment, one volume of NaCl‐free calcium‐imaging buffer was added to one volume of standard calcium‐imaging buffer for a final NaCl concentration of 70 mM. Cells were imaged every 10 seconds for 20 seconds prior to stimulation with hypotonic saline and then imaged every 10 seconds for an additional 2 minutes. Analysis of calcium levels over time were performed as previously described. 18 For GSK219 inhibitory dose–response measurements, GSK219 was added to the media at the time of transfection and to calcium imaging media at the indicated doses. For generation of dose–response curves and IC50 determination, we determined the Fura ratio elevation above background for each experimental replicate by subtracting the Fura ratio from empty vector transfected cells from the Fura ratio in each untreated experimental condition, either in the presence of varying concentrations of GSK219 (FuraGSK) or in untreated cells (FuraUntreated). Average FuraGSK values for each each mutant were then divided by the average FuraUntreated values for the same mutant and converted to a percentage using the following equation: . Dose–response curves were then generated using nonlinear regression analysis (Prism, GraphPad v9.2).
2.8. Cytotoxicity assays
HEK293T cells were transfected with TRPV4 plasmids as above for 24 hours. Cell death was assessed using an LDH release assay (Cytotoxicity Detection KitPLUS, Millipore Sigma) according to manufacturer protocols and as previously described. 14 Cell death was calculated as a percentage by subtracting the absorbance of a background control (normal medium) from the absorbance of the experimental samples (supernatant from transfected cells) and dividing by the absorbance of a high control (supernatant from cells treated with lysis buffer).
3. RESULTS
3.1. Novel R616G mutation causes severe neuropathy with vocal cord paralysis
A 2‐year‐7‐month‐old girl (31 months) was evaluated for a history of motor developmental delay and bilateral vocal cord palsy since birth (Fig. 1A). Throughout early infancy, she was noted to be hypotonic with diffuse joint laxity. She started sitting up independently at about 9 months of age, but did so in an odd manner by swinging her extended and abducted lower extremities. She started to crawl in a four‐point manner at around 12 months. Additionally, she was described as a “noisy” breather since birth, particularly during sleep.
Figure 1.
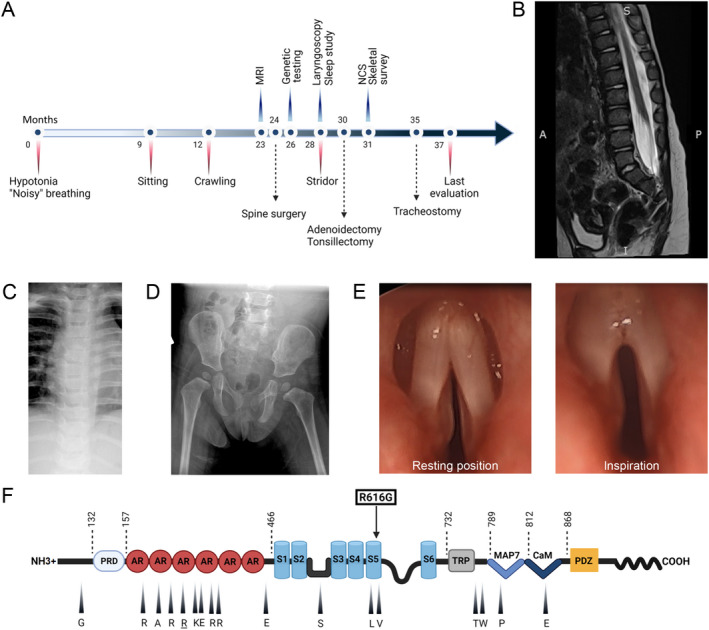
(A) Time line of clinical course showing most significant clinical events, diagnostic studies, and therapeutic interventions. (B) Lumbosacral spine MRI, T2 weighted sequence, shows evidence of tethered cord with thickening of the filum terminale, but without evidence of intrathecal mass. (C) Plain film of the chest shows no evidence of skeletal dysplasia. (D) Plain film of the pelvis and hips shows no abnormalities. (E) Direct laryngoscopy demonstrates vocal cords fixed in a paramedian position and no abduction during inspiration. (F) Representation of TRPV4 protein domains and mutations with corresponding clinical phenotypes. The ligand binding site and the pore region are located between S2‐S3 and S5‐S6, respectively. Amino acids that are mutated in mixed phenotype TRPV4 channelopathies are indicated with an arrowhead and corresponding symbol (WT residue shown). Arginine 269 (underlined) is the most commonly reported mutation site in the literature. The novel R616G missense mutation (squared) affects a residue in transmembrane S5 helix that has been previously associated with skeletal dysplasia. Abbreviations: NCS, nerve conduction studies; PRD, proline‐rich domain; AR, ankyrin repeat; S1 to 6, transmembrane domains; TRP, transient receptor potential; MAP7, microtubule associated protein 7 binding site; CaM, calmodulin binding site. [Colour figure can be viewed at wileyonlinelibrary.com]
On general exam, she had prominent inspiratory stridor present at rest (Video 1 , Segment A, B), straightening of normal spine curvature (Fig. 1B), mild equinovarus foot deformities, and clinodactyly of the fifth digits. There was also head tilt suggestive of torticollis. Neurological examination was notable for asymmetric proximal greater than distal upper extremity weakness, hypotonia, absent ankle reflexes, and sensorineural hearing loss. Examination of cranial nerves was otherwise normal. Sensation to light touch and vibration was preserved in the upper and lower extremities. There was mild developmental expressive language delay, but preserved comprehension.
MRI of the brain and whole spine demonstrated mild periventricular white matter volume loss and tethered lumbosacral spinal cord with thickening of the filum terminale (Fig. 1B). Note was also made of disc space narrowing at C4‐C5 and C5‐C6 with reduced anterior posterior diameter of the vertebral bodies, suggestive of Klippel‐Feil deformity. Plain films of the chest, pelvis, and hips as well as MRI of the spine did not demonstrate any signs of skeletal dysplasia (Fig. 1B‐D). Nerve conduction studies showed a length‐dependent sensory‐motor polyneuropathy (Table 1).
Table 1.
Nerve conduction studies in a 2‐year‐old patient with a novel TRPV4 R616G mutation.
| Peroneal | Tibial | Sural | Median | Ulnar | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
CMAP (>2 mV) |
MNCV (>39 m/s) |
CMAP (>2 mV) |
MNCV (>39 m/s) |
SAP (>5 μV) |
CMAP (>4 mV) |
MNCV (>49 m/s) |
SAP (>9 μV) |
CMAP (>4 mV) |
MNCV (>49 m/s) |
SAP (>9 μV) |
| NR | NR | NR | NR | NR | 3.6 | 41 | 9.4 | 2.8 | 47 | 7.4 |
Normal values are shown in parentheses. Peroneal, tibial, and sural nerves were bilaterally non‐recordable. For median and ulnar nerves, the recordings from the right side are shown. Abbreviations: CMAP, compound muscle action potential (distal stimulation site); MNCV, motor nerve conduction velocity; SAP, sensory action potential; NR, not recordable.
The stridor and respiratory difficulties were investigated with polysomnography, which demonstrated both obstructive and central sleep apnea, and direct laryngoscopy/bronchoscopy, which showed bilateral vocal cord paralysis and both tracheo‐ and bronchomalacia (Fig. 1E, Video 1 , Segment C, D). Whole exome sequencing identified a heterozygous missense mutation, c.1846C > G, in exon 12 of TRPV4, corresponding to R616G (CGA > GGA) substitution (Fig. 1F). Notably, the R616G mutation was only detected in 25% of the sequencing reads, suggesting the possibility of genetic mosaicism. The proband's mother and father were negative for this variant, consistent with a de novo mutation. There was no family history of neurological disorders and her 2‐month‐old brother was healthy without signs of neuromuscular disease. The R616G mutation has not been previously reported and is not present in healthy subjects in the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/) or the GnomAD database (https://gnomad.broadinstitute.org/). However, a different substitution of the same residue, R616Q, has been reported in families with isolated skeletal dysplasia and has been shown to cause gain of ion channel function. 20
The patient underwent several surgical procedures (Fig. 1A), including release of the tethered spinal cord, adenoidectomy, and tonsillectomy, although without clear benefit with respect to motor difficulties or respiratory status. Of note, some of these procedures were complicated by worsening work of breathing requiring prolonged intubation. Despite these interventions, her stridor and respiratory issues progressed, resulting in several acute apneic and hypoxic episodes and eventually prompting semi‐urgent tracheostomy tube placement. Repeat polysomnography postoperatively demonstrated improved obstructive sleep apnea, but persistent central sleep apnea.
3.2. Literature review of combined phenotypes
Given that the patient's R616G mutation was associated with a severe neuropathy phenotype, and a pure skeletal dysplasia phenotype was previously reported for the R616Q mutation in the same amino acid site, we reviewed the literature regarding TRPV4 mutations associated with mixed phenotypes. We did not uncover additional prior reports of different substitutions of the same amino residue separately causing isolated neuropathy and isolated skeletal dysplasia, as was the case in our patient. However, we identified 36 individual cases of mixed neuropathy and skeletal phenotypes arising from 19 different mutations. 10 , 11 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 , 32 , 33 , 34 This included nine affected families as well as several individuals with de novo mutations. We analyzed these reports with respect to genotype, age of onset, neurological and skeletal manifestations, electrophysiology, and other phenotypic manifestations (Table 2 ).
Table 2.
Systematic review of published literature of TRPV4 mutations associated with mixed neuropathy and skeletal dysplasia manifestations.
| Amino acid | Protein domain | Mutation | Patients (n) | Families (n) | Inheritance | Sex (male, n) | Mean age at onset in years (range) | Origin | Neuropathy phenotype | Progressive (yes) | Proximal UL motor | Distal UL motor | Proximal LL motor | Distal LL motor | Sensory symptoms | Vocal cord involvement | Diaphragmatic involvement | Scapular winging | Decreased CMAP UL | Decreased CMAP LL | Decreased SAP UL | Decreased SAP LL | Denervation | Skeletal dysplasia diagnosis | Additional features | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| G78W | N‐terminal | NR | 1 | 1 | De novo | 1/1 | 0 (congenital) | NR | CDSMA | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | MD | Contractures, fetal akinesia syndrome | 21 |
| R186Q | ARD1 | 557G > A | 1 | 1 | AD | NR | <5 | France | CSMAA | 1/1 | 1/1 | 1/1 | 1/1 | 1/1 | 0/1 | 0/1 | 0/1 | 0/1 | 1/1 | 1/1 | 0/1 | 0/1 | 1/1 | MD/brachyolmia | ‐ | 10 |
| A217S | ARD2 | 649G > T | 1 | 1 | De novo | 0/1 | 0 (congenital) | Argentina | SHSMA | NR | 1/1 | 1/1 | 1/1 | 1/1 | 0/1 | 0/1 | 0/1 | 1/1 | 0/1 | 0/1 | 0/1 | 0/1 | 1/1 | SMD‐K | Facial palsy, hyperlaxity | 22 |
| R232C | ARD2 | 694C > T | 3 | 2 | AD, de novo | 1/1 | 3 (0–5) | Greece, Italy | CDSMA (1/3), CMT2C (2/3) | 2/2 | ||||||||||||||||
| (1 NR) | 1/3 | 3/3 | 3/3 | 3/3 | 2/3 | 3/3 | 1/3 | 0/3 | 1/2 | 1/2 | 1/2 | 1/2 | 2/2 | MD/ brachyolmia | ‐ | 10,23 | ||||||||||
| R269H | ARD3 | 806G > A | 4 | 4 | AD, de novo | 1/3 | 1.3 (0–5) | Netherlands, France, Italy, USA | CDSMA/ CSMAA (3/4), SPSMA (1/4) | 3/3 (1 NR) | 2/4 | 2/4 | 2/4 | 4/4 | 1/4 | 1/4 | 1/4 | 2/4 | 1/2 | 1/2 | 0/3 | 0/3 | 4/4 | FDAB, MD/brachyolmia | Spina bifida | 10,24,25 |
| R269C | ARD3 | 805C > T | 1 | 1 | De novo | 1/1 | 0 (congenital) | UK | SPSMA | NR | 1/1 | 0/1 | 1/1 | 1/1 | 0/1 | 0/1 | 0/1 | 1/1 | NR | NR | NR | NR | 1/1 | MD | Dysphagia | 27 |
| K276E | ARD3 | NR | 1 | 1 | De novo | 1/1 | 0 (congenital) | Algeria | CDSMA | NR | NR | NR | 1/1 | 1/1 | NR | NR | NR | NR | NR | NR | NR | NR | 1/1 | MD | Contractures, fetal akinesia syndrome | 21 |
| E278K | ARD3 | 832G > A | 1 | 1 | AD | 0/1 | 0.5 | Korea | CMT2C | 1/1 | 0/1 | 0/1 | 1/1 | 1/1 | 0/1 | 0/1 | 0/1 | 0/1 | NR | 1/1 | NR | 0/1 | 1/1 | SMD‐K | ‐ | 22 |
| R315W | ARD4 | 943C > T | 1 | 1 | De novo | 0/1 | 1.5 | USA | CMT2C | 1/1 | 0/1 | 1/1 | 1/1 | 1/1 | 1/1 | 0/1 | 0/1 | 0/1 | NR | 1/1 | NR | 1/1 | 1/1 | SMD‐K | ‐ | 28 |
| R316C | ARD4 | 946C > T | 2 | 1 | AD | 2/2 | 2 (0–4) | USA | SPSMA | 2/2 | 2/2 | 2/2 | 2/2 | 1/2 | 0/2 | 0/2 | 0/2 | 1/2 | NR | NR | NR | NR | 2/2 | NR | Oculomotor abduction, facial palsy | 29 |
| E435K | pre‐TM1 | 1303G > A | 1 | 1 | De novo | 0/1 | 0 (congenital) | USA | Arthrogryposis multiplex | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | MD | Tethered cord, thrombocytosis | 30 |
| S542Y | Ligand binding | 1625C > A | 6 | 1 | AD | 2/6 | 11.5 (1–25) | USA | CMT2C | NR | 1/6 | 0/6 | 1/6 | 4/6 | 2/6 | 4/6 | 1/6 | 0/6 | 1/6 | 3/6 | NR | NR | 3/6 | MD | Sleep apnea, pupillary abnormality | 31 |
| L619P | TM5 | 1856_1857delinsCT, 1856 T > C | 2 | 2 | De novo | 0/2 | 1.5 (0–3) | Netherlands, Brazil | CMT2C, CDSMA | 2/2 | 1/2 | 2/2 | 1/2 | 1/2 | 0/2 | 2/2 | 1/2 | 0/2 | 1/1 | 1/1 | NR | NR | NR | Brachyolmia | *Central sleep apnea, tethered cord | 32 |
| V620I | TM5 | 1858G > A | 1 | 1 | De novo | 0/1 | 6 | Croatia | CMT2C | 1/1 | 0/1 | 0/1 | 0/1 | 1/1 | 0/1 | 0/1 | 0/1 | 0/1 | 0/1 | 1/1 | 0/1 | 1/1 | 1/1 | Brachyolmia | ‐ | 11 |
| T740I | C‐terminal | NR | 2 | 1 | De novo | NR | 0 (congenital) | NR | CDSMA | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | NR | MD | Contractures, fetal akinesia syndrome | 21 |
| W785C | C‐terminal | 2355G > T | 5 | 1 | AD | 2/5 | 2.8 (2–4) | China | CDSMA | 5/5 | 0/5 | 0/5 | 5/5 | 5/5 | 0/5 | 0/5 | 0/5 | 0/5 | 0/2 | 0/2 | NR | NR | 2/2 | NR | Scaly skin | 33 |
| P799R | C‐terminal | 2396C > G | 1 | 1 | De novo | 0/1 | 13 | Japan | CMT2C | 1/1 | 0/1 | 0/1 | 1/1 | 0/1 | 0/1 | 0/1 | 0/1 | 0/1 | NR | NR | NR | NR | NR | SEMD‐M | ‐ | 22 |
| E840K, N833S | C‐terminal | 2518G > A 2498A > G | 2 | 1 | Compound heterozygous | 1/2 | 0.3 (0.3–0.3) | Canada | CMT2C | 2/2 | NR | NR | NR | NR | NR | NR | NR | NR | 2/2 | 2/2 | 2/2 | 2/2 | NR | NR | Sensorineural hearing loss, retinopathy, intellectual disability | 34 |
Footnotes: Genetic, epidemiological, clinical, and electrophysiological characteristics are reported for each individual mutation. Data are expressed as fractions of observed cases/reported cases.
Abbreviations: CDSMA, congenital distal spinal muscle atrophy; SPSMA, scapuloperoneal spinal muscle atrophy; SHSMA, scapulo‐humeral spinal muscular atrophy; CSMAA, congenital spinal muscular atrophy and arthrogryposis; MD, metatropic dysplasia; SMD‐K, spondylometaphyseal dysplasia, Kozlowski type; SEMD‐M, spondyloepimetaphyseal dysplasia, Missouri type; FDAB, familiar digital arthropathy brachydactyly; NR, not reported; AD, autosomal dominant; UL, upper limbs; LL, lower limbs; CMAP, compound muscle action potential; SAP, sensory action potential. *multiple additional features were reported, including neurogenic bladder, pigmentary retinopathy, tongue fasciculations, thoracic meningomyelocele with syrinx, sensorineural and mixed hearing loss, left facial palsy, arthrogryposis multiplex congenital.
All identified mutations were missense, heterozygous mutations. There was incomplete penetrance reported in one family with the R186Q mutation, 10 and two individuals (two siblings) had compound heterozygous mutations. 34 The existence of biallelic TRPV4 disease remains controversial, and in the two above mentioned cases the authors argued that one missense variant (c.2498A > G, p.Asn833Ser), which was not pathogenic based on in silico analysis, could act in synergy with the pathogenic one (c.2518G > A, p.Glu840Lys). 34 Mutations causing mixed phenotypes most frequently occurred in the ARD (42%), but also in the extreme N terminus, transmembrane domains, and C terminus (Fig. 1F). TRPV4 mixed phenotypes were reported worldwide. Age of onset varied from congenital to young adulthood, with an average age of 2.7 ± 3.9 years. By comparison, the average age of onset for patients with isolated neuropathy in the case reports reviewed was 10.9 ± 16.0 years. For cases in which sex of the individuals was reported (n = 30), 12 (40%) were male. All three neuropathy phenotypes associated with TRPV4 mutations were represented: CDSMA in 14 of 36 (39%), CMT2C/HMSN2 in nine (25%), SPSMA in four (11%), and no specific neuromuscular phenotype in the remaining nine (25%).
Of the 29 individuals in which strength assessment was reported, weakness was noted in the distal lower extremities in 25 (86%), proximal lower extremities in 21 (72%), distal upper extremities in 12 (41%), and proximal upper extremities in 10 (34%). Sensory symptoms occurred in less than 20% of individuals. Although disease progression was not uniformly assessed, there was evidence of disease progression in 21 out of 23 patients (91%) for which longitudinal information was reported. Progression was evidenced by development of muscle atrophy, 25 worsening weakness, 25 gait impairment, 26 sensory deficits, 28 and/or was demonstrated with electrodiagnostic studies. 10 , 11 When reported, electrophysiological studies showed motor greater than sensory axonal neuropathy and evidence of denervation potentials on needle EMG.
Of the 36 individuals with a combined phenotype included in Table 2, 10 (28%) carried a formal diagnosis of skeletal dysplasia, including SMD‐K 22 , 28 (n = 3), MD 21 , 27 , 30 (n = 6), and SEMD‐M 22 (n = 1). These diagnoses occurred in individuals with all three TRPV4 neuropathy phenotypes. Individuals with MD were also more likely to be diagnosed with CDSMA than other neuropathy phenotypes. The diagnoses of SMD‐K and SEMD‐M occurred in individuals with CMT2C, or in phenotypes not included in the three neuropathy phenotypes associated with TRPV4 mutations. In 17 case reports, authors described skeletal abnormalities and suggested diagnoses of FDAB, 26 brachyolmia, 11 or mild forms of MD. 10 , 31 The remaining case reports described abnormalities of bone in the absence of a unifying skeletal dysplasia diagnosis. 29 , 33 , 34 These abnormalities were numerous and affected the head, neck, trunk, pelvis, and extremities (Table 3). Additional notable features were spinal dysraphisms (including tethered spinal cord, which was also noted in our case) (n = 3), facial weakness (n = 2), retinopathy 32 , 34 (n = 3), and central sleep apnea (one prior patient and the currently reported patient). 32 Of note, two patients with the L619P mutation developed a particularly severe and unusual mixed phenotype with severe skeletal abnormalities as well as giant cell lesions of the jaw and skull, cystic lesions of the long bones and vertebrae, cervical and thoracic vertebrae fusion, and facial and skull dysmorphisms. 32
Table 3.
Skeletal abnormalities described in patients with mixed phenotypes.
| Body region | Abnormalities described |
|---|---|
| Skull/face | Dolichocephaly, giant cell lesions of the jaw and skull, facial and skull dysmorphism |
| Trunk | Short stature (>2 SD below mean), shortened trunk, decreased thoracic circumference, short barrel‐shaped trunk |
| Vertebral bodies and spine | Reduced height of vertebral bodies, abnormal vertebral ossification, platyspondyly, anterior vertebral body abnormalities, cystic lesions of the vertebrae, spinal segmentation anomalies, narrowing of intravertebral discs, abnormal odontoid development, cervical and thoracic vertebrae fusion, thoracolumbar kyphoscoliosis, lumbosacral lordosis, sacrococcygeal tail |
| Pelvis | Flaring of iliac wings with flat acetabular roofs |
| Long bones/limbs | Short‐limbed at birth, short long bones, upper and lower limb asymmetry, metaphyseal expansion/widening/flaring, secondary demineralization, cystic lesions, osteonecrosis of femoral head, dysmorphic femoral head, short/wide/irregular femur neck, asymmetrical femoral neck‐shaft angles, dysplastic/flattened/irregular knee epiphyses |
| Joints | Cubitus valgus, genu valgum, dislocation of the femoral head, enlarged joints |
| Hands/feet | Clinodactyly, brachydactyly, camptodactyly, short phalanges, short metacarpal bones, metatarsal dysplasia, secondary demineralization, abnormal carpal ossification, hypoplastic carpal bones, unilateral reduced metacarpal span, medial deviation of digits, hypoplastic/dysplastic finger and toe changes |
In our review, we did not observe definitive genotype–phenotype correlation between specific TRPV4 mutations and combined neuropathy and skeletal dysplasia phenotypes, and we could not identify a pattern of neuropathic characteristics that predicted skeletal involvement. Skeletal disease associated with TRPV4‐mediated neuropathy occurred across a range of clinical severity and mutations, consistent with prior studies highlighting the heterogeneity of TRPV4 channelopathies. 3 However, there were some TRPV4 variants that appeared to be more likely to cause a mixed phenotype (S542Y and W785C) as well as some that were particularly severe (L619P and the novel R616G variant). We thus chose to study these variants in more detail using heterologous in vitro expression systems.
3.3. Functional analysis of expression levels and ion channel activity of selected TRPV4 mutants
To characterize the functional effects of TRPV4 mutations of interest, we expressed GFP‐tagged WT and mutant TRPV4 in cultured cells. We chose to examine the novel R616G mutant, the previously described R616Q mutant, a common neuropathy‐causing mutation within the ARD (R269C), three mutations causing mixed phenotypes (S542Y, L619P, and W785C), and a pure skeletal dysplasia mutant within the ARD (D333G) (Fig. 2A, Table 4). We first examined expression of TRPV4 in HEK293T cells by western blot. We noted mildly decreased expression of the D333G variant, but more dramatically decreased expression of R616G and L619P (Fig. 2B). To determine whether this was due to an inherent abnormality in expression or related to cytotoxicity, we cultured the cells in the TRPV4 antagonist GSK2193874 (GSK219). Addition of GSK219 increased TRPV4 expression of D333G, R616G, and L619P to levels comparable to WT TRPV4 (Fig. 2C), suggesting that the apparent reduced expression in the absence of antagonist was likely due to cell death. We next examined subcellular localization and surface expression of the various TRPV4 mutants in both MN‐1 cells (mouse motor neuron‐neuroblastoma fusion cell line 35 ) and MDCK cells (canine kidney epithelial cells 36 ) treated with GSK219, as both cell types readily demonstrate exogenous TRPV4 plasma membrane localization. All mutant forms of TRPV4 demonstrated expression at the cell surface and partial co‐localization with cortical actin in both MN‐1 (Fig. 2D) and MDCK (Fig. 2E) cells, indicating normal trafficking to the cell membrane.
Figure 2.
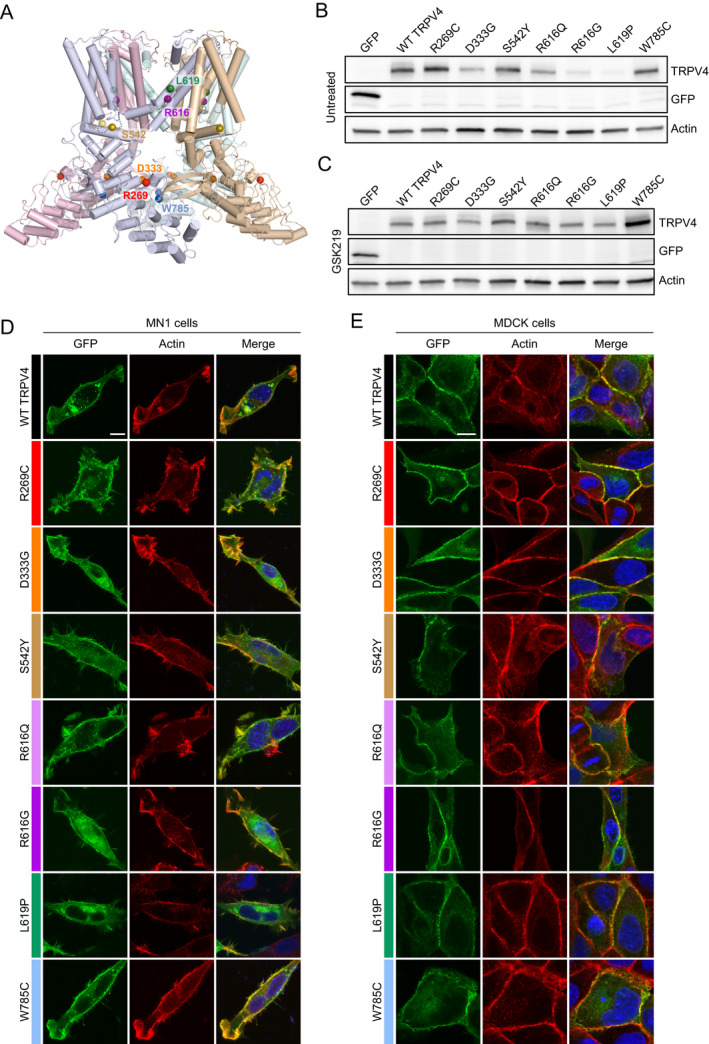
Expression and localization of selected TRPV4 mutants. (A) Cryo‐EM structure of the TRPV4 tetramer in an agonist‐bound state (PDB: 7AA5 47 ) depicting the location of mutated amino acid residues analyzed in vitro. Each monomer of the TRPV4 tetramer is displayed in a separate color. Colored spheres indicate the location of mutated amino acid residues within each monomer. (B) Immunoblot from HEK293T cells transfected with equal amounts of GFP‐tagged TRPV4 in the absence of TRPV4 antagonist. Expression of R616G and L619P appear reduced. (C) Immunoblot from HEK293T cells transfected with equal amounts of GFP‐tagged TRPV4 in the presence of 1 μM GSK219 TRPV4 antagonist. Expression of the individual mutants is normalized in the presence of antagonist. (D‐E) Immunohistochemistry of GFP‐tagged TRPV4 and actin (phalloidin) in MN‐1 cells (D) and MDCK cells (E) demonstrates normal trafficking to the cell membrane and co‐localization with cortical actin. [Colour figure can be viewed at wileyonlinelibrary.com]
Table 4.
TRPV4 mutations analyzed and summary of in vitro results.
| TRPV4 genotype | Clinical syndrome | Protein domain | In vitro results | |||
|---|---|---|---|---|---|---|
| Baseline calcium | Stimulated calcium | Cell death (%) | GSK219 IC50 (nM) | |||
| WT | — | — | 0.73 | 1.76 | 7.2 | 8.0 |
| R269C | Neuropathy with vocal cord weakness | ARD3 | 0.88 | 2.56 | 11.3 | 10.0 |
| D333G | Skeletal dysplasia | ARD5 | 1.12 | 2.36 | 10.7 | 20.7 |
| S542Y | Neuropathy with vocal cord weakness, short stature | Ligand binding | 1.19 | 2.74 | 12.2 | 55.3 |
| R616Q | Skeletal dysplasia | TM5 | 0.87 | 2.09 | 8.2 | 11.9 |
| R616G | Neuropathy with vocal cord weakness | TM5 | 1.83 | 1.99 | 26.3 | 215.6 |
| L619P | Severe neuropathy with vocal cord weakness, skeletal dysplasia, giant cell jaw lesions | TM5 | 2.17 | 2.37 | 28.5 | 198.4 |
| W785C | Mild neuropathy without vocal cord weakness, skeletal dysplasia, scaly skin | C terminal | 1.18 | 2.09 | 15.0 | 31.5 |
Baseline and stimulated calcium response values indicate Fura ratios (340/380). Abbreviations: ARD, ankyrin repeat domain; TM, transmembrane domain; IC50, half maximal inhibitory concentration.
As TRPV4 disease‐causing mutations generally result in increased ion channel activity and elevated intracellular calcium in vitro and in vivo2,14,15, we next assessed how the different TRPV4 mutants affected ion channel function. We chose to study hypotonic stress‐mediated calcium responses in MN‐1 cells as these cells do not express endogenous TRPV4 and do not respond robustly to hypotonic stress in the absence of exogenous TRPV4 18 (Fig. 3A). Whereas WT TRPV4 led to modest increases in calcium influx, the neuropathy mutant R269C and the skeletal dysplasia mutant D333G showed elevated baseline calcium and more marked response to hypotonic stress (Fig. 3A‐B). Notably, the baseline calcium elevation of the D333G mutant was greater than observed with the R269C mutant. We next compared baseline and stimulated calcium responses of R616G and R616Q TRPV4. Whereas the R616Q mutant showed mildly elevated baseline calcium and mildly augmented response to stimulation, the R616G mutant showed dramatic elevation of baseline calcium and only a transient and small increase following stimulation (Fig. 3C). We next evaluated the S542Y, L619P, and W785C mutants, which were associated with high rates of mixed phenotypic manifestations. Both the S542Y and W785C mutants showed marked baseline calcium elevations similar to D333G and greater than R269C, as well as elevated responses to hypotonic stress (Fig. 3D). In contrast, the L619P mutant demonstrated dramatically increased baseline calcium with blunted response to hypotonic saline (Fig. 3D), similar to what was observed with the R616G mutant. Comparison of baseline (Fig. 3E, Table 4) and stimulated calcium levels (Fig. 3F, Table 4) among all mutants did not reveal definitive patterns of channel function that uniformly distinguished neuropathy, skeletal dysplasia, and mixed phenotype mutants. However, mutations that caused phenotypes dominated by neuropathy manifestations (R269C and S542Y) tended to show more marked stimulated calcium responses, whereas skeletal dysplasia‐dominated phenotype mutants (D333G) and severe and/or mixed phenotype mutants (R616G, L619, W785C) tended to have more marked baseline elevations (Table 4). The R616Q mutation was an exception as its baseline calcium elevation was not as dramatic as other skeletal dysplasia/mixed mutants.
Figure 3.
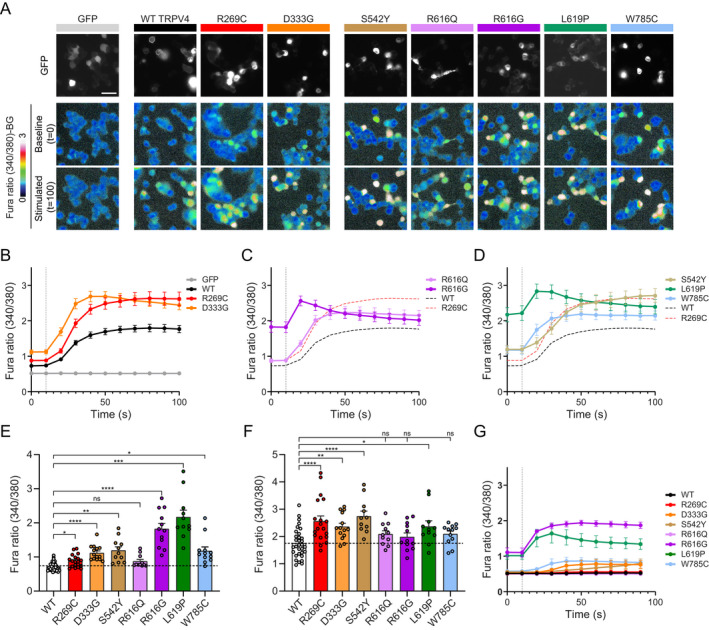
Severe and mixed phenotype mutants cause marked elevations of baseline calcium levels. (A) Representative images from ratiometric calcium imaging experiments. MN‐1 cells were transfected with GFP‐tagged TRPV4 plasmids and loaded with Fura‐2 AM calcium indicator. Baseline and hypotonic‐stimulated calcium responses were then measured over time. Pathogenic TRPV4 mutants lead to elevated baseline calcium compared to WT TRPV4, whereas baseline calcium levels are more markedly elevated in R616G and L619P TRPV4. (B‐D) Averaged calcium imaging traces before and after hypotonic stimulation, denoted by vertical dashed line. (B) R269C and D333G TRPV4 cause elevated baseline and stimulated calcium influx compared to WT TRPV4. (C) R616G causes marked baseline calcium elevation, whereas R616Q causes only mild elevations. (D) S542Y and W785C TRPV4 cause larger increases in baseline calcium compared to R269C TRPV4. L619P causes marked elevation of baseline calcium similar to R616G. (E) Comparison of baseline calcium levels across all tested TRPV4 mutants. Brown‐Forsythe and Welch ANOVA with Dunnett's post‐hoc test, n = 11–32 independent experiments per condition. (F) Comparison of maximum hypotonic stimulated calcium levels across all tested TRPV4 mutants. One‐way ANOVA with Dunnett's post‐hoc test, n = 11–32 independent experiments per condition. (G) Comparison of calcium imaging traces from cells treated with 50 nM GSK219 and then subjected to hypotonic saline. The R616G and L619P mutants demonstrate retained responses to hypotonic saline stimulation. Data are presented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. [Colour figure can be viewed at wileyonlinelibrary.com]
3.4. Analysis of cytotoxicity and sensitivity to pharmacological inhibition
Given the marked elevation of baseline calcium in the R616G and L619P mutants, we sought to further interrogate channel opening characteristics in these mutants by using low‐dose TRPV4 antagonist (GSK219, 50 nM) to suppress baseline calcium. While this dose of antagonist largely suppressed baseline calcium elevation in all other mutants, R616G and L619P mutants still demonstrated moderately elevated calcium levels (Fig. 3G). Both R616G and L619P mutants demonstrated clear increased calcium influx in response to hypotonic saline, indicating that these mutants were still able to respond to mechanical stimulation and that the blunted response in the absence of TRPV4 antagonist may be due to nearly maximal constitutive channel opening.
As gain of ion channel function with TRPV4 disease mutations is thought to be central to cytotoxicity and disease pathogenesis, we tested whether expression of the various TRPV4 mutants in HEK293T cells led to discernible cell death. Consistent with prior results, 14 we found increased cytotoxicity of R269C and D333G mutants (Fig. 4A). By comparison, cell death was similar with the S542Y mutant, more markedly elevated in W785C, and only mildly elevated in R616Q (Fig. 4A). In contrast, cell death was robustly elevated in the R616G and L619P mutants (Fig. 4A), consistent with the marked baseline calcium elevations.
Figure 4.
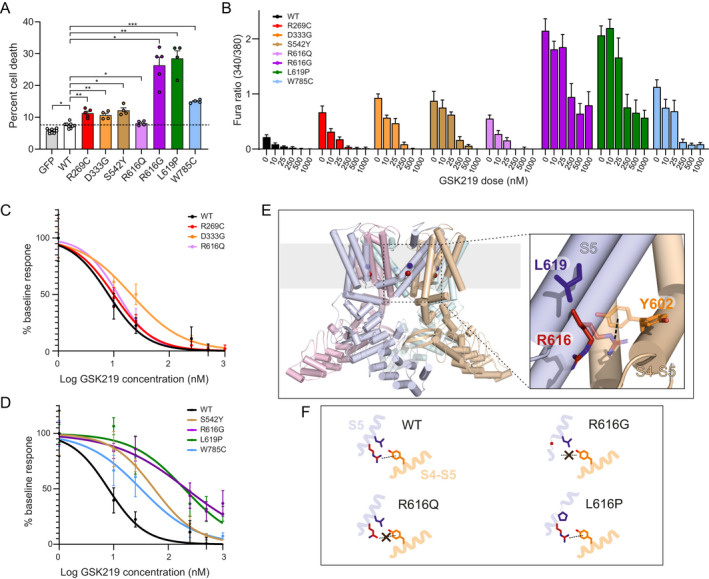
R616G and L619P cause increased cytotoxicity, reduced responsiveness to antagonism, and structural disruptions to the S5 transmembrane domain. (A) Cytotoxicity assay demonstrates increased toxicity with pathogenic mutants that is more pronounced with the R616G and L619P mutants. Repeated measures one‐way ANOVA with Dunnett's post‐hoc test, n = 4 independent experiments per condition. (B) Averaged intracellular calcium levels in TRPV4 mutants treated with escalating doses of GSK219 antagonist. (C) Inhibitory dose response curve comparing WT TRPV4 to R269C, D333G, and R616Q mutants. (D) Inhibitory dose response curve comparing WT TRPV4 to S542Y, R616G, L619P, and W785C mutants. (E) Location of R616 (red sphere) and L619 (blue sphere) in the S5 helix of TRPV4 shown on the cryo‐EM structure of the human TRPV4 channel in an agonist‐bound state (PDB: 7AA5 47 ). Each monomer of the TRPV4 tetramer is displayed in a separate color. On the right is a close‐up view of R616 (red) and L619 (blue) in the S5 helix as well as Y602 (orange) in the S4‐S5‐linker of a neighboring subunit with amino acid side chains shown in stick representations. R616 and Y602 side chain rotamers enabling a R616‐Y602 cation‐π interaction which presumably stabilizes the closed‐state were modeled as transparent sticks. In the structure of the agonist‐bound TRPV4 state, the R616 and Y602 side chains point away from each other such that the putative cation‐π interaction is prevented (opaque sticks). (F) Schematic representation of how the R616Q, R616G, and L619Q mutations may impact the TRPV4 structure. In the WT structure, the S5 helix and the R616‐Y602 cation‐π interaction are intact, whereas both are perturbed in the R616G and R616Q mutants. In the R616G mutant, the S5 helix is additionally perturbed due to the ability of glycine to act as a helix breaker. Likewise, substitution of proline in the L619P mutant can also structurally perturb the S5 helix. Breaking the S5 helix presumably disrupts the ion conducting pore and leads to uncontrolled ion flux. In contrast, the loss of the cation‐π interaction might lower the activation threshold by agonists. Data are presented as means ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. [Colour figure can be viewed at wileyonlinelibrary.com]
Next, given the variability of baseline calcium elevations and the lack of complete suppression by low‐dose antagonist in the R616G and L619P mutants, we tested antagonist dose–response characteristics of the various mutants (Fig. 4B). We examined baseline calcium levels in transfected MN‐1 cells at a range of doses of GSK219. Notably, R269C, D333G, and R616Q mutants showed similar IC50 concentrations as compared to WT TRPV4 (8 nM), indicating that these mutants are readily inhibited by this particular antagonist (Fig. 4C , Table 4). In contrast, the inhibitory curves were shifted for all of the mixed phenotype mutants, with the most dramatic effects seen in the R616G and L619P mutants, in which IC50 values were 216 nM and 198 nM, respectively (Fig. 4D, Table 4). In fact, baseline calcium levels were not fully suppressed even at doses of 1000 nM, over 100 times the IC50 for WT TRPV4. These data suggest that disease mutant forms of TRPV4 differ significantly in their sensitivity to ion channel inhibition by antagonist drugs.
3.5. Structural analysis of R616Q, R616G, and L619P TRPV4 mutants
The finding of marked differences in baseline calcium influx and cytotoxicity in R616G and R616Q mutants and the dramatic toxicity of the nearby L169P mutation led us to interrogate possible structural determinants of these functional effects. Residues R616 and L619 are both located in helix S5 in the TRPV4 transmembrane domain (Fig. 4E), which is preceded by the S4‐S5 linker, and both residues are involved in interdomain contacts to a neighboring subunit (Fig. 4F). 16 Importantly, both helix S5 as well as the linker have been described as crucial elements for ligand and lipid binding/regulation, 37 and therefore perturbation of the S4‐S5 linker or the S5 helix may have dramatic consequences for channel function. Previously, a cation‐π interaction between residue R616 in the S5 helix and residue Y602 of another subunit was identified in TRPV4 as a structural feature that stabilizes a closed gate, and the mutation of either residue (Y602C or R616Q) led to a gain of function phenotype. 38 Presumably, the disease‐causing R616Q mutation perturbs the cation‐π interaction (Fig. 4F). In contrast, the marked gain of function seen with R616G and L619P mutants suggests that they likely have more profound structural impacts. In fact, as both proline and glycine are helix breakers, the R616G and L619P mutations are predicted to disrupt the S5 helix structure, destabilizing the pore domain and leading to constitutive channel opening (Fig. 4F). In contrast, the R616Q mutation is unlikely to disrupt the S5 helix. Thus, we propose that the R616Q mutation results in disrupted cation‐π interaction of R616 with the conserved tyrosine residue, leading to altered fine‐tuning of the opening probability of TRPV4, whereas the R616G and L619P mutations disrupt the S5 helix and the closed pore conformation, leading to constitutive channel opening and markedly increased intracellular calcium influx.
4. Discussion
Despite recognition that gain of ion channel function is central to the pathogenesis of TRPV4 channelopathies, the basis of the highly variable skeletal and neuromuscular manifestations has remained enigmatic. Based on a detailed literature review, we identified specific mutations that more frequently cause severe, mixed phenotypes. Furthermore, in vitro and structural analysis suggests that these mutations cause a more dramatic gain of ion channel function and reduced sensitivity to small molecule antagonism.
4.1. The novel R616G mutation causes a severe neuropathy phenotype
We report a novel R616G mutation causing an asymmetric sensorimotor neuropathy with complete bilateral vocal cord paralysis and no definitive evidence of skeletal dysplasia. Additional features included mild white matter volume loss, central sleep apnea, and tethered spinal cord, none of which have been recognized as common features of TRPV4 neuropathy, although central sleep apnea and tethered spinal cord were recently described in patients with severe mixed phenotypes due to the L619P mutation. 32 Whether these features are a rare manifestation of TRPV4 channelopathies or may be underdiagnosed remains to be determined. The clinical significance of tethering of the spinal cord is difficult to discern given that clinical signs from tethered cord can be quite variable, but largely overlap with the sensory and motor manifestations seen with hereditary neuropathy. The occurrence of central sleep apnea is particularly interesting given the localization of respiratory nuclei to the rostral medulla immediately adjacent to the nucleus ambiguus, which innervates the vocal cords, and may imply focal brainstem pathology underlying both vocal cord and respiratory manifestations. The fact that our patient does not have evidence of skeletal dysplasia is somewhat surprising given the description of extensive skeletal manifestations in patients with the L619P mutation, which appears to have very similar functional and structural consequences. 32 This discrepancy may be yet another example of the marked phenotypic heterogeneity with TRPV4 mutations, as even family members harboring the same mutation can display a range of disease from severe to complete non‐penetrance. 10 , 11 An alternative explanation could be genetic mosaicism leading to reduced expression of the R616G mutant within relevant cell types of the skeletal system, although we have no direct evidence of this. In line with this possibility, a previously reported lethal L618P mutation 39 was found in a patient with a mild skeletal dysplasia phenotype and findings indicative of genetic mosaicism. 40 The authors hypothesized that decreased expression of mutant TRPV4 in the skeletal system could account for the reduced disease severity. Finally, we cannot rule out that additional and more significant skeletal abnormalities will appear in our patient during further follow‐up.
4.2. Mixed phenotypes are not uncommon
Our review of the literature of cases and families with mixed phenotypes identified at least 36 cases, demonstrating that mixed phenotypes may be more common than originally appreciated. We found a large spectrum of neuropathy and skeletal dysplasia manifestations in these patients. While motor manifestations most commonly affected the distal lower extremities, proximal upper and lower extremity weakness was also frequently reported, indicating a non‐length‐dependent pattern of motor involvement. This pattern is in contrast to most other forms of CMT and suggests that many patients with neuropathogenic TRPV4 mutations do not present with classical CMT phenotypes. Notably, the average age of onset in patients with mixed phenotypes was under 3 years of age, whereas those with isolated neuropathy experienced onset at an average age of 11 years. This early age of onset suggests that therapeutic intervention may be most efficacious if instituted very early in disease, as has been demonstrated in spinal muscular atrophy due to SMN1 deficiency. 41 We also noted evidence of disease progression in almost all patients with mixed phenotypes, including those with the CDSMA phenotype, which is typically believed to have a largely static course. We were not able to discern any clear or universal genotype–phenotype correlation regarding likelihood of developing a mixed phenotype with a given mutation. In fact, mixed phenotypes sometimes occurred in single family members whereas others with the same mutation had disease isolated to the neuromuscular system. Interestingly, just under half of all mixed phenotype cases occurred due to mutations within the ARD, which are most commonly associated with pure neuropathy phenotypes. In these cases, mixed phenotypes could be considered to represent “extension” of neuropathy phenotypes into the skeletal system, suggesting the need for more extensive screening for skeletal involvement in patients presumed to have isolated neuromuscular disease. As vertebral abnormalities are among the most common manifestations of TRPV4‐associated skeletal dysplasia, 42 we propose that all patients with known or suspected pathogenic TRPV4 mutations should undergo a skeletal survey to assess for skeletal disease. Notably, we found only two mutations that were previously associated with isolated skeletal dysplasia but also caused mixed phenotypes (V620I and P799R), but in neither was vocal cord weakness reported. 11 , 22 However, just as skeletal dysplasia may be unrecognized in patients with neuropathy manifestations, it remains possible that patients with apparently isolated skeletal manifestations could have undiagnosed neuropathy and/or vocal cord weakness.
Given the marked heterogeneity in phenotypes and clinical manifestations across all TRPV4 channelopathies, it is not surprising that we were unable to identify any universal correlations between specific mutations and the development of mixed phenotypes. However, in vitro characterization of some of the more frequent mutations associated with mixed phenotypes suggests that there may be some functional and structural correlations between specific mutations and the likelihood of mixed phenotypes, as discussed below.
4.3. Mutations frequently associated with mixed phenotypes result in marked gain of function
Our functional analysis demonstrated that mutations associated with mixed phenotypes and isolated skeletal dysplasia tended to have more marked baseline elevations of intracellular calcium. In contrast, the neuropathogenic R269C mutation and the S542Y mutation, which causes a predominant neuromuscular phenotype with only mild skeletal manifestations, resulted in more pronounced calcium influx in response to stimulation. These data suggest that there may be an association between constitutive channel opening and development of skeletal dysplasia on the one hand, and increased sensitivity to mechanical stimulation and development of neuropathy on the other hand. Of course, the distinction between these situations cannot be universal or absolute, and the novel R616G variant presents an exception to this pattern. Increased mechanical sensitivity in predominantly neuropathogenic mutants may relate to mutation‐dependent disruption of N‐terminal protein–protein or protein‐lipid interactions that are important for channel activation. 18 , 43 , 44
Importantly, our analysis of different pathogenic TRPV4 mutants uncovered differential sensitivity to inhibition by the TRPV4 antagonist GSK219. Mutants with more elevated baseline calcium, particularly R616G and L619P, caused dramatic rightward shifts in the inhibitory dose response curves for this antagonist. Our structural analysis suggests that these mutations are likely to disrupt the alpha helix within the S5 transmembrane domain of TRPV4, leading to alteration of the channel pore structure and resultant constitutive channel opening. In contrast, the R616Q mutation, which is not predicted to have such dramatic structural consequences, showed only mildly elevated baseline calcium and normal inhibition by GSK219.
As cellular systems and a fly model of TRPV4 neuropathy suggest that small molecule TRPV4 antagonists can reduce cytotoxicity and neurodegeneration, 2 , 5 , 14 , 15 , 45 TRPV4 inhibitors, such as GSK2798745, 46 warrant consideration for clinical trials in TRPV4 channelopathies. Our results suggest that prior to any such trials, careful dose–response analysis should be performed for specific pathogenic TRPV4 mutants, as individual mutations may differentially alter sensitivity to TRPV4 antagonism. In addition, the early age of disease onset suggests that pediatric patients may be ideal to demonstrate efficacy and maximize therapeutic benefit in clinical trials.
Conflict of Interest
The authors declare no conflicts of interest.
Supporting information
Video 1. Segment A. High‐pitched inspiratory stridor. Segment B. Respiratory distress with increased work of breathing and use of accessory muscles. Segment C. Fiberoptic laryngoscopy shows bilateral vocal cord palsy, evident as lack of abduction of the vocal folds during inspiration. Tracheomalacia is also evident as mild narrowing of distal trachea during expiration due to anterior prolapse of the trachealis muscle. Segment D. Direct bronchoscopy shows moderate bronchomalacia of the left main bronchus and visualization of the carina.
Acknowledgments
This work was supported by K08 NS102509 (B.A.M.), American Academy of Neurology Neuroscience Research Training Fellowship (B.A.M.), Max Planck Graduate Center (MPGC) (U.A.H.), Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2051 – Project‐ID 390713860 (U.A.H), NIH (NINDS) R35 NS122306, (C.J.S), and Muscular Dystrophy Association 629305 (C.J.S). We thank Natalia Nedelsky for manuscript editing and graphical assistance.
Funding Statement
This work was funded by American Academy of Neurology grant Neuroscience Research Training Fellowship; Deutsche Forschungsgemeinschaft grant 390713860; Max Planck Graduate Center; Muscular Dystrophy Association grant 629305; National Institute of Neurological Disorders and Stroke grants K08 NS102509 and R35 NS122306.
Contributor Information
Charlotte J. Sumner, Email: csumner1@jhmi.edu.
Brett A. McCray, Email: bmccray3@jhmi.edu.
References
- 1. McCray BA, Schindler A, Hoover‐Fong JE, Sumner CJ. In GeneReviews((R)) (eds Adam M. P. et al.). University of Washington, Seattle; 1993‐2022. [Google Scholar]
- 2. Landoure G, Zdebik AA, Martinez TL, et al. Mutations in TRPV4 cause Charcot‐Marie‐tooth disease type 2C. Nat Genet. 2010;42:170‐174. 10.1038/ng.512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nilius B, Voets T. The puzzle of TRPV4 channelopathies. EMBO Rep. 2013;14:152‐163. 10.1038/embor.2012.219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Auer‐Grumbach M, Olschewski A, Papic L, et al. Alterations in the ankyrin domain of TRPV4 cause congenital distal SMA, scapuloperoneal SMA and HMSN2C. Nat Genet. 2010;42:160‐164. 10.1038/ng.508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Deng HX, Klein CJ, Yan J, et al. Scapuloperoneal spinal muscular atrophy and CMT2C are allelic disorders caused by alterations in TRPV4. Nat Genet. 2010;42:165‐169. 10.1038/ng.509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vlam L, Schelhaas HJ, van Blitterswijk M, et al. Mutations in the TRPV4 gene are not associated with sporadic progressive muscular atrophy. Arch Neurol. 2012;69:790‐791. 10.1001/archneurol.2012.148 [DOI] [PubMed] [Google Scholar]
- 7. Andreucci E, Aftimos S, Alcausin M, et al. TRPV4 related skeletal dysplasias: a phenotypic spectrum ed byclinical, radiographic, and molecular studies in 21 new families. Orphanet J Rare Dis. 2011;6:37. 10.1186/1750-1172-6-37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nishimura G, Lausch E, Savarirayan R, et al. TRPV4‐associated skeletal dysplasias. Am J Med Genet C Semin Med Genet. 2012;160C:190‐204. 10.1002/ajmg.c.31335 [DOI] [PubMed] [Google Scholar]
- 9. Lamande SR, Yuan Y, Gresshoff IL, et al. Mutations in TRPV4 cause an inherited arthropathy of hands and feet. Nat Genet. 2011;43:1142‐1146. 10.1038/ng.945 [DOI] [PubMed] [Google Scholar]
- 10. Echaniz‐Laguna A, Dubourg O, Carlier P, et al. Phenotypic spectrum and incidence of TRPV4 mutations in patients with inherited axonal neuropathy. Neurology. 2014;82:1919‐1926. 10.1212/WNL.0000000000000450 [DOI] [PubMed] [Google Scholar]
- 11. Zimon M, Baets J, Auer‐Grumbach M, et al. Dominant mutations in the cation channel gene transient receptor potential vanilloid 4 cause an unusual spectrum of neuropathies. Brain. 2010;133:1798‐1809. 10.1093/brain/awq109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lawhorn BG, Brnardic EJ, Behm DJ. TRPV4 antagonists: a patent review (2015‐2020). Expert Opin Ther Pat. 2021;31:773‐784. 10.1080/13543776.2021.1903432 [DOI] [PubMed] [Google Scholar]
- 13. Lawhorn BG, Brnardic EJ, Behm DJ. Recent advances in TRPV4 agonists and antagonists. Bioorg Med Chem Lett. 2020;30:127022. 10.1016/j.bmcl.2020.127022 [DOI] [PubMed] [Google Scholar]
- 14. Sullivan JM, Zimanyi CM, Aisenberg W, et al. Novel mutations the key role of the ankyrin repeat domain in TRPV4‐mediated neuropathy. Neurol Genet. 2015;1:e29. 10.1212/NXG.0000000000000029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Woolums BM, McCray BA, Sung H, et al. TRPV4 disrupts mitochondrial transport and causes axonal degeneration via a CaMKII‐dependent elevation of intracellular ca(2). Nat Commun. 2020;11:2679. 10.1038/s41467-020-16411-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Deng Z, Paknejad N, Maksaev G, et al. Cryo‐EM and X‐ray structures of TRPV4 reveal insight into ion permeation and gating mechanisms. Nat Struct Mol Biol. 2018;25:252‐260. 10.1038/s41594-018-0037-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Phelps CB, Wang RR, Choo SS, Gaudet R. Differential regulation of TRPV1, TRPV3, and TRPV4 sensitivity through a conserved binding site on the ankyrin repeat domain. J Biol Chem. 2010;285:731‐740. 10.1074/jbc.M109.052548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McCray BA, Diehl E, Sullivan JM, et al. Neuropathy‐causing TRPV4 mutations disrupt TRPV4‐RhoA interactions and impair neurite extension. Nat Commun. 2021;12:1444. 10.1038/s41467-021-21699-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McEntagart M. TRPV4 axonal neuropathy spectrum disorder. J Clin Neurosci. 2012;19:927‐933. 10.1016/j.jocn.2011.12.003 [DOI] [PubMed] [Google Scholar]
- 20. Rock MJ, Prenen J, Funari VA, et al. Gain‐of‐function mutations in TRPV4 cause autosomal dominant brachyolmia. Nat Genet. 2008;40:999‐1003. 10.1038/ng.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Unger S, Lausch E, Stanzial F, et al. Fetal akinesia in metatropic dysplasia: the combined phenotype of chondrodysplasia and neuropathy? Am J Med Genet A. 2011;155A:2860‐2864. 10.1002/ajmg.a.34268 [DOI] [PubMed] [Google Scholar]
- 22. Cho TJ, Matsumoto K, Fano V, et al. TRPV4‐pathy manifesting both skeletal dysplasia and peripheral neuropathy: a report of three patients. Am J Med Genet A. 2012;158A:795‐802. 10.1002/ajmg.a.35268 [DOI] [PubMed] [Google Scholar]
- 23. Fiorillo C, Moro F, Brisca G, et al. TRPV4 mutations in children with congenital distal spinal muscular atrophy. Neurogenetics. 2012;13:195‐203. 10.1007/s10048-012-0328-7 [DOI] [PubMed] [Google Scholar]
- 24. Fleury P, Hageman G. A dominantly inherited lower motor neuron disorder presenting at birth with associated arthrogryposis. J Neurol Neurosurg Psychiatry. 1985;48:1037‐1048. 10.1136/jnnp.48.10.1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Biasini F, Portaro S, Mazzeo A, et al. TRPV4 related scapuloperoneal spinal muscular atrophy: report of an Italian family and review of the literature. Neuromuscul Disord. 2016;26:312‐315. 10.1016/j.nmd.2016.02.010 [DOI] [PubMed] [Google Scholar]
- 26. Fleming J, Quan D. A case of congenital spinal muscular atrophy with pain due to a mutation in TRPV4. Neuromuscul Disord. 2016;26:841‐843. 10.1016/j.nmd.2016.09.013 [DOI] [PubMed] [Google Scholar]
- 27. Evangelista T, Bansagi B, Pyle A, et al. Phenotypic variability of TRPV4 related neuropathies. Neuromuscul Disord. 2015;25:516‐521. 10.1016/j.nmd.2015.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Faye E, Modaff P, Pauli R, Legare J. Combined phenotypes of Spondylometaphyseal dysplasia‐Kozlowski type and Charcot‐Marie‐tooth disease type 2C secondary to a TRPV4 pathogenic variant. Mol Syndromol. 2019;10:154‐160. 10.1159/000495778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. DeLong R, Siddique T. A large New England kindred with autosomal dominant neurogenic scapuloperoneal amyotrophy with unique features. Arch Neurol. 1992;49:905‐908. 10.1001/archneur.1992.00530330027010 [DOI] [PubMed] [Google Scholar]
- 30. Thom CS, Brandsma E, Lambert MP. Thrombocytosis in an infant with a TRPV4 mutation: a case report. Platelets. 2021;32:429‐431. 10.1080/09537104.2020.1755644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chen DH, Sul Y, Weiss M, et al. CMT2C with vocal cord paresis associated with short stature and mutations in the TRPV4 gene. Neurology. 2010;75:1968‐1975. 10.1212/WNL.0b013e3181ffe4bb [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ragamin A, Gomes CC, Bindels‐de Heus K, et al. De novo TRPV4 Leu619Pro variant causes a new channelopathy characterised by giant cell lesions of the jaws and skull, skeletal abnormalities and polyneuropathy. J Med Genet. 2021. 10.1136/jmedgenet-2020-107427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu Y, Yan X, Chen Y, He Z, Ouyang Y. Novel TRPV4 mutation in a large Chinese family with congenital distal spinal muscular atrophy, skeletal dysplasia and scaly skin. J Neurol Sci. 2020;419:117153. 10.1016/j.jns.2020.117153 [DOI] [PubMed] [Google Scholar]
- 34. Thibodeau ML, Peters CH, Townsend KN, et al. Compound heterozygous TRPV4 mutations in two siblings with a complex phenotype including severe intellectual disability and neuropathy. Am J Med Genet A. 2017;173:3087‐3092. 10.1002/ajmg.a.38400 [DOI] [PubMed] [Google Scholar]
- 35. Salazar‐Grueso EF, Kim S, Kim H. Embryonic mouse spinal cord motor neuron hybrid cells. Neuroreport. 1991;2:505‐508. 10.1097/00001756-199109000-00002 [DOI] [PubMed] [Google Scholar]
- 36. Gaush CR, Hard WL, Smith TF. Characterization of an established line of canine kidney cells (MDCK). Proc Soc Exp Biol Med. 1966;122:931‐935. 10.3181/00379727-122-31293 [DOI] [PubMed] [Google Scholar]
- 37. Teng J, Loukin SH, Anishkin A, Kung C. A competing hydrophobic tug on L596 to the membrane core unlatches S4‐S5 linker elbow from TRP helix and allows TRPV4 channel to open. Proc Natl Acad Sci USA. 2016;113:11847‐11852. 10.1073/pnas.1613523113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Teng J, Anishkin A, Kung C, Blount P. Human mutations highlight an intersubunit cation‐pi bond that stabilizes the closed but not open or inactivated states of TRPV channels. Proc Natl Acad Sci USA. 2019;116:9410‐9416. 10.1073/pnas.1820673116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Camacho N, Krakow D, Johnykutty S, et al. Dominant TRPV4 mutations in nonlethal and lethal metatropic dysplasia. Am J Med Genet A. 2010;152A:1169‐1177. 10.1002/ajmg.a.33392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weinstein MM, Kang T, Lachman RS, et al. Somatic mosaicism for a lethal TRPV4 mutation results in non‐lethal metatropic dysplasia. Am J Med Genet A. 2016;170:3298‐3302. 10.1002/ajmg.a.37942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Finkel RS, Mercuri E, Darras BT, et al. Nusinersen versus sham control in infantile‐onset spinal muscular atrophy. N Engl J Med. 2017;377:1723‐1732. 10.1056/NEJMoa1702752 [DOI] [PubMed] [Google Scholar]
- 42. Nemec SF, Cohn DH, Krakow D, Funari VA, Rimoin DL, Lachman RS. The importance of conventional radiography in the mutational analysis of skeletal dysplasias (the TRPV4 mutational family). Pediatr Radiol. 2012;42:15‐23. 10.1007/s00247-011-2229-6 [DOI] [PubMed] [Google Scholar]
- 43. Takahashi N, Hamada‐Nakahara S, Itoh Y, et al. TRPV4 channel activity is modulated by direct interaction of the ankyrin domain to PI(4,5)P(2). Nat Commun. 2014;5:4994. 10.1038/ncomms5994 [DOI] [PubMed] [Google Scholar]
- 44. Goretzki B, Glogowski NA, Diehl E, et al. Structural basis of TRPV4 N terminus interaction with syndapin/PACSIN1‐3 and PIP2. Structure 2018;26:1583‐1593 e1585. 10.1016/j.str.2018.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Klein CJ, Shi Y, Fecto F, et al. TRPV4 mutations and cytotoxic hypercalcemia in axonal Charcot‐Marie‐tooth neuropathies. Neurology. 2011;76:887‐894. 10.1212/WNL.0b013e31820f2de3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Goyal N, Skrdla P, Schroyer R, et al. Clinical pharmacokinetics, safety, and tolerability of a novel, first‐in‐class TRPV4 Ion Channel inhibitor, GSK2798745, in healthy and heart failure subjects. Am J Cardiovasc Drugs. 2019;19:335‐342. 10.1007/s40256-018-00320-6 [DOI] [PubMed] [Google Scholar]
- 47. Botte M, Ulrich AKC, Adaixo R, et al. Cryo‐EM structural studies of the agonist complexed human TRPV4 ion‐channel reveals novel structural rearrangements resulting in an open‐conformation. bioRxiv. 2020;2010(2013):334797. 10.1101/2020.10.13.334797 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video 1. Segment A. High‐pitched inspiratory stridor. Segment B. Respiratory distress with increased work of breathing and use of accessory muscles. Segment C. Fiberoptic laryngoscopy shows bilateral vocal cord palsy, evident as lack of abduction of the vocal folds during inspiration. Tracheomalacia is also evident as mild narrowing of distal trachea during expiration due to anterior prolapse of the trachealis muscle. Segment D. Direct bronchoscopy shows moderate bronchomalacia of the left main bronchus and visualization of the carina.